Генетические аномалии человека Ч.2 Автор:
реклама

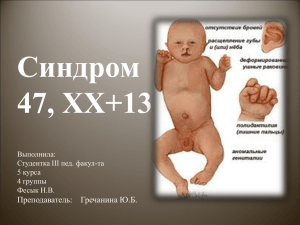
Ч.2 Генетические аномалии человека Автор: ДОРОДНИЦЫНА Лариса Васильевна учитель биологии высшей категории МАОУ «Гимназия №3» г. Саратов Синдром Эдвардса Частота рождаемости - 1:3300(у девочек), 1:10000(у мальчиков) Синдром Эдвардса – хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Причиной синдрома Эдвардса является наличие дополнительной 18-й хромосомы (трех вместо двух в норме для диплоидного набора) в кариотипе зиготы. Дети с синдромом Эдвардса рождаются с низким весом. При этом длительность беременности - нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой. Осложнения.Дети с синдромом Эдвардса умирают в раннем возрасте (90% - до 1 года) от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность). Диагностика.Наиболее значимыми в диагностике синдрома Эдвардса являются изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки развития сердечно-сосудистой системы. Лечение.Способа исправить хромосомные нарушения нет. Профилактика. Предотвратить рождение ребенка с синдромом Эдвардса можно, проведя пренатальную диагностику во время беременности или предимплантационную диагностику до наступления беременности.. Синдром Патау Частота рождаемости – 1:14000 Синдром Патау — тяжелое неизлечимое хромосомное заболевание, поражающее практически все органы. Оно характеризуется наличием в клетках дополнительной хромосомы 13. Трисомия 13 впервые описана Эразмусом Бартолином в 1657. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960. Заболевание названо в его честь. Синдром Патау также был описан для племен с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе. Синдром Патау представляет собой наследственное заболевание, частота его встречаемости среди новорожденных составляет 1 на 5000-7000. Соотношение мальчиков и девочек при данном заболевании составляет 50 на 50%. При синдроме Патау наблюдаются следующие тяжелые врожденные пороки: неправильное развитие костей мозгового и лицевого черепа; со стороны опорно-двигательной системы выявляются неправильное анатомическое строение кистей и стоп, часто встречается многопалость, как на кистях, так и на стопах, чаще симметричная; патология со стороны пищеварительной системы представлена незавершенным поворотом кишечника; патология сердечно-сосудистой системы представлена пороками развития крупных сосудов; пороки развития центральной нервной системы представлены недоразвитием основных мозговых структур, могут отсутствовать или быть сильно недоразвитыми некоторые нервные тракты, может быть отсутствие глазного яблока, помутнение хрусталика; всегда имеют стойкие нарушения интеллекта; Такое количество внутренних пороков быстро развивает многогранную недостаточность. Более 90% детей погибают в течение первого года жизни. Но некоторые доживают до 5 и даже до 10 лет. Продление жизни обеспечивается оперативным устранением пороков развития, тщательным уходом, полноценным питанием. Синдром Клайнфельтера Частота рождаемости 1:1400 В 1942 году доктор Harry Klinefelter собрал 9 случаев клинических наблюдений за пациентами мужского пола, общим у которых был высокий рост, слабое оволосение тела, яички маленького размера, бесплодие, общий женоподобный вид и гинекомастия (см. рисунок). Такой комплекс симптомов получил название синдрома Клайнфельтера (Klinefelter's Syndrome). Это проявление неправильного распределения хромосом, при котором к нормальному мужскому (46,ХY) набору хромосом присоединяется дополнительная Ххромосома (47,ХХY) во всех или в большинстве клеток организма. Причины Это хромосомное нарушение происходит в результате неправильного распределения хромосом со стороны яйцеклетки или сперматозоида во время образования зародышевых клеток. Синдром Клайнфельтера является крайне распространенной патологией - на каждые 500 новорожденных мальчиков приходится 1 ребенок с данным синдромом. Клинические проявления В основе заболевания лежит недостаточная продукция яичками мужского полового гормона тестостерона (гипогонадизм). Поэтому основные клинические проявления и жалобы будут обусловлены именно гипогонадизмом. Синдром Клайнфельтера обычно клинически проявляется лишь после полового созревания и, поэтому, диагноз ставится относительно поздно. Но, тем не менее, при внимательном подходе, на разных этапах полового созревания можно заподозрить этот синдром по наличию ряда характерных признаков. До начала полового развития удается отметить только отдельные физические признаки, такие как длинные ноги, высокая талия, высокий рост. Кроме того, некоторые дети с данным синдромом могут испытывать трудности в учебе и в