ПОИСК ДОСТУПНОГО МЕТОДА СИНТЕЗА ФОСФАТА
реклама

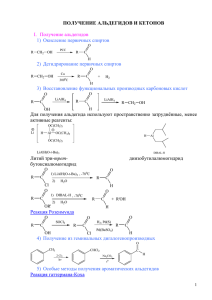
СИНТЕЗ 1,7,7-ТРИМЕТИЛБИЦИКЛО[2.2.1]ГЕПТАН-2-ИЛ НИТРОКАРБАМАТА И ЕГО КАЛИЕВОЙ СОЛИ ВЗАИМОДЕЙСТВИЕМ БОРНЕОЛА С ДИНИТРОМОЧЕВИНОЙ целевой продукт (3) обработкой хлороводородом. (Схема 1) Me Me Me Me H Me O H T O Me C NK + CO + H2O + N2O OK NO2 Me Me H Me ЗАКЛЮЧЕНИЕ Me Me ложения (схема 2), о чем свидетельствует исчезновение характерного пика на УФспектрах и интенсивное газовыделение. Выделенный осадок по своим физикохимическим характеристикам полностью соответствует исходному борнеолу. O T O C NH H Me + CO + H2O + N2O OH NO2 Схема 2 Сравнение ИК-, ЯМР-спектров очищенных продуктов (3, 4) со спектрами исходных реагентов (1, 2) позволило идентифицировать полученные соединения. Многократное упаривание маточного раствора и выкристаллизация полученных затем продуктов не дает дополнительного выхода соединений (3, 4). Термическая обработка целевых продуктов (3, 4) в условиях реакции позволили доказать, что во время синтеза идет параллельная реакция их раз- Таким образом показано, что при взаимодействии борнеола с динитромочевиной при температуре от 40°С до 60°С образуется 1,7,7-триметилбицикло[2.2.1]гептан-2-ил нитрокарбамат с выходом 53 %. Структура полученного соединения доказана методами ИК-, ЯМР-спектроскопии и элементным анализом. СПИСОК ЛИТЕРАТУРЫ 1. Рудаков, Г.А. Химия и технология камфары – М.: Лесная промышленность, 1976. – 208 с. 2. Лобанова А.А., Сатаев Р.Р., Попов Н.И., Ильясов С.Г. // Журн. орг. химии. – 2000. – Т. 36, вып. 2. – С. 188-191. ПОИСК ДОСТУПНОГО МЕТОДА СИНТЕЗА ФОСФАТА ОСЕЛЬТАМИВИРА Калашников А.И., Сысолятин С.В., Сонина Е.Г., Сурмачева И.А. Учреждение Российской академии наук Институт проблем химико-энергетических технологий Сибирского отделения РАН (ИПХЭТ СО РАН) Рассмотрены известные способы получения фосфата осельтамивира. Наиболее доступными признаны методы на основе (-) шикимовой кислоты. Проведена экспериментальная проверка вариантов синтеза осельтамивира через О-триметансульфонилпроизводное этилового эфира шикимовой кислоты. Установлены основные закономерности процесса и получен продукт с выходом 16,9 %. Ключевые слова: фосфат осельтамивира, (3R,4R,5S)-4-ацетиламино-5-амино-3-(1этилпропокси)-1-циклогексен-1-карбоксиэтиловый эфир, шикимовая кислота, азиридин. Во всем мире проявляют озабоченность к потенциальной возможности пандемии гриппа Н5N1. Коэффициент смертности от этого вируса (птичий грипп) превышает 50 %. По данным Всемирной организации здравоохранения в настоящее время единственным способом защиты от пандемии этого вируса являются лекарственные средства - занамивир и осельтамивир фосфат. Эти препараты ингибируют вирусный фермент – нейраминидазу и тем самым предотвращают проникновение вируса в клетки человека. Наибольшее распространение получил осельтамивир фосфат (торговая марка «ТаПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 мифлю»). Хотя молекула этого соединения относительно мала, разработка практического синтеза является сложной задачей. В литературе приводится около 30 способов и различных модификаций получения осельтамивира. Описанные способы исходят из различного сырья и включают от 8 до 25 технологических стадий. Выход продукта зависит от стадийности синтезов, использованного сырья и составляет от 5 % до 57 %. Основные способы получения продукта приведены в таблице 1. 151 КАЛАШНИКОВ А.И., СЫСОЛЯТИН С.В., СОНИНА Е.Г., СУРМАЧЕВА И.