Филогенетическое дерево и его реконструкция
реклама

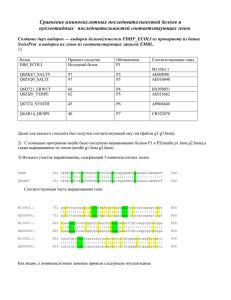
Филогенетическое дерево и его реконструкция. 1. Изображение дерева На основе имеющейся скобочной формулы (((А:35,В:35):5,(С:35,D:35):15):50,(Е:40,F:40):60); посредством программы было построено изображение дерева. 2. Описание дерева как разбиения множества листьев. Предложенное в задании дерево (см. рис.) имеет 9 ветвей, шесть из которых являются внешними, а три внутренними. Внешними ветвями называются те «линии», которые отходят от какого-либо листа, т.е. попросту соединяют его с другими элементами дерева. А внутренние ветви образуют более сложную организацию листьев, формируют так называемые кластеры (множества листьев, расположенные с одного или другого конца этой ветви). На рисунке все три внутренние ветви выделены разными цветами. Например, красная ветвь делит множество всех листьев на кластеры A,B и C,D,E,F (все остальные), а синяя – на E,F и A,B,C,D. Таким образом, топологию дерева можно представить в виде таблицы, где столбцы есть ничто иное, как листья, а строки – это ветви. Причем в нем можно оставить лишь те строки, которые соответствуют внутренним ветвям, поскольку внешние ветви – информации не несут. ABCDEF ..**** **..** ****.. 3. Получение искуственных мутантных последовательностей. Мы можем создать искусственные последовательности, которые будут соответствовать листьям и узлам дерева, считая, что в корне находится последовательность гена белка alf_ecoli. Длина последовательности этого гена составляет 1080bp. Поскольку в качестве расстояний между элементами даны количества мутаций на 100 оснований, то для перехода к абсолютным значениям мутаций (имеются в виду исключительно замены) нужно воспользоваться следующей формулой: X=(1080N)/100, где X – абсолютное значение числа мутаций, N – расстояние, приведенное в скобочной формуле, 1080 – длина последовательности гена. Воспользовавшись данной формулой, мы можем пересчитать все расстояния. Далее для получения «мутантов» обратимся к программе msbar пакета EMBOSS. Запущенный скрипт имел следующий вид: msbar alf_ecoli_gene.fasta junc_EF.fasta -point 4 -count 648 -auto msbar junc_EF.fasta E.fasta -point 4 -count 432 -auto msbar junc_EF.fasta F.fasta -point 4 -count 432 -auto msbar alf_ecoli_gene.fasta junc_ABCD.fasta -point 4 -count 540 -auto msbar junc_ABCD.fasta junc_AB.fasta -point 4 -count 54 -auto msbar junc_ABCD.fasta junc_CD.fasta -point 4 -count 162 -auto msbar junc_AB.fasta A.fasta -point 4 -count 378 -auto msbar junc_AB.fasta B.fasta -point 4 -count 378 -auto msbar junc_CD.fasta C.fasta -point 4 -count 378 -auto msbar junc_CD.fasta D.fasta -point 4 -count 378 -auto 4. Реконструкция дерева алгоритмами UPGMA, Neighbor-joining и максимального правдоподобия. Сравнение деревьев. Полученные последовательности были записаны в один файл. При этом «мутанты» были вручную переименованы. Алгоритм максимального правдоподобия (неукорененное дерево). fdnaml mutants.fasta -ttratio 1 -auto Полученное изображение: +--------F +------------------------------4 +--1 +---------E | | | +-------B | | +------D 3----2 | +-------C | +-------A Предварительно посчитаем попарные расстояния между последовательностями программой fdnadist: fdnadist mutants.fasta -ttratio 1 -auto Полученный файл в формате *.fdnalist используется далее. Алгоритм Neighbor-joining (неукорененное). fneighbor mutants.fdnadist -auto +-------C +---2 ! +-------D ! ! +-------B 3-4 ! ! +----------E ! +--------------------------------1 ! +--------F ! +--------A Реконструкция алгоритмом UPGMA (укорененное дерево!) fneighbor mutants.fdnadist -treetype u -outfile mutants_upgma -auto +----------------A +---2 ! +----------------B +-------------------------------4 ! ! +--------------C ! +-----1 --5 +--------------D ! ! +------------------E +---------------------------------3 +------------------F ABCDEF . . **** ** . . ** **** . . . * . . ** Исходное дерево модели + + + - Максимальное правдоподобие + + + Neighborjoining + + + UPGMA + + + - Единственный метод, построивший верное дерево в данной ситуации, - это метод, основанный на алгоритме UPGMA. 5. Бутстреп-анализ множественного выравнивания и построение консенсусного дерева. Последовательно использовались следующие команды: fseqboot mutants.fasta -auto [создается 100 бутстреп-реплик выравнивания] fdnaml mutants.fseqboot -ttratio 1 -auto [полученные 100 выравниваний подаются на вход программе fdnaml. В выходном файле (.treefile) получаем 100 скобочных формул, соответствующих реконструкциям, сделанным по каждому из выравниваний] fconsense mutants.treefile Косенсусное дерево выглядит следующим образом: +------F +100.0-| | +------E +-99.0-| | | +------A +------| +-58.0-| | | +------B | | | +--------------------D | +---------------------------C Топология полученного консенсусного дерева описывается следующим образом: ..**.. ..**** ....** 100.00 99.00 58.00 Топология реального дерева (см. пункт 1): ABCDEF ..**** **..** ****.. Эти цифры справа (бустреп-значения) показывают, в скольки деревьях из ста «случайных» встретилась эта ветвь, таким образом характеризуя статистическую надежность, биологическую достоверность соответствующих ветвей. Несмотря на невысокое значение у одной из ветвей, она есть и в реальном дереве. Т.е. топология реального и консенсусного деревьев полностью совпадает. 6. Создание изображения дерева программой fdrawtree. Использовалась команда: fdrawtree mutanty.treefile