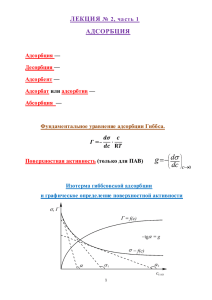
раствор (киселев)
реклама
")
Федеральное агентство по образованию Московская государственная академия тонкой химической технологии им. М.В.Ломоносова Кафедра коллоидной химии Кафедра информационных технологий Киселев В.Я., Комаров В.М. Адсорбция на границе раздела «твердое тело – раствор» Учебное пособие Москва 2005 год УДК 541.183 ББК 24.6 Рецензент: Д.х.н., профессор Поленов Е.А. Киселев В.Я., Комаров В.М. Адсорбция на границе раздела «твердое тело – раствор» Учебное пособие М.: МИТХТ им. М.В.Ломоносова, 2005. Утверждено Библиотечно-издательской комиссией МИТХТ им М.В.Ломоносова в качестве учебного пособия Поз. Данное учебное пособие соответствует программе курсов «Коллоидная химия» и «Поверхностные явления и дисперсные системы» для направления бакалавриата и магистратуры по направлению 550800 «Химическая технология и биотехнология» очной формы обучения. В нем подробно излагаются закономерности адсорбции индивидуальных жидкостей, растворов, ионов, поверхностно-активных веществ и высокомолекулярных соединений на твердом теле. ©МИТХТ им. М.В.Ломоносова, 2005 г. 2 1.ВВЕДЕНИЕ ............................................................................................................................................................. 4 2. АДСОРБЕНТ И АДСОРБТИВ ........................................................................................................................... 4 2.1. АДСОРБЕНТ....................................................................................................................................................... 5 2.2. АДСОРБТИВ ...................................................................................................................................................... 9 3. ПРИРОДА АДСОРБЦИОННЫХ СИЛ ........................................................................................................... 20 4. МЕХАНИЗМ АДСОРБЦИИ .............................................................................................................................. 25 5. ТЕОРИЯ АДСОРБЦИИ ИЗ БИНАРНЫХ РАСТВОРОВ ........................................................................... 28 6. АДСОРБЦИЯ НИЗКОМОЛЕКУЛЯРНЫХ НЕСМЕШИВАЮЩИХСЯ И СМЕШИВАЮЩИХСЯ ЖИДКИХ СИСТЕМ .................................................................................................................................................................... 33 7. АДСОРБЦИЯ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ НА ТВЕРДЫХ ПОВЕРХНОСТЯХ ....... 38 7.1. АДСОРБЦИЯ НЕИОНОГЕННЫХ ПАВ ............................................................................................................... 39 7.2. АДСОРБЦИЯ ИОНОГЕННЫХ ПАВ .................................................................................................................. 41 7.3. ПЛЕНКИ ЛЕНГМЮРА-БЛОДЖЕТТ ................................................................................................................... 50 8. АДСОРБЦИЯ ЭЛЕКТРОЛИТОВ (ИОНОВ) .................................................................................................. 53 9. АДСОРБЦИЯ ПОЛИМЕРОВ НА ТВЕРДОМ ТЕЛЕ. ................................................................................... 64 10. МЕТОДЫ ИССЛЕДОВАНИЯ АДСОРБЦИИ НА ГРАНИЦЕ РАЗДЕЛА РАСТВОР - ТВЕРДОЕ ТЕЛО ..................................................................................................................................................................................... 77 10.1. ТЕРМОДИНАМИЧЕСКИЕ МЕТОДЫ ........................................................................................................... 77 10.2. МЕТОДЫ, СВЯЗАННЫЕ СО СПЕКТРОСКОПИЕЙ И РАССЕЯНИЕМ ............................................................ 77 10.3. ГИДРОДИНАМИЧЕСКИЕ МЕТОДЫ .................................................................................................................. 78 10.4. ЭЛЕКТРО-ХИМИЧЕСКНЕ МЕТОДА .................................................................................................................. 78 10.5. МЕТОДЫ РАЗЪЕДИНЯЮЩЕГО ДАВЛЕНИЯ..................................................................................................... 79 11. СПИСОК ЛИТЕРАТУРЫ ................................................................................................................................ 80 3 1.Введение Адсорбцией называют процесс выравнивания химических потенциалов между поверхностью и объемом, приводящий к изменению концентрации компонента на поверхности. Адсорбция из раствора на твердой поверхности процесс более сложный, чем адсорбция газов и индивидуальных жидкостей. Она лежит в основе важнейших явлений коллоидной химии, как получения, так и разрушения лиозолей. Изучение адсорбции сжиженных газов, низкомолекулярных индивидуальных жидкостей на твердых поверхностях показало, в основном, применимость уравнений Генри, Бедеккера-Фрейндлиха, Ленгмюра и БЭТ. Однако, присутствие в системе, как минимум, третьего компонента во многом усложняет математическое описание и теоретическую трактовку адсорбции из смеси. Это связано, во-первых, с наличием взаимодействия между растворителем и растворенным веществом, во-вторых, значительной конкуренции за активные центры поверхности и, в-третьих, замедлением процесса диффузии растворенного вещества к поверхности из-за вязкости среда и созданием граничного слоя. Уплотненный граничный слой, состоящий из растворителя и растворенного вещества, препятствует обмену компонентов на поверхности и значительно замедляет установление равновесия в системе. Явление адсорбции из растворов классифицируют по свойствам адсорбента на адсорбцию неэлектролитов (молекулярную) и электролитов (ионную). 2. Адсорбент и адсорбтив Адсорбент - тело, на котором происходит концентрация вещества, может быть жидкостью, аморфным или твердым. Адсорбтив - множество молекул, ионов с сольватными оболочками, отдельные макромолекулы или их агрегаты, коллоидные частицы, движущиеся во всех направлениях. Рассмотрим поведение одной выбранной молекулы или агрегата. Сталкиваясь в объеме с себе подобными она меняет скорость и направление движения и может встретить на своем пути поверхность, например, стенку сосуда. В большинстве случаев молекула, ударившаяся о поверхность, на некоторое время остается на ней. Продолжительность пребывания зависит от множества факторов: от места на поверхности, природы взаимодействия между поверхности и поверхностью и самой молекулы, молекулой и специфики самими сил молекулами, кинетической энергии молекулы. При этом столкновении концентрация молекул на 4 поверхности несколько отличается от объема. Де Бур предложил расчет количества молекул газа, ударяющихся за одну секунду об 1 см2 поверхности при давлении 1 атм при комнатной температуре: для водорода - 11.1023 , для азота - 2,9.1023, для кислорода - 2,7.1023. Воздух с влажностью 10%, давление водяного пара порядка 1,75 мм рт. ст. способствует соударению 8,5.1020 молекул воды с 1 см2 поверхности в 1 секунду. А для покрытия мономолекулярным слоем 1 см2 поверхности достаточно 1015 молекул воды, то полное равновесие достигается за одну миллионную долю секунды. Следовательно, в растворе даже большие по размеру молекулы, агрегаты, ассоциаты, коллоидные частицы, фракталы, наночастицы способны за достаточно короткое время образовывать монослой, а динамическое равновесие зависит от множества факторов и зачастую не может быть описано единой теорией. 2.1. Адсорбент Адсорбция молекул из жидкой фазы на твердом теле определяется интенсивным силовым полем, зависящим от состава и структуры. Поверхность всегда неровна (рис.1), и разные грани обладают различной адсорбционной способностью, являясь активными центрами адсорбции. Не все атомы на поверхности находятся в одинаковых условиях. Атомы на острых выступах и шероховатостях намного богаче энергией и обладают избыточной поверхностной энергией и подвижностью. Наряду с гранями, выступами большое значение имеют дефекты структуры реальных кристаллов, подробно изучаемых в курсе физики твердого тела (шероховатости, трещины, дислокации, грани, впадины, ребра, дефекты, выщербены, сколы). Рис. 1. Шероховатость поверхности и активные центры (по степени активности) 1>2>3>6>5>4 5 Поверхностное натяжение твердого тела в отличие от поверхностного натяжения жидкости неоднозначная величина. Это связано с тем, что при образовании новой поверхности атомы поверхностного слоя перегруппировываются и перемещаются в конечное равновесное положение в течение длительного времени. Образующаяся при этом грань кристалла принимает такую форму, чтобы суммарная свободная энергия кристалла данного объема была наименьшей. Кристаллы с ковалентными связями (например, алмаз) обладают поверхностной энергией плоскости (111) порядка 5650 мДж/м2, а для плоскости (100) эта величина около 9800 мДж/м2. Ионные кристаллы имеют значительно меньшие величины поверхностной энергии и несимметричность иона вблизи поверхности. Расчеты для хлористого натрия (NaCl) тех же граней (111) - 350 мДж/м2, (100) - 170 мДж/м2. Молекулярные кристаллы, к которым относится большинство твердых тел и все органические соединения, в том числе и полимеры, характеризуются еще меньшими значениями поверхностной энергии, от 18 до 80 мДж/м2. Дефекты кристаллических решеток значительно сказываются на свойствах кристалла и поверхностной энергии. Наличие точечных дефектов в результате образования избыточных, внедренных атомов или ионов, а также дефекты, вызванные недостатком атомов в решетке - вакансии, приводят к появлению дислокации (совокупности дефектов). Дислокации образуются на поверхности в виде ступенек и находятся в состоянии высокого поверхностного напряжения. Химический потенциал такой дислокации настолько велик, что молекулы вещества передвигаются на поверхности, оставляя пространство в виде полости, являясь центром адсорбции. В зависимости от природы твердого тела число линий дислокаций, приходящихся на единицу поверхности кристалла, может изменяться в широких пределах от 106 до 1016 дислокаций на м2. Выступы, ребра, грани и вершины кристаллических решеток обладают значительным числом ненасыщенных химических связей, большой суммарной поверхностной результате энергией, являются близкодействующих центрами хемосорбции, электростатических сил возникающей в притяжения. А дальнодействующие межмолекулярные силы, наоборот, увеличиваются по мере возрастания числа окружения соседей. Таким образом, центры физической адсорбции, в основном, находятся в трещинах, зазорах, впадинах. Следовательно, микротопография поверхности, наряду с химической природой и структурой, позволяет с большим приближением рассчитать средние значения поверхностной энергии с применением современных методов исследования поверхности, таких как: 1. Микроскопия: a) оптическая - огранка поверхности и развитие дислокации, b) электронная - поверхность с разрешением до 10Å, 6 c) сканирующая с рентгеновским анализатором – поверхность с разрешением до 100 Å и анализом состава, d) эллипсометрия - измерение толщины пленок по изменению поляризации светового пучка для пленок толщиной до 1 Å, 2. Дифракция и рассеяние электронов и ионов: дифракция медленных (ДМЭ-I-5ООЭВ) и быстрых (ДБЭ-18-25 КЭВ) a) электронов, b) автоэлектронная (30-50 Å) и автоионная (0-0,5 Å) эмиссия, c) рассеяние ионов, 3. Спектроскопия: инфракрасная (ИКС) и комбинированное рассеяние (КРС), в том a) числе НПВО и МНПВО, b) электронноспиновый (ЭПР) и ядерно-магнитный резонанс (ЯМР), c) ОЖЕ-спектроскопия, d) фотоэлектронная (ЭСХА), e) Масс-спектроскопия вторичных электронов (МСВЭ), 4. Поверхностная проводимость (для полупроводников) и другие. Наличие дефектов на поверхности, а также в объеме приводят к увеличению истинного значения удельной поверхности твердого тела. Гладкие (идеальные) поверхности встречаются очень редко. К ним можно отнести стекла, полимеры, керамику и специально приготовленные поверхности. В основном твердые тела обладают значительной пористостью. К ним относятся уголь и торф, изделия из древесины, ткани, зерно, кожа, почва и грунты, губчатые изделия, пенопласты и многие другие. Пористые тела обладают различной адсорбционной способностью по отношению к раствору или растворителю, механической прочностью, удельной поверхностью, селективностью и другими специфическими свойствами. Наиболее широкое применение в технике имеют активированные угли, сажи, силикагели и алюмогели, цеолиты (алюмосиликат со строго регулярной кристаллической структурой), пористые стекла, мембраны, целлофан. Согласно классификации по Дубинину с учетом размеров пор и механизма протекающих в них адсорбционных процессов пористые тела делятся на: 1. Макропористые - поры с радиусом более 100-200 нм и удельной поверхностью от 0,5 до 2,0 м2/г. В основном играют роль транспортных каналов и адсорбцией в них пренебрегают. 2. Переходнопористые - поры с радиусом от 2,0 до 100 нм и удельной поверхностью от 10 до 500 м2/г - силикагели, алюмогели, алюмосиликагели. 3. Микропористые - поры с радиусом от 0,5 до 2,0 нм и удельной 7 поверхностью от 500 до 1000 м2/г - активированные угли, цеолиты. 