А. Таблица 1 1 [1] 2 3 4 5 [2,3] [2,4] 6 7 [5] [6] Исходный реагент 10 (-) шикимовая кислота 12 (-) хинная кислота 17 (-) хинная кислота 15 (-) шикимовая кислота 12 (-) хинная кислота 10 (-) шикимовая кислота 10 этиловый эфир (-) шикимовой кислоты Выход Кол-во стадий № п/п Лит. Способы получения фосфата осельтамивира % Использование на отдельных стадиях труднодоступных реа21 гентов. Первый промышленный вариант. 4,4 Лабораторный метод, недостаточный выход Доступные реагенты, очистка промежуточных соединений кри9-10 сталлизацией, реализован на опытном производстве. 30- Доступные реагенты, очистка промежуточных соединений кри32 сталлизацией, реализован в производстве Доступные реагенты, очистка промежуточных соединений кри16 сталлизацией, реализован на опытном производстве. 38 12 20 8 [7] 13 (-) шикимовая кислота 40,5 9 [8] 9 (-) шикимовая кислота 47 9 фуран и этилакрилат 3,2 10 [9] 11 14 1,6-диметоксифенол, 1-этилпропил метансульфонат 1,3-бутадиен, 2,2,2трифторэтилакрилат N-нозил-3-гидрокси-2пиридон, этилакрилат 7-(3,5-динитробензо-ил)7-азабицикло-[4.1.0]гепт3-ен 1-(триметилсилокси)-1.3бутадиен, фумарил хлорид 12 [10] 11 13 [11] 7 14 [12] 20 15 [13] 12 16 [14] 25 L-серин 17 [15] 14 пиридин 18 [16] 16 D-ксилоза 19 [17] 11 (1S,2S)-3-бром-3,5-циклогексадиен-1,2-диол 20 [18] 14 этиловый эфир циклогексадиеновой кислоты 21 [19] 8 22 [20] 9 23 [21] 18 D-маннит 24 [22] 18 L-метионин 152 6-оксабицикло[3.2.1] окт3-ен-7-он (1-этилпропокси) ацетальдегид, третбутилнитроакрилат Примечания Доступные реагенты, очистка промежуточных соединений кристаллизацией, реализован в производстве Доступные реагенты, очистка промежуточных продуктов препаративной хроматографией. Доступные реагенты, очистка промежуточных соединений кристаллизацией. Доступные реагенты, очистка всех промежуточных продуктов колоночной жидкостной хроматографией. Короткий процесс, доступные реактивы, выделение большинства промежуточных соединений с применением препаративной хроматографии Циклизация Дильса-Алдера, применение энзимов при разделении энантиомеров, жесткие требования по соблюдению температурного режима и рН среды. Сравнительно доступные реагенты, применение энзимов для 28 несимметричного гидролиза, каталитическое гидрирование при давлении 100 атм. Циклизация Дильса-Алдера в присутствии энантоселективного 27 катализатора, сравнительно доступные реагенты. Применение препаративной хиральной хроматографии для 11 разделения смеси рацематов. Труднодоступные реагенты, большое количество стадий, ме16 тод не стереоселективен. Сравнительно доступные реагенты, применение препаратив16 ной хиральной хроматографии для разделения смеси рацематов. Сравнительно доступные реагенты, большое количество ста8 дий и длительность, очистка большинства промежуточных продуктов препаративной хроматографией. Циклизация Дильса-Алдера в присутствии энантоселективного 5,6 катализатора, доступные реагенты, полупродукты легко очищаются кристаллизацией. Сравнительно доступные реагенты, большое количество ста14 дий и длительность, очистка большинства промежуточных продуктов препаративной хроматографией. Малодоступные реагенты, очистка большинства промежуточ26 ных продуктов препаративной хроматографией. Малодоступные реагенты, очистка отдельных промежуточных продуктов препаративной хроматографией. Малодоступные реагенты, использование платиновых и ро30 дийорганических катализаторов. 5 Выход приведен на осельтамивир-основание. Малодоступные 57 реагенты, выделение промежуточных соединений с применением препаративной хроматографии Труднодоступные реагенты, многостадийность, простая очист7,5 ка полупродуктов. 