h = σ
реклама

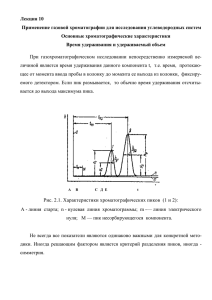
МФ-2. Методические рекомендации к лабораторной работе 13(Хроматография»). Качественный и количественный анализ смеси алканов методом газовой хроматографии Хроматография широко применяется в медицине и фармации. Её используют в медико-биологических исследованиях, в клинической практике для диагностики различных заболеваний, при анализе сложных лекарственных препаратов, для определения чистоты лекарств и изучения их метаболизма в организме. Препаративную хроматографию используют при производстве лекарств. В основе хроматографии лежит явление адсорбции, а именно различная адсорбционная способность веществ. По технике эксперимента различают бумажную, тонкослойную, колоночную и капиллярную хроматографию. По характеру фаз различают газовую и жидкостную хроматографию. Хроматография - физико-химический метод разделения и анализа сложных смесей, при котором компоненты смеси распределяются между двумя фазами: неподвижной, с большой поверхностью контакта и подвижной в виде потока газа или жидкости, проходящего через неподвижный слой. Анализируемые вещества растворены в подвижной фазе. Перемещаясь вдоль неподвижной фазы, помещенной в трубку, которая называется хроматографической колонкой, они взаимодействуют с ней и поэтому скорость их движения изменяется. Чем больше время нахождения молекул данного вещества в сорбированном состоянии, тем меньше их скорость движения вдоль колонки с потоком газа-носителя в случае газовой хроматографии. Невзаимодействующее с неподвижной фазой вещество выходит быстрее (время выхода t0 зависит от длины колонки и плотности набивки хроматографической фазы). Результаты хроматографического разделения смеси веществ регистрируются в виде хроматограммы, которая представляет зависимость сигнала детектора, пропорционального концентрации компонента смеси в газе-носителе, от времени. Хроматограмма показывает последовательность выхода компонентов и их количества, пропорциональные площади пиков S (рис.1). Стрелкой обозначен момент ввода пробы в поток газа-носителя у входа в колонку. Вещество А не сорбируется поверхностью стационарной фазы. Рис.1. Хроматограмма смеси трёх веществ и определение площади пика S h Принципиальная схема газового хроматографа показана на рис.2. Пробу вводят шприцем или через дозатор 2 в поток газа-носителя, который поступает в колонку 1, в который компоненты пробы распределяются вдоль слоя сорбента. Детектор 3 фиксирует концентрации выходящих из колонки компонентов в потоке газа-носителя, его сигнал регистрируется самописцем или дисплеем 6. Рис. 2. Схема газового хроматографа. 1 - хроматографическая колонка; 2 - дозатор; 3 - детектор; 4 - баллон с газом-носителем; 5а вентиль тонкой регулировки; 5б - манометр; 5в - пенометр; 6 -регистрация; 7 - термостат. Качественный анализ. Основными характеристиками являются время удерживания вещества tu и удерживаемый объем V R . Время удерживания ( t u ) - время от момента ввода пробы до момента регистрации максимума пика на хроматограмме (см. рис.1). Оно складывается из времени движения вещества с газом-носителем t o вдоль колонки и суммарного времени нахождения в сорбированном состоянии t R (исправленное время удерживания). tu to t R (1) , VR t R w (2) Заметим, что t o одинаково для всех веществ смеси и определяется как время выхода неадсорбирующегося газа, например, воздуха, тогда как t R при постоянных условиях опыта (температуре и скорости газа-носителя) зависит от природы анализируемого вещества и неподвижной фазы. Компоненты смеси, различающиеся по энергии взаимодействия с данной неподвижной фазой, будут иметь различные значения t R . Удерживаемый объем V R - это объем газа-носителя, прошедшего через колонку за время удерживания t R . Его значение рассчитывают по формуле (2), где w объемная скорость газа-носителя (объем газа, проходящий через колонку за единицу времени). Величина V R зависит от объёма неподвижной фазы в колонке Va и Са ( Ca концентрация С вещества в неподвижной фазе, C в газе-носителе. следовательно, V R . распределения вещества между двумя фазами VR Va Удерживаемый объём