Министерство образования и науки Российской Федерации
Федеральное государственное бюджетное
образовательное учреждение высшего
профессионального образования
«Московский государственный университет тонких
химических технологий имени М.В. Ломоносова»
Кафедра
Химии и технологии
основного органического
синтеза
И.В. Ошанина, Л.Г. Брук, А.В. Зейгарник, О.Н. Темкин
«Физико-химические основы реакционных
процессов органического синтеза»
(часть 2)
Москва
2014
УДК 541.121;541.124
ББК 24.53 24.54
Ошанина И.В., Брук Л.Г., Зейгарник А.В., Темкин О.Н.,
«Физико-химические
основы
реакционных
процессов
органического синтеза» (часть 2),
М. :МИТХТ им. М.В.Ломоносова, 2014, с.88.
Рекомендовано к изданию кафедрой ХТООС
(протокол № 3 от 13 ноября 2014г).
Утверждено Библиотечно-издательской комиссией
МИТХТ им. М.В. Ломоносова в качестве учебного пособия
по курсу «Теория химико-технологических процессов
органического синтеза». План 2014 г (поз. 166).
Конспект
лекций по курсам «Теория химикотехнологических процессов органического синтеза» и
«Катализ в органическом синтезе» предназначен для
студентов 3 и 4 курса, обучающихся по направлению
бакалавриата
240100.62
«Химическая
технология»
профессиональный профиль «Химическая технология
органических веществ». В этой части пособия рассмотрены
следующие
лекционные
разделы:
классификация
химических реакций, промежуточные соединения и
частицы в некаталитических и каталитических реакциях,
кинетика и механизмы гомолитических и гетеролитических
реакций, использование корреляционных уравнений для
изучения механизмов реакций.
Рецензент:
проф. каф. ОХТ,д.х.н.
Е.А.Кацман
© МИТХТ им. М.В.Ломоносова, 2014 г.
2
Содержание
1. Промежуточные соединения и частицы в
некаталитических и каталитических реакциях
1.1.Карбокатионы
1.1.1. Номенклатура и устойчивость
1.1.2. Способы генерирования ионов карбения
1.1.3. Реакции ионов карбения
1.1.4. Методы регистрации ионов карбения
1.2. Карбанионы
1.2.1.Устойчивость
1.2.2. Методы генерирования
1.2.3. Свойства
1.2.4. Методы регистрации
1.3. Радикалы
1.3.1. Устойчивость углеродных радикалов
1.3.2. Методы генерирования
1.3.3. Основные реакции
1.3.4. Методы регистрации
1.4. Карбены
1.4.1. Методы генерирования
1.4.2. Основные реакции
1.5. Катион-радикалы и анион-радикалы
1.6. Молекулярные комплексы
1.7. Металлоорганические и координационные
соединения
1.7.1. Строение металлорганических соединений
1.7.2. Реакции координационных соединений
2. Классификация реакций
3
5
5
5
9
12
13
13
14
16
18
19
19
19
21
22
22
23
24
25
26
28
28
28
33
34
3. Кинетика и механизмы гомолитических реакций
3.1. Стадии радикально-цепных процессов
3.1.1. Зарождение цепи
3.1.2. Продолжение (развитие) цепи
3.1.3. Обрыв цепи
3.2. Уравнения Поляни–Семенова
3.3. Кинетика неразветвленных цепных реакций
3.4. Правило длинных цепей
3.5. Кинетика разветвленных цепных реакций
4. Кинетика и механизмы гетеролитических реакций
4.1. Механизмы реакций
4.1.1. Нуклеофильное замещение
4.1.2. Электрофильное замещение в
ароматических соединениях
4.1.3. Элиминирование
4.1.4. Присоединение
4.2. Теории кислот и оснований
4.3. Теоретические основы теории реакционной
способности
4.3.1. Количественные характеристики
заместителей в органических молекулах
4.3.2. Уравнение Бренстеда
4.3.3. Уравнение Гаммета
4.3.3. Уравнение Тафта
4.3.4. Использование корреляционных уравнений
для изучения механизма реакций
Литература
4
35
36
37
41
41
43
47
48
54
61
61
61
64
66
69
74
76
78
80
80
83
84
87
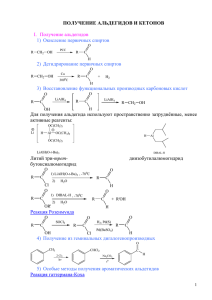
Промежуточные соединения и частицы
в некаталитических и каталитических
реакциях
1.
1.1 Карбокатионы
1.1.1. Номенклатура и устойчивость
Карбокатион – это катион, в котором положительный
заряд находится на атоме углерода. Этот термин включает
ионы карбения (карбениевые ионы) и ионы карбония
(карбониевые ионы):
R
R
R C+
R C+
R
Карбениевые ионы
(формально
трехкоординированный
углерод)
R
H
R
Карбониевый ион
(формально
пятикоординированный углерод)
Термин «карбониевый ион» часто по традиции
используют неверно, имея ввиду ион карбения. Адекватное
употребление этого термина означает, что карбокатион
содержит пятикоординированный
углеродный атом.
Структура иона карбония не может быть представлена
5
двухэлекронными двухцентровыми связями. Структуру
ионов карбония часто изображают следующим образом:
+
H
H
H C
H
H
с трехцентровой связью между катионом водорода и С-Н
связью метана или между молекулой Н-Н и катионом СН3+.
Особым
случаем
карбониевого
иона
является
протонированный циклопропан
H+
CH2
CH2
CH2
строение,
которого
можно
представить
следующими
мезомерными структурами:
H
H
H
H
H
C
C +
C
H
H
CH2
+ H
CH2
CH2
C H2
C H2
6
H
+
C H2
Ионы карбения - это карбокатионы, в которых
имеется
трехкоординированный
углеродный
атом,
содержащий 6 электронов в валентной оболочке атома и
свободную sp3- орбиталь.
Протонирование ароматического ядра приводит к
образованию иона карбения:
+
+ H
H
В
таких
делокализован
H
карбокатионах
положительный
по
π-электронов
системе
заряд
кольца.
Устойчивость ионов карбения всегда возрастает, если
заряд соседствует с кратной связью или системой
сопряженных кратных связей. В этом случае возникает
множественность резонансных (мезомерных) структур
(резонансная стабилизация):
H
H
H
H
H
H
+
+
+
+
+
=
7
+
Такой же эффект дает соседство с гетероатомом,
имеющим неподелённую электронную пару:
+ ..
R C O
. . Me
R
+
R C=O
.. Me
R
+ ..
+
R C=O
R C=O
..
..
После образования иона карбения весьма вероятны
его
превращения
стабильных
в направлении
структур.
образования
Заряженный
углеродный
более
атом
частично смещает на себя электронную плотность и с
соседних алкильных групп. В случае простых алкильных
карбокатионов устойчивость уменьшается в следующем
ряду:
третичные > вторичные > первичные.
При этом третичные ионы карбения настолько
устойчивы, что константа равновесия реакции
СН3СН2СН+СН3
К298
≈1010
изомеризации
и
(СН3)3С+
определяет
парафинов
и
направление
скелетной
олефинов
кислотном
в
катализе.
Миграция гидридной, алкильной и арильной групп в
ионах карбения происходит в направлениях, которые
приводят к какой-либо стабилизации иона карбения.
8
Ниже показан пример, в котором миграция арильной
группы происходит так, что появляется возможность для
стабилизации
карбокатиона
сопряжением
с
атомом
кислорода:
+
OH OH2
OH
H3C
H3C
C
C CH3
-H2O
+
C CH3
C
сдвиг
..
HO
+
C
C CH3
H3C
H+
- H+
OH OH
H3C
C
C
CH3
O
C
C CH3
H3C
1.1.2. Способы генерирования ионов карбения
1. Гетеролитический распад связи С-Х, где Хустойчивый анион
Ph3CCl
Ph3C+ + Cl¯.
Как правило, такие эндотермические процессы
протекают под действием растворителя:
RX
Rsolv+ + Xsolv¯
9
Степень диссоциации существенно увеличивается,
если в раствор добавляют вещества (апротонные кислоты),
способные образовывать анионные комплексы с X¯.
Например,
[SnCl5] ¯ + R+
RCl + SnCl4
Под
действием
суперкислот
(например,
смеси
фторсульфоновой кислоты и пентафторида сурьмы) даже
гидрид-анион может быть отщеплен от углеводорода через
промежуточное образование иона карбония:
R+ + SbF5(FSO3) ¯ + Н2
RH + FSO3H + SbF5
Спирты и галоидные алкилы реагируют в среде
суперкислот еще более охотно:
R+ + [SbF6] ¯ + H2SO4
ROH + FSO3H + SbF5
2. Гетеролитический распад диазокатионов по связи
C-N.
Диазокатионы
диазотирования
обычно
получают
по
реакции
первичных аминов действием нитрита
натрия в присутствии HCl или эфиров азотистой кислоты.
Нестабильные диазокатионы самопроизвольно разлагаются
с выделением азота и образованием иона карбения.
RNH2 + NaNO2 + 2HCl
NaCl + 2H2O + R–N≡N+Cl¯
R–N≡N+
N2 + R+
10
3. Присоединение
протона
или
других
электрофильных катионных частиц к кратным связям.
+
+ H
H
+ H+
C=C
4. Окисление
+
алкильных
H
C C
H
радикалов
солями
переходных металлов
C4H9• + Cu(II)
C4H9+ + Cu(I)
Один из возможных механизмов этой реакции
включает стадии присоединения R• к соли металла с
последующим
двухэлектронным
восстановительным
элиминированием R+ из полученного интермедиата:
R• + Cu(II)
R+ + Cu(I)
RCu(III)
Обсуждая реакции ионов карбения и методы их
образования, следует иметь ввиду, что свободных ионов R+
так же, как и свободных ионов
практически
нет.
Только
в
очень
Н+, в растворах
слабодонорных
растворителях типа SO2(ж) могут существовать ионы R+, с
sp3-орбиталью незанятой парой электронов основания
11
(растворителя). Так же, как и в случае водных кислот, где
Н+ связан с двумя молекулами Н2О
[H2O
+
OH2] ,
H
карбокатион взаимодействует с любым основанием В с образованием аддукта RB
+
+
+
+
ROH2, R2OR`, RXCH3, R(H2SO4)+ .
Поэтому все реакции переноса иона карбения на
разные молекулы и внутримолекулярные превращения
сольватированного иона карбения происходят как реакции
замещения.
RB+ + S
RS+ + B
1.1.3. Реакции ионов карбения
1. Перегруппировка - миграция Н¯, R¯, Ar¯ к
положительному центру.