7,5 Малодоступные реагенты, многостадийность. ПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 ПОИСК ДОСТУПНОГО МЕТОДА СИНТЕЗА ФОСФАТА ОСЕЛЬТАМИВИРА Среди множества известных способов наибольший интерес представляют методы №№7, 9 (таблица 1) которые отличаются использованием доступных реагентов, наименьшим количеством стадий и приемлемым выходом. Именно эти способы были рассмотрены при разработке метода получения фосфата осельтамивира. Первоначально была рассмотрена схема синтеза на основе шикимовой кислоты, содержащая 9 химических стадий и позволяющая получить наибольший выход продукта (схема 1) [8]. Использование природной шикимовой кислоты гарантирует получение нужного стереоизомера и позволяет избежать использования препаративной хроматографии с труднодоступными хиральными сорбентами. В большинстве случаев исследователи пытаются исходить из природных соединений со строго определенной конфигурацией таких как: шикимовая кислота, хинная кислота, Lсерин, D-ксилоза, D-маннит, L-метионин. Такой подход облегчает получение необходимого рацемата. Однако есть немало работ по синтезу препарата из широкодоступных соединений таких как: 1,3-бутадиен, фуран, пиридин, 1,6-диметоксифенол. Эти методы основываются на циклизации Дильса-Алдера в присутствии энантоселективного катализатора. Полупродукты, образующиеся в этих синтезах, как правило, представляют собой смесь рацематов с преобладанием нужного изомера. Промышленное применение нашли только способы, разработанные компаниями «Gilead Scinces Inc.» и «Roshe» (№№1-6, таблица 1). O O HO Ms HO OH EtOH, H O + MsCl, Et3N, AcOEt OH O N Ac 3-пентанол , BF3.Et2O O Et Et O O Et O Ms 6 Ms NaN3 NH Et Ms Et NH 7 N3 8 (Ph)3P Ms Et O O Ac O 3 O Et Ac O Et Et O O O HN 4 O Ms 5 Et Et O O O O O O Et Et Ac Ac2O Et Ms O Ms 2 Et3N, H2O Et O 1 (Ph)3P, O Et HO OH O N3 O NaN3 Et HO O O O NH Ac NH2 9 NH 10 NH2 * H 3PO4 Ms = CH3SO2- (метансульфонил) Рисунок 1. Схема синтеза осельтамивира фосфата Этиловый эфир шикимовой кислоты (2) образуется при нагревании шикимовой кислоты(1) (ШК) в этаноле (содержание воды 0,5÷0,8 %) в присутствии кислотного катализатора. Наибольшее качество и выход продукта обеспечивает использование в качестве катализатора этерификации катионита КУ2-8 и проведение синтеза с удалением воды из реакционной массы азеотропной отгонкой либо цеолитами. Поскольку катализатор находится в твердой фазе, длительность процесса составляет 24-30 часов. Степень конверсии ШК достигала 98÷99 %, а чистота полученного эфира 2 – 98,5÷99,0 %. Использование тионилхлорида, хотя и сокращает время реакции (до 20-24 часов), требует применения спирта с содержанием влаги не более 0,1 %. При использовании коммерчески доступного абсолютного этанола (0,5 % воды) остаточное содержание ШК в эфире 2 составило 3,0 %. Использование толуолсульфокислоты загрязняет полученный эфир, и вызывает значительные потери при очистке продукта. Степень конверсии ШК составляла 94-96 %. ПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 Триметилсульфонилпроизводное 3 получали обработкой этилового эфира 2 хлорангидридом метансульфокислоты. Процесс проводили в присутствии каталитического количества 4-диметиламинопиридина. Полученный этил (3R,4S,5R)-3,4,5-о-триметансульфонил шикимат (3) легко кристаллизуется при обработке метанолом, изопропиловым спиртом или толуолом. Выход очищенного продукта составил 81-82 %. Этил (3S,4R,5R)-3-азидо-4,5-диметилсульфонилоксациклогекс-1-ен-1-карбоксилат (4) получают обработкой триметансульфонил- производного 3 азидом натрия. При этом происходило стереоселективное замещение только одной из метансульфонильных групп (в положении 3) и обращение конфигурации углерода в положении 3 с R на S. В известном методе [8] реакция проводится в водном ацетоне (ацетон : вода = 5:1) при температуре не выше 0 °С. Воспроизведение этой методики показало образование продукта с содержанием около 10 % примесей 11, 12 ароматического характера. 