определяется температурой и природой вещества, являясь более воспроизводимой характеристикой вещества по сравнению с t u и t R . Для веществ одного гомологического ряда график зависимости lnVR от числа атомов углерода в молекуле nc обычно представляет собой прямую линию (рис.3). Наклон прямой определяется природой анализируемых веществ и неподвижной фазы, а также температурой колонки. Рис.3. Зависимость логарифма удерживаемого объема от числа атомов углерода в молекуле для различных гомологических рядов: 1 - алканы; 2 - спирты; 3 - алкилацетаты. Линейная зависимость lnVR от числа атомов углерода н-алканов используется для качественного анализа веществ, принадлежащих к разным классам. Н-алканам приписываются значения индексов, равные числу углеродных атомов в молекуле, умноженному на 100 (индексы Ковача I ). Например, для метана I =100, для пропана I =300, для декана I =1000 и т.д. Индекс Ковача I x любого другого вещества для данной неподвижной фазы при заданной температуре колонки имеет промежуточное значение между значениями индексов Ковача н-алканов. Количественный анализ Площадь пика i-го компонента S i на хроматограмме пропорциональна его количеству qi в смеси, введенной в колонку qi K i Si , где K i - поправочный коэффициент, зависящий от природы вещества, который учитывает разную чувствительность детектора по отношению к компонентам смеси. Видно, что на хроматограмме эквимолярной смеси пики веществ, взятых в равных количествах, отличаются по площади. Зная K i можно рассчитать долю i-го компонента смеси x i по формулам (5) или (6), в которых суммирование производится по всем пикам xi K i S i / K i S i i (3) , ( Ki / K st ) Si Ki* Si xi ( Ki / K st )Si Ki*Si i i (4). Широко используется метод внутреннего стандарта, в котором определяются относительные поправочные коэффициенты Ki* Ki / K st . Для определения K i* составляют смесь известного состава (например, эквимолярную смесь) анализируемых веществ, включая стандарт. Из хроматограммы такой смеси * определяют площади пиков S и рассчитывают величины K i для всех веществ. С * помощью мольных коэффициентов K i определяют мольные доли, а с помощью весовых - весовые доли веществ в смеси. Порядок работы. 1. Проверить линейность зависимости lnVR от числа атомов н-алканов С6-С9, 2. Провести качественный и количественный анализ контрольной смеси углеводородов. Условия опыта: Хроматограф ЛХМ-80. Сорбент хроматографической колонки - Апьезон, нанесенный на Хроматон. Газ-носитель - гелий. Температура колонки- 125о С. Детектор - катарометр. Ток моста детектора 60 mA. Температура детектора- 150о С. Температура испарителя- 150о С. 1.Рассчитать объёмную скорость газа-носителя в колонке при 125 оС по формуле (5) с учетом того, что измерение потока газа производится при 25 оС wcor w Tkol 398 w Tk 298 (5), здесь Tkol и Tk - температуры колонки и пенометра, 0К. 2.Определить время выхода t u (мин.) веществ эквимолярной смеси по расстоянию от ввода пробы до максимума пика l см, пользуясь масштабом. Рассчитать время удерживания tR ( to 0,5мин. ), V R и ln VR . Данные записать в табл.1. Построить график зависимости lnVR от nc . Убедиться в линейности этой зависимости. 3.Определить площади пиков эквимолярной смеси и рассчитать относительные коэффициенты Ki* Sst / Si , взяв за стандарт гексан. Данные записать в табл.2. Качественный состав смеси определяется по совпадению времен удерживания компонентов эквимолярной смеси и контрольной смеси. Количественный состав смеси углеводородов в мольных процентах проводится на основании определения площади пиков методом треугольника S h , где h высота пика, а - ширина его на половине высоты. Отсчет высот пиков производится от положения базовой («нулевой») линии. Все расчёты, а также выводы записываются в тетради. Таблица 1. Времена выхода t u и удерживаемые объемы н-алканов эквимолярной смеси вещество l см nc tu tR мин. VR мин. VR lnVR гексан гептан октан нонан Таблица 2. Определение коэффициентов чувствительности и состава контрольной смеси. Эквимолярная смесь вещество гексан гептан октан нонан Выводы. h см см Si см2 Контрольная смесь K i* 1 h см см Si см2 xi