4. Супермикропористые - размер пор меньше 0,5 нм - дефекты и дислокации кристаллических решеток. Для характеристики пористых тел недостаточно знать только размер пор или капилляров, которые отличаются количеством на единицу объема, размером и длиной капилляра. Поэтому принято характеризовать пористость структуры тела величиной пористости - отношением объема пор (Vп) к общему объему тела (Vобщ). П Vп (1) Vобщ Однако классификация тел не учитывает возможность одновременного присутствия открытых, закрытых и тупиковых пор с различной длиной капилляра. Закрытые, внутренние поры, дефекты кристаллической решетки и дислокации внутри кристалла не могут быть определены обычными методами, а применение томографии не позволяет однозначно рассчитать пористость. Поэтому пористость складывается из трех составляющих: Побщ П З ПО ПТ (2) где Пз, По, Пт - объемы соответственно закрытых, открытых и тупиковых пор в единице объема твердого тела. При адсорбции следует учитывать не только количество тех или других пор, но и кривизну капилляра, сужение и расширение, а также распределение пор по размерам (рис.2). Рис. 2. Примеры пор в твердом теле 1) тупиковая, 2) открытая, 3) закрытая, 4) открытая с утолщением внутри, 5) открытая с утоньшением внутри, 6) бутылочная, 7) гребенчатая, 8) поверхностная. Кроме плоских поверхностей в большинстве случаев в процессе адсорбции участвуют нити, имеющие незначительную удельную поверхность и, главным образом, высокодисперсные твердые частицы. По размеру частиц дисперсные системы делятся на: 8 1) высокодисперсные - 10-7-10-9м (наночастицы), 2) микрогетерогенные - 10-5-10-7м, 3) грубодисперсные-10-3-10-5м (макрогетерогенные). Тонкодисперсные твердые частицы получаются как измельчением грубодисперсных систем - диспергирование, так и конденсацией. В последнем случае свободнодисперсные системы: порошки или суспензии в большинстве случаев обладают заданным комплексом свойств - состав, удельная поверхность, форма и размер частиц, текучесть, насыпная плотность и другие. Введение зародышей кристаллизации позволяет получить системы с заданной полидисперсностью. Дисперсность частиц определяет адсорбционную способность твердого тела и хорошо иллюстрирует влияние энергии ребер дробленого хлорида натрия при условии, что кубическая решетка при дроблении не искажается (табл. 1). Таблица 1. Зависимость удельной поверхностной энергии от размера частиц хлорида натрия. Длина ребра, м Суммарная площадь, *104, м2 Суммарная длина ребер, *102, м Поверхностная энергия, мДж/кг Энергия ребер, мДж/кг 0,77*10-2 3,6 9,3 540*10-3 2,8*10-2 0,1*10-2 10-3 28 550 4,2 1,7*10-1 0,01 10-4 280 5,5*104 42 1,7*10-4 0,001 10-5 2,8*103 5,5*106 4,2*102 1,7*10-2 0,0001 10-6 (1мкм) 2,8*104 5,5*108 4,2*109 1,7 0,000001 10-8 (100Å) 2,8*106 5,5*1012 4,2*1011 1,7*104 2.2. Адсорбтив Адсорбтив, в отличие от твердого тела, состоит либо из отдельных молекул, либо смесей молекулярных растворителем молекул. Концентрационная образований и во растворенным многом конформация зависят веществом. от и конфигурация взаимодействия Отдельными между представителями являются растворы электролитов, хотя ионогенные ПАВ, мицеллообразующие системы, значительно отличаются от неорганических электролитов. Растворители, широко применяемые 9 в промышленности, делятся на неорганические и органические жидкости. К неорганическим, прежде всего, относится вода, кислоты и щелочи, сжиженные газы Н2, О2, N2, NH3, SO2, CO2 и другие. Особенно широко представлены синтетические органические растворители, отличающиеся природой функциональных групп, их количеством и молекулярной массой. Необходимость классификаций широкого спектра растворителей привела к созданию разнообразия по подразделению их по тому или иному признаку. 1. По физическим свойствам 1) По диэлектрической проницаемости ( ε ): a) с низкой (1,9-12) - гептан, гексан, фторуглероды... b) со средней (12-50) - органические кислоты, спирты, c) с высокой (> 50) - вода, глицерин, неорганические кислоты, основания. 2) По дипольному моменту 3) По вязкости a) низковязкие (< 2 спз), Па . с . 10-3 b) средневязкие (2-10 спз), Па . с . 10-3 c) высоковязкие (> 10 спз), Па . с . 10-3 4) По температуре кипения a) низкокипящие (< 100°С), b) среднекипящие (100-150°С), c) высококипящие (> 150°С). 5) По их способности к испарению (за 1 принято испарение этилового эфира при 20°С и относительной влажности 65- 70%) a) легколетучие (< 10), b) средне летучие (10-35), c) труднолетучие (> 35). 2. По химическим свойствам. Помимо известной классификации - кислоты, спирты, углеводороды, галогенамиды и т.д., и по кислотно-основному взаимодействию - протонногенные (кислые), протоннофильные (основные) и амфипротонные. Наиболее применима и разработана классификация растворителей по способности к образованию Н-связи. I класс: жидкости, способные к образованию объемной трехмерной сетки Нсвязи (вода, муравьиная кислота, гликоли) и имеют значительные величины ε и хорошо растворяются друг в друге. II класс: жидкости, в которых возникают двухмерные сетки Н-связи (фенолы, одноатомные спирты и кислоты) - содержание одну ОН -группу. III класс: жидкости, имеющие в составе молекулы атомы азота, кислорода, 10 серы, фтора и другие, которые способны образовывать Н -связи с протоном партнера (эфиры, амины, кетоны и другие органические соединения. IV класс: жидкости, молекулы которых имеют атом водорода, способный к участию в Н - связи, но не имеют атомов, которые могли бы быть акцепторами протона (хлороформ, дихлорэтан). V класс: жидкости, молекулы которых не способны к образованию Н - связи ни в качестве доноров, ни в качестве акцепторов (четыреххлористый углерод). Наиболее часто встречаемая жидкость со стандартными свойствами - вода, особенности свойств которой определены, прежде всего, строением ее молекул. Молекула воды имеет четыре полюса зарядов, расположенных в вершинах тетраэдра с расстоянием положительных зарядов до центра атома кислорода порядка 0,99 Å (рис. 3). Рис. 3. Структура молекулы воды Ажурность структуры воды объясняется в значительной мере образованием водородных связей между молекулами и определяет особенности диффузии ее молекул. В этом случае активированный переход отдельных молекул не требует "дырок", так как они имеются в самой структуре воды. Изучение самодиффузии молекул воды и ионов в разбавленных растворах солей показало, что ионы в растворе не только гидростатируются, но и, главным образом, изменяют структуру самих молекул воды. Коэффициент самодиффузии воды в растворах солей значительно выше, чем в чистой воде, что связано с образованием гидратной оболочки и разрушением структуры прилегающих слоев воды. Происходит, с одной стороны, как бы "замораживание" молекул воды, а с другой, как бы плавление прилегающих слоев воды. Проведенные исследования показали, что ион лития Li+ (r=0,68Å), обладающий высокой поляризуемостью, способен значительно упорядочить структуру воды, включенную в гидратную оболочку, по сравнению со свободной "несвязанной" водой. Ион цезия Cs+ (r=1,65Å) незначительно изменяет структуру воды в гидратной оболочке, но зато в сильной степени изменяет структуру воды в прилегающих к 11 гидратированному иону слоях воды. Следовательно, из-за нарушения структуры воды вокруг иона вязкость чистой воды значительно падает, поэтому подвижность ионов Cs+ становится больше по сравнению с подвижностью ионов Li+ вместе с сольватной (гидратной) оболочкой. При этом самодиффузия чистой воды в разбавленных растворах Cs+ выше, чем в растворах Li+. Гидратацию рассматривают не как связывание ионами того или иного числа молекул воды, а как влияние ионов на трансляционное движение ближайших молекул воды. Эксперимент показал, что некоторые ионы уменьшают подвижность ближайших молекул воды, а около других подвижность молекул воды значительно возрастает по сравнению с чистой водой, что определяет отрицательную гидратацию. Расчет показал, что полная энергия взаимодействия иона с молекулами воды необычайно велика и намного больше взаимодействия между молекулами воды друг с другом, и процесс обмена с ближайшими молекулами воды не связан с энергией удаления воды из сольватной оболочки. При этом частота обмена (коэффициент диффузии) определяется потенциальным барьером, отделяющим молекулы, связанные гидратной оболочкой, от молекул, находящихся в свободном состоянии. Следовательно, если обмен затруднен, то гидратация велика и, наоборот, по мере возрастания частоты обмена гидратация ослабляется. Однако концентрированные растворы ионов электролита характеризуются структурами близкими координационное к число структурам ионов соответствующих соответствует кристаллогидратов, координационному а числу кристаллогидратов. Таким образом, наблюдается переход от структуры чистой воды к структуре кристаллогидрата по мере повышения концентрации иона в растворе. Существование обеих структур при повышенных концентрациях и низких температурах приводит к образованию "квазиэвтектических" растворов, Льюис (1905г.) для характеристики поведения реальных систем предложил понятие активности - концентрация, которую имела бы исследуемая жидкая система, если бы она обладала свойствами идеальной системы. а с где (3) а - активность системы, с - концентрация, γ - множитель (коэффициент активности). Так как работа идеальной системы (Аид) в связи с изменением концентрации от с1 до с2 может быть рассчитана по уравнению Аид RT ln c1 (4) c2 то для расчета работы реальной системы (А следующий вид 12 реал) это уравнение приобретает Aреал RT ln a1 (5) a2 Химический потенциал μ вещества в идеальной и реальной смешанной фазе описываются следующими уравнениями ид 0 RT ln c (6) реал 0 RT ln a (7) Из этого следует, что уравнение, ранее пригодное для идеальных систем, становится пригодным и для реальных систем. "Активность" как аргумент может рассматриваться как функция от термодинамических свойств растворов ln a ид0 (8) RT При соответствующих подстановках получим: реал 0 RT ln c 0 RT ln c RT ln ид RT ln (9) тогда коэффициент активности имеет вид: ln реал ид (10) RT где lnγ - безразмерная величина, определяющая разность химических потенциалов в реальной и идеальной системах, и чем больше эта разница, тем больше энергии надо затратить, чтобы перевести реально существующую систему из реальной в идеальную (мера работы переноса одного моля растворенного вещества из реальной системы в идеальную). Семенченко (1939 г.) вывел понятие средней потенциальной энергии одного моля (Un) с учетом межмолекулярного взаимодействия между растворенным веществом и растворителями, что осуществляется равенством Un (11) RT тогда RT ln Un (12) а коэффициент активности равен Un е RT (13) Наблюдающееся отличие коэффициента активности от концентрации вызваны изменением концентрации растворенного вещества в связи с образованием различных продуктов присоединения и сольватов, приводящих к уменьшению числа частиц в растворе, и значительному отличию энергии растворенных частиц, связанную с их взаимодействием между собой и с молекулами растворителя. Это особенно ярко проявляется у сильных электролитов, что определяется наличием 13 значительного электростатического взаимодействия между ионами. В концентрированных растворах изменение коэффициента активности связывают с изменением свободной энергии из-за гидратации ионов. Рис. 4. Зависимость средних коэффициентов активности от концентрации галогенидов щелочных металлов: 1 – LiF; 2 – LiCl; 3 – CsJ; 4 - CsCl Как видно из рис.4 и 5, взаимодействие между растворенным веществом и средой (вода) во многом зависит от ионного радиуса и валентности катиона и аниона, а в неводных растворах еще накладывает определенное влияние диэлектрическая проницаемость среды. Таким образом, рассматривая состояние растворов электролитов в реальных растворителях необходимо учитывать следующее: 1) равновесный состав и реакции взаимодействия в растворе, приводящие к этому составу; 2) характеристику сил, определяющих взаимодействие частиц растворенного вещества и молекул растворителя; 3) количественную характеристику зависимости между энергией сольватации ионов и молекул и энергией их взаимодействия в растворах; 4) общие закономерности, образующихся растворов позволяющие на основании присутствующих в растворах веществ. 14 охарактеризовать данных о свойства свойствах Рис. 5. Зависимость средних коэффициентов активности от концентрации электролитов: 1 - MgCl2; 2 - СаCl2; 3 - BaCl2; 4 - Li2SO4; 5 - Na2SO4, K2SO4. Адсорбция между твердым телом и растворителем подчиняется известным законам и описывается элементарными уравнениями для двухфазных систем, поэтому в нашем случае не рассматривается. Другими представителями, находящимися в виде молекул в водной среде и одновременно растворяющих молекулы воды, являются простейшие органические спирты и кислоты, которые изучены различными исследователями и представляют собой типичные модельные системы. Взаимные смеси различных органических растворителей сильновязкие системы и сложны для описания. Существенным вопросом является ориентация молекул на поверхности раздела "жидкость - жидкость" или "жидкость - газ", зависящая от интенсивности силового поля, которая в свою очередь зависит от полярности и особенностей структуры жидкости. На границе раздела фаз молекулы ориентируются таким образом, чтобы обеспечить постепенный переход из одной фазы в другую, и чтобы энергия их взаимодействия была максимальной. Рассмотрим ориентацию молекул этанола на поверхности воды (рис. 6). Согласно Ленгмюру (1924 г.), поверхностная свободная энергия молекулы аддитивно складывается из локальной свободной энергии этанола и воды. В первом случае поверхность покрыта гидроксильными группами, поверхностная энергия которых составляет порядка 190 мДж/м2. Во втором случае поверхностная энергия равняется поверхностной энергии углеводородов около 50 мДж/м2. 