Модуль 2
реклама

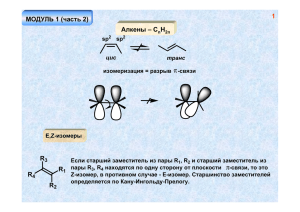
МОДУЛЬ 2 Алкены – СnH2n E,Z-изомеры R3 R4 R2 Если старший заместитель из пары R1, R2 и старший заместитель из R1 пары R3, R4 находятся по одну сторону от плоскости -связи, то это Z-изомер, в противном случае - Е-изомер. Старшинство заместителей определяется по Кану-Ингольду-Прелогу. 1 2 Методы синтеза алкенов основание NaOEt + Cl OEt 2 1 1 - продукт элиминирования 2 - продукт замещения Увеличение силы основания и повышение температуры реакции увеличивает выход 1 за счет снижения выхода 2 NaOH Cl EtOH > > Правило Зайцева: В реакции элиминирования в большей степени образуется более замещенный алкен Транс-изомера обычно образуется больше, чем цис-изомера вследствие большей термодинамической выгодности первого. 3 НО! Для протекания реакции элиминирования, как правило, необходима такая конформация молекулы, в которой уходящие группы находятся в одной плоскости и связи направлены в противоположные стороны (антиперипланарное расположение). H Br H Ph H Br H H H Ph NaOEt Ph Ph Br диастереомеры Br Br H H H Ph H NaOEt H H Ph Ph Br Ph диастереомеры дают разные продукты! (конфигурационный контроль реакции) 4 Правило Гофмана: При термолизе четвертичных аммонийных оснований преимущественно образуются менее замещенные алкены. t N > -N(CH3)3 HO- 2. Дегидратация спиртов OSO3H t H2SO4 I2 t OH KHSO4, t (Al2O3, 400o) > H+ t > 5 + H < OH + - H+ H+ OH2 - H2O A B A, B - карбокатионы, В - устойчивее, чем А Превращение А в В – катионоидная перегруппировка, происходит всегда, когда существует возможность образования более устойчивого катиона из менее устойчивого! Ряд устойчивости карбокатионов: третичный > вторичного >> первичного Причина устойчивости алкильных карбокатионов – гиперконьюгация – сопряжение связывающей орбитали сигма-связи С-Н с вакантной р-орбиталью, приводящее к уменьшению величины заряда на катионном центре за счет перераспределения заряда на атомы водорода (делокализация заряда). 6 Гиперконьюгация 7 Другой, еще более эффективный способ стабилизации карбокатионного центра – р- -сопряжение и р-n-сопряжение (донорный мезомерный эффект заместителя) р- сопряжение -система как мезомерный донор электронов H2 C H C CH2 H2C H C CH2 аллильный катион CH2 CH2 CH2 бензильный катион CH2 8 p-n-сопряжение гетероатом с неподеленной парой электронов - сильный мезомерный донор O O A B более значимая структура В - у каждого атома октет электронов Перегруппировки карбокатионов OH2 H H CH2 CH3 - H2O H H H А миграция атома водорода с парой электронов, образующих связь С-Н, к карбокатионному центру - гидридный сдвиг Первичные алкильные катионы (например, А) настолько неустойчивы, что не образуются вовсе. В данном случае гидридный сдвиг происходит одновременно с отщеплением молекулы воды. CH3 OH2 H H H синхронный процесс миграция метильной группы OH H+ -H+ - H2O вторичный катион третичный катион > 9 10 OH H+ этот процесс не идет неустойчивый первичный катион! - H2O H+ OH2 - H+ H миграция связи С-С гибридизация sp2, валентный угол ~ 90o гидридный сдвиг этот процесс не идет неустойчивый, хотя и третичный катион! 