153 КАЛАШНИКОВ А.И., СЫСОЛЯТИН С.В., СОНИНА Е.Г., СУРМАЧЕВА И.А. Азиридин 5 (этил (3S,5R,6S,)-5-метансульфонилокси-7-азабицикло[4.1.0]гепт-2-ен3-карбоксилат) получают по реакции Штаудингера [8] последовательной обработкой азидодиметансульфонилпроизводного 4 трифенилфосфинома и водным триэтиламином. Обработка малоустойчивого азиридина 5 уксусным ангидридом в избытке триэтиламина приводит к ацетилпроизводному 6. Ряд проведенных экспериментов показал крайне плохую воспроизводимость этого метода. Реакция проходит в основной среде, так как трифенилфосфин является слабым основанием (фосфорный аналог трифениламина), а водный триэтиламин является еще более сильным основанием. Это вызывает (как и на предыдущей стадии) образование ароматических соединений. В результате из реакционной массы удалось выделить только оксид трифенилфосфина и этиловый эфир мета-аминобензойной кислоты (15). Кроме того, зафиксирован еще целый ряд неидентифицированных продуктов. O O Ms O N3 O O Et 11 Et 12 При использовании других растворителей: метанола, этанола, диоксана – содержание этих примесей находилось в пределах 812 %. Применение в качестве растворителя N,N-диметилформамида приводит к образованию 11 и 12 в качестве основного продукта. Результаты показывают, что образование ароматических соединений определяется использованием достаточно основного азида натрия, а в случае использования основного растворителя (N,N-диметилформамида) побочная реакция становится доминирующей. Замена азида натрия на азид аммония (получают из азида натрия и хлорида аммония) и проведение реакции в метаноле позволяет снизить содержание побочных продуктов до уровня 5 %. Полученный продукт 4 может быть использован на последующей стадии без дополнительной очистки. Ph O N3 Ph P O (Ph)3P, THF Et Ms O Ms O Ms Замена рекомендуемого растворителя (тетрагидрофуран) на диоксан или ацетонитрил изменений состава продуктов не вызывала. Аналогичные результаты получены при добавлении в реакционную массу небольших количеств уксусной кислоты. Попытка проацетилировать образующийся, малоустойчивый азиридин 5 уксусным ангидридом без выделения из реакционной массы также усPh O Et Ms O (Ph)3P, AcOH Ph P O Et O O 13 Ms 154 O LiAlH4, CH2Cl2 O Ph O Et -Ph3PO Ms Ms В литературе известно получение производных азиридина на основе соединений, содержащих азидную и метилсульфоксогруппы в соседних положениях [23, 24]. Азидную группу восстанавливают до аминогруппы алюмогидридом лития и после выдержки происходит образование азиридинового фрагмента. N3 15 O O Ph H N O Ph Ac NH Ms 4 Ph + Ph P пеха не принесла. Разделить ацетилпроизводное 6 и оксид трифенилфосфина не удалось даже с помощью препаративной жидкостной хроматографии. Проведение реакции в среде уксусной кислоты приводит к другим продуктам реакции (16, 17). При этом выход продукта с азиридиновым циклом 17 не превышал 5 %. N -N2 Ms O Et 14 13 O O O Et 4 N3 H2N H2O Ph - (C2H5)3HN+OMs O O N Ph P (C2H5)3N, H2O Et Ms O Ph O Ph -N2 O O N O + Ph Ph P O N Et O Ms 16 O Ms 65 % 17 O Ms около 5 % Поэтому была исследована возможность получения азиридина 5 путем восстановления азидной группы 4 до аминной с использованием более доступных восстановителей. Были проверены боргидрид натрия в среде диоксан–вода, формамидинсульфиновая кислота в среде диоксан–вода, а также восстановление водородом в присутствии палладиевого катализатора. Продукт восстановления 18 переводили в среду хлористого метилена, добавляли ПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 ПОИСК ДОСТУПНОГО МЕТОДА СИНТЕЗА ФОСФАТА ОСЕЛЬТАМИВИРА триэтиламин, выдерживали и обрабатывали уксусным ангидридом. Для разделения смеси использовали препаративную жидкостную хроматографию. O 1.NaBH4 N3 O Et Ms O O 2 CH4N2SO2 H2N 3 H2, Pd/C O Et -N2 Ms O O 4 Ms O Ms O Ac NH O O Et Et Ms O Ms 18 Ac2O O HN аминогруппы проходит достаточно успешно, но образования азиридинового фрагмента в этих условиях не происходит. Таким образом, все попытки получить этил (3S,5R,6S,)-5-метансульфонилокси-7азабицикло[4.1.0]гепт-2-ен-3-карбоксилат (5) с приемлемым выходом оказались безуспешными. Поэтому, в связи с малой устойчивостью азидодиметансульфоксипроизводного шикимовой кислоты 4 в основной среде, был рассмотрен другой путь получения (3S,4S,5R)-4ацетамидо-3-(1-этилпропокси)-5-метансульфонилоксициклогекс-1-ен-1-карбоксилата. Для восстановления азидной группы вместо основного трифенилфосфина использован триэтилфосфит (триэтиловый эфир фосфористой кислоты) [6]. Пятичасовое кипячение раствора приводит к образованию фосфорилпроизводного азиридина 20. O 5 O Ms 19 Во всех экспериментах основным продуктом реакции был этил 3-ацетамидо-4,5диметансульфонилоксацикло-гекс-1-ен-1карбоксилат (19). Это указывает на то, что реакция восстановления азидной группы до O O Et N3 O Et O (EtO)3P O -N2 Et Ms O O Ms O P N 3-пентанол , BF3.Et2O Et O O Et O 4 Et Ms Et O O P Et Et Et Et O O O NaN3 O Et Ac NH Ms 7 8 На первой стадии процесса обработкой 7 азидом натрия в среде водного этанола замещали метансульфонилокси- группу на азидо-группу. Процесс также сопровождается инверсией атома С-5 с R-конфигурации на SПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 O Et Ac NH O Et 22 Ms O Ms 7 Et Et O O O O O H3PO4 O Et Et Ac N3 O O Замену фосфорильного остатка аминной группы на ацетильный проводили в две стадии. Вначале в среде этилового спирта и серной кислоты проводили гидролиз фосфориламидной группы. Полученный амин 22 (этил (3S,4S,5R)-4-амино-3-(1-этил-пропокси)5-метансульфонилокси-циклогекс-1-ен-1-карбоксилат) обрабатывали (после нейтрализации реакционной смеси содой) уксусным ангидридом. В результате с выходом 76,1 % (в пересчете на 22) получали кристаллический этил (3S,4S,5R)-4-ацетиламино-3-(1-этилпропокси)-5-метансульфонилокси-циклогекс1-ен-1-карбоксилат (7). Фосфат осельтамивира получали в результате последовательного проведения трех стадий процесса. (Ph)3P NH Ac2O O 21 Ms Et Et O Et Et Ac O Et O H2N O Et O O Et H2SO4, EtOH O NH Et 20 Полученный этиловый эфир (1S,5R,6S)7-(диэтокси-фосфорил)-5-метан-сульфонилокси-7-аза-бициклоцикло[4.1.0]гепт-2-ен-3карбоновой кислоты (20, фосфорилпроизводного азиридина) представляет собой жидкий продукт и может быть очищен с помощью препаративной жидкостной хроматографии. Однако присутствующие примеси не мешают прохождению следующей стадии процесса. Поэтому полученный продукт без очистки обрабатывали эфиратом трехфтористого бора в среде 3-пентанола. В результате происходило раскрытие азиридинового цикла и получался этил (3S,4S,5R)-4-(диэтокси-фосфориламино)-3-(1-этил-пропокси)-5-метансульфонилокси-циклогекс-1-ен-1-карбоксилат (21). Выход продукта составил 56,6 % в пересчете на исходный азид 4. Et O O NH Ac NH2 9 NH NH2 * H3PO4 10 конфигурацию. На следующей стадии процесса обработкой трифенилфосфином восстанавливали азидную группу 8 до аминогруппы. В результате получали смесь этил (3S,4R,5S)-4155 КАЛАШНИКОВ А.И., СЫСОЛЯТИН С.В., СОНИНА Е.Г., СУРМАЧЕВА И.