15 Разность между представленными величинами составляет 140 мДж/м2 или 3.10-16 Дж/молекулу. Это убедительно доказывает преимущественную ориентацию органических молекул по типу 2 (рис. 6), ОН Н Г Н С Н Г Н С Н Ж Н С Н Ж Н С Н Н ОН 1 2 Рис. 6. Расположение молекулы этилового спирта на поверхности воды. Это хорошо согласуется с величинами поверхностного натяжения большинства органических жидкостей и расчетов работ когезии и адгезии и описываются уравнением Гиббса. Г2 = - (dσ/dμ2)T, (14) где Г2 - избыток компонента 2 (этанол) в поверхностном слое площадью 1 м2 по сравнению с числом молей компонента в объеме раствора, содержащем такое же Г, 106моль/м2 число молей растворителя, что и поверхностный слой (рис. 6). 6 4 2 0 0 0,5 Вода-этанол 1 Рис. 7. Изменение поверхностных избытков этанола в зависимости от состава раствора. Таким образом, наличие дифильных молекул спирта приводит к неаддитивному распределению молекул этанола и воды друг в друге. Чем больше длина углеводородного радикала, тем хуже растворимость, взаимодействие между 16 молекулами, тем интенсивнее расслаивается система. Это подтверждается известным правилом Траубе. Увеличение длины углеводородного радикала на СН 2 группу способствует увеличению поверхностной активности растворенного вещества в 3,2 раза. Это правомерно для гомологических рядов простейших органических соединений (С1 – С5) по отношению к воде и обратно для их растворов в органических растворителях. Эти вещества называются поверхностноактивными (ПАВ). В случае, когда длина углеводородного радикала превышает 10-20 единиц групп - СН2 , содержит большое количество функциональных групп, не диссоциирует на ионы (неионогенные), принято говорить о коллоидных ПАВ. При малых концентрациях эти ПАВ находятся в растворителе в виде истинного раствора (отдельные молекулы) и к ним применимо правило Траубе, но, начиная с некоторой малой концентрации (критической концентрации мицеллообразования - ККМ) образуются в результате "гидрофобного взаимодействия" ассоциаты - мицеллы. Форма и размер мицелл с увеличением концентрации коллоидных ПАВ значительно отличается от сферической (рис. 8). Рис. 8. Переход коллоидных ПАВ из молекулярного состояния (1) в глобулярное (2 – по Гертли) и цилиндрическое (3 – по Мак Бену). Поэтому форма и размер мицелл коллоидных ПАВ должны привносить и значительные изменения в их адсорбционную способность. ПАВ классифицируют следующим образом: Поверхностно-активные вещества 8 млн. т/в год Ионогенные 70%(катионые, анионые, Неионогенные 30% амфолитные,катионо-анионые) 1) Катионноактивные 60% 2) Анионноактивные >10% 17 3) Амфолитные 4) Катионо – анионоактивные 0,2% Классификация: 1) Катионноактивные - растворимы в кислой среде а) октадициламин - C18H37NH2 октадицилхлорамин б) четырехзамещенный NH3 и пиридиновые основания, и их соли алкилтриметиламмонийфторид RN (CH3)3CI, цетазол- Сl C16H33N 2) Анионактивные - растворимы в щелочной среде а) карбоновые кислоты и их щелочи (мыла) C17H35СОО-Na+ (олеат Nа) RCОOMe б) алкиларилсульфонаты R SO3H Na (70%) - очень дешевы в) алилсульфонаты R – SO3Na (C15 – 20) г) алкилсульфонаты (более дорогие) ROSO3H (C12 – 14) д) алкенсульфонаты RНSО3 (C15 – 20) Ценон Т – С17Н35 - С - О - N(CH3) - C2H4 - SO3Na 4) Амфолитные - диссоциируют в обеих средах, но Цетиламиноуксусная кислота C16H33NHCH2-СООН катионоактивный анионоактивный в кислой среде - NH2+ в щелочной среде СОО- 4) Катионо-анионоактивные Дибутилнафталинсульфонат¯ (диметилфенилбензил]+ аммония CH3 C4H9 C4H9 N (CH3)2 SO3 CH3 18 по-разному 5) Неионогенные а) октилфенолы ОП нонилфенолы NP - 8 Продукты полиоксиэтилирования к спиртам или фенолам (RO)2 – P – (OC2H4)OH алкилортофосфорная кислота б) плюроники - растворимы в воде блоксополимеры окиси этилена (носитель гидрофобный) и окиси пропилена (носитель гидрофобный) HO(С2Н4O)п - х(С3Н6О)м(С2Н4О)хН в) маслорастворимые растворимы в органике Эфиры жирных кислот и многоатомных спиртов дистеарат сульфат Классификация ПАВ по молекулярному механизму действия (по Ребиндеру). I группа: В системе вода воздух - не структурирующие, слабое пенообразование, слабые смачиватели - высшие спирты, амины, карбоновые кислоты. II группа: В системе вода - масло не структурообразующие - диспергаторы, эмульгаторы, селективно смачивающие в системе твердое тело / жидкость. III группа: структурообразователи, гидрофилизируют, стабилизируют, при малых концентрациях защищают коллоидные частицы от коагуляции, при больших концентрациях - пластификаторы - глюкозида, белки, полисахара. IV группа: моющие вещества - обладают всеми свойствами трех предыдущих групп, а также обладают мицеллообразованием и солюбилизацией (включение растворенных молекул в объем мицеллы). Следующими типичными представителями молекулярных растворов безусловно являются растворы высокомолекулярных соединений (ВМС). Обладая длинными и гибкими органическими макромолекулами с большой адсорбционной способностью, они хорошо растворяются в органических растворителях и образуют ассоциаты при высоких концентрациях. Однако, коренным отличием молекул ВМС от других вышеперечисленных растворенных веществ безусловно является чувствительность неионогенных молекул ВМС к природе растворителя. Растворители по отношению к макромолекулам делятся на "хорошие", "плохие" и θ-растворители - условное обозначение, 19 нашедшее подтверждение по соотношению параметров растворимости и описываемое следующим уравнением р2 п2 (15) где- β - параметр совместности, (МДж/м3)1/2, δр - параметр растворимости растворителя, (МДж/м3)1/2, δп - параметр растворимости полимера,(МДж/м3)1/2. При β ≈ 0 - θ-растворитель, макромолекула представляет из себя гибкую цепочку (рис. 9, а), при 0 < β < 0,2 - "хороший" растворитель, и макромолекула ВМС выглядит в виде эллипсоида вращения (рис. 9, б), при 0,2 < β < 0,5 - "плохой" растворитель, и форма макромолекул наиболее выгодная - глобула - близкая к шарообразной (рис. 9, в). а б в Рис. 9. Форма макромолекул ВМС в различных растворителях: а - в θ-растворителе, б - в "хорошем", в - в "плохом". Совершенно по-другому ведут себя макромолекулы полимеров в концентрированных растворах. При этом концентрированные растворы делятся на: 1) разбавленные (1-3 мас.%), 2) умеренноконцентрированные (5-10 мас.%), 3) концентрированные (10-25 мас.%) и 4) высококонцентрированные (25-65 мас.%). С увеличением концентрации размер макромолекулярных образований проходит через максимум, но плотность упаковки индивидуального ассоциата увеличивается. 3. Природа адсорбционных сил Взаимодействие молекул адсорбтива с твердой поверхностью может осуществляться либо за счет химических близкодействующих сил (хемосорбция), либо за счет физических, дальнодействующих сил притяжения (Ван-дер-Ваальса). Наличие у поверхности активных центров, которые характеризуются высотой локализованных вблизи поверхности электронных энергетических уровней. В зависимости от расстояния между центром и адсорбированной молекулой может при сближении происходить отталкивание в результате индуцирования диполей взаимоориентированных вдоль электрического поля, либо сближение, приводящее к перекрытию электронных уровней молекул адсорбтива и адсорбата. примером 20 может служить адсорбция воды на кремнезёме (рис. 10). H H O OH HO O OH HO H2O H2O Si O Si Si O Si Si O Si Физическая Химическая Дегидратация адсорбция (1) адсорбция (2) адсорбата (3) Рис. 10. Адсорбция воды на кремнеземе Физически адсорбированная вода удаляется при температурах близких к 1ОО°С, а хемосорбированная требует повышения температуры до 300-400°С, дальнейшее увеличение температуры более 500°С приводит к деформации силиконовых групп и пасификации поверхности. При хемосорбции идет изменение как свойств твердого тела (на поверхности), так и адсорбтива. При этом взаимодействие осуществляется электростатическими силами, описываемыми законом Кулона для точечных зарядов: f e1 e 2 ξ r2 (16) где f - сила: взаимодействия между ионами, e1 и e2 - величина заряда иона, ξ - диэлектрическая проницаемость, r - расстояние между ионами. Рис. 11. Кривые потенциальной энергии неактивированной (а) и активированной (б) хемосорбции 21 По потенциальной энергии образования прочного адсорбционного комплекса химические силы в основном можно расположить в следующий ряд: ионные - с увеличением валентности возрастают, обратно пропорциональны квадрату расстояние (дальнодействующие), порядка 150 - 300 Дж/моль ковалентные или координационные - короткодействующие с небольшой энергией межмолекулярного взаимодействия (10-40 Дж/моль). В одном из наиболее успешных подходов хемосорбцию нередко рассматривают просто как реакцию, протекающую с образованием между твердым телом и адсорбтивом реакцию энергетически подобную образованию двухатомной молекулы. При адсорбции ВМС, ПАВ или растворителей на твердых поверхностях наиболее существенный вклад оказывают физические силы (Ван-дер-Ваальсовые). Молекулярные физические силы взаимодействия получили свое название потому, что они обуславливают отклонение газов от идеального состояния, характеризующегося постоянной а в известном уравнении Ван-дер-Ваальса: (p a )( b) RT V2 (17) где a - поляризуемость молекул, ν – частота колебаний атома. К ван-дер-ваальсовым силам относятся: 1) Взаимодействие молекул с постоянным жестким диполем (ориентационный эффект) (силы Кеезома) описываемое уравнением uор где 21222 1 3r 6 kT (18) uор - потенциальная энергия электростатического взаимодействия двух постоянных диполей, μ1 и μ2- дипольные моменты обеих молекул, r - расстояние между центрами диполей, kT - энергия микроброуновского движения (постоянная Больцмана и абсолютная температура. В зависимости от дипольного момента контактирующих молекул ориентационный эффект привносит до 50 % в общий вклад в межмолекулярное взаимодействие: чем меньше дипольный момент молекулы, тем меньше величина прочности связи. Это взаимодействие осуществляется на близких расстояниях (1-5 А) и энергия ориентационных сил порядка 5-10 кДж/моль. 2) Взаимодействие молекул с постоянным 22 диполем и молекул с индуцированным диполем (индукционный эффект) (силы Дебая) описываемое уравнением uин где 2a112 a222 r6 (19) UИН - потенциальная энергия взаимодействия двух различных молекул, μ1 и μ2- дипольные моменты молекул, а1 и а2 - поляризуемость молекул, r - расстояние между диполями. Поскольку температура не влияет на поляризуемость, индукционное взаимодействие в отличие от ориентационного не зависит от температуры. Энергия индукционного взаимодействия не превышает 5% от общего вклада ван-дерваальсовых сил и поэтому всегда имеет знак минус. Расстояние, при котором возрастает индукционное взаимодействие, не превышает 1-2 А, а энергия связи мала и составляет не более 1-4 ккал/моль. В ряде случаев молекулы, не имеющие постоянный или индукционный диполь, также способны взаимодействовать друг с другом за счет мгновенного дипольного момента. Даже между неполярными молекулами возникают силы межмолекулярного взаимодействия (дисперсионный эффект) (силы Лондона) и описываемые уравнением: 3 a2h uдес 6 0 4 r где (20) uдес - потенциальная энергия взаимодействия двух неполярных атомов, a - поляризуемость атома, h - постоянная Планка, ν0 - частота дисперсионного спектра колебаний атома, r - расстояние между атомами. Дисперсионные силы возникают из-за флуктуации электронной плотности в атоме адсорбата, которая индуцирует подобные флуктуации в атоме адсорбтива. Резонанс подобных флуктуации приводит к значительному снижению общей энергии системы в целом, приводящее к взаимному притяжению контактирующих неполярных молекул. Дисперсионное взаимодействие универсально и возникает у любых молекул , поэтому их доля от 50 до 100% в общем вкладе в прочность адсорбционного взаимодействия между молекулами и атомами. Дисперсионные силы не зависят от температуры и всегда вызывают только притяжение, поэтому имеют отрицательный знак. Наиболее полно взаимодействие молекулы неполярного адсорбтива с атомом на поверхности неполярного твердого тела описывается уравнением Леннарда- 23 Джонса u C B 12 , 6 r r (21) где u - общая энергия межмолекулярного взаимодействия двух молекул начиная с расстояния между ними равного нулю, С - константа уравнения описывающая все три возможных варианта взаимодействия, B - силы броуновского отталкивания, имеющие электростатическую природу и действующие на очень близких расстояниях. 2 4 4 2a2 2 h 0a2 3kT 3 с Дисперсионные силы являются (22) аддитивными, суммирование их дает значительный эффект дальнодействия. Энергия притяжения молекул адсорбтива поверхностью адсорбата уменьшается с расстоянием гораздо медленнее, чем энергия притяжения молекул адсорбтива в системе. Из этого следует, что максимальное действие сил притяжения проявляется в углублениях, впадинах, трещинах твердого тела. Это можно объяснить тем, что силовое поле, образуемое взамопротивоположными стенками поры, щелей, углублений, накладывается друг на друга, и таким образом значительно усиливается. Расстояние, на котором действуют дисперсионные силы, порядка 5-100 А, и энергия взаимодействия их превышает вышеперечисленные и составляет 10-30 кДж/моль. При изучении алюмосиликатного адсорбции типа молекул адсорбционное на поверхности взаимодействие силикатного и объясняется возникновением водородных связей. Это объясняется тем, что у атомов 2-го периода таблицы Д.