11 3. Дегалоидирование Zn Cl или Mg цис-элиминирование (син-элиминирование) Cl H H Cl Cl мезоформа H Zn Zn H Cl Cl d,l-форма 12 4. Восстановление алкинов H2 Pd/BaSO2 "отравленный катализатор" цис-изомер Na NH3 (ж.) 2NH3 +e -2NH2более устойчивый транс-дианион транс-изомер 5. Крекинг алканов (смесь продуктов) CnH2n+2 500-700o алюмосиликаты алканы + алкены 13 Свойства алкенов Электрофил – частица, способная принять пару электронов (кислота Льюиса) Нуклеофил – частица, способная отдать пару электронов (основание Льюиса) 14 Возможность взаимодействия алкена с электрофилом 15 Невозможность взаимодействия алкена с электрофилом (запрет по симметрии) 16 Реакции электрофильного присоединения а. Галогеноводороды X HX H продукт присоединения (по правилу Марковникова) XH+ H+ H H вторичный катион неустойчивый первичный катион (не образуется) Правило Марковникова: Электрофильное присоединение к алкенам происходит через стадию образования более устойчивого катиона 17 E переходное состояние (активированный комплекс) промежуточный продукт (катион) энергия активации продукт присоединения координата реакции Первая стадия – присоединение протона, является лимитирующей, поэтому реакционная способность изменяется в следующем ряду: HI > HBr > HCl > HF Более замещенная кратная связь (большее число донорных алкильных заместителей) более реакционноспособна по отношению к электрофилу, поскольку, к тому же, образует более устойчивый (более замещенный) катион. < < < < вторичный катион третичный катион < 18 Серная кислота H2SO4 OSO3H HSO4- кислый алкилсульфат вода + - H OH - + спирты RO H с алкенами не реагируют (нет протонов) Реакция происходит в присутствии сильных кислот, например, серной R ROH H+ ROH O R = Н, алкил Н+ - катализатор (не расходуется!) OR H - H+ 19 б. Галогены (Br2, Cl2) Br Br2 H H Br2 H Br H -комплекс H Br H H Br H H Br H -Brбромониевый катион поляризация связи Br-Br H Br большой размер атома брома H позволяет ему эффективно координироваться с обоими атомами углерода бромониевый катион 20 Стереоспецифическое электрофильное транс-присоединение брома к алкену (цис-бутену-2) H Br H Br Br H H H H Br Br - Br H H Br энантиомеры, образуются в равных количествах (рацемат) H Brнуклеофильная атака равновероятна по двум направлениям Br H H Br H Br d,l-форма Br 21 Br Br2 Br HH H Br H H H Br Br - Br H Br Br H H H Br H H Br Br- нуклеофильная атака равновероятна по двум направлениям Br мезоформа 22 Br Br2 Br + Br Br2 H2O Br d,l-форма (рацемат) H2O OH OH + Br -H + Br Br d,l-форма (рацемат), трео-форма Аналогично происходит реакция с хлором. С иодом алкены не реагируют, реакция с фтором происходит со взрывом, продукт электрофильного присоединения выделить невозможно 23 в. Другие электрофилы типа А+Б- H Br CN HO Br NO Cl H - + - + + + - H A B + - Hg(OCOCH ) 3 2 I N3 A A A H H -B B B рацемат (трео-форма) B- H + - A A B -B- H + - H H H A A H H H + H H B- H B B рацемат (эритро-форма) 24 В случае несимметричных алкенов присоединение происходит по правилу Марковникова + - A B - -B A B A A B- A 1 A 2 Структура 1 более значима,чем 2 положительный заряд на вторичном атоме углерода. Поэтому присоединение нуклеофила идет по этому положению. Ацетоксимеркурирование CH3 CO2 H = AcOH Hg(OAc)2 HgOAc HgOAc + AcOH H3O+ OAc AcOH -H 1.NaBH4 OH 2. HO- 25 Hg(OAc)2 OCH3 NaBH4 OCH3 HgOAc CH3OH CH3OH H+ Преимущества ацетоксимеркурирования по сравнению с кислотно-катализирумым присоединением воды (спиртов) – реакция происходит по тому же направлению, но без образования карбокатионов, способных, в частности, перегруппировываться, и вступать в другие реакции. Реакция происходит стереоселективно. 