А. ацетамидо-5-амино-3-(1-этилпропокси)-циклогекс-1-ен-1-карбоксилат (9, осельтамивироснование) с оксидом трифенилфосфина. Обработка полученного раствора спиртовым раствором ортофосфорной кислоты приводила к выпадению из раствора фосфата осельтамивира (10). Продукт был выделен фильтрованием и многократно промыт ацетоном. Это позволило практически полностью отмыть остатки оксида трифенилфосфина и получить продукт с содержанием основного вещества 98,6 %. Средний выход продукта на каждой из десяти стадий синтеза составил 83,7 %, а выход фосфата осельтамивира в пересчете на ШК составил 16,9 %. Это свидетельствует о конкурентноспособности рассмотренного метода и возможности создания на его основе технологии получения продукта. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ ИК-спектры соединений записывали на приборе «Инфралюм ФТ-801» в таблетках с KBr. Спектры ЯМР 1Н и 13С регистрировали на приборе «Bruker AM-400», 400.13 (1Н) и 100.61 Мгц (13С). ВЭЖХ проводилась на приборе «Agilent 1200» с УФ-детектором; колонка 2.1×150 мм, сорбент – «Zorbax SB-18», фр. 5 μm. Элюент MeCN– вода подкисленная 0,2 % H3PO4. Элементный анализ проводили на элементном C, H, N, O анализаторе FlashEATM 1112. Этиловый эфир шикимовой кислоты (2). Смесь 5,7 ммоля г ШК, 40 мл абсолютного этанола, 10 г катионита КУ-2-8 кипятили на установке с аппаратом Сокслета заряженным 32 г цеолита NaA и обратным холодильником в течение 30 часов. Полученный раствор охлаждали, фильтровали, катионит промывали Спиртом и упаривали под вакуумом. Остаток от упаривания обрабатывали 60 мл эфира и после фильтрования получали 93,7 % 2 с Тпл=97,8-99,0 °С и содержанием основного вещества 98,8 % (ВЭЖХ). ИК спектр, см-1: 3354, 2911, 1717, 1654, 1456, 1372, 1238, 1092,1053, 932, 873, 837, 749. Спектр ЯМР 1Н ((CD3)2SO, δ, м.д.): 7,13-7,12 (1Н, s, СН), 4,75-4,74 (1Н, s, СН), 4,10-4,8 (1Н, m, СН), 3,95 (2Н, m, СН2), 2,99-2,93 (1Н, d, СН2), 2,59-2,54 (1Н, d, СН2), 1,70-1,66 (3Н, t, СН3). Спектр ЯМР 13C ((CD3)2СO, δ, м.д.): 166,9 (С=O); 139,5 (CH); 128,8 (C); 66,3 (CH); 60,6 (CH2); 30,5 (CH2); 14,4 (CH3). Этил (3R,4S,5R)-3,4,5-о-триметилсульфонил шикимат (3). Получали по [8]. Выход 82 %, Тпл=97-98 °С (изопропанол). ИК спектр, см-1: 3033, 2942, 1714, 1658, 1348, 1260, 1180, 1105. Спектр ЯМР 1Н (CDСl3, δ, м.д.): 6,78-6,76 (1Н, s, СН), 5,47-5,44 (1H, t,CH), 5,09-5,02 (2H, m, CH), 4,96-4,92 (2H, m, CH), 4,23-4,16 (2H, m, CH2), 3,18-3,03 (1H, m, CH2), 2,67-2,61 (1H, m, CH2), 1,28-1,24 (3H, t, CH3). Спектр ЯМР 13C ((CD3)2СO, 156 δ, м.д.): 164,3 (C=O) 132,8 (C); 129,8 (CH); 74,4 (CH2); 72,8 (2 CH); 61,6 (CH2); 38,5 (CH3); 30,2 (CH2); 13,9 (CH3). Этил (3S,4R,5R)-3-азидо-4,5-диметилсульфонилоксацикло-гекс-1-ен-1-карбоксилат (4). В смесь 13,8 ммоля 3, 40 мл метанола и 31,2 ммоля хлорида аммония при температуре 0 °С небольшими порциями загружали 20,8 ммоля азида натрия. Реакционную массу выдерживали 4 ч без охлаждения, разбавляли 40 мл толуола и упаривали под вакуумом. Остаток обрабатывали 10 мл воды и 50 мл толуола. Органическую фазу после сушки упаривали под вакуумом. Получали 90 % 4 с содержанием основного вещества 95,1 % (ВЭЖХ). Найдено (%): C 37,25; H 4,58; N 8,93; S 15,05. C11H17N3O8S2. Вычислено (%): C 34,46; H 4,47; N 10,96; S 16,73. ИК спектр, см-1: 2940, 2108, 1717, 1661, 1446, 1358, 1253, 1176, 1075,1016. Спектр ЯМР 1Н (CDСl3, δ, м.д.): 6,67-6,65 (1Н, s, СН), 4,73-4,89 (1Н, m, СН), 4,71-4,65 (1Н, m, СН), 4,32-4,29 (1Н, s, СН), 4,19-4,11 (2Н, m, СН2), 3,14-3,06 (3Н, ss, СН3), 1,25-1,14 (3Н, m, СН3). Спектр ЯМР 13C (CDCl3, δ, м.