2. Элиминирование протона или R+ :
+
C C
C=C
H
+ H+
3. Присоединение к двойной связи:
C=C
4.
+ R+
+
C C
R
Взаимодействие иона карбения с частицей,
имеющей электронную пару (OH¯, Cl¯ и др.), и с
12
соединениями, содержащими гидрид-ионы (BH4¯, HSiR3,
HMn(CO)5 и др.). Например,
R+ + HMn(CO)5
R-H + Mn(CO)5+
1.1.4. Методы регистрации ионов карбения
Доказательство существования ионов карбения в
растворе и установление их структуры осуществляется
методами
спектроскопии
ЯМР
и
комбинационного
рассеяния
(Раман-спектроскопия).
карбокатионы
можно
исследовать
с
Стабильные
помощью
ИК-
спектроскопии и УФ- спектрометрии.
Для
доказательства
образования
карбокатионов
используют вещества-ловушки (например, тетрагидротиофен или азид-ион N3¯).
1.2. Карбанионы
Карбанионы – анионы, содержащие четное число
электронов (8) с неподелённой парой электронов на атоме
углерода (например, R3C¯, Cl3C¯ или HC≡C¯):
R
R C :R
13
Структура
карбанионов
зависит
от
природы
заместителей:
H
C
H
C
-
C
H
-
C
пирамидальное
строение
C
плоский карбанион - четыре атома
углерода, расположены в одной
плоскости
1.2.1.Устойчивость
Устойчивость карбанионов – менее ясный вопрос, чем
устойчивость ионов карбения, но кое-что известно.
1.
Стабилизация простых алкиланионов увеличивается в
следующем ряду:
третичные < вторичные < первичные
То
есть
ряд
устойчивости
карбанионов
противоположен ряду устойчивости ионов карбения.
Устойчивость карбанионов коррелируется, в основном, с СН кислотностью молекул RH. Поэтому величина pKa
кислотной диссоциации RH в воде может служить мерой
устойчивости
R¯.
Чем
выше
pKa,
тем
меньше
Ka
диссоциации RH, тем более сильным основанием является
R¯ и тем он менее устойчив.
14
pKa
СН4 <
C6H5CH3 <
(C6H5)3CH <
45-48
41
31
Таким
образом,
ион
C2H2 < HCN
25
является
NС¯
10
наиболее
устойчивым и самым простым карбанионом.
2. Сопряжение неподеленной пары и кратной связи
повышает устойчивость карбанионов за счет резонансной
стабилизации:
R-CH=CH-CH2
-CH
R-CH-CH=CH2
-
CH2
2
Карбанионы
CH2
CH2
очень
Ph3C¯
устойчивы,
благодаря
наличию трех фенильных заместителей. Очень устойчив
циклопентадиенильный анион C5H5¯, имеющий ароматическую природу.
-
-
H
3. Повышение s-характера карбанионного углерода
определяет возрастающую устойчивость карбанионов:
15
R2C-CH2 < Ar ~~ R2C=CH < RC
3.
Электроноакцепторные
C
.
заместители
положении к карбанионному центру
в
α-
также повышают
стабильность карбаниона:
–Hal < –CN < –SO2 < –CO2R < –COR < –NO2.
1.2.2. Методы генерирования
1. Одним
из
способов
получения
карбанионов
является ионная диссоциация в полярных растворителях:
R¯ + E+
R–E
Группа
E+
уходит
без
электронной
пары.
Диссоциация может иметь место, если отрицательный
заряд в карбанионе делокализован, а уходящая группа –
стабильный катион. Сольватация играет существенную
роль, повышая стабильность образующихся ионов. Часто
E+ – протон, который оказывается сольватированным.
Отщеплению протона способствует присутствие основания
в среде, где протекает реакция. В водных, спиртовых и
водно-спиртовых средах такие реакции протекают, если
Rsolv¯ в этих условиях более слабое основание, чем OH¯ и
RO¯.
16
RH + NaOH
Rsolv¯ + Nasolv+ + H2O
RH + NaOR′
Rsolv¯ + Nasolv+ + R′OH
Если исходное вещество анион, то уходящая группа
может быть нейтральной:
.. C C O....
O
Возможна
генерация
-
C .. + CO2
карбанионов
через
σ-
металлоорганические соединения (соединения со связью
металл–углерод):
CH3CN + NaNH2
NaCH2CN + NH3
OCH3
OCH3
CH
CH
H
Li
+ 1/2H2
+ Li
В полярных растворителях (например, в тетрагидрофуране) протекает диссоциация металлоорганического
соединения на ионы (металл становится уходящей группой
E+). Происходит это постепенно (через ряд стадий):
R–M
(R¯ M+)solv
(R¯ || M+)solv
R solv¯ + Msolv+
где (R¯ M+)solv – тесная ионная пара, (R¯ || M+)solv – рыхлая
ионная пара.
17
2. Присоединение нуклеофилов Y¯ к кратной углерод
-углеродной связи:
-C C Y
C C + Y-
Y¯ может быть и карбанионом.
1.2.3. Свойства
1.
Рекомбинация
с
положительно
заряженными
частицами или замещение группы у насыщенного атома
углерода:
R- + C-X
R-C +X -
2. Присоединение по двойным связям.
C C + R-
-C C R
3. Отщепление Н+ от R′H или XH:
R¯ + XH
RH + X¯
18
1.2.4. Методы регистрации
УФ спектроскопию используют для определения
тесных и рыхлых ионных пар, ЯМР-спектроскопию - для
изучения строения карбанионов.
1.3. Радикалы
Радикалы – незаряженные частицы с неспаренным
электроном.
Неспаренный
электрон
может
быть
локализован на атоме углерода, кислорода, азота и т.д.
Чаще всего радикалы неустойчивы, но есть и устойчивые
радикалы (например, NO, NO2, нитроксильные радикалы
RR′N-O• и др). Углеродные радикалы R• имеют 7
электронов в валентной оболочке атома углерода, на
котором локализован неспаренный электрон.
1.3.1. Устойчивость углеродных радикалов
Устойчивость радикалов определяется, как и в случае
ионов
карбения,
возможностью
делокализации
электронной плотности и наличием резонансных структур.
Устойчивость увеличивается в следующих рядах:
CH3• < C2H5• < (CH3)2HС• < (CH3)3С•
CH3CH2 < H
.
< H3C
.
. CH3
< Ph C
CH3
19
<
< Ph
.C
Ph
.
Ph
.
CH=CH2
< Ph C
< Ph C
C2H5
Ph
CH=CH2
Трифенилметильный радикал настолько устойчив,
что лишь частично димеризуется в растворе при комнатной
температуре.
Для
оценки
термодинамической
устойчивости
можно использовать энергию диссоциации связей R–H в
соответствующих нерадикальных частицах. Устойчивость
радикала
возрастает
по
мере
уменьшения
энергии
диссоциации D (ккал/моль) связи R-H:
D
Ph•
CH3•
C2H5•
HCO•
PhCH2•
109
104
98
87
85
Кинетическая стабильность свободных радикалов
("пониженная"
реакционная
способность)
связана
со
структурными особенностями радикалов и выражается в
повышенной энергии активации реакций их превращения.
Чаще
всего
стабильность
стерические
затруднения
(делокализация
также играет роль).
20
обеспечивают
электронной
плотности
1.3.2. Методы генерирования
1. Термическое
разложение
органических
или
металлорганических соединений (пиролиз):
2RCOO•
RCOO–OOCR
O
R C
OOR'
O
R C
.
O
' .
+ RO
2R• + Hg
R2Hg
2. Фотолиз:
(CH3)2CO
hν
2CH3• + CO
2Cl•
Cl2
3.
2CO2 + 2R•
Окислительно-восстановительные реакции.
Ионы металлов переменной валентности в реакциях с
различными
пероксидами
способны
генерировать
радикалы:
RO• + OH¯ + M(n+1)+
ROOH + Mn+
Распад
гидропероксидов
может
происходить
каталитически:
ROOH + Cu+
RO• + ОH¯ + Cu2+
ROOH + Cu2+
RO2• + H+ + Cu+
Cu+
2ROOH
RO• + RO2• + H2O
21
и
1.3.3. Основные реакции
1.
Рекомбинация радикалов:
R• + R′•
R–R′
(два радикала)
Na + R•
Na–R
(радикал и металл)
R• + •O–O•
R–O–O•
(радикал и бирадикал)
2. Радикальное замещение:
R–X + R′•
R• + X–R′
3. Присоединение к кратной связи:
H2С=СH2 + R•
H2С•СH2R
и обратная реакция (β-элиминирование):
H2С•СH2R
H2С=СH2 + R•
R3Sn–CH2–CH=CH2 + X•
R3Sn–CH2–CH•–CH2X
R3Sn• + CH2=CHCH2X
4. Диспропорционирование:
C2H5• + C2H5•
C2H6 + C2H4
1.3.4. Методы регистрации
Важнейшее свойство радикалов – парамагнетизм
регистрируется методом ЭПР. Для обнаружения радикалов
используют
образующие
«ловушки»
более
радикалов
стабильные
–
соединения
радикалы
при
взаимодействии с R• без потери неспаренного электрона,
22
которые легко обнаружить методами ЭПР- и
ИК-
спектроскопии
.
(СH3)3C-N-O.
R
(СH3)3CNO + R
.
Замещенные фенолы, связывая активные радикалы,
превращаются в стабильные радикалы, поэтому их также
используют в качестве ловушек:
X
X
.
OH + R
X
.
O
RH + X
X
X
,
где Х =(СН3)3С.
Кроме радикалов, есть еще бирадикалы, в которых
есть несколько радикальных центров. Бирадикалы ведут
себя в основном также, как и радикалы. Простейшими
бирадикалами являются молекула О2 и карбены.
1.4 Карбены
Карбены – незаряженные частицы R2C:, содержащие
два электрона у одного атома углерода и образующиеся в
качестве
интермедиатов
в
некоторых
процессах.
В
карбенах у углерода 6 электронов, 4 из них участвуют в
образовании двух σ-связей (как в :СH2) или одной σ- и
23
одной
π-связи
(как
в
:С=CH2),
а
2
электрона
–
несвязывающие. Два электрона могут находиться на одной
орбитали (либо s, либо p) – такое состояние называют
синглетным, или на разных орбиталях – такое состояние
называют триплетным. Таким образом, могут быть три
конфигурации (s2, p2, sp).