26 г. Взаимодействие алкенов с алканами H+ H + + H+ = AlCl3, BF3, H2SO4, HF и т.д. д. Реакция Принса H OH OH H2O H OH H+ CH2O CH2O OH - H+ H3O+ OCH2 OH O O устойчивый катион! 27 е. Синтез Реппе H CO, H2 H2C CH2 Co2+, t, p H O O Реакции окисления Цис-гидроксилирование CH2 KMnO4 CH2 HO- CH2 O O Mn CH2 O- O H2C H2C O O HO Mn O- - H2O O MnO2 KMnO4 HOOH OH цис-гликоль (мезоформа) OH OH гликоль HO H H OH OsO4 H H2O2 28 H H OH H OH цис-гидроксилирование (направление атаки реагента равновероятно "сверху и снизу") R2 + R1CO2H O R3 R2 KMnO4 R1 + H d,l-форма (рацемат) OsO4 R3 H2O2, NaIO4 Cr6+, H+ Эпоксидирование O2 / Ag или RCO3H R2 O O эпоксид (оксиран) + O R1 H R3 29 транс -гидроксилирование OH O RCO3H H3O+ OH HCO3H d,l-форма (HCO2H + H2O2 / H2O) Озонолиз R2 R1 O3 O O O R3 R1 O O R3 R2 O R2 O R1 R3 O R3 O озонид R1 мольозонид Zn H2O H2O H2O2 O O R1CHO + R 2 R2 R3 восстановительное расщепление R1CO2H + R 2 окислительное расщепление R3 30 Реакции восстановления 1. Каталитическое гидрирование а. Гетерогенный катализ H2 RCH=CHR1 катализатор RCH2CH2R1 катализатор = Pt, Pd, Ni-Ренея H2 цис-присоединение, мезоформа Pt б. Гомогенный катализ RhCl3 + PPh3 (Ph3P)3RhCl катализатор Уилкинсона условия гидрирования - комнатная температура, атмосферное давление цис-присоединение скорость гидрирования: менее замещенные алкены селективно гидрируются быстрее более замещенных 31 в. Диимид N2H4 R1 R H2O2 / Cu2+ или никель Ренея R1 R диимид (NH)2 N2H4 или никель Ренея H H R1 R1 R R цис-присоединение мезоформа 32 Герберт Браун, нобелевская премия, 1950 г. г. Гидроборирование NaBH4 BF3.OEt2 B2H6 BH3 присоединение воды "против правила Марковникова" H2O2 H - B BH + RCO2H 3B BH2 триалкилборан + B(OCOR)3 O H HO- 2 BH3 3 OH O R H цис-присоединение "водорода" 33 Радикальные реакции алкенов Br BrH BrH R Br только HBr! ROOH RO ROOH RO + HBr Br• C• H Br ROH + Br• цепной радикальный процесс BrH Br - Br•- вторичный радикал CCl4 RO Cl CCl3 34 Радикальное замещение по аллильному положению Cl2 Cl 300o Cl N O O аллильное хлорирование цепной радикальный процесс N-хлорсукцинимид t Cl2 Cl 2Cl Cl Cl2 Cl -HCl CH2 аллильный радикал -Cl Cl хлористый аллил SR 35 O2 RSH OOH RO NH3, O2 катализатор CN акрилонитрил окислительный аммонолиз Изомеризация алкенов 500o Al2O3 более замещенные алкены более устойчивы h транс -изомер более устойчив 36 Полимеризация алкенов 1. Гетеролитическая (катионная и анионнная) катионная полимеризация H+ n полиизобутилен Полимеризация идет «голова к хвосту» 37 анионная полимеризация Bu CN Bu CN CN Bu CN устойчивый анион + Li Bu C N Bu n CN CN CN делокализация заряда 38 2. Радикальная полимеризация H2C CH2 H2 H2 C C F2C CF2 F2 F2 C C H2 H C C Cl полиэтилен n n n тефлон поливинилхлорид Cl инициаторы полимеризации - источник радикалов: O NC O Ph Ph O перекись бензоила N N O CN азобисизобутиронитрил 39 3. Координационная полимеризация AlEt3 + TiCl4 + H+ HML3 катализаторы Циглера-Натта (нобелевская премия, 1963) HML3 - ML3 n + L3M ML3 n n атактические полимеры изотактические полимеры синдиотактические полимеры n 