д.): 164,08 (C=O); 131,9 (CH); 130,1 (C); 78,9 (CH); 73,8 (CH); 61,6 (CH2); 39,2 (CH3); 38,8 (CH3); 30,9 (CH2); 14,0 (CH3). Этиловый эфир (3R,4S,5R)-4-(диэтоксифосфориламино)-3-(1-этил-пропокси)-5метансульфонилокси-циклогекс-1-ено-вой кислоты (21). К 20,0 ммолям 4 в 86 мл толуола дозировали 4,2 мл триэтоксифосфита, выдерживали 3 ч при комнатной температуре и 5 ч при кипении. Реакционную массу упаривали под вакуумом. Получали 20 в виде темного масла. Полученный продукт растворяли в 110 мл 3-пентанола и при температуре 0÷3 °С обрабатывали 3,1 мл эфирата трехфтористого бора. Реакционную массу оставляли на ночь без охлаждения, разбавляли 200 мл этилацетата и промывали 2×380 мл воды. Органический слой после сушки упаривали под вакуумом и обрабатывали небольшим количеством метилтретбутилового эфира (МТБЭ). Получали 56,6 % 21 c Тпл=102-104 °С. Найдено (%): C 45,98; H 7,34; N 2,74; S 6,35. C19H36NO9PS. Вычислено (%): C 47,00; H 7,47; N 2,88; O 29,66; P 6,38; S 6,60. ИК спектр, см-1: 3238, 2978, 2936, 1714, 1460, 1386, 1356, 1264, 1247, 1175, 1136, 1101, 1032, 913, 805. Спектр ЯМР 1H (CDCl3, δ, м.д.): 6,76-6,75 (1H, s, CH), 4,99-4,98 (1H, s, CH), 4,15-4,10 (2H, q, CH2), 4,02-3,96 (5H, m, CH2, CH3), 3,61-3,58 (1H, m, CH), 3,39-3,30 (2H, m, 2CH), 3,05-3,03 (3H, s, CH3), 2,70-2,68 (2H, s, CH2), 2,34 (1H, s, CH), 1,521,38 (4H, m, 2CH2), 1,26-1,19 (9H, q, 3CH3), 0,860,80 (6H, m, 2CH3). Спектр ЯМР 13C (CDCl3, δ, м.д.): 165,4 (C=O); 135,1 (CH); 128,2 (C); 81,2 (CH); 77,7 (CH3); 77,2 (CH2); 73,3 (2 CH); 62,4 (CH2); 60,8 (CH2); 53,3 (CH); 38,0 (CH3); 28,5 (CH2); 25,9 (CH2); 25,5 (CH2);15,9 (CH3); 15,8 (CH3); 9,4 (CH3); 9,1 (CH3). Этил (3S,4S,5R)-4-ацетиламино-3-(1-этилпропокси)-5-метансульфонилокси-циклогекс1-ен-1-карбоксилат (7). Смесь 14,6 ммоль 21, 34 мл этилового спирта, 6,6 мл серной кислоты ПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 ПОИСК ДОСТУПНОГО МЕТОДА СИНТЕЗА ФОСФАТА ОСЕЛЬТАМИВИРА выдерживали 16 часов при 70 °С и разбавляли 34 мл этилацетата и 7 мл воды. Реакционную массу нейтрализовывали 25 % раствором соды до рН=6,5-7,0 и добавляли 1,5 мл уксусного ангидрида. Затем в течение 2 ч постепенно добавляли еще 1 мл уксусного ангидрида, поддерживая содовым раствором рН=6,5-7,0. Массу выдержали 2 часа и разбавили 20 мл этилацетата. Органический слой сушили над Na2SO4 и упарили под вакуумом. Остаток кристаллизовали из смеси МТБЭ – EtOAc (5,5:1) и получали 76,1 % 7 с Тпл=138-139 °С. Найдено (%): C 51,90; H 7,40; N 3,49; S 8,10. C17H29NO7S. Вычислено (%): C 52,16; H 7,47; N 3,58; O 28,61; S 8,19. ИК спектр, см-1: 3306, 2976, 2959, 2878, 1716, 1650, 1537, 1346, 1254, 1177, 1099, 1067, 1018, 920, 798. Спектр ЯМР 1H (CDCl3, δ, м.д.): 6,61 (H, s, CH), 4,99-4,97 (1H, m, CH), 4,90-4,86 (1H, t, CH), 4,82-4,81 (1H, s, CH), 4,52 (2H, s, CH2), 4,23-4,17 (2H, q, CH2), 3,16-3,09 (7H, m, CH3, CH3, CH2), 2,75-2,65 (1H, m, CH), 1,98 (3H, s, CH3), 1,30-1,26 (3H, t, CH3). Спектр ЯМР 13C (CDCl3, δ, м.д.): 171,8 (C=O); 165,0 (C=O); 136,1 (CH); 127,6 (C); 78,2 (2 CH); 75,1 (2 CH);61,2 (CH2); 50,1 (CH3); 48,7 (CH2); 47,8 (CH2); 38,3 (CH3); 38,0 (CH3); 30,0 (CH2); 22,0 (CH3); 13,6 (CH3). Этил (3S,4R,5S)-4-ацет-амидо-5-азидо-3(1-этилпропокси)цик-логекс-1-ен-1-карбоксилат (8). К 7,2 ммоля 7 в 35 мл этанола добавляли раствор 28,0 ммоля азида натрия в 5,5 мл воды и выдерживали 24 ч при кипении. Реакционную смесь упаривали под вакуумом, обрабатывали 35 мл этилацетата и 10 мл воды. Органический слой промывали 7 мл 20 %-ного NaCl и после сушки упаривали под вакуумом. Остаток растворяли при кипении в 14,5 мл МТБЭ и добавляли 14,5 мл гексана. После охлаждения и фильтрации получали 69,9 % 8 с Тпл.