И. Менделеева (фтор, кислород, азот, а также у атомов серы и хлора) из-за малого радиуса ионов и заметному сродству к электрону появляется способность оттягивать электрон от соседствующего с ними атома водорода. Атом водорода в этом случае приобретает свойства протона Н+ и взаимодействует с электронами других элементов за счет во водородной связи. Примером является образование водородной связи и айсберговая структура ассоциатов воды и льда H H O H O H H O H 24 Подобные ассоциаты могут образовываться и у органических молекул R H O H O H O R R Большое количество молекулярных соединений, которые могут образовывать водородную связь принято выделять в особый класс ассоциирующих веществ. К ним относятся углевода, белки и ВМС, "Гидрофобное взаимодействие". Термин "гидрофобное взаимодействие" прежде всего обозначает усиление связи между двумя частицами в присутствии растворителя, если взаимодействие типа растворитель - растворитель сильнее, чем взаимодействие растворитель - частица. Примером может служить следующий эксперимент: 2 металлические пластинки, покрытые кремнеорганической жидкостью (гидрофобная) помещены в полярную жидкость - воду. При их соприкосновении они удерживаются друг около друга за счет "стягивающего" (гидрофобного) усилия, а те же пластинки, но не обработанные органической жидкостью, разделяются самопроизвольно под действием силы тяжести пластинки. Выигрыш энергии, обусловленный сближением двух модифицированных металлических поверхностей, связан с действием ван-дер- ваальсовых(дальнодействующих) сил притяжения, а именно – дисперсионных согласно Eизд 1 А 12 r 2 (23) подробно описанное в предыдущих разделах. С учетом наличия поверхности (1), гидрофобизирующэй жидкости (2) и среды (3) дисперсионное взаимодействие будет определяться константой Гамакера с учетом вклада всех сил А*123 = А*12 – ( А*13 + А*23 + А*33 ) (24) Следовательно дисперсионные силы А*123 значительно больше А*12 при условии, что А*33 > А*13 + A*23, что и наблюдается в нашем случае. 4. Механизм адсорбции Наиболее предпочтительным подходом к изучению механизма адсорбции является исследование изотермы. Важнейшими характеристиками адсорбции безусловно являются 25 1) скорость адсорбции (константа скорости), 2) форма изотермы, 3) наличие плато на изотерме, 4) степень адсорбции растворителя, 5) тип адсорбции (мономолекулярная или полимолекулярная), 6) ориентация адсорбированных молекул, 7) влияние температуры на изотерму адсорбции, 8) природа взаимодействия между раствором и растворенным веществом, адсорбатом и адсорбентом. Различные формы рассматриваемых изотерм адсорбции представлены на рис. 11 по классификации Гильса. Исходя из формы начального участка были выделены 4 характерные класса изотерм, а деление изотерм на отдельные типы внутри класса связано с последующим изменением их формы при более высоких концентрациях. 1. Класс S - начальный участок изотерм класса S выгнут относительно оси концентраций и при увеличении концентрации вещества получается точка перегиба, которая придает изотерме характерную S -образную форму. 2. Класс L (класс Ленгмюра) с наиболее общей и в начальной стадии изотермы этого класса характеризуются вогнутой линией относительно оси концентрации ( L 1 в L2). По мере увеличения концентрации адсорбция достигает насыщения и приводит к образованию плато (L3) и протеканию процесса полимолекулярной адсорбции до достиже ния второго и последующих плато ( L4 ). L5 характерно для адсорбции ассоциатов ПАВ и красителей, но в чистом вида невозможен по термодинамическим причинам. 3. Класс Н - наблюдается при чрезвычайно сильной адсорбции при низких концентрациях. Обеспечено высоким сродством адсорбата и адсорбтива. 4. Класс C (постоянное распределение) наблюдается на микропористых телах. Теоретический анализ различных типов изотерм адсорбции позволяет получить большую массу полезной информации о механизме адсорбции. Константа в уравнениях адсорбции (Ленгмюра, БЭТ и др.) в основном связана с энергией активации удаления растворенного вещества с поверхности. Если взаимодействие между молекулами растворителя и растворенного вещества мало, и мы можем им пренебречь, то энергия активации не зависит от степени заполнения поверхности, что приводит к изотермам типа L или H. Если сила взаимодействия между адсорбированными молекулами больше силы взаимодействия между растворенным веществом и твердым телом, энергия активации возрастает, а совместная адсорбция описывается, S-образной изотермой. В этом случае молекулы растворенного вещества стремятся расположиться на поверхности в виде цепей или 26 кластеров. Подобному их расположению способствует сильная адсорбция растворителя и монофункциональный характер растворенного вещества. Рис. 12. Схема различных форм изотерм адсорбции по классификации Гильса Теоретический анализ различных типов изотерм адсорбции позволяет получить большую массу полезной информации о механизме адсорбции. Константа в уравнениях адсорбции (Ленгмюра, БЭТ и др.) в основном связана с энергией активации удаления растворенного вещества с поверхности. Если взаимодействие между молекулами растворителя и растворенного вещества мало, и мы можем им пренебречь, то энергия активации не зависит от степени заполнения поверхности, что приводит к изотермам типа L или H. Если сила взаимодействия между 27 адсорбированными молекулами больше силы взаимодействия между растворенным веществом и твердым телом, энергия активации возрастает, а совместная адсорбция описывается, S-образной изотермой. В этом случае молекулы растворенного вещества стремятся расположиться на поверхности в виде цепей или кластеров. Подобному их расположению способствует сильная адсорбция растворителя и монофункциональный характер растворенного вещества. Примером различного типа изотерм адсорбции может послужить адсорбция фенола и резорцина на оксида алюминия (полярный) и вода (полярный). В первом случае процесс описывается изотермой S-типа, а во втором L-типа. Параллельная ориентация молекул резорцина приводит к занятию активных центров при малых концентрациях и поэтому приводит к изотермам типа L. Красители и неионогенные ПАВ образуют в растворе агрегаты, и их адсорбция (циановые красители в водных растворах на галогенидах серебра) описывается изотермами S-типа. Изотермы сопровождается H -типа наблюдаются образованием в химического тех случаях, соединения когда адсорбция (хемосорбция). Так адсорбция стеариновой кислоты из бензола на сильнополярных порошках металлов Микропористые адсорбенты в основном характеризуются изотермой С -типа, так как число активных адсорбционных центров из-за их недоступности остается постоянным в широкой области концентраций. По мере заполнения молекулами адсорбтива одних активных центров появляются новые и доступная для адсорбции поверхность увеличивается пропорционально количеству адсорбированного из раствора вещества. В основном, между адсорбцией из растворов твердых (при комнатной температуре) и умеренно растворимых соединений, а также адсорбцией жидких ограниченно растворимых или твердых неограниченно растворимых соединений различия не делаются, тогда как на соответствующих изотермах наблюдаются характерные особенности. В первом случав уже при низких концентрациях адсорбция достигает некоторой предельной величины, после чего практически не изменяется (L2) Изотермы адсорбции жидких растворенных веществ часто имеют Sобразную форму - адсорбция быстро увеличивается по мере достижения предела растворимости (S3). 5. Теория адсорбции из бинарных растворов Адсорбцию из растворов следует рассматривать как процесс концентрирования на поверхности одного из двух адсорбирующихся компонентов. Исходя из фундаментального уравнения Гиббса d Г1d1 Г 2d 2 (25) 28 где Г1 и Г2 - величина Гиббсовой адсорбции компонентов, μ1 и μ2 - химический потенциал компонентов, чтобы связать Гиббсову адсорбцию с концентрацией компонента, необходимо воспользоваться уравнением Гиббса-Дюгема x1d1 x2d2 0 (26) где х1 и х2 - мольная доля компонентов. Тогда: 1 х2 d2 х1 (27) подставим в уравнение Гиббса (25) и получим: d ( Г 2 Г1 х2 )d2 х1 (28) Сделав допущение, что общее число молей до и после адсорбции остается неизменным, получим, что при адсорбции одного компонента равномерно увеличивается количество другого. Тогда Г1 =Г2, х2 + х1=1. Из этого получается: Г 2 (1 х2 ) и заменив d d2 (29) μ2 на с2 получим уравнение, подобное адсорбционному уравнению Гиббса для разбавленных растворов Г 2 (1 х2 ) с2 d RT dc2 (30) Типичным примером зависимости Гиббсовой адсорбции от состава бинарных смесей представлена на рис. 13. Точка пересечения отвечает такому состоянию системы компонентов в целом, при котором одинаковы составы раствора и поверхностного слоя. Это возможно только при следующем допущении: во-первых, первый компонент поверхностно-инактивен для второго и наоборот и, во-вторых, приравняв в поверхностном слое одного компонента равняется убыли второго компонента в этом слое. Г2 Г2 Г1 Х=1 Г1 Рис. 13. Зависимость Гиббсовой адсорбции от состава бинарных растворов 29 При адсорбции ПАВ из раствора на поверхность твердого тела соблюдается правило Траубе и его обращение в зависимости от природы твердой поверхности и растворителя (рис. 14). Газ Воздух Газ бензол вода бензол вода (неполярный) (полярная) Граница раздела фаз «газ – жидкость» Уголь Двуокись кремния (неполярный) (полярная) Граница раздела фаз «жидкость - твердое тело» Рис. 14. Схема ориентации ПАВ на границе раздела фаз "газ - жидкость" и "жидкость - твердое тело" При адсорбции из бинарных растворов наблюдается перераспределение компонентов х1 и х2 между объемом и поверхностным слоем. В общем виде подобный обмен можно представить в виде квазихимической обменной реакции с константой обмена Ка а2s a1 a1s a2 (31) где аis и аi - активности компонентов в поверхностном слое и в объеме. Выразив активность а через мольные доли и коэффициент активности Кf получим: s s x 2 x1 f f x2 x Ka s 1 2 Kf s 1 Kf K x1 x2 f1 f2 x1 x2 (32) где К коэффициент, учитывающий изменение коэффициента активности. Заменив мольные доли первого компонента на мольные доли второго, исходя из выражения (х1 + х2 = 1), получим: 30 K x2s (1 x2 ) (1 x2s ) x2 (33) Относительно мольной доли второго компонента выражение принимает следующий вид: x2s K x2 1 ( K 1) x2 (34) Это уравнение называют общим уравнением изотермы адсорбции из бинарных растворов с компонентов константой обмена. (концентрационная Константа константа) К - коэффициент равняется разделения термодинамической константе Ка (31) только в том случае, когда Кf = 1. х2 1 3 2 х2 Рис. 15. Изотермы адсорбции из раствора по уравнению (34) Проведем анализ уравнения (34) (рис. 15). Если х2 = 0, то при любом значении К можно пренебречь величиной (К-1)х2, и изотерма адсорбции принимает вид закона Генри : x2s K x2 (35) Таким образом отклонение от линейной зависимости x 2s от х2 наблюдается при К > 1 (кривая 1), и при К < 1 (кривая 2). С ростом концентрации и значительном отличии К от 1 ( К>>I и К<< I ) можно не учитывать влияние изменения Кf. Если К>>1, то выражение К-1 имеет положительный знак и уравнение принимает вид уравнения Ленгмюра: x2s K x2 , К''=К 1 K x2 (36) Если К<<1, то выражение К-1 приобретает отрицательный знак и уравнение (34) принимает следующее соотношение: x2s Наиболее K x2 1 K '' x2 интересной (37) представляется зависимость изменения знака величины (К-1) по мере изменения состава раствора при адсорбции бинарных 31 систем. Может наблюдаться такой случай, когда при малых концентрациях изотерма проходит выше диагонали (кривая 3 рис. 15) начала координат, а при повышенных концентрациях - ниже диагонали и имеет точку пересечения при К=1. Это соответствует проявлению адсорбционной азеотропии. Природа твердого тела, растворителя и растворенного вещества во многом определяют избирательность (селективность) адсорбции, которая характеризуется коэффициентом разделения - отношением коэффициентов распределения компонентов на поверхности и в объёме D где cs cv (38) сs - концентрация компонентов на поверхности, сv - концентрация компонентов в объеме. Тогда коэффициент разделения β для бинарных систем имеет следующее выражение: 1 / 2 и показывает, во сколько D1 c1s c2 D2 c2s c1 раз (39) различаются коэффициенты распределения разделяемых компонентов. Коэффициенты распределения и разделения не зависят от соотношения между количеством адсорбента и раствора и поэтому не могут характеризовать распределение массы вещества между этими распределение массы вещества целесообразно фазами. В этом характеризовать случае степенью извлечения и степенью разделения. Чем меньше степень извлечения вещества, тем лучше их разделение в поверхностном слое и тем хуже их разделение в объеме раствора. Для оценки адсорбируемости веществ широко используется правило полярности П.А.