40 Качественные реакции (алканы, циклоалканы, алкены) нет эффекта Br2 / H2O (бромная вода) обесцвечивание (медленно) обесцвечивание (быстро) нет эффекта KMnO4 / HO-, H2O нет эффекта обесцвечивание, выпадение бурого осадка MnO2 41 Диены H2C сопряженный диен изолированный диен C CH2 кумулированный диен Методы получения а. 1,3-диены 1. Синтез бутадиена по Лебедеву EtOH MgO-ZnO 400-500o 2. Синтез Реппе H2 CH2O Cu+ HO OH Ni OH H3PO4 (CH2)4 t OH NaPO3 O t тетрагидрофуран (ТГФ) 42 3. Реакция Принса O OCH2+ CH2O -H+ H+ O OH 2 4. Cu+ H2 NH4Cl Pd/BaSO4 H+ t Al2O3/Cr2O3/CuO t 5. Дегидрирование Аллен – синтез и гидратация Br Br2 Br Br2 Br EtO- Br Br 300o Br CH2 аллен CH2 OH3+ C+ CH3 H2O более устойчивый катион!!! H C+ HH O Zn 43 Сопряженные диены (1,3-диены) сопряжение затрудняет вращение вокруг одинарной связи С-С s-транс-изомер s-цис-изомер 44 Электрофильное присоединение к сопряженным диенам E E+ E+ E E аллильный катион не образуется Nu- Nu- E E Nu Nu 1,2-присоединение 1,4-присоединение энергия активации обратной реакции энергия активации прямой реакции исходные соединения продукты термодинамического контроля (1,4-присоединение) продукты кинетического контроля (1,2-присоединение) 45 Присоединение брома Br Br Br 80% Br Br Br2 Br2 t < 0o t = 40o 20% Br Br Br 20% 80% 40o Присоединение HBr HBr Б Br HBr катион А устойчивее, чем Б! А Br - кинетический контроль BrBr термодинамический контроль 46 Радикальное присоединение происходит преимущественно по типу 1,4-присоединения (термодинамический контроль) Восстановление H2 Pd восстановление только одной связи невозможно (результат сопряжения - изолированная кратная связь восстанавливается легче сопряженной) Na NH3 (ж.) транс-изомера больше, чем цис-, изолированные кратные связи в этих условиях не восстанавливаются Синхронные процессы. Реакция Дильса-Альдера Отто Дильс Курт Альдер Нобелевская премия, 1950 г. + диен диенофил 47 Процесс контролируется орбитальной симметрией 48 Невозможность димеризации этилена реакция не происходит закрепленная s-транс-конформация диена Синхронные процессы происходят с высокой стереоселективностью d,l-форма мезоформа В случае взаимодействия ВЗМО диена с НСМО алкена (наиболее распространенный вариант), донорные заместители в диене и акцепторные в алкене ускоряют реакцию. В случае акцепторно-замещенного диена и донорно-замещенного алкена реализуется взаимодействие НСМО диен и ВЗМО алкена. В реакцию могут вступать гетеродиены и гетеродиенофилы. При этом гетеродиенофилы менее реакционноспособны, чем обычные алкены: + O O O O диен O + O диенофил N + N N N + C CN 49 50 Алкины могут выступать в качестве диенофилов; диины и енины не могут выступать в качестве диенов (не достигается требуемая для реакции s-цис-конформация) O O + не могут выступать в роли диена Полимеризация диенов (анионная, радикальная, координационная) Радикальная полимеризация R R R R изопрен R менее устойчив, не образуется R более устойчивый радикал 51 R + R R Z-изомер - гуттаперча n Е-изомер - каучук (катализатор Циглера-Натта) 52 Вулканизация Sn Sn n S8 n Sn n Sn n резина, эбонит Анионная полимеризация + Na Ph стирол n Ph бутадиен-стирольный каучук ("буна-S") Терпены 53 + лимонен O OH карен пинен ментол камфора 54 Алкины CnH2n-2 Методы получения 1. Гидролиз