выражении своих мыслей. С началом полового созревания синдром чаще всего проявляется недоразвитием яичек. Недостаточная продукция тестостерона собственными яичками приводит к задержке или отсутствию полового развития, к отсутствию или скудному оволосению на лице и теле, увеличению грудных желез (гинекомастии). В постпубертатном периоде наиболее частой причиной обращения к врачу пациентов с синдромом Клайнфельтера является бесплодие и нарушение половой функции. В последующем после детального изучения этот симптомокомплекс был расширен следующими признаками: Неполная маскулинизация (недостаточное развитие по мужскому типу) Снижение либидо Остеопороз (разрежение костной ткани) Тауродонтизм (особая патология зубов) Венозная патология Проблемы психического развития Аутоиммунные системные проявления Низкий уровень энергетики Низкая самооценка Трудности общения Эта патология встречается довольно часто и обусловлена патологией хромосом – у этих пациентов одна лишняя X-хромосома (в норме у мужчин одна X и одна Y хромосома, тогда как у этих пациентов две X-хромосомы). Хромосомная формула у таких людей может быть представлена как XXY. Диагностика Для постановки диагноза необходимым является гормональный анализ крови (определение уровня гонадотропинов), исследование хромосомного набора (кариотипа), подтверждающее наличие дополнительной Х-хромосомы. Лечение Единственным методом лечения является назначение заместительной терапии препаратами тестостерона. Данная терапия у таких пациентов проводится пожизненно. Начинать заместительную терапию необходимо как можно раньше, чтобы предотвратить появление симптомов и тяжелых последствий недостатка тестостерона таких, например, как остеопороз. Гормональная терапия устраняет все клинические проявления гипогонадизма, кроме бесплодия. Синдром Шерешевского – Тернера. Частота рождаемости – 1:3000 Для синдрома характерно отсутствие в кариотипе половой Х-хромосомы. Частота встречаемости 1:3000, среди девочек, страдающих олигофренией – 1:1500. Частота синдрома возрастает среди низкорослых женщин с недоразвитием вторичных половых признаков и аменореей. Большинство больных с синдромом Шерешевского–Тернера имеют нормальный или близкий к норме интеллект, но умственная отсталость у них встречается чаще, чем в общей популяции. Интеллектуальные нарушения обычно сочетаются с недоразвитием эмоционально-волевой сферы: больные повышенно внушаемы, несколько некритичны, упрямы, часто эйфоричны. Диагностика синдрома возможна уже в период новорожденности: Девочки рождаются с низкой массой тела и небольшого роста; Отмечается отечность кистей и стоп; Низкий рост волос на шее, шея короткая с крыловидными складками, идущими от сосцевидных отростков к плечам; Характерна чрезмерная подвижность кожи на шее. Отмечаются множественные аномалии развития: Эпикант, антимонголоидный разрез глаз; Низко расположенные ушные раковины; Гипомимия («лицо сфинкса»); Микроретрогнатия, высокое небо, аномалии зубов. Важными диагностическими признаками являются также врожденные пороки сердца, низкий рост (в 98% случаев), половой инфантилизм с первичной аминореей, часты гипоплазия или гипертрофия ногтевых пластинок, гиперпигментация кожи. Наблюдаются дефекты зрения (22%) и слуха (52%) Характерны разнообразные скелетные нарушения «Щитообразная» широкая грудная клетка; Гипоплазия или сращение I и II шейных позвонков; Широкие кисти с короткими IV и V пальцами; Синдром Джакобс (синдром дубль-Y). Частота рождаемости - 1:1000 Высокорослость, умственная отсталость легкой степени, позднее развитие речи, повышенная агрессивность, иногда бесплодие. Заболевание ращвивется в результате геномного нарушения, когда в кариотипе присутствует одна лишняя половя хромосом –Y(то есть генотип по половым хромосомам - XYY). Трисомия по X-хромосоме. Частота рождаемости - 1:1000 Высокорослость, умственная отсталость легкой степени, позднее развитие речи, эпилепсия, иногда бесплодие. Заболевание развивается в результате геномного нарушения, когда в кариотипе присутствует одна лишняя половая хромосома X (XXX в классическом варианте, но встречаются и более редкие кариотипы: XXXX, XXXXX). Используемые источники информации: http://www.vospitanie-detey.ru/geneticheskiezabolevaniya/sindrom-laquo-koshachego-krikaraquo.html http://www.vospitanie-detey.ru/geneticheskiezabolevaniya/sindrom-laquo-koshachego-krikaraquo.html http://www.krugosvet.ru/enc/medicina/DAUNA_SIND ROM.html http://downsyndrome.at.ua/publ/12-1-0-120 http://www.pedlib.ru/Books/4/0472/4_047210.shtmhtml http://www.rost507.ru/Biologiy/11kl/GenAnam.gifр http://www.nevromed.ru/content/part_1/nekotorie_g eneticheskie_bolezni/