Ребиндера (1927г.). Это правило заключается в том, что вещество может сорбироваться на поверхности раздела фаз, если в результата его адсорбции будет уравниваться разность полярностей этих фаз. Таким образом по полярности адсорбируемое вещество должно занимать промежуточное положение между твердой поверхностью и растворителем. Наиболее характерными свойствами, определяющими в большинства случаев полярность компонентов является величина диэлектрической проницаемости ξ. (В настоящее время существуют более чувствительные характеристики, особенно для органических веществ. Поэтому адсорбция поверхностно-активного вещества, имеющего диэлектрическую проницаемость ξПАВ возможна в следующих случаях: ξтв > ξПАВ > ξр-ля или ξтв < ξПАВ < ξр-ля 32 Из правила уравнивания полярности следует, что чем больше разность полярностей между растворимым веществом и раствором, то есть, чем меньше растворимость вещества, тем лучше оно будет адсорбироваться. Поэтому дифильные молекулы ПАВ в водном растворе (рис. 14) адсорбируются таким образом (частокол Ленгмюра), чтобы выравнить полярность твердого тела и растворителя. 5. Адсорбция низкомолекулярных несмешивающихся и смешивающихся жидких систем В основном при изучении адсорбции из растворов на границе раздела "твердое тело - жидкость" экспериментально измеряется изменение концентрации раствора. Так как раствор содержит более одного компонента изотерма изменения концентрации, по существу, является изотермой изменения состава раствора и для двухкомпонентной системы описывается следующим соотношением: n0 где x n1s (1 x ) n2s x m (40) Δх- уменьшение мольной доли компонента, n0 - мольная доля исходного раствора, приходящаяся на m граммов адсорбента, n1s и n2s - число молей компонента 1 и 2 в расчете на 1 грамм адсорбента. Обозначения, символы и терминология приняты ИЮПАК. Так как в уравнение (40) входят две неизвестные величины, следовательно, индивидуальные изотермы могут быть получены только с использованием дополнительной информации. В этом случае возможны два варианта: Вариант 1. Если растворы сильно разбавлены и если растворимость растворенного вещества мала даже при большом n2s произведение n2sx < n 0 x m и n1s(1-x) = n2s (пример: стеариновая кислота в бензоле). Изотерма изменения состава раствора преобразуется в индивидуальную изотерму адсорбции растворенного вещества. Вариант 2. Если же изотерма адсорбции имеет максимум при мольной доли растворенного вещества более низкой, чем предел растворимости, что также возможно. В этом случае, несмотря на низкие значения x растворенного вещества, изотерма изменения состава раствора явля ется изотермой адсорбции только растворенного вещества. Можно предположить, что монослой заполняется при относительно низкой концентрации растворенного вещества и 33 n 2s = 0. Уравнение изотермы адсорбции приобретает следующий вид: n0 x n1s (1 x ) m (41) Следовательно, изотерма изменения состава раствора после завершения образования монослоя адсорбатом линейно падает с изменением концентрации. Определение количества растворенного вещества, адсорбированного при определенных условиях - задача несложная, но установить механизм адсорбции и влияние на него возможных переменных факторов намного сложнее и не всегда учтено. Известно, что на величину адсорбции на твердом теле из растворов первостепенное влияние оказывает поверхность, природа растворенного вещества, его химическое строение и взаимодействие с растворителем, природа самого растворителя, природа взаимодействия между поверхностью и адсорбированным из раствора веществом, структура адсорбированного слоя, его толщина и плотность упаковки, и, конечно, температура. Четкое представление о природе поверхности исследуемого образца, получение одинаковых поверхностей в различных условиях, характеристика основных свойств твердой поверхности - наиболее трудная задача. Загрязнение под действием окружающей среды оказывается достаточным, чтобы вызвать необратимые изменения в свойствах поверхности. Методы подготовки образцов, хотя и весьма разнообразные, но и они не дают возможность получить поверхности хотя бы с близкими свойствами. Наиболее доступным адсорбентом с наиболее воспроизводимыми свойствами поверхности являются графитовые сажи, а их использование дает надежную информацию при исследовании адсорбции из раствора. Химические свойства других модификаций углерода значительно отличаются, что связано с наличием на поверхности ряда кислотных и основных групп. Диоксид кремния в результате температурной обработки образует огромное количество пористых и непористых модификаций, отличающихся типом и числом гидроксильных групп. А диоксид титана образует известные модификации - рутил, анатаз и др., имеющие те или иные анионные группы, сказывающиеся на свойствах поверхности. Так например, при исследовании адсорбции двухосновных кислот (от щавелевой до себациновой) из водного раствора на графитовой саже - сферон 6 (по мере увеличения температуры прокаливания в вакууме и удаления кислотных соединений, определяющих сродство к полярному растворителю) было обнаружено увеличение адсорбции в ряду сферон 6 (2700°С) > графон > сферон 6 (1000°С) > сферон 6 (без обработки). 34 Фенол и нитробензол хуже адсорбируются на графоне по сравнению с полициклическими углеводородами в циклогексане, бензол и нафталин сильнее адсорбируются на сфероне 6, чем на менее полярном графоне. Данные по адсорбции полярных адсорбатов из растворов в углеводородах на поверхности диоксидов убедительно показывают насколько важно знать предысторию образца. Так, на непористых силикогелях с различной степенью гидроксилирования (2,8; 4,2; 7,9 ммоль ОН/м2) показано, что адсорбция алифатических спиртов из разбавленных растворов в тетрахлоруглероде (ССl4) повышается по мере увеличения степени гидроксилирования. Неоднократно отмечалось, что адсорбированная вода на поверхности диоксида кремния и титана в значительной мере способствует уменьшению адсорбции кислот и спиртов из разбавленных растворов в углеводороде. При анализе данных по адсорбции из растворов пористыми твердыми телами необходимо учитывать как химические, так и физические характеристики доступности поверхности. В ряде случаев, преимущественной адсорбцией обладают молекулы с малым размером, при этом степень и внутренней поверхности определяется отношением размеров пор и размеров молекул. Близость поверхности стенок пор, а также селективность адсорбции, определяемая химией поверхности твердого тела, в основном является следствием более сильной адсорбции внутри пор по сравнению с адсорбцией на внешней поверхности. Разделить оба фактора не всегда предоставляется возможным, поэтому в большинстве случаев интерпретация полученных данных адсорбции из разбавленных растворов пористых твердых поверхностях является до некоторой степени неопределенной. Особенности индивидуального строения молекул растворенного вещества такие как, наличие полярных трупп, длина цепи, структура кольца, физическое состояние в растворе, дипольный момент, постоянная Гаммета, растворимость вещества в растворителе оказывают существенное влияние на адсорбцию их на поверхности твердого тела. Соблюдение известного правила Траубе наблюдается только в случае простейших жирных кислот на угле из водных растворов в разбавленных концентрациях. Попытка установить закономерность адсорбции от длины углеводородного радикала спиртов, количества ароматических колец в молекуле, дипольного момента на широком спектре адсорбтивов не увенчалась успехом. Величина адсорбции молекул растворенного вещества определяется доступностью поверхностных групп, образованием "упорядоченного" слоя сольватированных углеводородных цепей и, в основном, не зависит от их длины. Растворитель в значительной мере определяет адсорбцию растворенного вещества на твердом теле так как, во-первых, их совместная растворимость и 35 отклонение от идеальности зависит от взаимодействия растворенного вещества в растворе, во-вторых, взаимодействием с адсорбатом, зависящим от их химической природы и структуры и, наконец, взаимодействие с растворенным веществом в адсорбционном слое вблизи границы раздела "твердое тело - раствор". Так как при исследовании адсорбции растворов в большинстве случаев не учитывается адсорбция растворителя на поверхности твердого тела, особенно когда равновесная концентрация раствора велика. Изотерма изменения состава раствора, которая может принимать как положительные, так и отрицательные зависимости, существенно отражает конкурирующую адсорбцию обоих компонентов. Существует пять классов изотерм (рис. 15). Рис. 16. Классификация изотермы изменения состава по Шаю и Надю. Ниже приведена типичная изотерма адсорбции, рассчитанная по экспериментальным данным при условии, что адсорбция монослойна, площадь, приходящаяся на моль компонента в адсорбционном слое, не зависит от концентрации адсорбата в растворе, и компоненты раствора не содержатся в твердой фазе (рис. 17). 36 Рис. 17. Изотерма адсорбции в системе этанол (1) и ксилол (2) (рутил при 298 К) и изотерма изменения состава раствора (3) В настоящее время благодаря хорошей экспериментальной базе, которая позволяет количественно оценить параметры жидкости на молекулярном уровне, стало возможным рассчитать и построить модель границы раздела "твердое тело раствор", базирующихся либо на решетчатой модели поверхности и раствора, либо на модели однородной "поверхностной фазы", находящейся в равновесии с бесструктурной жидкостью. Для расчета параметров решеточной модели используют метод Монте-Карло, который в значительной мере углубил наши представления о жидком состоянии. Применяемый в методе Монте-Карло подход требует, чтобы полная внутренняя энергия любого расположения молекул (конфигурации) в системе может быть вычислена или уже известна. Затем конфигурация молекул меняется (здесь применимы строгие правила учета состояния) и рассчитывается разность энергий между новой и старой конфигурациями, и полученная разница анергии позволяет судить о приемлемости этой новой конфигурации и так далее. Таким образом, совокупность принятых конфигураций образует некоторую цепь (цепь Маркова), которая позволяет рассчитать механические свойства каждой принятой конфигурации. Цепь обрывается в тот момент, когда среднее значение рассматриваемых механических свойств не зависит от некоторой выбранной произвольной конфигурации. Чтобы осуществить расчет по методу Монте-Карло, необходимо знать потенциальную энергию данной конфигурации системы, Если система стационарна, то ее внутреннюю энергию можно разложить, во-первых, на потенциальную энергию взаимодействия с гравитационным и другими внешними полями, во-вторых, кинетическую энергию теплового движения, молекул и, втретьих, потенциальную энергию взаимодействия между самими молекулами. Средняя, энергия первых двух вкладов получается непосредственно, а внутренняя энергия третьего типа слагается из 37 энергии отталкивания при малых межмолекулярных расстояниях, всегда существующей энергии дисперсионного взаимодействия моментов и электростатического (заряда, диполей), вклада Выражения постоянных потенциальных электрических функций парных межмолекулярных взаимодействий для атомов инертных газов достаточно хорошо известны, а простейший потенциал Леннарда-Джонса удовлетворительно выражает объединенное взаимодействие и определяется следующим образом, представленным при изучении природы адсорбционных сил (21). Кроме макроскопических свойств системы (давление, плотность, сжимаемость и энергия) методом Монте-Карло рассчитывают и микроскопические свойства - функцию распределения частиц по размеру. Однако, полученные модельные расчеты по методу Монте-Карло значительно отклоняются от реальной структуры жидкости, но разработанная и действующая "решеточная модель жидкости" в настоящее время позволила рассчитать модели жидкой фазы с учетом межмолекулярного взаимодействия, что и было сделано Гуггенгеймом. Другая модель, основанная на том, что граница раздала фаз может быть аппроксимирована тонким однородным слоем жидкости. Выделенный тонкий слой с одной стороны ограничен твердой поверхностью, а с другой стороны - объемом гомогенного раствора. В этом случае для расчета полной энергии межмолекулярного взаимодействия необходимо знать только соответствующие одночастичные или парные функции распределения, которые существуют в так называемом "липком жестком слое". Рассчитанный потенциал не обладает физическим смыслом, но учитывает действие сил притяжения и отталкивания и позволяет получить реальные макроскопические величины, например, второй вариальный коэффициент. Сравнение последнего с методом Монте-Карло единственная возможность проверить полученные результаты еще раз, чтобы сказать о перспективном начале термодинамической оценки состояния адсорбционного слоя. 7. Адсорбция поверхностно-активных веществ на твердых поверхностях Поверхностно-активные вещества (ПАВ) ведут себя в водных растворах иначе, чем большинство других, растворенных в воде веществ. Необходимо напомнить, что ПАВ при очень низких концентрациях находятся в свободном молекулярном состоянии (истинный раствор), а начиная с некоторой критической концентрации (ККМ) происходит агрегация, случай когда индивидуальные молекулы и мицеллярные кластеры существуют в динамическом равновесии. Дальнейшее увеличение концентрации ПАВ в водном растворе мицеллы различного размера и формы концентрируются в характерные