карбидов CaC2 H2O HC 2. Крекинг CH Mg2C3 O2 CH4 + H2O 1400 o O2 C2H6 1200 H2O + H2O o 3. Синтез из элементов (Бертло) C + H2 äóãà 4. Из алкенов X2 R1CH=CHR2 EtOR1CH-CHR2 X X t R1 R2 + диен 55 5. Из дигалогенидов X X EtO-, t R1 (NaNH2) R1 Свойства 1. СН-кислотность (ацетилениды) CH3MgI HC C MgI HC HC - CH4 CH + NaOH CH HC NaNH2 -NH3 HC C Na C Na + H2O кислотность: CH4 << NH3 < HC CH << H2O Реактив Толленса – качественная реакция на терминальные алкины [Cu+] HC C Cu R CH Ag(NH3)2OH HC C Ag 56 Реакции ацетиленидов как метод построения углеводородного скелета HC C RX R CH NaNH2 HC C R1X R1 R O 1. 2. H2O R R 1 R RX, RX1 - первичные (вторичные) алкилгалогениды HO реакция Фаворского ряд активности: RI > RBr > RCl Реакции электрофильного присоединения X H R R1 CH R CH HX R CH2 винильный катион XR CH2 Низкая устойчивость винильного катиона – результат отсутствия факторов, стабилизирующих его вакантная sp2-гибридная орбиталь вакантная р-орбиталь 120o R R sp sp2 X X R CH HX H+ - X R CH2 малоустойчивый катион HX = галогеноводород R R CH3 X XR CH2 X X R CH3 CH3 устойчивый катион 57 58 Низкая устойчивость винильного катиона - причина меньшей реакционной способности алкинов по сравнению с алкенами к электрофильному присоединению Следовательно: а. Первая стадия присоединения НХ к алкинам происходит медленнее, чем вторая. б. При наличии в составе молекулы несопряженных двойной и тройной углерод-углеродных связей электрофильное присоединение происходит в первую очередь по двойной связи. HX (1моль) X X HX (1моль) образование сопряженной диеновой системы Присоединение HBr против правила Марковникова HBr R R R Br 59 R R Br2 Br R Br Br Br2 R R Br R Br Br Реакция Кучерова (синтез кетонов) OH H3 O+ R Hg2+ R O R присоединение воды по правилу Марковникова Присоединение воды «против правила Марковникова (синтез альдегидов) HBR2 R1 R1 R= BR2 H2O2 HO- O OH R1 R1 60 Другие электрофилы R1CO2H OCOCH3 OCOCH3 2+ Hg OH H2O HO- n виниловый эфир SR n поливинилацетат SR RSH RSH BF3 R AsCl3 Cl AsCl2 поливиниловый спирт HCN CuCl2/NH3 люизит ROt,p CN акрилонитрил CO + EtOH Ni(CO)4 OEt OR единственый пример нуклеофильного присоединения к алкинам (виниловые эфиры) O Реппе этилакрилат 61 Реакции восстановления Na H2 Pd/BaSO2 NH3 (ж.) транс-изомер H2 Pt H2 Ni/ZnCl2 цис-изомер 1. HBR2 2. RCO2H «Полимеризация» 3 C 400o Ph3PNi(CO)2 мезитилен 62 циклооктатетраен 4 Ni(CN)2 AlCl3 гексаметилбензол Дьюара AlEt3+TiCl4 2 R Cu+ NH4Cl R R Cu+ NH3/O2 R R H2 Pd/BaSO4 R R 63 R Cu+ R C C Cu R NaOCl R1 0o R C C Cl R1 Ходкевич Ацетилен-алленовая перегруппировка R EtONa, t или NaNH2 (0.1 M), t R C NaNH2 (изб.) t В обоих направлениях реакция идет в условиях термодинамического контроля интернальный алкен термодинамически выгоднее терминального, солеобразование же сдвигает равновесие в сторону соли - ацетиленида! R 64 Ароматические соединения 1825 г. Фарадей открыл бензол 1865 г. Кекуле предложил современную структуру бензола H2 3 C2H4 3 C2H6 + Q1 Pt H2 + Q2 Pt Q1 - Q2 = 36.5 ккал/моль - энергия сопряжения 1. O3 2. Zn/H2O O 2 O O O O 1. O3 2. Zn/H2O + O O + 2 O 65 2 1. O3 2. Zn/H2O O O O + 2 O O + 3 O Молекулярные орбитали бензола 66 67 Правило Хюкеля: Ароматическими являются циклические, плоские, сопряженные - системы, содержащие 4n+2 электрона. Циклические, плоские, сопряженные - системы, содержащие 4n электрона, называются антиароматическими. Бензол – ароматическое соединение с n = 1 (6 -электронов) H H 10 -электронов неароматическое соединение (не может быть плоским!) 