H
H
C
C
H
H
синглет
триплет
1.4.1. Методы генерирования
1. Термический распад соединений:
R2C=Z
R2C: + :Z
CH2N2
N2 + :CH2
CCl3–SiCl3
SiCl4 + :CCl2
2. Фотолиз кетена
СH2=C=O
hv
3. α-Элиминирование
24
:CH2 + CO
-
HCCl3
+
B(OH ), -BH
Cl– + :CCl2
CCl3¯
Процесс протекает в две стадии.
1.4.2. Основные реакции
1. Присоединение к кратным связям
C(R)H
CR2=CR2 + :C(R)H
CR2
CHCl
+ :CHCl
CR2
-
+
+ Cl
В последней реакции образуется цикло-гептатриенилкатион (катион тропилия).
2. Внедрение в одинарные связи
X
A
.
. X
BC
A +
Y
B + :C
Y
A
X
C
B
A
X
C
B
Y
3. Димеризация (обычно не успевает протекать)
R2C: + :CR2
CR2=CR2
25
Y
4. Перегруппировки
H
CH3-CH2-CH-CH
CH3-CH2-CH=CH2
R-C-CH
O
O=C=CH-R
CH
R
RCH=C=CHR
R
5. Отрыв водорода или другого атома от молекулы
(триплетные карбены)
•
CH3 + •CH2CH3
:CH2 + CH3CH3
6. Реакции с радикалами
:CH2 + •CH3
CH2=CH2 + H•
1.5. Катион-радикалы и анион-радикалы
Эти
частицы
окислении
или
образуются
при
восстановлении
одноэлектронном
органических
и
неорганических молекул. Наиболее известным анионрадикалом является кислородный супероксидный анионрадикал,
.
O2
играющий важную роль в реакциях
окисления в живой природе. Присоединение одного
26
электрона к бирадикалу О2 приводит к появлению одного
неспаренного электрона и одного отрицательного заряда в
молекуле кислорода.
В органической химии анион-радикалы получаются
при
взаимодействии
ненасыщенными
щелочных
молекулами.
металлов
Так,
с
растворение
металлического натрия в растворе нафталина (Napt) в
тетрагидрофуране приводит к образованию голубого
раствора соли анион-радикала нафталина Napt
соединение
является
.
инициатором
с Na+. Это
анионной
полимеризации олефинов и диенов и эффективным
одноэлектронным восстановителем. Инициатор ионной
полимеризации диенов получают при растворении Naмет в
1,3-бутадиене также через промежуточный анион-радикал.
Катион-радикалы возникают при одноэлектронном
окислении
органических
ненасыщенных
молекул
сильными окислителями. Например,
CH3 + Co
Неустойчивый
+.
3+
катион-радикал
CH3 + Co
в
2+
полярном
растворителе выбрасывает протон, образуя бензильный
радикал, принимающий участие в различных реакциях:
27
+.
.CH
CH3
2
+ H+
1.6. Молекулярные комплексы
Среди
активных
упоминались
промежуточных
π-комплексы
частиц
уже
электрофилов
с
ароматическими соединениями. В органических реакциях
принимают участие и различного типа незаряженные
молекулярные
комплексы,
образуемые
за
счёт
π-
электронов донора и вакантных σ- или π- орбиталей
акцептора – донорно-акцепторные комплексы. Например,
комплексы С6Н6→I2 и тринитробензол←тетраметилфенилендиамин, комплексы, образуемые за счет водородных
связей n-бензохинон– гидрохинон (хингидрон), димеры
(ROOH)2, димеры уксусной кислоты и другие ассоциаты.
1.7. Металлоорганические и координационные
соединения
1.7.1. Строение металлорганических соединений
В
органической
химии
в
некаталитических
и
каталитических реакциях важнейшую роль играют σ- и πметаллоорганические соединения, которые в большинстве
случаев являются координационными соединениями.
28
Классическими
σ-металлорганическими
соедине-
ниями, широко используемыми в органическом синтезе для
образования связей С-С, являются соединения ряда
непереходных металлов: LiR, NaR, KR, RMgX, AlR3, BR3,
ZnR2, RHgCl, PbR4, SnR4, SnR5. Кроме простых полярных
σ- связей M-R, соединения непереходных металлов
образуют
димеры
и
многоядерные
ассоциаты
с
мостиковыми группами R, например, (LiR)n, Al2(CH3)6, в
которых имеет место образование электронодефицитных
трехцентровых двухэлектронных связей.
Мостиковыми являются алкилиденовые группы (МСН2-М) и даже атом углерода в соединении С(HgCl)4. В
ионной решётке CaC2 содержатся дианионы С22-.
Различные
органические
группы
или
молекулы
являются типичными лигандами в координационных
соединениях переходных и постпереходных металлов (Cu,
Ag, Au). Комплексы металлов с различными лигандами –
типичные катализаторы и интермедиаты в каталитической
химии, прежде всего в гомогенном металлокомплексном
катализе.
Рассмотрим
комплексов
несколько
металлов,
чтобы
примеров
типичных
продемонстрировать
разнообразие лигандов (атомов, фрагментов молекул и
29
молекул) и показать, что фактически любая молекула или
частица (то есть любой участник каталитической реакции)
может находиться в координационной сфере металла. В
таблице
приведены
некоторые
типичные
лиганды
(подробнее см. Дж. Коллмен). Q – это формальный заряд
лиганда, n – число донируемых электронов.
K[PtCl3(η2-C2H4)] – первое соединение, содержащее
молекулу этилена, связанную с атомом металла за счет
пары π-электронов этилена (соль Цейзе).
Лиганды
Q
n
M–X (σ- или η1-лиганды):
(X = H, Cl, Br, I, F, CN, OR, NR2, CH3,
C2H5, –СR=O)
–1
2
M–X–M (µ-лиганды, то есть мостиковые
лиганды) (X = H, CH3, C2H5)
–1
2
–1
4
0
2
0
2
–1
2
M–X–M (µ-лиганды): X = Cl, Br, I, F, CN,
OR, NR2, PR2
ML (σ- или η1-лиганды):L- H2O, NH3, PR3,
AsR3, SnCl2, CO, N2
ML (σ- или η2-лиганды): L- H2, CH4
(σ- или η -лиганды):
1
M
M
M
30
Лиганды
Q
n
0
2
–1
4
0
4
–1
6
0
6
–1
8
0
2
–2
4
–3
6
(η2-лиганды):
O
M
N N
M
M
M
(η3-лиганды):
M
M
(η4-лиганды):
M
(η5-лиганды):
M
(η6-лиганды):
M
(η7-лиганды):
M
M←:CCl2, M←:CPh2
(карбены Фишера) и
M=C=O, PR3, P(OR)3, M=NR
M=CHR, M=C=CR2
(карбены Шрока) и M=O,
O
O
M
M≡CR, M≡N
31
Степень окисления металла в комплексах может быть
положительной (PdCl42–, Ni(H2O)62+, HPtCl(PR3)2, K2ReH9,
R3W(CH3)6),
равняться
нулю
(Ni(CN)44–,
Ni(CO)4,
Cr(C6H6)2) или даже отрицательной (Na2Fe(CO)4).
К фундаментальным открытиям последнего времени
следует
отнести
синтезы
стабильных
комплексов
с
молекулярным водородом, в которых молекула Н–Н
связана с металлом за счет своей σ-пары электронов (без
разрыва связи Н–Н, η2 - лиганд) (аналогично связи CH3+ c
H2
в
ионах
карбония
и
BH3
c
BH3
в
B2H6):
W(CO)3(PR3)2(H2), Ir(H)2(H2)2(PR3)2+.
Помимо комплексов с одним центральным атомом
металла известны комплексы с мостиковыми лигандами:
M
O
M
M
M
Cl
C
M M
H
M
M
CH3
M
R
M
M
O
C M
M
и комплексы со связями М–М (кластеры металлов). Связи
металл-металл в кластерах имеют различную кратность.
Двойные и тройные связи между атомами металла (M=M,
M≡M)
близки
органических
по
свойствам
(C2H4,
C2H2)
к
и
кратным
связям
неорганических
молекулах. Есть примеры и связи M M (Re2Cl82-).
32
в
(N2)
В отличие от иона H+ ионы переходных металлов
могут координировать до 9 лигандов, то есть связывать
несколько
молекул
и
фрагментов
молекул
в
координационной сфере металла. При этом атом или ион
переходного металла может быть не только акцептором
пары электронов, но и донором неподеленных пар dэлектронов на лиганд, что очень важно для активации
координированной частицы.
1.7.2. Реакции координационных соединений
Известны
три
типа
реакций
координационных
соединений металлов:
1. Реакции переноса электронов:
M1Lnp+ + M2Lmq+
M1Ln(p-1)+ + M2Lm(q+1)+
2. Реакции замещения или присоединения лигандов:
MXn
MXn-1 + X
MXn + Y
MXnY
3. Реакции координированных лигандов.
Этот тип реакций будет рассмотрен в разделе
«Металлокомплексный
катализ».
Промежуточные
соединения на поверхности металлов и оксидов будут
рассмотрены в разделе «Гетерогенный катализ».
33
2. Классификация реакций
Известно
несколько
классифицировать
признаков,
химические
реакции
позволяющих
следующим
образом:
− по типу разрыва связей в реагентах реакции делят
на гомолитические и гетеролитические;
− по области протекания выделяют гомогенные
(протекают в объеме фазы) и гетерогенные
(протекают на поверхности раздела фаз) реакции;
− использование катализаторов позволяет разделять
реакции на каталитические и некаталитические.
По фазовому состоянию реагентов системы делят на
гомофазные (все реагенты в одной фазе) и гетерофазные
(реагенты в разных фазах).
Так, например, каталитическая гидратация пропилена в
водном растворе кислоты – гомогенная реакция,
протекающая в гетерофазной системе. Если ту же реакцию
проводить в водном растворе, содержащем катионит, то
реакция гетерогенная. Гидрирование этилена в растворе
металлокомплексного катализатора – гомогенная реакция,
протекающая в гомофазной системе. Гидрирование
34
этилена в воде, содержащей металлический катализатор, –
гетерогенная реакция.
3. Кинетика и механизмы гомолитических
реакций
К гомолитическим относят реакции с участием
свободных радикалов. Важное место среди радикальных
реакций
занимают
радикально-цепные
процессы,
протекающие и в газовой, и в жидкой фазах.