симметричные структуры и образуют лиотропные изоморфные фазы. В результате адсорбции ПАВ существенно изменяют свойства границы раздела фаз при весьма низких концентрациях и в 38 следствие кооперативной самоассоциации они агрегируются с образованием мицелл. Существенно отличается теория адсорбции неионогенных и ионогенных ПАВ на твердом теле. 7.1. Адсорбция неионогенных ПАВ Рассмотрим наиболее простую адсорбцию неионогенных ПАВ, которую относят к представителям классической физической адсорбции. Неионогенные ПАВ от другого широкого круга ПАВ отличаются тем, что даже малые изменения концентрации, температуры, молекулярной структуры, природа поверхности твердого тела оказывают существенное влияние на их адсорбцию. Это, прежде всего, связано с тем, что взаимодействие “ПАВ – ПАВ” и “ПАВ – растворитель” вызывает агрегацию растворенного вещества в объеме или приводит к изменению ориентации и упаковки ПАВ на поверхности. На рис. 18 показана схема вероятных изменений ориентации молекул неионогенных ПАВ при адсорбции из водного раствора на твердом теле по мере увеличения концентрации. На первой стадии адсорбции (а), когда число активных центров и количество адсорбированных молекул ПАЗ очень мало, взаимодействием между молекулами ПАВ можно пренебречь. Адсорбция в основном определяется вандерваальсовыми силами, в большинстве случаев дисперсионными. Взаимодействие между твердым телом и гидрофобной группировкой ПАВ приводит к расположение молекул параллельно поверхности, так как последние адсорбируются положительно. Если молекулы при адсорбции располагаются параллельно поверхности, то энергия адсорбции увеличивается равными порциями с каждым дополнительным атомом в углеводородной цепи. При этом правило Траубе соблюдается для большинства неионогенных ПАВ и твердых поверхностей. По мере насыщения к монослойному насыщению горизонтально расположенными молекулами (б) наблюдается постепенное уменьшение наклона изотермы адсорбции. Можно предположить, что ПАВ в этом случае вытеснит большую часть молекул растворителя с поверхности, но все таки некоторое его количество остается на активных центрах твердого тела. Все последующие стадии адсорбции определяются главенствующей ролью взаимодействия молекул ПАВ друг с другом за счет гидрофобного взаимодействия, которая зависит от природы твердой поверхности и гипофильно-липофильного баланса ПАВ (соотношение полярной и неполярной составляющих молекулы). 39 Рис. 18. Модель адсорбции неионогенных ПАВ по мере увеличения концентрации ПАВ (а, б, в, г, д) и изотермы адсорбции (в) Например, в случае 1 мы имели неполярный адсорбент и ПАВ с короткой цепью (низкое ГЛБ). В случае 3, наоборот, преобладание длинноцепочечной молекулы ПАВ с высоким ГЛБ на сильнополярной поверхности. Вариант 2 некоторое промежуточное состояние. Третья стадия адсорбции (в), когда концентрация ПАВ в растворе приближается к ККМ, способствует агрегации цепей адсорбированных молекул. Это вызывает 40 вертикальную ориентацию. При этом наблюдается резкое увеличение количества адсорбированных молекул, некоторая десольватация, вытягивание скрученных молекул за счет сжатия головной группы и, в результате этого, значительная плотноупакованность монослоя. На неполярных адсорбентах наблюдали образование полумицелл на поверхности при концентрациях более низких, чем ККМ в объеме раствора. Это объясняется тем, что энергия адсорбции метиленовой группы почти равняется ее энергии мицеллообразования. Однако, на полярных адсорбентах, где полярная головная группа может довольно сильно связывается с поверхностью, а углеводородная цепь отделена от поверхности твердого тела на значительное расстояние, для достижения плотной упаковки понадобится значительно большая концентрация ПАВ в растворе, чем необходимо для ККМ. Дальнейшее повышение концентрации неионогенного ПАВ в растворе (г, д) приводит к тому, что взаимодействия в адсорбционном слое подобны взаимодействиям в объеме раствора. При этом изменение энтальпии обусловлено установлением равновесия между усиливающимися углеродно-углеродными взаимодействиями или головными группами и процессом разрушения сольватных оболочек. Отмечено, что теплота адсорбции становится постоянной, хотя с увеличением температуры адсорбция растет, так как головные группы становятся более компактными из-за меньшей сольватизируемости. Проведенные немногочисленные исследования показали, что поверхность твердого тела покрыта плотноупакованными полумицеллами, т.е. агрегатами такого размера и формы, которые можно получить, разделив находящуюся в объеме мицеллу пополам. В ряде случаев отмечалось, что в системах «неионогенное ПАВ полимерный адсорбент» при концентрациях ниже ККМ адсорбция отсутствует, а молекулы ПАВ удерживаются вблизи твердой поверхности дальнодействующими ван-дер-ваальсовыми силами через толстую прослойку сольватизированного слоя растворителя. Адсорбция в этом случав не происходит до тех пор, пока преобладает влияние поверхностной агрегации. Здесь требуется уточнение. Исходя из того, что большинство неионогенных ПАВ содержит полярную группу, которая достаточно сильно адсорбируется полярными поверхностями, например, за счет водородных связей, так что отсутствие адсорбции при концентрации ниже ККМ является кажущимся и может быть объяснено нечувствительностью эксперимента при низких концентрациях ПАВ. 7.2. Адсорбция ионогенных ПАВ Адсорбция ионогенных ПАВ на границе раздела «твердое тело – раствор» имеет огромное значение, так как определяет устойчивость коллоидных, систем и регулирует смачиваемость. Способность ПАВ в создании устойчивых коллоидных 41 систем лежит в фармацевтических основе и разработки красящих дисперсий, детергентов и эмульсионной антикоагулянтов, полимеризации и флотации. Смачивание играет важную роль при очистке, диспергировании и флотации, крашении и печатании, при использовании пестицидов и гербицидов. В процессе адсорбции в случае ионогенных ПАВ участвуют как минимум 4-х компонентная система, а в случае высоких концентраций ПАВ количество отдельных систем может достигать 8-ми, обладающих индивидуальными свойствами. Изменение смачиваемости жидкостью твердой поверхности при добавлении ПАВ определяется не только адсорбцией молекул самого ПАВ, но и адсорбцией двух других, смачивающихся в системе, границ раздела «газ - твердое тело» и «газ – жидкость». Из-за сложностей взаимодействий, которые происходят в подобных многокомпонентных системах, несмотря на большое количество исследований по этому вопросу, единой теории, объясняющей хотя бы удовлетворительно процесс адсорбции ионогенных ПАВ на твердом теле, к настоящему времени окончательно еще не выработано. Основным фактором, определяющим величину адсорбции ионов ПАВ на любой границе раздела фаз является электрическое взаимодействие в области двойного слоя. Таким образом, объяснить процесс адсорбции можно только в случае, когда известно, чем обусловлено возникновение поверхностного заряда, величины электрокинетического заряда и, конечно, структура двойного электрического слоя (ДЭС). Существует несколько механизмов образования поверхностного заряда, хотя общий поверхностный заряд из-за наличия в растворе противоионов и электронейтральности системы в целом равен нулю. Наиболее простейшими считаются модели AgI, BaSO4, где вследствие преимущественного растворения одного типа ионов решетки на поверхности кристалла адсорбируются потенциалопределяющие ионы, не влияющие на химический потенциал фазы. Грэм предложил модель двойного электрического слоя для границы раздела твердое тело - жидкость, которая приведена на рис. 19. 42 Рис. 19. Модель ДЭС по Грэму Общая плотность заряда σв внешней плоскости Гельмгольца и задней составляет σd, а дифференциальная емкость этой диффузной области равна Cd и параметры двойного слоя связаны между собой следующим комплексом уравнений: К1 = σ0 . (φ0 – φβ), (42) К2 = - σd . (φβ – φA), (43) К-1 = К1-1 + К2-1, (44) Cd = -dσd/dφA, (45) σ0 + σв + σd = 0. (46) В случае симметричного электролита 2kT zed sinh ze 2kT d (47) где ε - абсолютная диэлектрическая проницаемость объема растворителя, γ - обратная величина ионной силы раствора Дебая-Гюккеля, которая составляет при Cd 2 2z 2 e 2C d N A kT (48) z - валентность иона, е - заряд иона. Исходя из электрохимического равновесия между числом ионов на поверхности твердого тела и в объеме раствора, выведенного Нернстом, имеем 43 Δφ = NA(pX), где (49) Δφ - изменение внутреннего потенциала твердой фазы с изменением концентрации потенциалопределяющего иона типа X с зарядом z, N = - 2,3 (kT/ze), (50) Заменив Δφ на φ0, который характеризует изменение поверхностного потенциала вблизи поверхности твердого тела, связывающей ориентированные диполи воды (жидкости) внутри твердой фазы d 0 N d px (51) В изоэлектрической точке φd = 0 получим уравнение d 0 1 Cdk 1 d d (52) d d d px (53) Вводя выражение S1 и подставляя его в уравнение (52), получим: NS1-1 = 1 + C d0 k-1. (54) Так как φd нельзя измерить непосредственно, то для проверки уравнения используют легко экспериментально доступный ζ - электрокинетический потенциал, который с некоторыми допущениями позволяет рассчитать адсорбцию ионов на твердом теле, тогда уравнение приобретает следующий вид NS-1 = (1 + Cd0k-1) exp (æΔ1). (55) В изоэлектрической точке φd = ζ = 0 оценивать Δ1 нет никакой необходимости. При построении зависимости NS-1 от Cd0 для частиц AgJ в растворах KNO3 имеет вид прямой, отсекающей от оси ординат отрезок единичной длины, соответствующий Δ1, и по расчетам не превышает 0,5 Нм. Это позволяет считать справедливым применение равенства φΔ и ζ при объяснении межфазных явлений, в том числе включая адсорбцию ионогенных ПАВ. Также предполагают, что механизм возникновения заряда на поверхности неорганических оксидов SiO2, MgO, TiO2, Al2O3 и других определяется присутствием способных к ионизации групп, какими могут быть амфотерные гидроокисные группы. Механизм возникновения заряда, хотя и значительно идеализированный, можно себе представить следующим образом: H+ H+ MeОН2+ ↔ MeОН ↔ MeO¯. 44 В этом случае подобные межфазные поверхности заряжаются потенциалопределяющими ионами Н+ или 0Н¯, что справедливо для ионных решеток, которые подчиняются уравнению Нернста (51). В этом случае линейная зависимость NS-1 от Cd0 для TiO2 и SiO2, позволяет рассчитать, что Δ1 меньше 0,5 нм. Это еще раз подтверждает правильность предложенной модели и для поверхностей оксидов, а также позволяет количественно оценить поверхностные группы, способные к ионизации. Монофункциональные группы, такие как R–C–SO3¯, R–C–NH2¯ и другие, в результате включения их в процесс полимеризации латексов и присутствии на поверхности вызывают ионизацию, что в свою очередь приводит к возникновению поверхностного заряда. Поверхностный заряд газовой сажи связан с присутствием различных комплексов кислорода, химически связанных с атомом углерода базисных плоскостей и действующих как слабые кислоты. Другим механизмом объясняют образование заряда на поверхности глин и слюды. Эти слоистые силикаты построены из двумерных сеток кремнекислородных тетраидов и вследствие замещения некоторых атомов Si+4 на Al+3, или Al+3 на Mg+2 кристалл приобретает некоторый отрицательный заряд. Особенно это проявляется на ребрах чешуи. Исходя из построенной модели, число молей адсорбированного ПАВ, приходящихся на единицу поверхности (Г1) зависит от объемной концентрации раствора С1. Форма изотермы не всегда является правильным выводом о характере адсорбции. Для расчета применяют метод конгруэнтности (подобия) адсорбции по отношению к заряду или потенциалу. При этом адсорбция конгруэнтна к заряду, если электрическая составляющая свободной энергии (σ0),выраженная через заряд, является постоянной и в графической зависимости Г1 от С1 наблюдаются параллельные друг другу графики. Составляющие свободной энергии адсорбции в основном зависят от степени заполнения поверхности θ. Наиболее широко применимо описание изотермы адсорбции по уравнению Штерна-Ленгмюра 1 0 адс C1 exp 55,51 kT (56) где Δσ0адс - стандартная свободная энергия, составляющая энергию адсорбции, не зависящая от степени заполнения. При низких степенях заполнения (θ → 0) Грем вывел аналогичное уравнение 0 адс Г1 2rC1 exp kT (57) где r - радиус адсорбированного иона. Объяснение различных механизмов адсорбции разнообразных ионогенных ПАВ 45 и адсорбентов включает в себя то, что свободная энергия адсорбции состоит из нескольких составляющих Δσадс = Δσэл + Δσспец (58) Δσэл - электрическое взаимодействие, которое в свою очередь складывается из энергии раствора Δσр-ра и дипольного члена Δσдип: Δσэл = Δσр-ра + Δσдип (59) Δσэл = z1eφl + ∑Δni μi ES, (60) или где Δni - изменение числа адсорбированных диполей i с дипольным моментом μi, ES - напряженность электрического поля в плоскости адсорбированных частиц. Принято считать, что Δσдип обусловлен обменными процессами между ионами ПАВ в объеме раствора и в адсорбированном состоянии, происходящих в результате десорбции молекул воды с поверхности. Если пренебречь Δσдип, то можно рассчитать Δσэл во всех возможных вариантах. I ВАРИАНТ. Если заряд иона ПАВ противоположен по знаку общему объемному заряду (σ0 + σβ), то z1 и φβ имеют противоположные знаки, тогда Δσр-ра < 0 и электрическое взаимодействие способствует процессу адсорбции, соответственно, при низких степенях заполнения отрицательное для катионоактивных ПАВ и положительное для анионоактивных ПАВ. II ВАРИАНТ. Если заряд иона ПАВ имеет тот же заряд, то z1 и φβ имеют тот же знак, а Δσр-ра > 0. В этом случае электрическое взаимодействие препятствует процессу адсорбции. Однако при высоких степенях заполнения в силу преобладания специфической адсорбции происходит перезарядка в изоэлектрической точке. При этом анионоактивные ПАВ придают поверхности отрицательный заряд, а катионоактивные - положительный. Ш ВАРИАНТ. Δσэл равна нулю, тогда адсорбция определяется наличием специфической адсорбции Δσспец. Проведенные эксперименты показали, что ионогенные ПАВ в этом случае не адсорбируются на некоторых одноименнозаряженных поверхностях. На рис. 20 показано изменение адсорбции додецилсульфоната натрия на Al2O3 от рН раствора. Адсорбция уменьшается по мере понижения величины положительного поверхностного заряда. Следует заметить, что выше изоэлектрической точки кривые ζ -потенциала совпадают, что говорит об отсутствии процесса адсорбции. 