10 -электронов ароматическое соединение (гомоароматика) Циклобутадиен - плоская сопряженная система с 4 -электронами антиароматическая структура (бирадикал) 68 Br EtO- O O O EtO- 2 C2H2 -240 oC h -263 oC аргон O А высокий дипольный момент (структура А - ароматическая) Ph Ph Ph OH Ph HClO4 - H2O Ph ClO4Ph 69 - H+ циклопентадиен неароматическое соединение O циклопентадиенильный анион - ароматическое соединение (6 -электронов) O тропон - высокий дипольный момент плоский циклопентадиенильный катион - антиароматическое соединение (4 -электронов) 70 _ + азулен - высокий дипольный момент циклооктатетраен - был бы антиароматическим, если бы был плоским, но он не плоский, устойчивое соединение, неароматика 2+ 6 -электронов - 2e + 2e 2- 10 -электронов 71 Гетероароматичность Полициклические ароматические соединения нафталин антрацен фенантрен 72 бензо[b]пирен гелицен 24 -электрона (4n), но ароматическое соединение - две независимые -системы аннулен[14] аннулен[18] 73 Методы получения 1. Выделение из каменноугольной смолы после коксования угля 2. Прямая перегонка нефти или крекинг нефти (нефтепродуктов) Pt, 300o или Cr2O3/Al2O3, t 3. Синтез из алкинов C2H2 R C 400o R (PPh3)2Ni(CO)2 R R Реппе R R R R R AlEt3/TiCl4 Циглер R R R 74 R O Pt H2SO4 + R R R Реакции ароматического электрофильного замещения E+ E+ E+ -комплекс H E H E -комплекс E H H E Nu H Nu- E + H E -H+ E -комплекс E E промежуточный продукт ( -комплекс) -комплекс -комплес координата реакции 75 76 Протонирование (дейтерообмен) D2SO4 PhH C6D6 PhD реакция неселективная Галоидирование AlCl3 Cl2 Cl Cl Cl+ AlCl4 - AlCl3 электрофил H Cl Cl+ PhH - AlCl4- Fe -HAlCl4 HCl + AlCl3 катализатор (не расходуется, берут HAlCl4 PhH PhCl 0.1 М) X2 Fe X2 PhX X = Cl, Br FeX3 катализатор (кислота Льюиса) PhH I2/HNO3 или ICl PhI 77 Алкилирование (реакция Фриделя-Крафтса) PhH CH3Cl PhCH3 AlCl3 + H3C Cl толуол AlCl3 катализатор, используется 0.1 М AlCl3 электрофил Cl PhH H AlCl3 H Cl AlCl3 - AlCl4- Катион (электрофил) можно генерировать и по-другому: OH H+ H+ или OH лимитирующая стадия OH PhH Ph H+ H3O+ t Реакция обратима лимитирующая стадия - распад -комплекса Кинетический изотопный эффект H C6H6 E+ - H+ D C6D6 E PhE E KH/KD <1 E+ -D + C6D5E лимитирующая стадия распад -комплекса 78 79 Ацилирование (реакция Фриделя-Крафтса) O AlCl3 RCOCl R H PhH Cl AlCl3 R электрофил O RCOCl PhCOR HAlCl4 O O Ph R PhCOR HCl + AlCl3 O O AlCl3 R H2O AlCl3 катализатор, но его требуется 1 моль O O AlCl3 AlCl3 -HAlCl4 H HCOCl не существует! R AlCl4- COR AlCl3 R O R O AlCl3 R R RCO2AlCl3электрофил При ацилировании ангидридами карбоновых кислот 1 моль AlCl3 расходуется на образование электрофила, и ещё один моль связывается в комплекс с продуктом реакции - кетоном. Общий расход катализатора - 2 моля. 80 PhH RCO2H PhCOR HF PhCOR Zn/Hg H3O+ O O PhH POCl3, PCl3, ZnCl2 PhCH2R Клеменсен O RCO2H 1. N2H4 PhCOR 2. HO- синтез н-алкилбензолов Кижнер-Вольф O O Zn/Hg PhH AlCl3 Ph CO2-AlCl2 H3O+ O Zn/Hg H3O+ Pt, t HF Ph CO2H 81 Синтез ароматических альдегидов (реакция формилирования) Гаттерман