=137-138 °С. ИК спектр, см-1: 3270, 2972, 2105, 1716, 1660, 1563, 1375, 1332, 1254, 1079. Спектр ЯМР 1H (CDCl3, 24°C): δ = 7,15-7,13 (1H, d, CH), 6,7 (1H, s, CH), 4,30-4,28 (1H, d, CH), 4,14-4,11 (2H, m, CH2), 3,89-3,86 (1H, m, CH), 3,63-3,60 (1H, q, CH), 3,303,27 (1H, q, CH), 2,78-2,72 (1H, q, CH2), 2,25-2,1 (1H, m, CH2), 1,97 (3H, s, CH3), 1,44-1,38 (4H, m, CH2, CH2), 1,24-1,20 (3H, t, CH3), 0,83-0,78 (6H, m, CH3, CH3). Спектр ЯМР 13C (CDCl3, δ, м.д.): 171,2 (C=O); 165,7 (C=O); 138,1(CH); 127,8 (C); 82,1 (CH); 74,3 (CH); 60,9 (CH2); 58,0 (CH); 56,3 (CH); 30,2 (CH2); 26,0 (CH2); 25,4 (CH2); 23,3 (CH3); 14,0 (CH3); 9,3 (CH3); 9,0 (CH3). Осельтамивира фосфат (10). К смеси 9,5 ммоля 8, 60 мл тетрагидрофурана, 11 мл воды добавляли 9,9 ммоля трифенилфосфина и выдерживали 10 ч при 50 °С. Реакционную массу упаривали под вакуумом, остаток растворяли в 15 мл сухого этанола. Полученный раствор обрабатывали 0,59 г ортофосфорной кислоты в 14 мл этанола при температуре 50-55 °С. Суспензию охлаждали до 10 °С, фильтровали, осадок промывали 4×5 мл ацетона. Получали 81,2 % 10 с Тпл.= 203-204 °С и содержанием основного веПОЛЗУНОВСКИЙ ВЕСТНИК № 4-1 2010 щества 98,8 % (ВЭЖХ). ИК спектр, см-1: 3351, 3160, 2967, 2939, 2878, 1721, 1660, 1552, 1374, 1337, 1294, 1246, 1129, 1069, 1032, 951, 875, 731. Спектр ЯМР 1H (CDCl3, δ, м.д.): 6,82-6,80 (1H, s, CH), 4,29-4,26 (1H, d, CH), 4,24-4,19 (2H, q, CH2), 4,00-4,19 (H, m, CH), 3,63-3,57 (1H, m, CH), 3,423,37 (1H, m, CH), 3,02-2,98 (1H, m, CH2), 2,50-2,44 (1H, m, CH2), 2,20-2,00 (3H, s, CH3), 1,57-1,50 (4H, m, 2CH2), 1,33-1,28 (3H, t, CH3), 0,94-0,86 (6H, m, 2CH3). Спектр ЯМР 13C (CDCl3, δ, м.д.) δ: 174,8 (C=O); 166,5 (C=O); 139,0 (CH); 128,0 (CH); 83,3 (CH); 75,3 (CH); 61,7 (CH2); 53,8 (CH); 49,4 (CH); 29,3 (CH2); 26,6 (CH2); 26,0 (CH2); 23,4 (CH3); 14,3 (CH3); 9,6 (CH3); 9,3 (CH3). СПИСОК ЛИТЕРАТУРЫ 1. Rohloff J.C. et al. // J.Org.Chem.- 1998.- 63.- p.p. 4545 – 4550. 2. Federspiel M. et al. // Org. Process Res. Dev.1999.- 3.- p.p. 266–274. 3. Karpf M., Trussardi R., J. // Org. Chem.- 2001.- 66.p.p. 2044–2051. 4. Harrington P.J., Brown J.D., Foderaro T., Hughes R.C. // Org. Process Res. Dev.- 2004.- 8.- p.p. 86 – 91. 5. Пат. США US2009076296 USA C07C / R. Trussardi– Fiel. Date 18.09.2008; Pub. Date 19.03.2009 11 с. 6. Karpf M., Trussardi R. // Angew. Chem. Int. Ed.2009.- 48.- p.p. 5760–5762. 7. Nie L.D., Shi X.X. // Tetrahedron: Asim.- 2009.- 20.p.p. 124–129. 8. Nie L. D., Shi X.X., Ko K.H., Lu W.D. // J. Org. Chem.- 2009.- 74.- p. 3970. 9. Abrecht S., et al. // Chimia.- 2004.- 58.- p. 621 10. Yeung Y.Y., Hong S., Corey E.J. // J. Am. Chem. Soc.- 2006.- 128.- p. 6310. 11. Okamura H., Kina K., Hamada T., Iwagawa T. // Org. Lett.- 2008.- 10.- p. 815. 12. Mita T., et al. // Org. Lett.- 2007.- 9.- p. 259. 13. Yamatsugu K., Kamijo S., Suto Y., Kanai M., Shibasaki M. // Tetrahedron Lett.- 2007.- 48.- p. 1403. 14. Yamatsugu K., Yin L., Kamijo S., Kimura Y., Kanai M., Shibasaki M. // Angew. Chem., Int. Ed.- 2009.48.- p. 1070. 15. Cong X., Yao Z.J. // J. Org. Chem.- 2006.- 71.- p. 5365. 16. Satoh N., Akiba T., Yokoshima S., Fukuyama T. // Angew. Chem., Int. Ed.- 2007.- 46.- p. 5734. 17. Shie J.J. et al. // J. Am. Chem. Soc.- 2007.- 129.p. 11892. 18. Shie J.J., Fang J.M., Wong C.H. // Angew. Chem., Int. Ed.- 2008.- 47.- p. 5788. 19. Bromfield K.M., et al. // Chem. Commun.- 2007.p. 3183. 20. Matveenko M., Banwell M.G., Willis A.C. // Tetrahedron Lett. 2008.- 49.- p. 7018. 21. Ishikawa H., Suzuki T., Hayashi Y. // Angew. Chem., Int. Ed.- 2009.- 48.- p. 1304. 22. Mandai T.; Oshitari T. // Synlett.- 2009.- p. 783. 23. Eric F.V. Scriven, K.T. // Chemical Reviews.1988.- v. 88.- № 2.- p.p. 58–60. 24. Poch M., et al. // Tetrahedron Letters.- 1991.v.32.- №47.- p.p. 6935-6938. 157