1. Заместительное фторирование и хлорирование:
R3C–H + F2
CH4
Cl2
-HCl
CH3Cl
CHCl3
R3C–F + HF
Cl2
CH2Cl2
-HCl
Cl2
Cl2
-HCl
CCl4
-HCl
2. Аддитивное хлорирование:
C6H6 + 3Cl2
C6H6Cl6
3. Дегидрохлорирование:
ClCH2CH2Cl
CH2=CHCl + HCl
4. Окисление:
а) Получение гидропероксида
RH + O2
ROOH
35
- важная стадия во многих процессах. Например,
в
процессе
совместного
синтеза
фенола
и
ацетона:
CH3
CH3
C
CH3
O2
C
H
H
CH3
OOH
+
OH + CH3COCH3
б) Ацетальдегид окисляется под действием
кислорода в уксусную кислоту:
CH3CHO + 1/2O2
CH3COOH
5. Термический крекинг (пиролиз) углеводородов:
СН3CH2CH2CH2R
CH4 + CH2=СНCH2R
6. Дегидрирование:
C2H6
H2 + C2H4
7. Теломеризация:
CCl4 + 2C2H4
CCl3(CH2)4Cl
8. Полимеризация олефинов, например, этилена
nC2H4
–(CH2CH2)n–
3.1. Стадии радикально-цепных процессов
1. Зарождение цепи (стадии образования радикалов).
36
2. Продолжение или развитие цепи (стадии, в
которых расходуется и образуется одинаковое
число радикалов).
3. Разветвление цепи
- стадии, приводящие к
увеличению
концентрации
элементарной
стадии
радикалов
радикалов
(в
образуется
больше, чем расходуется).
Дополнительный
источник
активных
радикалов – образование двух радилов из одного:
или
H• +O2
HО• + O:
образование
радикалов
при
распаде
стабильных молекул:
RО• + HО•
ROOH
4. Обрыв цепи (исчезновение радикалов).
Пример механизма радикально-цепного процесса:
CH3CHO
CH3• + HC•O
CH4 + CH3C•O (развитие цепи)
CH3• + CH3CHO
CH3C•O
CH3• + CH3•
(зарождение цепи)
CH3• + CO
C2H6
3.1.1. Зарождение цепи
1. Термическое инициирование
37
(развитие цепи)
(обрыв цепи)
Результатом
инициирования
цепи
является
гомолитическая диссоциация связи в одном из реагентов.
Энергия диссоциации D – положительная величина, а
реакция диссоциации – эндотермический процесс. При
нагревании
становится
Прочность
(энергия
температуру,
при
возможным
диссоциации)
которой
разрыв
связи.
связи
определяет
необходимо
проводить
термическое инициирование. Чем прочнее связь, тем выше
энергия активации и тем выше необходимая температура.
Высокотемпературные процессы:
2Cl•
Cl2
T=400-4800C
2CH3•
CH3CH3
T=7000C
Некоторые процессы не мономолекулярны, например,
O2 + HBr
H-O-O• + Br•
Здесь энтальпия реакции определяется не только
разрывом связи H–Br и разрывом π-связи в O2, но и
образованием связи Н-О.
2. Химическое инициирование
Инициаторами
называют
вещества,
способные
расщепляться с образованием свободных радикалов при
более низкой температуре, чем сами реагенты. Очевидно,
что, вводя инициаторы в реакционную массу, можно
38
существенно ускорить зарождение цепи и провести
процесс при более низкой температуре.
CH3
CH3
0
Т=80-90 С
2CN-C .
N2 +
NC-C-N=N-C-CN
CH3
CH3
CH3
CH3
Т=90-1000С
C-O-O-C
. + 2CO2
2
O
O
CH3
CH3
Т=1500С
CH3
2 O-C-CH3
CH3-C-O-O-C-CH3
CH3
.
CH3
CH3
В высотемпературных реакциях инициатором может
служить хлор, азотная кислота или оксиды азота:
HONO2
Распад
HO• + NO2
RH + NO2
R• + HNO2
инициаторов
(стадии
инициирования)
приводит к образованию радикалов и к появлению стадий
реиницирования. На стадии реиницирования происходит
образование
радикалов,
участвующих
в
стадиях
продолжения цепи.
Inc2
2Inc•
Inc• +Cl2
39
IncCl+ Cl•
3. Фотохимическое инициирование (видимым светом,
УФ)
Молекулы поглощают кванты энергии и переходят из
основного в возбужденное состояние. При достаточной
энергии кванта происходит разрыв связей и образование
свободных радикалов. Скорость распада при этом не
зависит от температуры, а определяется интенсивностью
облучения. При этом рвется наиболее слабая связь и
образуются радикалы.
4. Каталитическое инициирование
Выше уже было замечено (раздел 1.3), что генерация
радикалов может происходить в результате катализа
комплексами металлов переменной валентности распада
пероксидов
Обнаружены
(ROOH).
и
органические
катализаторы зарождения цепи в процессе окисления
углеводородов.
Так,
например,
N-гидроксилфталимид
(NHPI) выступает в роли такого катализатора реакции
2R• + H2O2
2RH + O2
2
CO
NOH + O2
CO
2
40
CO
.
NO + H2O2
CO
CO
.
NO + 2RH
CO
2
2
CO
.
NOH + 2R
CO
3.1.2. Продолжение (развитие) цепи
В неразветвленных процессах в стадиях продолжения
цепи один радикал превращается в другой радикал.
Например,
Заместительное хлорирование:
Cl• + RH
HCl + R•
R• + Cl2
RCl + Cl•
или пиролиз:
C2H5•
CH2=CH2 + H•
H2 + C2H5•
H• + C2H6
3.1.3. Обрыв цепи
Квадратичный обрыв:
Cl• + Cl•
(рекомбинация)
Cl2
R• + R•
R2
(рекомбинация)
Cl• + R•
RCl
(перекрестный обрыв,
рекомбинация)
(диспропорционирование)
2C2H5•
CH2=CH2 + C2H6
41
Стадии рекомбинации и диспропорционирования R•
являются основными стадиями образования продуктов в
реакции радикальной полимеризации.
Линейный обрыв (на стенке):
Cl• + стенка
{Cl• стенка}
R• + стенка
{R• стенка}
Для обрыва цепи можно использовать ингибиторы –
вещества, выводящие активные радикалы из цепного
процесса. Ингибиторами могут быть: а) соли металлов
переменной
валентности
способные
образовывать
(Cu2+,
Fe2+);
стабильные
б)
вещества,
радикалы
или
молекулярные продукты (замещенные фенолы, хиноны,
амины, нитроксильные радикалы, оксиды азота):
.
NO - нитроксильный радикал
Энергия
активации
рекомбинации
(но
не
диспропорционирования) двух радикалов близка к нулю.
Активационный барьер линейного обрыва очень низкий.
Предэкспоненциальные множители разных, но близких по
типу, реакций рекомбинации примерно равны между
собой. Таким образом,
константы скорости примерно
42
одинаковы и поэтому можно судить о преимущественном
протекании того или иного типа обрыва цепи, оценив тип
радикала с наибольшей концентрацией.
3.2 Уравнения Поляни–Семенова
Корреляционные
используются
для
уравнения
приближенной
Поляни-Семёнова
оценки
энергии
активации радикальных реакций по величине теплового
эффекта (см.
раздел 4.3 и ниже). Уравнение Поляни-
Семенова применимо, главным образом, к реакциям
взаимодействия радикала и молекулы
X• + YZ → XY + Z•,
X• + CH2=CH2 → XCH2C•H2,
которые часто являются стадиями продолжения цепи. Этот
метод может быть использован для оценки вероятности
протекания той или иной стадии продолжения цепи и для
выбора стадий обрыва.
Обычно используют уравнение Поляни-Семенова со
следующими коэффициентами:
Ea = 11,5 + b|∆H|, ккал/моль,
где значение b = – 0.25 для экзотермических реакций и
b = + 0.75 для эндотермических реакций.
43
Изменение энтальпии можно оценивать по разности
энергий рвущихся и образующихся связей. Точность
расчетов по методу Поляни–Семенова ±3 ккал/моль.
Примеры
использования
уравнений
Поляни–
Семенова
Предположим,
необходимо
решить
вопрос
о
преимущественном направлении реакции продолжения
цепи в радикально-цепном бромировании изопентана.
Возможны варианты реакции с отщеплением первичного,
вторичного и третичного атомов водорода от изопентана:
(1) Br• + CH3CH2CH(CH3)2
HBr + •CH2CH2CH(CH3)2
(2) Br• + CH3CH2CH(CH3)2
HBr + CH3•CHCH(CH3)2
(3) Br• + CH3CH2CH(CH3)2
HBr + CH3CH2C•(CH3)2
Для
того,
Поляни–Семенова
чтобы
воспользоваться
необходимо
задаться
уравнением
какой-либо
температурой (пусть Т = 554 К) и знать энергии связей,
чтобы рассчитать энтальпии реакций. Энергия разрыва
связи H–Br равна 87.5 ккал/моль, а энергии разрыва С–Н
связи для первичного, вторичного и третичного атомов
водорода равны 94, 89 и 85 ккал/моль, соответственно.
Тогда энтальпия реакции
44
∆Н = DС–Н – DH–Br
и, следовательно,
∆Н1 = 94 – 87.5 = 6.5 ккал/моль,
∆Н2 = 89 – 87.5 = 1.5 ккал/моль,
∆Н3 = 85 – 87.5 = –2.5 ккал/моль.
Первые две реакции эндотермические, для них
Ea1 = 11.5 + 0.75 × 6.5 = 16.4 ккал/моль,
Ea2 = 11.5 + 0.75 × 1.5 = 12.6 ккал/моль,
а третья реакция экзотермическая. Поэтому
Ea3 = 11.5 – 0.25 × 2.5 = 10.9 ккал/моль.
Учитывая, что выражения для скоростей стадий по
закону
действия
масс
одинаковые,
а
предэкспо-
ненциальные множители для сходных реакций примерно
равны, получаем
W3 / W1 = k3 / k1 = ( A3 / A1 )e( Eа 1 − Eа 3 ) / RT =
= e ( Eа1− Eа3 ) / RT = exp[( Eа1 − Eа 3 ) /RT] = 147
W2/W1 = exp([Ea1 –Ea2]/RT) = 31
W3 : W2 : W1 = 147 : 31 : 1
Последнее
выражение
показывает
скоростей третьей, второй и первой реакций.
45
соотношение
Теперь рассмотрим реакции разного типа, имеющие
разные предэкспоненты, т.е. разные члены с энтропией
активации e ∆S
≠
/R
в выражении для константы скорости в
соответствии с теорией абсолютных скоростей реакций.
Например,
.
k1
Cl + CH2=CH2
.
Cl + CH2=CH2
.