CO, HCl PhH AlCl3, CuCl PhCHO CO + HCl AlCl3 [HCOCl] Гаттерман-Кох PhH HCN, HCl ZnCl2 PhCH=NH H3O+ PhCHO HCl HCN ZnCl2 NH HC Cl Реакция хлорметилирования CH2O H+ CH2OH электрофил PhH CH2O HCl ZnCl2 PhCH2OH LiAlH4 Cl- H+ PhCH2 PhCH2Cl PhCH3 селективный синтез толуола 82 Взаимодействие с альдегидами OH H+ RCHO электрофил R H PhH OH RCHO H+ Ph H+ PhH R -H2O Ph R -H+ Ph R Ph Cl PhCl Cl CCl3CHO H2 SO4 ДДТ CCl3 1,5-3 млн. человек в год погибает от малярии 83 Сульфирование, сульфохлорирование OO H S O H2SO4/SO3 PhSO3 H PhH t ± H+ электрофил SO3 PhH HSO3 Cl t H3O+ t PhH реакция обратима, вторая стадия - лимитирующая, присутствует кинетический изотопный эффект PhSO2Cl SO2Cl2 AlCl3 Нитрование O HO N H2SO4 OPhH -H2O NO2+ электрофил HNO3 H2SO4 NO2+BF4- PhNO2 84 Реакция замещенных производных бензола с электрофилами X X X H орто- H H X X X X E E E пара- E+ E E H H E X X X H мета- E H H H E E Выводы: 85 1. В случае электрофильной атаки в орто- и пара-положения положительный заряд оказывается на атоме углерода, связанном с имеющимся в кольце заместителе. При атаке в мета-положение заряд на этот атом углерода не попадает. 2. Донорные заместители стабилизируют сигма-комплексы, образующиеся в результате электрофильной атаки в орто- и пара-положения. Акцепторные заместители напротив – в наибольшей степени дестабилизируют сигма-комплексы, образующиеся в результате атаки в орто- и пара-положения. 3. Предпочтительными направлениями электрофильной атаки в случае донорно-замещенных производных бензола являются орто- и пара-. В случае акцеторного заместителя атака преимущественно происходит в мета-положение. 4. Донорные заместители повышают реакционную способность замещенных бензолов, акцепторные заместители – снижают. Донорные заместители – алкил (индуктивный эффект), фенил, гетероатомы, несущие неподеленную пару электронов (мезомерный эффект). орто-пара-ориентанты заместители "первого рода":алкил, арил, OH, OR, NH2, NR2 , NHCOR, галоген X X E E H Ph H E E H H мета-ориентанты заместители "второго рода": CN, CO2H, CO2R, CHO, NO2, SO3H, NR3+ Заместители «первого рода» активируют кольцо к реакциям электрофильного замещения (за исключением галогенов) по сравнению с бензолом. Заместители «второго рода» кольцо дезактивируют. При наличии более одного заместителя, обладающих разным и несогласованным действием, направление реакции определяется донорным заместителем. В случае нескольких донорных заместителей направление реакции определяется заместителем с большим эффектом. Пара-замещение обычно преобладает над орто-замещением, и тем в большей степени, чем больше объем заместителя и/или электрофила (стерический фактор) 86 87 CH3 CH3 CH3 NO2 NO2 E+ E NO2 > согласованное влияние заместителей E Cl Cl согласованное влияние заместителей E+ HO3S HO3S E OH OH OH E+ CF3 OH E E >> + CF3 CF3 CF3 E несогласованное влияние заместителей 88 E+ E стерический фактор OCH3 OCH3 E+ OCH3 > E NHCOCH3 E NHCOCH3 NHCOCH3 E+ >> NO2 NO2 E E NO2 89 Реакции электрофильного замещения, которые могут протекать только с ароматическими субстратами, не менее реакционно-способными, чем хлорбензол: 1. Реакции Фриделя-Крафтса (алкилирование, ацилирование) 2. Реакции с карбокатионами, генерируемыми тем или иным способом 3. Реакция хлорметилирования 4. Реакции формилирования (Гаттермана и Гаттермана-Коха) 5. Кислотно-катализируемые реакции с альдегидами Реакции нитрования, сульфирования, хлорирования и бромирования могут происходить с практически любыми ароматическими субстратами Выводы: 1. Практически сложно провести селективно реакцию алкилирования (реакционная способность продукта выше, чем у исходного бензола). 