46 Рис. 20. Зависимость ζ - потенциала Al2O3 от рН раствора додецилсульфоната натрия при ионной силе раствора 2.10-3 моль/дм3. Концентрация ПАВ, моль/дм3: 1 - 1.10-5; 2 - 3.10-5, 3 - 1.10-4; 4 - 2,5.10-4 Специфическая адсорбция Δσспец включает в себя все другие вклады в свободную энергию, зависящей от неэлектрической природы системы Δσспец = Δσцц + Δσцт + Δσпт + … где Δσцц - изменение свободной энергии при притяжении «цепь – цепь», Δσцт - то же, «цепь - твердое тело», Δσпт - то же, «полярная группа - твердое тело». Если «цепь – цепь» можно объяснить гидрофобным связыванием, то «цепь твердое тело» взаимодействиями. объясняется Характер Ван–дер–Ваальсовыми взаимодействия во многом дисперсионными определяется особенностями структуры ионогенного ПАВ вблизи поверхности твердого тела, поэтому имеет смысл говорить, что Δσцц и Δσцт включают как гидрофобное, так и дисперсионное взаимодействие. При этом Δσцт в значительной мере зависит от природы поверхности твердого тела, ассоциированной структуры адсорбированной воды и от того разрушается или не разрушается эта структура гидрофобными цепями. Иллюстрацией этого может послужить адсорбция ацетатов алкиламмония, отличающихся длиной углеводородной цепи при рН = 6÷7, приведенная на рис. 21. 47 Рис. 21. Изменение ζ - потенциала кварца при рН = 6÷7 от концентрации ацетатов алкиламмония. Как видно из рисунка адсорбция происходит на поверхности сильно гидратированного кварца и основной вклад в специфическую адсорбцию вносит взаимодействие «цепь – цепь». Эта составляющая приобретает первостепенное значение, так как адсорбирующиеся ионогенные ПАВ стремятся освободиться от водного окружения с образованием на поверхности твердого тела двухмерных агрегатов (полумицелл). До точки, соответствующей ККМ практически не происходит изменение ζ - потенциала поверхности кварца, однако, во всех без исключения случаях адсорбция резко увеличивается вследствие образования полумицелл и резкое изменение ζ - потенциала. Значение вклада взаимодействия «цепь - твердое тело» Δσцт играет существенную роль на неполярных адсорбентах, которые не вызывают структурирования приповерхностного слоя воды (может быть за исключением самого первого). В то же время, вклад взаимодействия полярная группа - адсорбент наиболее четко проявляется в тех случаях, когда преобладает химическое взаимодействие. Применение модели двойного электрического слоя для адсорбции ионогенных ПАВ и описание изотермы адсорбции согласно выражениям (56) и (57) возможно только при низких степенях заполнения. Так же как выставленные условия: 1) специфически адсорбируются ионы только ПАВ, 2) один ион ПАВ замещает одну молекулу растворителя, т.е. они имеют одинаковый размер, 48 3) поверхность считается однородной, применимы в случае длинноцепочечных молекул ионогенных ПАВ и расстояние как внутри адсорбционного слоя Гельмгольца, так и от слоя Гельмгольца до границы скольжения Δ1 может быть равным только в том случае, когда концентрация ПАВ мала и низка степень заполнения. При этом соблюдается правило Траубе, что показано в табл. 2. Таблица 2 Величина Δσспец и ККМ анионогенных ПАВ в водном растворе (рН = 2) на золе нейлона 6-6 ПАВ - Δσспец / кТ ККМ, моль/дм3 Децилсульфонат С10 6,4 4,4 . 10-2 Додецилсульфонат С12 8,4 9,8 . 10-3 Гексадецилсульфонат С16 12,9 8,0 . 10-4 (500С) Из таблицы видно, что инкремент в величине Δσспец на одну СН2 - группу в гомологическом ряду увеличивается на 1кТ, что отражает взаимодействие «цепь твердое тело» при низких степенях заполнения. Вблизи ККМ изотерма адсорбции ионогенных ПАВ в основном достигает плато, а дальнейшее концентрирование раствора вызывает только рост количества мицелл, которые в этом случае уже не являются поверхностно-активными. Адсорбционный максимум объясняется неоднородностью твердой поверхности, вымыванием из адсорбента легко растворимых веществ, которые солюбилизируются (поглощаются) мицеллами, а также наличием незначительных количеств твердых частичек (центров адсорбции ПАВ) в растворе. Состав мицелл в ряде случаев значительно отличается от состава объема смеси ПАВ, состоящих из молекул различной длины. Плотноупакованный слой на границе раздела фаз в зависимости от силы взаимодействия «монослой – поверхность» может быть достигнут при концентрациях ионогенного ПАВ как выше, так и ниже ККМ. В случае поверхности углеродного типа плотноупакованный слой молекул ПАВ представляет собой полумицеллу с малой кривизной с вертикальной ориентацией молекул. Однако на поверхности раздела с высокой энергией (сильнополярные) адсорбция углеводородных радикалов вряд ли возможна. Отмечается, что хотя анионогенные ПАВ адсорбируются на твердом теле, но их плотность упаковки адсорбированного слоя определяется электростатическим взаимодействием, ориентацией молекул ПАВ вглубь полислоя, а не полярностью поверхности раздела. 49 7.3. Пленки Ленгмюра-Блоджетт Заслуга в установлении мономолекулярной природы слоев на границе раздела «жидкость – газ» принадлежит лауреату Нобелевской премии Ленгмюру, который первый перенес монослои поверхностно-активного вещества с поверхности воды на твердые подложки. В дальнейшем Блоджетт усовершенствовала методику получения моно- и мультимолекулярных слоев ПАВ и разработала технологию переноса их на твердую поверхность. Taк родилось «молекулярное зодчество», которое в настоящее время широко используется при молекулярном (надмолекулярном) конструировании, «молекулярной инженерии», молекулярной и биоподражательной электронике, создании "синтетических" организмов, а также создании молекулярного ансамбля с заранее заданной архитектурой для определенных электрических и оптических характеристик. В качестве таких молекул широко используются жирные кислоты и их соли (мыла), эфиры, фосфолипиды для получения биослойных мембран, красители, ароматические соединения слоистой структуры и автокомплексы. Суть метода создания пленок Ленгмюра-Блоджетт на водной поверхности состоит в том, что с помощью плавучего барьера происходит уменьшение площади поверхности, занимаемой ПАВ. При этом дифильные молекулы ПАВ структурируются из газообразных пленок (I стадия) в жидкие (2 стадия) и жидкокристаллические (3 стадия) (рис. 23) Стадия 1 2 Стадия 2 1 3 Стадия III 5 50 Рис. 23. Ориентация молекул ПАВ на поверхности воды в зависимости от площади, занятой монослоем (метод Ленгмюра) 1-барьер; 2-блок; 3-груз; 4-механизм подъема и опускания; 5-поверхность. Последовательность фазовых переходов или полиморфизм монослоев исследуют, наблюдая за соотношением между величиной поверхностного давления (π) и площадью (S), приходящейся на одну молекулу монослоя. На рис. 24 показан сложный пример полиморфизма азокрасителя, при этом конформационное состояние молекулы меняется по мере увеличения поверхностного давления. Дальнейшее увеличение поверхностного давления приводит к “сминанию” монослоев или коллапсу и может образоваться двух, трех и четырехслойная структура. π, мН/м - азофрагмент - эфирные группы - углеводородная цепочка Рис. 24. Полиморфизм монослоев ПАВ азокрасителя Перемещением (мм в минуту) твердой поверхности (5) (рис. 23, стадия 3) через 51 поверхность монослоев опусканием или подниманием обеспечивается различная ориентация ПАВ, изображенная на рис. 25. Обычно различают три типа слоев: два полярных X и Z и один симметричный, неполярный слой Y. X Y Z Рис. 25. Типы мультислоев ПАВ на твердой поверхности. Рентгеновские и электроннографические методы контроля показали, что ленгмюровские пленки на твердых поверхностях зачастую имеют твердокристаллическую структуру заранее заданной толщины и ориентации. Качество пленок зависит от чистоты взятых компонентов, значения рН буферного раствора, температуры эксперимента и состояния подложки. Стабильные мономолекулярные слои на твердой подложке получают методом адсорбции ПАВ из жидкой фазы, а также из смеси ПАВ, красителей методом "вымывания" селективными растворителями из монослоя молекул красителя с целью получения скелетизированного монослоя (рис. 26 ) Смешанный монослой Монослой из ПАВ + растворитель, вымывающий краситель Рис. 26. Схема создания скелетизированного монослоя Moно- и мультислои зачастую получают контактным методом с использованием гибких полимерных пленок (рис. 25). 52 Рис. 27. Иллюстрация принципа отрыва (1) и контактного (2) нанесения монослоя. В зависимости от природы ПАВ, его размера, формы, катиона металла, плотности упаковки, количества мультислоев наблюдаются изменения электропроводности, диэлектрических характеристик, туннельный эффект, волновые свойства, показатель преломления, дихроизм красителей, пиро- и пьезоэффекты, фотопроводимостъ, фото ЭДС, фотохимические реакции, которые в настоящее время находят широкое применение в электронной промышленности. 8. Адсорбция электролитов (ионов) Адсорбция электролитов требует специального рассмотрения, так как адсорбент, имеющий различные по активности функциональные группы, может по различному адсорбировать малые по размеру ионы, на которые диссоциируют молекулы низкомолекулярных веществ в растворе. Ионы электролита в растворе являются носителями электрического заряда, поэтому при переходе ионов из объемной фазы на твердую поверхность в результате адсорбции происходит перераспределение зарядов и возникновение двойного электрического поля (ДЭС) в области поверхностного слоя. Исходя из электронейтральности адсорбции ионов, перенос ионов из объема к поверхности происходит только как электронейтральное объединение нескольких ионов, не сопровождающееся каким-либо изменением заряда. Поэтому (ДЭС) всегда электронейтрален и раствор, в котором произошла адсорбция ионов. Электрохимический процесс образования ДЭС безусловно является адсорбционным, так как концентрация ионов в поверхностном слое меняется. При этом следует выделить две возможности процесса перехода ионов на поверхность: 1) из раствора электролита на поверхность твердой фазы для уравнивания поверхностного натяжения на границе раздела - адсорбция; 2) из твердой фазы в жидкую, что приводит к адсорбции растворителя и сольватации ионогенных групп твердой фазы – поверхностная диссоциация. Рассмотрим оба процесса на конкретных примерах. Наиболее типичным примером ионной адсорбции является образование мицелл иодида серебра в избытке одного из компонентов реакции. AgNO3 + KJ = AgJ↓ + KNO3 Введем дополнительные обозначения 53 □∆ + ○ ↓ٱ□ = ٱ+ ○∆ В избытке AgNO3 получим следующую электронейтральную мицеллу, состоящую из агрегата AgJ и адсорбированных на ее поверхности ионов (рис. 28). поверхность граница межмицеллярная скольжения жидкость Рис. 28. Схема строения двойного электрического слоя для мицеллы AgJ с избытком AgNO3 Сольватированные ионы (□) Ag+ в результате специфического взаимодействия прочно удерживаются на нескомпенсированных валентностях J- ()ٱ, которые с избытком находятся на поверхности твердой частицы AgJ, имеющей размер порядка 40-70 нм. В данном случае ионы Ag+ (□) следует называть потенциалопределяющими (ПОИ), так как при исследовании электрокинетических свойств (электрофорез) данной коллоидной частицы она движется к аноду. В то же время, вблизи поверхности в результате дальнодействия адсорбционных сил, несмотря на тепловое движение и наличие значительного диффузионного слоя, простирающегося на расстояние 400-1600 А, располагаются противоионы NO3- (∆), делающие мицеллу электронейтральной, а также электронейтральной остается и межмицеллярная жидкость. При рассмотрении образования ДЭС за счет поверхностной диссоциации вещества твердой фазы следует отнести типичный пример образования силановых групп в морской воде, способствующей лучшей биологической очистке морской воды по сравнению с речной (рис. 29). Таким образом в результате диссоциации SiO2 с образованием в воде иона гидроксония H3O- поверхность твердого тела заряжается отрицательно, а в растворе образуются противоионы положительного знака для придания системе в целом электронейтральности. 