ClCH2CH2
.
k2
HCl + CH2=CH
Отношение скоростей стадий в этом случае равно
отношению констант скорости
k1/k2=A1/A2exp([Ea2–Ea1]/RT) = P1/P2exp([Ea2–Ea1]/RT),
≠
≠
где P1 = e ∆S1 / R , P2 = e ∆S 2 / R .
Для реакций присоединения P1= 10-3- 10-4, для
реакций замещения P2= 10-1. Энергия разрыва двойной
связи С=С до одинарной равна 40 ккал/моль, энергия
диссоциации связи С–Cl составляет 80 ккал/моль. Энергия
диссоциации связи С-Н в
алкене равна 104 ккал/моль,
энергия диссоциации связи НCl составляет 102 ккал/моль.
Тогда энтальпии реакций:
∆Η1 ≅ 40−80=−40 ккал/моль,
∆Η2 ≅104−102=2 ккал/моль.
Энергии активации:
46
Ea1 = 11.5 – 0.25 × 40 = 1.5 ккал/моль,
Ea2 = 11.5 + 0.75 × 2 = 13.0 ккал/моль.
k1/k2=10-4/10-1ехр([13.0 –1.5]/RT) и
при Т=600 К k1/k2≈10, а при Т=840 К k1/k2≈1.
Роль отношений предэкспонентов возрастает с ростом
температуры.
3.3 Кинетика неразветвленных цепных реакций
Неразветвленные цепные реакции включают стадии
инициирования, продолжения цепи и обрыва. Как было
отмечено
выше,
неразветвленных
в
стадиях
цепных
продолжения
реакциях
не
цепи
в
происходит
увеличения числа радикалов.
Рассмотрим реакцию заместительного хлорирования:
(1) Cl2
2Cl• (зарождение цепи)
Wи1
(2) Cl• + RH
HCl + R• (развитие цепи)
Wпрод2
(3) R• + Cl2
RCl + Cl• (развитие цепи)
Wпрод3
и возможные варианты обрыва цепи в этой реакции:
(4) Cl• + Cl•
Wо4
Cl2
(5) R• + R•
R2
Wо5
(6) Cl• + R•
RCl
Wо6
(7) Cl• + ст
{Cl•ст}
Wо7
47
(8) R• + ст
{R•ст}
Wо8
В дальнейшем будем рассматривать механизмы,
состоящие из стадий 1, 2, 3 и одной из стадий обрыва,
предполагая квазистационарное протекание реакции. Для
упрощения кинетического анализа процесса используют
правило длинных цепей.
3.4. Правило длинных цепей
Длиной кинетической цепи γ называется отношение
скорости стадии продолжения цепи Wпрод, в которой
образуется продукт, к скорости обрыва Wо или скорости
инициирования Wи. Длина кинетической цепи показывает,
сколько происходит актов образования молекул продукта
на каждый акт инициирования цепи, в котором происходит
генерация радикала (ведущего цепь).
γ = Wпрод/Wо = Wпрод/Wи
Правило длинных цепей состоит в том, что если цепи
длинные (то есть Wпрод > Wо, Wи), то Wо = Wи и скорости
разных последовательных стадий маршрута продолжения
цепи также равны. В нашем случае Wпрод2 = Wпрод3. При
этом линейный радикально-цепной процесс протекает
стационарно. В цепных процессах цепи обычно являются
длинными (γ > 10).
48
Вернемся к рассмотрению реакции заместительного
хлорирования и рассмотрим первый вариант обрыва.
Квадратичный обрыв Cl• + Cl•
2Cl•
Cl2
Wи1
Cl• + RH
HCl + R•
Wпрод2
R• + Cl2
RCl + Cl•
Wпрод3
Cl• + Cl•
Cl2 (обрыв)
Wо4
Условия квазистационарности:
d[R•]/dτ = Wпрод2 – Wпрод3 = 0
⇒
Wпрод2 = Wпрод3
d[Cl•]/dτ = – Wпрод2 + Wпрод3 + 2 Wи1 – 2 Wо4 = 0
Wи1 = Wо4
Тогда имеем:
kи1[Cl2] = kо4[Cl•]2
[Cl• ] =
kи1
[Cl 2 ]
kо4
d[RH]
= –Wпрод2
dτ
d[RCl]
= Wпрод3
dτ
A
A
E
A
E
Wпрод2 = Wпрод3
–
d[RH] d[RCl]
k
=
= kпрод2 и1 [Cl2 ] [RH]
dτ
dτ
kо4
A
A
E
A
A
E
49
⇒
Квадратичный перекрестный обрыв Cl• + R•
2Cl•
Cl2
Wи1
Cl• + RH
HCl + R•
Wпрод2
R• + Cl2
RCl + Cl•
Wпрод3
Cl• + R•
RCl
Wо6
d[R•]/dτ
= Wпрод2 – Wпрод3 – Wо6 = 0
d[Cl•]/dτ = 2Wи1 – Wпрод2 + Wпрод3 – Wо6 = 0
В приближении длинной кинетической цепи имеем
Wпрод3 = Wпрод2 >> Wо6,
Wпрод2 = Wпрод3
2Wи1 = Wо6.
⇒ kпрод2[Cl•][RH] = kпрод3[R•][Cl2] ⇒
•
[R ] =
2Wи1 = Wо6 ⇒
kпрод2 [RH][Cl• ]
kпрод3[Cl2 ]
2kи1[Cl2] = kо6[R•][Cl•]
2kи1[Cl2] = kо6[Cl•]2
[Cl•] =
–
E
A
A
k прод2 [RH]
k прод3 [Cl 2 ]
2kи1kпрод3
kпрод2 kо6 [RH]
d[RH] d[RCl]
=
= Wпрод2 =
dτ
dτ
[Cl2]
2kи1kпрод2 kпрод3
A
E
50
⇒
kо6 [RH]
[Cl2]
Линейный обрыв Cl• + стенка
2Cl•
Cl2
Wи1
Cl• + RH
HCl + R•
Wпрод2
R• + Cl2
RCl + Cl•
Wпрод3
Cl• + ст
{Cl• ст}
Wо7
d[R•]/dτ = Wпрод2 – Wпрод3 = 0
d[Cl•]/dτ =2 Wи1 – Wпрод2 + Wпрод3– Wо7 = 0
⇒
2Wи1 = Wо7 ⇒
2kи1[Cl2] = kо7[Cl•]
[Cl•] =
–
⇒
2kи1
[Cl2]
kо7
2k k
d[RH] d[RCl]
=
= Wпрод2 = и1 прод2 [Cl2][RH]
dτ
dτ
kо7
A
A
E
A
A
E
В итоге для трех рассмотренных случаев имеем
следующие выражения:
Квадратичный обрыв Cl•+Cl•
d[RCl]/dτ = kпрод2
k и1
[Cl 2 ] [RH] = kнабл[Cl2]0.5[RH]
k о4
Квадратичный перекрестный обрыв Cl•+R• с учетом
приближения длинных цепей
51
d[RCl]/dτ =
2kи1kпрод2 kпрод3
kо6 [RH]
[Cl2]= kнабл[RH]-0.5[Cl2]
Линейный обрыв на стенке Cl• + стенка
d[RCl]/dτ =
По
2kи1kпрод2
[Cl2][RH] = kнабл[RH][Cl2]
kо7
порядкам
реакции
можно
судить
о
преимущественном протекании той или иной реакции
обрыва цепи. Выбор преимущественной стадии обрыва
цепи можно провести и до кинетических исследований,
используя уравнение Поляни - Семенова. Из равенства
скоростей стадий продолжения цепи следует, что
kпрод2
kпрод3
=
[R • ][Cl2 ]
[Cl• ][RH]
При равенстве [Cl2] ≅ [RH] и предэкспонентов
[R•]/ [Cl•] = exp([Eпрод3 –Eпрод2]/RT)
Рассчитав Eпрод3 и Eпрод2 по Поляни - Семёнову,
получим
при
заданной
температуре
соотношение
концентраций радикалов. Обычно обрыв происходит на
том радикале, концентрация которого
больше. При
близости [R•]/[Cl•] к 1 обрыв может происходить на всех
радикалах. Для наблюдаемых констант скорости можно
52
говорить о наблюдаемых энергиях активации. Структуру
наблюдаемых
поскольку
энергий
все
активации
константы
легко
скорости
установить,
подчиняются
уравнению Аррениуса. Например, для квадратичного
обрыва Cl• + Cl•:
kнабл = Aнаблexp(–Eнабл/RT) = kпрод2kи10.5kо4–0.5 =
=Aпрод2exp(–Eпрод2/RT)[Aи1exp(–Eи1/RT)]0.5[Aо4exp(–Eо4/RT)]–0.5=
= Aпрод2Aи10.5Aо4–0.5exp(–[Eпрод2 + 0.5Eи1 –0.5Eо4]/RT)
Откуда получаем, что
Eнабл = Eпрод2 + 0.5Eи1 – 0.5Eо4
Учитывая, что Eо4 ≈ 0
Eнабл = Eпрод2 + 0.5Eи1
Аналогично
можно
вывести
выражения
для
наблюдаемой энергии активации для остальных случаев:
Для квадратичного перекрестного обрыва Cl•+R• с учетом
приближения длинных цепей
Eнабл = 0.5(Eпрод2 + Eпрод3 + Eи1),
а для линейного обрыва на стенке Cl• + стенка
Eнабл = Eпрод2 + Eи1 – Eо7
(Eо7 ≠ 0, но очень мала).
53
3.5. Кинетика разветвленных цепных реакций
Рассмотрим реакцию окисления водорода:
(1) H2 + O2
HO2• + H•
(2) H• + O2
(3) O•• + H2
(4) HO• + H2
Wи1
HO• + O••
(зарождение
цепи),
(разветвл.),
HO• + H•
(разветвл.),
Wразв3
(прод. цепи),
Wпрод4
H2O + H•
Wразв2
(лин. обрыв
Wо5
на стенке),
(6) H• + O2 + M
HO2• + M* (лин. обрыв в Wо6
объеме),
HO2• – малореакционноспособный радикал, М –
(5) H• + ст
{H•ст}
молекула, которая забирает выделяющуюся энергию.