2. Реакции ацилирования и формилирования могут приводить только к продуктам монозамещения (реакционная способность продукта гораздо ниже, чем у исходного бензола). Реакции электрофильного замещения, протекающие только с очень активными ароматическими субстратами: OH OH HO- CHCl3 PhOH HO- Реймер-Тимман CHO CHCl2 HO- CHCl3 90 CCl2 дихлоркарбен - электрофил OH PhOH 1. POCl3 / ДМФА - 2. HO , H2O O N Вильсмайер-Хаак OHC POCl3 C H ДМФА O Cl2OP N C H O Cl2OP N C H электрофил OPOCl2 PhOH POCl3 / ДМФА N N - OPOCl2- H2O HOOHC OH 91 OH N HO Ph PhN2+ N PhOH N CO2, p,t HOКольбе-Шмидт азосочетание Ph CO2- N Ph N N соль фенилдиазония Обратимость реакции алкилирования PhH CH3Cl AlCl3 PhCH3 CH3Cl t + AlCl3 AlCl3 кинетический контроль термодинамический контроль 92 H PhCH3 CH3Cl + AlCl3 H H H H H H H H H самый устойчивый -комплекс 93 Использование защитных групп в электрофильном ароматическом замещении PhCH3 CH3COCl > AlCl3 COCH3 COCH3 OH H+ H3 O+ t CH3COCl AlCl3 практически единственный изомер COCH3 единственный изомер 94 PhCH3 HNO3 CH3COCl NO2 NO2 SO3 H2SO4 AlCl3 + H2SO4 H3O+ t HNO3 HO3S практически единственный изомер H2SO4 HO3S NO2 единственный изомер 95 Электрофильное замещение в ряду нафталина H E H E H E атака в -положение менее значимые резонансные структуры E+ H H атака в -положение E E менее значимые резонансные структуры Более устойчивый сигма-комплекс образуется при атаке в альфа-положение. Если реакция необратима, то основным является продукт замещения в альфаположении. Если реакция обратима и проводится в условиях термодинамического контроля, образуется продукт замещения в бета-положение. 96 H дестабилизирующее взаимодействие в случае -атаки X O O CH3COCl AlCl3 CS2 CH3COCl AlCl3 PhNO2 HNO3 NO2 H2SO4 100o SO3 H SO3H + H 160o 97 Br H Br2 Br Br H Br 1,4-присоединение Реакции восстановления PhH H Br- t -HBr H Br H2 Pt (Pd) 98 Восстановление по Берчу X X X EtOH -EtO- Na NH3 (жид.) EtOH H H X X H или H H H H X = акцептор Na NH3 (жид.) EtOH H X EtOH -EtO- или +e или H H X X Na NH3 (жид.) EtOH Х = донор X X X Na 99 Na NH3 (жид.) EtOH NH3 (жид.) EtOH X = акцептор Х = донор Реакции окисления O O O O2 KMnO4 V2O5 HO-, t малеиновый ангидрид X O O X O2 O2 V2O5 (Cr6+,H+,t) V2O5 (Cr6+,H+,t) O фталевый ангидрид Х = донор Х = акцептор X=H CO2H CO2H X O O H2O CO2H CO2H O 100 Свойства алкилбензолов Cl Cl Cl Cl2 Ph R h Ph Ph R R Ph R R =H PhCCl3 устойчивый бензильный радикал R PhCH2Cl HOH2O PhCHCl2 Ca(OH)2 H2O PhCCl3 H2SO4 H2O, t R R R PhCH2OH PhCHO PhCO2H PhCH2R MnO4- HO , t (O2, Co2+) (H2Cr2O7) PhCO2H 101 CO2H CO2H O2 O2 Co2+ Co2+ CO2H CO2H O2 O2 Co2+ Co2+ O O2 Co2+ 102 Кумольный способ получения фенола и ацетона OOH H+ O2 Co2+ кумол O Ph O OH2 Ph H2O PhO OH H+ -H+ - H2O гидроперекись кумила H PhO OH OH OH O -H+ - PhOH фенол 1. уротропин ацетон Соммле 2. H3O+ MnO2 PhCH2Cl H2O ДМСО N O PhCHO CrO2Cl2 Этар S PhCH3 ДМСО N N N уротропин NH3 CH2O