Российским ученым Н.Н. Семеновым было сделано
предположение о том, что в разветвленных реакциях
концентрации одних радикалов могут быть стационарными
(HO• и O••), а других (H•)– нестационарными. Тогда,
применив
принцип
Боденштейна
к
концентрациям
радикалов HO• и O••, получим:
d[OH•]/dτ = Wразв2 + Wразв3 – Wпрод4 = 0
d[O••]/dτ = Wразв2 – Wразв3 = 0
⇒ Wразв2 = Wразв3
d[H•]/dτ = Wи1 – Wразв2 + Wразв3 + Wпрод4 – Wо5 – Wо6
54
Сложив выражения для d[OH•]/dτ и d[H•]/dτ и учтя,
что Wразв2 = Wразв3, получаем выражение:
d[H•]/dτ =Wи1 + 2Wразв2 – Wо5 – Wо6 =
=Wи1 + 2kразв2 [H•][O2] – kо5[H•] – kо6[H•][O2][M] =
=Wи1 + [H•](2kразв2[O2] – kо5 – kо6[O2][M]) =Wи1 + [H•]ξ
где ξ ≡ 2kразв2[O2] – kо5 – kо6[O2][M].
Так
как
парциальное
давление
кислорода
и
концентрация М меняются незначительно, считаем ξпостоянной величиной.
d[H•]/dτ =Wи1 + [H•]ξ
Проинтегрируем последнее выражение:
[H]
∫
0
d [H • ]
1
=
•
Wи1 + ξ [H ] ξ
[H]
∫
0
τ
d (Wи1 + ξ [H • ])
= ∫ dτ
Wи1 + ξ [H • ]
0
1/ξ (ln{Wи1 + [H•]ξ} – lnWи1) = τ
Преобразуем последнее выражение:
(Wи1 + [H•]ξ)/ Wи1 =eξτ
⇒
[H•]= (eξτ -1)Wи1/ξ
Если скорость стадий разветвления больше скорости
обрыва, то ξ>0 и при больших значениях τ eξτ >>1. Тогда
для радикально цепного процесса:
[H•]= eξτ∙Wи1/ξ
55
W
f=g
f>g
f<g
f<<g
τ
Рисунок 3.1 Динамика разветвленных цепных
процессов
при
разных
соотношениях
скорости
разветвления (f=2kразв2[O2]) и суммы скоростей обрыва (g =
kо5 + kо6[O2][M]).
Если
предположить,
что
в
какой-то
момент
концентрация H• стационарна, то
d[H•]/dτ = Wи1 + [H•]ξ = 0
[H•] = – Wи1/ξ =
Приближение
kи1[O 2 ][H 2 ]
kо5 + kо6 [O 2 ][М[ - kразв2[O 2 ]
квазистационарности
позволяет
объяснить появление пределов взрыва в смеси H2 и О2
равенством
знаменателя
нулю
при
определенных
давлениях (концентрациях) реагентов. Взрыв возникает,
56
когда знаменатель стремится к нулю: kо5 + kо6[O2][M] =
2kразв2[O2]. В этом случае [H•] → ∞. В области малых
давлений обрыв в основном идет на стенках. Тогда kо5 =
2kразв2[O2]. При больших давлениях обрыв протекает
преимущественно в объеме: kо6[O2][M] = 2kразв2[O2]. Этим
определяется появление двух пределов взрыва – верхнего и
нижнего.
Концентрации
[O2]
и
[M]
пропорциональны
давлению. Следовательно, функции A = kо5 + kо6[O2][M] и B
= 2kразв2[O2] могут быть изображены в координатах от A, B
от P (рисунок 3.2.).
В пределах между критическими значениями Р
происходит протекание реакции со взрывом. Чем выше
температура, тем шире область взрыва. На следующих
рисунках приведена область взрыва в координатах W-P
(рис. 3.3 ) и Р-Т (рис. 3.4).
57
А
A, B
В
P
P2
P1
Рисунок 3.2 Взаимосвязь скоростей обрыва (А) и
продолжения цепи (В) при варьировании давления.
P1 и P2 – нижний и верхний пределы взрываемости.
Скорость
реакции
взрыв
Р1
Р2
Р
Рисунок 3.3 Изменение скорости разветвленного
цепного процесса при варьировании давления.
58
Р
Область
взрыва
Т1 Т2
Т
Рисунок 3.4. Зависимость пределов взрываемости от
температуры.
Образование двух радикалов из ROOH обнаружено в
реакциях
жидкофазного
соединений
окисления
(«вырожденное
органических
разветвление
цепи»).
Например, в случае термического
R•O2 + R•O + H2O
2ROOH
или каталитического (см. раздел 1.3.)
2ROOH
Cu+
RO + RO2 + H2O
распада ROOH.
Механизм жидкофазного окисления RH до ROH, в
котором ионы металла переменной валентности играют
59
роль катализатора реакции зарождения цепи (сумма стадий
(1) и (2) 2RH+O2
2R•+H2O2) и катализатора стадии
разветвления цепи (5), представлен на схеме:
R• + Co2+ + H+
(1) RH + Co3+
(2) 2Co2+ +O2 +2H+
(3) R• + O2
(6) R•O +RH
R• + ROOH
Co
цепи)
(прод. цепи)
R•O2
(4) R•O2 + RH
(5) 2ROOH
2Co3+ +H2O2
(зарождение
(прод. цепи)
2+
R•O + R•O2 + H2O (разветвление)
ROH + R•
(прод. цепи)
(7) Обрыв цепи – рекомбинация радикалов R• и R•O2.
60
4. Кинетика и механизмы гетеролитических
реакций
К гетеролитическим относят реакции, в которых при
разрыве связи оба электрона остаются на одном из
фрагментов. Поэтому гетеролитические реакции всегда
протекают с участием ионов или ионных пар.
Наиболее
изучены
следующие
типы
гетеролитических реакций:
Реакции замещения, S (substitution)
электрофильное SE RX + Y+
RY + X+
нуклеофильное SN RX + Y¯
RY + X¯
Реакции присоединения, Ad (addition)
электрофильное AdE
нуклеофильное AdN
Реакции элиминирования, E (elimination)
4.1. Механизмы реакций
4.1.1. Нуклеофильное замещение
RX + Nu¯
RNu + X¯
Nu– – нуклеофил
X– – уходящая группа
Одностадийное (синхронное) замещение SN2:
61
=
Nu- +
C
X
Nu
C
X
Nu
C
+ X-
В результате реакции наблюдается полное обращение
конфигурации атома углерода.
Скорость бимолекулярной элементарной реакции
описывается уравнением второго порядка:
R = k[RX][Nu¯ ]
В
присутствии
механизм
ментально
останется
большого
избытка
бимолекулярным,
определенная
кинетика
нуклеофила
но
реакции
экспериможет
описываться уравненим псевдопервого порядка:
R = k[RX]
Двухстадийное (диссоциативное) замещение SN1:
k1
RX
k-1
R+ + Nu¯
R+ + X ¯
k2
RNu
В этом случае образуется рацемат, оптическая
активность субстрата RX теряется, если он был хиральным.
В стационарном процессе:
62
d [R + ]
= k1[RX] – k–1[R+][X ¯] – k2[R+][Nu¯] = 0
dτ
[R+] =
R=
k1[RX]
k-1[X - ] + k 2[Nu - ]
k 2 k1[RX ][Nu - ]
k-1[X - ] + k 2[Nu - ]
При k–1[X¯ ] << k2[Nu¯ ], то есть когда первая стадия
практически необратима, уравнение для скорости реакции
упрощается:
R = k1[RX]
Поскольку кинетические исследования не всегда
позволяют провести дискриминацию механизмов SN1 и SN2
(см. выше),
необходимы дополнительные исследования,
например, сравнение различных нуклеофилов, поскольку
скорость
реакции
SN1
при
высокой
реакционной
способности нуклеофила не зависит от его природы, а
скорость реакции SN2 всегда зависит.
Примеры:
SN1
(СH3)3CBr +H2O → (СH3)3COH + HBr
(СH3)3CBr +OH¯ → (СH3)3COH + Br¯
SN2
СH3Br +H2O → СH3OH + HBr
СH3 Br +OH¯ → СH3OH + Br¯
63
В случае механизма SN2 замена воды, сопряженным
основанием ОН¯ приводит к увеличению скорости реакции
в 5000 раз. Для реакций, протекающих по механизму SN1,
эта замена не скажется на скорости реакции.
4.1.2. Электрофильное замещение в ароматических
соединениях
Среди многих реакций этого типа в основном
органическом синтезе большое значение имеют реакции
алкилирования,
сульфирования,
хлорирования
и
нитрования.
ArH + E+
ArE + H+
E+ – электрофил
Н+ – уходящая группа
Механизм SE2
Механизм реакции включает следующие стадии:
образование π-комплекса; превращение π-комплекса в σкомплекс; взаимодействие σ-комплекса с основанием,
приводящее к отрыву протона и образованию продукта
замещения.
64
+ E
k1
+
E
B
k2
k-1
π -комплекс
E
H
σ-комплекс
+ BH
E
При выводе кинетического уравнения быструю
равновесную
стадию
образования
π-комплекса
не
учитывают, поскольку его концентрация обычно очень
мала и не влияет на материальные балансы по ArH и Е+.
Такая квазиравновесная стадия не влияет и на вид
кинетического
уравнения.
Тогда
скорость
процесса,
включающего две стадии
+
ArH + E
k1
ArHE+
k-1
k2, B
ArE + BH+,
с использованием допущения о квазистационарности и при
условии [ArHE+] <<E0+ (E0+ - суммарная концентрация
электрофила) описывается уравнением:
R=
k 2 k1 [B][ArH][E + ]
k -1 + k 2 [B]
Пример – реакция нитрования бензола, катализируемая протонной кислотой НА (например, Н2SO4):
H2NO3+ + A¯
HA + HNO3
(протонирование азотной кислоты)
65
+
H2NO3+
H2O + NO2+
(NO2+ -ион нитрония, электрофил)
[ C6H6–NO2]+
C6H6 + NO2+
[C6H6–NO2]+ + A¯
C6H5NO2 + HA
4.1.3. Элиминирование
Реакции
элиминирования
(отщепления)
в
промышленности
основного
органического
синтеза
используют
получения
ненасыщенных
веществ
для
(процессы дегидрохлорирования, дегидратации и др.) и
гетероциклических соединений (получение оксида этилена
из этиленхлоргидрина и эпихлоргидрина из изомерных
дихлоргидринов глицерина).
В зависимости от условий проведения реакций и
строения реагирующих веществ 1,2-отщепление может
протекать по разным механизмам.
В механизме Е1 разрыв связи С-Х приводит к
образованию карбокатиона, который при взаимодействии с
основанием отщепляет протон с образованием олефина:
k1
H-C-C-X
H-C-C+ + X-
k-1
+
H-C-C + B
k2
66
BH+ + C=C
Кинетическое уравнение для этого механизма
k1k 2 [S][B]
k-1[X - ] + k 2 [B]
R=
где S – субстрат H-C-C-X .
Первая стадия механизма Е1 в точности совпадает с
первой стадией механизма SE1. Вторая стадия отличается
тем, что основание (растворитель) отрывает протон
быстрее,
чем
происходит
атака
по
положительно
заряженному атому углерода. То есть реакции замещения и
отщепления конкурируют между собой.
По
механизму
Е1
могут
протекать
реакции
дегидратации третичных спиртов, дегидрохлорирования
третичных алкилгалогенидов. Причем, увеличение объема
заместителей увеличивает долю продуктов отщепления по
отношению к продуктам замещения. Так, выход олефина в
реакции сольволиза алкилхлоридов
этиловом спирте
R2R′CCl в 80%
меняется при варьировании замести-
телей следующим образом:
R
R′
Выход
олефина.,
СН3
СH3
СН3
С2H5
16
33
СН3
С2H5
С2H5
CH(СH3)2 CH(СH3)2 C(СH3)3
62
%
67
80
90
В
присутствии
сильных
оснований
реакции
элиминирования протекают по механизму Е2. В этом
случае разрыв связей С-Н и С-Х происходит синхронно в
процессе бимолекулярной элементарной реакции:
=
k
B...H...C C ...X
-
:B + H-C-C-X
BH + C=C + X
Скорость этого процесса описывается уравнением
второго порядка:
R = k[S][B¯ ]
Наличие
приводит
к
электроноакцепторных
повышению
заместителей
кислотности
отщепляемого
водорода. В этом случае реакция начинается со стадии
отщепления протона, то есть с образования сопряженного
субстрату основания (механизм Е1cb – conjugated base).
Первая стадия - быстрая равновесная реакция, за которой
следует
медленная
стадия
распада
карбаниона
с
отщеплением Х¯:
Z
-
:B + H-C-C-X
K
- BH
Z
Z
C C X
k2
C=C + X -
Выразив концентрацию карбаниона через константу
равновесия
и
концентрации
68
реагентов
первой
квазиравновесной стадии, получим выражение для расчета
скорости процесса:
R = k2K [S][B¯ ]
В качестве примера рассмотрим механизм реакции
получения винилхлорида из дихлорэтана в присутствии
щелочи:
ClCH2CH2Cl + NaOH → H2O + NaCl + CH2=CHCl
-
H + OH
Cl-C-CH2Cl
H
Cl-C-CH2Cl + H2O
H
Cl-C-CH2Cl
H
Cl-C=CH2 + ClH
4.1.4. Присоединение
К реакциям нуклеофильного присоединения (AdN)
относят реакции присоединения
к ненасыщенным С-С
связям, к карбонильной группе, к α-оксидам и другим
гетероциклическим
присоединения
соединениям.
HCN
к
Например,
реакция
карбонильной
группе
ацетальдегида.
СН3СHO + HCN
CH3CH(OH)CN
69
Реакция образования нитрила молочной кислоты идет
в присутствии NaOH или NaCN по следующему механизму
(нуклеофильный катализ):
δ−
δ+ O
CH3-C
H
+ CN
HCN
CH3 C O-
-
H
H
CH3 C OH + CN
CN
CN
В присутствии NaOH идет присоединение воды по
двойной связи акрилонитрила и образуется нитрил βгидроксипропионовой кислоты:
- H2O
...
[HOCH2 CH CN]
CH2=CHCN + OH¯
HOCH2CH2CN + OH¯
В
альдольной
конденсации,
протекающей
по
механизму специфического основного катализа, вторая
стадия AdN-типа:
CH2CHO + H2O
CH3CHO + OH
CH2CHO + CH3CHO
CH3CHCH2CHO
OCH3CHCH2CHO + OH
CH3CHCH2CHO + H2O
O-
OH
70
-
Механизм
двойным
нуклеофильного
присоединения
к
С=С связям можно представить как реакцию,
обратную Е1cb отщеплению (карбанионный механизм):
C=C + Y
Z
k1
HY
k2
Y C C
Z
Y-C-C-H + Y
Z
-
или как реакцию обратную Е2- отщеплению (синхронный
процесс):
-
C=C + Y + HY
Z
=
- ...
k
Y...C C...H...Y
Z
Y-C-C-H + YZ
Реакции нуклеофильного присоединения характерны
для алкинов. Алкены вступают в реакцию с нуклеофилом
только при наличии элекроакцепторного заместителя
(-СOOR, -NO2, -CF3 и др.).
К реакциям электрофильного присоединения (AdE)
относят реакции аддитивного хлорирования и гидратации
олефинов и алкинов, реакции присоединения кислот и
других веществ к ним, реакции катионной полимеризации
олефинов.
Галогенирование:
C=C + Br2
Br C C
71
+
+ Br-
Br C C
+
+ Br-
Br-C C-Br
Гидрогалогенирование:
H+ + X¯
HX
RCH=CH2 + H+
RCH+–CH3
RCH+–CH3 + X¯
RCHXCH3
(согласно правилу Марковникова).
Реакция электрофильного присоединения протона
или другого электрофила по двойной связи не является
одностадийной – присоединению всегда предшествует
образование
π-комплекса
электрофила
с
олефином.
Дальнейшее превращение π-комплекса в карбокатион
происходит по медленной мономолекулярной реакции (или
при действии Nu-):
C=C
+H
+
K1
H C C
Общая
H+
скорость
C=C
+
k2
H C C
+
NuH-C-C-Nu
k3
процессов
электрофильного
присоединения описывается кинетическим уравнением
второго порядка:
R=K1k2[HA][>C=C<],
72
так как ни первая стадия образования π-комплекса, ни
последующее
взаимодействие
карбокатиона
с
нуклеофилом не является лимитирующим.
Присоединение некоторых электрофильных реагентов
(Cl2, Br2, HHal) к алкенам идет хорошо и не требует
введения
катализаторов.
Присоединение
слабых
электрофилов (H2O, H2S, ROH, RCOOH) проводят в
присутствии кислотных катализаторов: Н2SO4, H3PO4,
галогенводородных кислот
или апротонных кислот
Льюиса (см. ниже). Сила кислот меняется в ряду
HF<HCl<HBr<HI.
Использование кислотных катализаторов приводит к
двум возможным механизмам: а) первичное присоединение
протона по двойной связи алкена с образованием иона
карбения и б) первичным активированием реагента с
образованием более сильного электрофила, способного
присоединятся по двойной связи олефина.
Например, скорость процесса хлорирования алкенов
в присутствии хлорида железа
FeCl3 + Cl2
+
K1
-
Cl+ [FeCl4 ]
ClC-C + FeCl4-
k2
Cl+[FeCl4]
-C=C-
ClC-CCl + FeCl3
73
-
описывает следующее кинетическое уравнение:
R=K1k2[FeCl3] [Cl2][>C=C<].
4.2 Теории кислот и оснований
1. Теория Бренстеда–Лоури – носитель кислотных
свойств протон:
–
кислота (AН) – донор H+
–
основание (В) – акцептор H+ .
Бренстедовскую
кислотность
органических
соединений обычно определяют по отношению к воде и
оценивают константой кислотной диссоциации этого
соединения (Kа):
AH (кислота) + H2O (основание)
А¯ + H3O+
Каждой кислоте (AH) соответствует сопряженное
основание (A¯ ), а каждому основанию (В) – сопряженная
кислота (ВН+).
2. Теория Льюиса:
–
кислота
–
присоединить
вещество,
которое
неподеленную
может
электронную
пару атома другой молекулы (акцептор пары
электронов);
74
–
основание
вещество,
–
обладающее
неподеленной электронной парой, которая
может быть использована акцептором (донор
пары электронов).
AlCl3 (кислота) + Cl¯ (основание)
AlCl4¯
3. Теория Усановича:
–
кислота – вещество, способное отдавать
катионы и присоединять анионы;
–
основание – вещество, способное отдавать
анионы и присоединять катионы.
HI (к-та) + N(CH3)3 (основание)
CH3I (к-та) + HN(CH3)2 (основание)
Обе кислоты по Усановичу (HI и
I¯ + HN(CH3)3+
I¯ + HN(CH3)3+
CH3I) образуют
соль при нейтрализации.
4. Теория Пирсона:
–
кислота – электрофил (акцептор электронов);
–
основание – нуклеофил (донор электронов).
Основания и кислоты бывают жесткие и мягкие.
Жесткие
кислоты
имеют
тенденцию
реагировать
с
жесткими основаниями, а мягкие кислоты с мягкими
основаниями.
75
Кислоты
Основания
Мягкие
Cu+, Hg2+, Pd2+
I¯, CN¯,CO, C2H4
Жесткие
H+, R+, Na+
OH¯, F¯, NO3¯, CH3OH, H2O
4.3. Теоретические основы теории реакционной
способности
Теория химических реакций не позволяет пока в
общем случае предсказывать скорость протекания того или
иного химического взаимодействия на основе данных об
основных свойствах реагирующих веществ и среды. Тем не
менее, было сделано предположение о существовании
линейной зависимости между свободными энергиями
активации (∆G≠) и свободными энергиями реакции (∆G0).
Основой
для
линейности
появления
свободных
этого
принципа
(принципа
явилось
уравнение
энергий)
Бренстеда (k = k0 Kaα см. п.4.3.2) в форме:
∆G≠акт= β +α∆ G0
Этот принцип позволил
корреляционных
кинетические
уравнений,
характеристики
характеристиками реакции:
76
предложить несколько
позволяющих
с
связать
термодинамическими
а) величины энергии активации Ea с тепловым
эффектом (уравнение Поляни-Семенова, вытекающее из
принципа Белла-Эванса-Поляни, рис. 4.1,
рассматри-
валось выше в разд. 3.2. и 3.4.);
Ea
E a2
∆H2
E a1
∆H1
χ
Рисунок 4.1. Взаимосвязь энергий активации и
энтальпий реакции при варьировании заместителей.
б) величины констант скорости с константами
равновесия и характеристиками заместителей в реагентах
(Бренстед, Гаммет, Тафт).
Корреляционные уравнения (уравнения Бренстеда,
Гаммета, Тафта)
строением
устанавливают
замещенных
соотношение
органических
между
соединений
и
изменением их реакционной способности по сравнению со
стандартным реагентом для определенной реакционной
серии. Очевидно, что введение различных заместителей
приводит к изменению электронной плотности в молекуле,
77
меняет
ее
геометрическое
строение.
Первое
корреляционное уравнение было предложено Бренстедом в
1924 г. В те годы не было методов, позволяющих измерить
распределение электронной плотности в молекуле, и
большая
часть
исследований
была
выполнена
при
изучении влияния заместителей на кислотно-основные
свойства органических соединений. Современные методы
исследования и квантово-химические расчеты согласуются
с данными корреляционного анализа, которые в свою
очередь позволяют получать ценную информацию о
механизмах
простых
корреляционных
реакций.
уравнений
для
Использование
дискриминации
механизмов сложных реакций нецелесообразно.
4.3.1. Количественные характеристики
заместителей в органических молекулах
На изменение скорости реакции при варьировании
заместителя
влияют разные характеристики – объем
заместителя, эффекты его электронного взаимодействия с
реакционным
центром.
Последние
подразделяют
на
индуктивный эффект, эффект поля, эффект сопряжения и
сверхсопряжения.
78
Индуктивный
эффект
(I)
–
эффект
передачи
электронной плотности через σ- связи. Заместители
отталкивающие (притягивающие) электроны
повышают
(понижают)
электронную
σ- связей
плотность
на
реакционном центре.
Эффект поля (F) действует не через связи, а через
пространство или молекулы растворителя и зависит от
геометрии молекулы.
Эффект сопряжения (С) проявляется в тех случаях
когда заместитель имеет π-связи, или р-электроны и
находится при атоме углерода в sp2 или sp-гибридизации.
H
..
CH3-O CH
Эффект
CH2=CH C O
CH2
сверхсопряжения
эффект
–
π,
σ-
сопряжения, который заключается во взаимодействии
электронов
σ-связи
с
незаполненной
или
частично
заполненной р-орбиталью. Сильнее всего этот эффект
проявляется
в
случае
С-Н-связей
при
соседнем
с
реакционным центром атоме углерода.
Стерический эффект: большой объем заместителя
может препятствовать сближению молекул и замедлять
реакцию.
79
4.3.2. Уравнение Бренстеда связывает силу кислоты
(константу кислотной диссоциации) с каталитическими
свойствами этой кислоты (величиной константы скорости
кализируемой кислотой реакции).
lgk = α lgKa + lgk0
где k – константа скорости реакции, катализируемой
кислотой, Ka–константа кислотной диссоциации кислоты,
k0- постоянная величина. Константа α определяется
экспериментально и
ее значение характеризует степень
переноса протона в переходном состоянии.
Для
реакций,
катализируемых
основаниями,
предложено аналогичное уравнение:
lgk = βlgKb + lgk0,
В большинстве случаев значения α и β и лежат в
интервале
от 1
до
0.
Уравнение
Бренстеда
чаще
применяется для описания связи между константами
скорости и равновесия одной и той же стадии (реакции).
4.3.3.
Уравнение
Гаммета
описывает
влияние
заместителей в мета- и пара-положениях ароматических
соединений
(м-
и
п-ХС6Н4Y)
по
отношению
к
реакционному центру на скорость реакции с участием
этого центра.
80
lg(k/ko) = σρ
где ko – константа скорости или константа равновесия при
Х = Н; k – соответствующая константа при замещении
водорода группой Х; ρ - константа, характеризующая
чувствительность данной реакции в данных условиях к
свойствам заместителя; σ - константа, характеризующая
заместитель Х.
В
качестве
стандартной
выбрана
реакция
диссоциации бензойной кислоты в воде при 25 оС. Для этой
реакции величина ρ принята равной 1,00. Значения σ,
экспериментально
полученные
для
мета-
и
пара-
замещенных бензойных кислот ( ХС6Н4СООН), приведены
в таблице 4.1.
Таблица 4.1 Значения коэффициентов σ для разных
заместителей бензойной кислоты.
Заместитель
H
n-OH
n-CH3
n-F
n-Cl
м- Br
Ka*105
6.5
2.62
4.33
7.2
1.03
1.55
81
σ
0
-0.37
-0.17
+0.062
+0.227
+0.391
Величина ρ для других реакционных серий зависит от
их чувствительности к влиянию заместителей. Если, как
при диссоциации бензойных кислот, понижение электронной плотности на реакционном центре ведет к повышению
скорости, то ρ>0. В противоположном случае ρ<0.
Используя
значения σ, можно найти ρ других
реакций, экспериментально определив скорости реакций
только для двух Х-замещенных соединений (с известными
значениями σ групп Х). На практике для получения более
точных результатов
используется не менее четырех
величин с подходящим интервалом значений. Определив
величину ρ и зная величины σ для других групп, можно
предсказывать скорости реакций до их проведения.
Величины σ являются мерой суммарных электронных
эффектов (эффекта поля и эффекта сопряжения) группы Х,
связанной с бензольным кольцом. К соединениям с ортозаместителями подход обычно неприменим из-за сильного
влияния стерических факторов. В справочниках имеются
величины ρ примерно для 200 разнообразных реакций, для
которых определены константы скорости.
Расчеты
по
удовлетворительные
уравнению
результаты
82
Гаммета
(точность
дают
±15
%)
независимо
от
того,
атакуется
электрофильным,
ли
субстрат
нуклеофильным
или
свободнорадикальным реагентом; важно только, чтобы в
пределах данной серии реакций механизм их был
одинаковым.
4.3.3. Уравнение Тафта используется для описания
реакций
алифатических
соединений.
Это
уравнение
предполагает аддитивность вкладов различных эффектов
при переходе от метильного заместителя к заместителю Х:
ΔEa = ΔEI+F +ΔEстер.+ΔE cв.с. +ΔEС или
lg(kx/ko) = ρ* σ* + δEcS + h(n-3) + ψ
где ko и kx константы скорости реакции с участием
алифатических веществ, имеющих в качестве заместителей
метильную группу (принята за стандарт) и заместитель Х.
ρ*
-
константа,
характеризующая
чувствительность
реакции к индуктивному эффекту заместителя; σ* константа,
характеризующая
заместителя;
δ
чувствительность
-
константа,
реакции
заместителя;
EcS
стерический
эффект
-
индуктивный
к
характеризующая
стерическому
константа,
заместителя;
эффект
эффекту
характеризующая
член
h(n-3)
характеризует эффект сверхсопряжения (n – количество α83
С-Н связей); величина ψ характеризует вклад сопряжения
какой-либо
π-электронной
системы
с
реакционным
центром. Она почти не зависит от типа π-электронной
системы и от реакционной серии и равна:
ψ = ± 2,3RT(6 ± 1).
Константы уравнения Тафта известны примерно для
80
реакций
и
позволяют
рассчитывать
скорость
с
точностью примерно ±10 %.
4.3.4. Использование корреляционных уравнений
для изучения механизма реакций
Знак и абсолютное значение констант ρ в уравнениях
Гаммета
и
Тафта
позволяют
установить
наиболее
вероятный механизм реакций. Так, для реакции
XC6H4CH=NCl + OH-
XC6H4C
N + H2O + Cl-
можно предложить два механизма процесса. Первый
механизм предусматривает диссоциацию по связи N-Cl.
Второй - отрыв протона от исходной молекулы и
образование карбаниона.
84
-
XC6H4CH=NCl
-Cl-
+ OH
XC6H4C
XC6H4CH=N
-H2O
OH-
XC6H4CH=NCl
XC6H4C=NCl
-Cl-H2O
XC6H4C
N
N
Положительный знак ρ (ρ= +2,24) свидетельствует о
том, что реакция облегчается при введении электроноакцепторных
заместителей
в
ароматическое
ядро.
Следовательно, наиболее вероятным является второй
вариант.
Реакция
кислотнокатализируемой
изомеризации
арилпропенилкарбинолов
характеризуется
большим
отрицательным значением
ρ (ρ = -6,08). Это свиде-
тельствует об образовании карбкатионного интермедиата.
ArCH-CH=CH-CH3
H+
ArCH-CH=CH-CH3
+
OH
+
Ar-CH-CH=CH-CH3
OH 2
+H2O
-H+
-H2O
Ar-CH=CH-CH-CH3
OH
85
В тоже время малые абсолютные значения ρ в
реакциях Дильса-Альдера позволяют считать, что реакция
идет не через образование цвиттер-ионов,
+
CO
O
CO
+
Ar
Ar
а как синхронный согласованный процесс.
86
CO
O
,
CO
Общая
Литература
1. Лебедев Н.Н., Манаков М.Н., Швец В.Ф. Теория
химических процессов основного органического и
нефтехимического синтеза. М.:Химия, 1984, с. 375.
2. Потехин В.М., Потехин В.В. Основы теории химических
процессов технологии органических веществ и
нефтепереработки: Учебник для вузов.- СПб:
ХИМИЗДАТ, 2005, с. 912.
Активные частицы и промежуточные соединения
1 Дж. Марч. Органическая химия. Реакции, механизмы и
структура. В 4-х т., Пер с анг. М. Мир, 1987.
2 Днепровский А.С., Темникова Т.И., Теоретические основы
органической химии., Л., Химия, 1991, с.560.
3. Дж. Коллмен, Л. Хигедас, Дж. Нортон, Р. Финке.,
Металлоорганическая химия переходных металлов. М.: Мир,
1989. т.1, с.396.
4. Эмануэль Н.М., Кнорре Д.Г. Курс химической кинетики,
Москва, Высшая школа, 4е изд., 1984., с.463.
5. Семенов Н.Н., О некоторых проблемах химической кинетики
и реакционной способности». АН СССР, 1958, с.354.
6. Денисов Е.Т., Саркисов О.М., Лихтенштейн Г.И.,
Химическая кинетика, М. Химия, 2000, с.567.
7. Темкин О.Н. Введение в металлокомплексный катализ
(Катализ и координацинная химия).М.: МИТХТ, 1980, с.185.
Гетеролитические и гомолитические реакции
1. Днепровский А.С., Темникова Т.И., Теоретические основы
органической химии., Л., Химия, 1991, с.560.
2. Семенов Н.Н., О некоторых проблемах химической кинетики
и реакционной способности». АН СССР, 1958, с.354.
3. Денисов Е.Т., Саркисов О.М., Лихтенштейн Г.И.,
Химическая кинетика, М. Химия, 2000, с. 567.
87
Издание учебное
И.В. Ошанина, Л.Г. Брук, А.В. Зейгарник, О.Н. Темкин
«Физико-химические основы реакционных процессов
органического синтеза» (часть 2)
Учебно- методическое пособие
Подписано в печать ____________Формат 60х84 1/16.
Бумага писчая. Отпечатано на ризографе. Усл. печ. листов.
Тираж 100 экз. Заказ №_______
ФГБОУ ВПО «Московский государственный университет
тонких химических технологий имени М.В.Ломоносова».
Издательство МИТХТ
119571, Москва, пр. Вернадского, 86.
88