54 Si OH Si O Si Si O OH Si O Si O H Si H3O O H3O O Si O H O OH O O Si O H O H3O Рис. 29. Схема строения двойного электрического слоя для частицы SiO2 в воде И в первом и во втором случаях возможно изменение знака заряда поверхности за счет потенциалопределяющего иона на противоположный. Это наблюдается для системы AgJ при избытке KJ и для SiO2, когда в избытке находятся ионы ОН-. Используя при изучении адсорбции ионов модель Грэма (рис. 18) можно увидеть, что если охарактеризовать размеры ионов величиной ионного радиуса ( r ), то внутренняя неподвижная часть ДЭС (слой Штерна) представляет собой адсорбированный моноионный слой толщиной не менее двух ионных радиусов, порядка 1-5 А для неорганических ионов. В развитии представлений о ДЭС Штерн попытался учесть специфическую адсорбцию и ее влияние на электрический потенциал, обусловленный действием дополнительных ковалентных сил к действующим электростатическим силам. Общая плотность заряда ДЭС складывается из плотности заряда слоя Гельмгольца и диффузного слоя d общ d (62) При этом, согласно Штерну, заряд слоя Гельмгольца складывается из заряда ионов, адсорбированных за счет электростатического адсорбционного потенциала ( F z φ ) и за счет некоторого дополнительного потенциала специфической адсорбции ( Ф ). Если представить некоторую квазихимическую реакцию при условии, что на одном активном центре может адсорбироваться один потенциалопределяющий ион, то константа этой реакции приобретает следующий вид: k exp( где Fz Ф ) RT F - число Фарадея (96500 кулонов на моль), z - валентность иона, φ - электроионный потенциал, 55 (63) Это позволило Штерну рассчитать плотность поверхностного заряда в слое Гельмгольца с учетом специфической адсорбции по уравнению: где 1 1 Fz Ф exp( ) с0 RT (64) - предельная плотность заряда для данных противоионов с учетом гидротации в слое Гельмгольца, c 0 - концентрация противоионов. Так как теория Штерна учитывает наличие плотного адсорбционного слоя ионов на поверхности, то таким же образом можно выяснить и влияние размера гидратированного иона на плотность заряда. При изучении влияния лиотропного ряда (ряды Гофмейстера ) на ионную адсорбцию было установлено, что при одинаковой валентности наибольшей адсорбцией обладает тот ион, который имеет больший размер. Причина этого заключается как в большей поляризуемости этих ионов, так и меньшей толщине сольватной (гидратной) оболочки. Наличие гидратной оболочки в значительной мере снижает электрическое взаимодействие, а, следовательно, и способность иона притягиваться поверхностью. На рис.30 в качестве примера приведена гидратация ионов щелочных металлов. h=7,44 h=6,85 h=6,07 h=5,22 Рис. 30. Гидратация ионов щелочных металлов Таким образом в лиотропном ряду по возможности адсорбироваться катионы одновалентных щелочных металлов располагаются в следующий ряд: 56 Li+ < Na+ < K+ < Rb+ а, щелочноземельные двухвалентные в аналогичный ряд по мере увеличения величины ионного радиуса катиона: Be2+ < Mg2+ < Ca2+ < Sr2+ < Ba2+ r,А Одновалентные 0,54 анионы 0,78 1,06 располагаются 1,28 в 1,36 следующий ряд по мере возрастающей адсорбционной способности: F- < Cl- < Br- < NO3- < J- < NCSr,А 1,33 1,81 1,96 – – 2,20 Наибольшее влияние на адсорбцию ионов оказывает валентность иона. Так с увеличением валентности на единицу адсорбционная способность иона возрастает как минимум на порядок. Это приводит к полной компенсации потенциалопределяющих ионов в плотном слое Гельмгольца и в результате этого противоионы могут выступать в роли новых потенциалопределяющих с противоположным знаком противоионов. Это явление называется перезарядкой и характеризуется сменой противоионов в диффузном слое на ионы с зарядом другого знака. По адсорбционной способности ковалентные катионы располагаются в следующий ряд: Изотерма адсорбции ионов отвечает квазихимической реакции и описывается кривыми С-типа (рис.11). Поэтому действие лиотропных рядов объясняется взаимодействием между ионами различного размера и представлено на рис. 31. - - + сильное слабое + слабое сильное Рис. 31. Схема взаимодействия ионных пар, отличающихся размером Тенденция этого взаимодействия в объеме раствора хорошо демонстрируется показаниями коэффициента активности, а качественное объяснение можно увидеть, признав существование сильного электростатического притяжения вследствие электростатического поля, возникающего между малыми катионами и анионами. В случае больших катионов и анионов связь между ними осуществляется сильным гидрофобным взаимодействием. 57 В зависимости от заряда поверхности в лиотропных рядах при адсорбции наблюдается обратная зависимость действия ионного радиуса. Это объясняется наличием отрицательной адсорбции, присущей в большей степени ионной адсорбции. Если при обычной адсорбции концентрация ионов уменьшается, то при отрицательной адсорбция адсорбции, вне области модифицированным пропорциональна наоборот, увеличивается. диффузионного уравнением слоя Ленгмюра, концентрации с , так может то как Положительная быть отрицательная в этом случае ионная описана адсорбция совместно адсорбирующиеся ионы характеризуются неспецифической адсорбцией (рис. 32). Отрицательная адсорбция, г/г Концентрация г/г Рис. 32. Отрицательная адсорбция (∆) SO 42 и ( ) PO43 на AgJ, полученная методом меченых атомов. Используя отрицательную адсорбцию как способ определения площади поверхности удалось получить идеальные характеристики поверхности, так как у ионов отсутствует агрегация или какое-либо поперечное сечение молекулы. Этот метод применим к очень низким поверхностям порядка 1 м2/г. Большое значение в технологии, биология и земледелии имеет обменная адсорбция, которая протекает как на поверхности твердого тела, так и, намного замедленная, в объеме его. В результате обмена между нонами вследствие обменной адсорбции твердая поверхность поглощает из объема определенное количество ионов одного заряда и при этом диффундирует в объем эквивалентное количество ионов того же знака, чтобы система в целом была электронейтральна. В обмене участвует не только чужеродные ионы, адсорбированные поверхностью, но в ионы самого адсорбента в результате диссоциации его молекул даже из объема твердого тела, если доступ растворителя возможен. Обменная адсорбция специфична - в обмене участвуют не все ионы, присутствующие в объеме, а только строго определенные. На обменной адсорбции сказывается природа твердой поверхности, 58 плотность и заряд двойного электрического слоя, образующегося за счет селективной адсорбции, а также природа иона, участвующего в обменной адсорбции. Специфичность обменной адсорбции, по своей природе приближаемой к химической реакции, зависит от химической природы поля и подразделяется на кислотную (обмен катионов) и основную (обмен анионов). В природе существует большое количество адсорбентов, характеризующихся амфотерными свойствами поверхности, которые в одних условиях обменивают катионы, а в других - анионы. Из-за специфического взаимодействия ионов с твердой поверхностью обменная адсорбция не всегда обратима, и, в отличие от молекулярной, ионная адсорбция протекает медленно. Это связано с тем, что необходимо затратить большое количество времени, чтобы был вытеснен ион, находящийся в глубине адсорбента, так как проникновение иона размером 5 А на глубину 10 А (в зазор 10 А) потребуется 103 секунд, а обратная диффузия вытесненного иона происходит еще медленнее. Огромное значение при обменной адсорбции играет рН среды, и наиболее существенное влияние оказывает при адсорбции из водного раствора, когда в обмене участвуют Н+ или OН- ионы. При адсорбции происходят замена катиона на водородный ион (Н+) адсорбент Н+ + Nа+ + Сl- → адсорбент Nа+ + Н+ + Clв силу большего электростатического притяжения ионов Сl- происходит уменьшение рН (кислая среда) и адсорбент ведет себя как катионит. Наоборот, если адсорбент меняет анион на ион гидроксония (ОН-), то адсорбент ведет себя как анионит, при этом рН среды увеличивается адсорбент ОН- + Nа+ + CI- → адсорбент Сl- + Nа+ + OH-. Никольский (1927) предложил следующее уравнение, связывание содержание обменивающихся ионов 1 и 2 (°С) на твердой поверхности с их активностью (а) и валентностью (z) 0 1 z1 0 1 z2 с1 с2 где k 1 z1 a1 1 z2 a2 (65) k - константа обменной адсорбции при одинаковой валентности; z – валентность иона; a – активность иона. При небольших концентрациях ( a ≈ c ) уравнение, связывающее адсорбционную способность ионов с их концентрацией, имеет следующий вид: 59 с10 c k 1 0 с2 c2 где (66) k’- константа обменной адсорбции с учетом коэффициентов активности ионов, участвующих в химической реакции. Наиболее широко обменная адсорбция протекает в почве, где поглощающий комплекс, состоящий из алюмосиликатов и органоминеральных соединений, участвуют в обмене необходимых для растений ионов К+ и NH3+ на Са2+ и Mg2+ Типичной необратимой обменной адсорбцией является процесс умягчения воды пермутитами и глауконитами - минералами, содержащими алюмосиликаты: пермутит 2-2Na+ + Ca2+ + SO42- → пермутит 2-Ca2+ + 2Na+ + SO42После обработки полученного соединения концентрированным раствором NaCl происходит регенерация пермутита и он вновь может участвовать в процессах очистки воды. В последнее время широкое применение нашли ионообменные смолы. Другим типичным адсорбентом, на поверхности которого происходит обменная адсорбция (широко применяемым в быту) являются древесные и животные угли. Согласно Фрумкину в период изготовления углей и при его активации на поверхности может адсорбироваться как водород, так и кислород, выступающие донорами или акцепторами электронов. угольН2 → уголь 2- 2Н+ Взаимодействуя с парами или вода угольО2 → уголь неустойчивые 4+ ионы 2О2O2- образуют гидроксильные ионы уголь 4+ + 2О2- + Н2О → уголь 2- + 2ОНАктивированный уголь выступает в роли катионита или аммонита, изменяя рН среды. По Шилову при активизации на поверхности угля образуются тончайшие слои окислов, прочно связанные с кристаллической решеткой адсорбента. В одних условиях получения эти окислы при взаимодействии с парами воды образуют карбоксильные группы, способные обменивать водород на катион, а в других условиях получения - на поверхности угля образуются гидроксильные группы, способные заменять ОН- группы на анион. Часть углей, получаемых в некотором промежуточном режиме обладают амфотерными свойствами, так как на их поверхности находятся одновременно группы первого и второго типа. Приведенные примеры обменной адсорбции относятся к природным ионитам (ионообменники). В настоящее время наиболее широкое применение находят синтетические ионообменные смолы, обладающие 60 хорошей способностью поглощать ионы и, в отличие от природных, высокой химической стойкостью. Обменная адсорбция с помощью синтетических смол может происходить во всем объеме вещества, так как растворенные ионы свободно проникают внутрь решетчатой структуры ионита. Ионообменные смолы получаются как методом конденсации, так и методом полимеризации из мономеров, содержащих активные функциональные группы с заранее заданными свойствами. Причем смолы, полученные методом полимеризации, обладают регулированной сетчатой структурой с определенной степенью поперечной связи для ограничения набухания. Типичный пример получения ионообменных смол с заранее заданными свойствами методом полимеризации путем введения функциональных групп: CH СН CH СН2 CH2 CH2 NH3 Сl CH2 O CH3 CH2NH2 N(CH3)3 CH2Cl CH CH2 CH2 N(CH3)3 Cl Для увеличения контакта между ионами и смолами, последние дробят до размера зерен 0,5-2 мм, так как дальнейшее увеличение их дисперсности хотя и способствует росту обменной адсорбции, но и в значительной мере способствует повышению гидродинамического сопротивления слоя. Основными свойствами ионообменных смол являются: 1. Высокая обменная емкость - число грамм-эквивалентов извлекаемых с помощью I грамма ионита. Ёмкость определяется числом активных функциональных групп. При этом необходимо различать: а) статическую обменную емкость (СОЕ) (теоретически рассчитанная), причем природные иониты имеют малую СОЕ - 0,2-0,5 Мэкв/г, а синтетические 3-10 Мэкв/г; б) динамическую обменную емкость (ДОЕ), которая значительно ниже СОЕ, так как в ионном обмене участвуют не все функциональные группы, 61 содержащиеся в ноните, а также от размера ионообменной колонки, скорости движения раствора, плотности упаковки зерна и другие. Кислотно-основные: 2. ИОНИТ Катиониты Амфотерные 1. Сльнокислотные Аниониты 2. Сильноосновные (-SO3H) (- N+(CH3)3Cl-) 2. Слабокислотные 2. Слабоосновные (-ОН, -СООН, -SiOH) (-NH2, =NH2, N) 3. Набухаемость. Синтетические иониты набухают на 200-600% (в ряде случаев более чем на 2000%), что связано с получением гидрофильных функциональных групп, и увеличение расстояния между функциональными группами способствует участию этих групп в обменной адсорбции. Набухание характеризуют степенью набухания где m m0 m0 (67) m0 - вес ионита до контакта со средой, m - вес набухшего образца, и константой скорости набухания, зависящей от термодинамической активности растворителя, гидротации противоионов, разбавления раствора и степени сшивки полимера 1 k ln max max (68) Кинетика набухания ионитов, способствующая увеличению обменной емкости по мере раздвижения полярных функциональных групп, происходит в пятистадийном процессе, характеризующемся собственной скоростью: I) диффузия адсорбирующихся ионов из объема к поверхности твердого тела (самая быстрая); 2) диффузия внутрь зерна ионита (медленная); 3) ионный обмен; 4) диффузия замещенных ионов к поверхности ионита (самая медленная); 5) диффузия от 62 поверхности зерна в объем. В зависимости от природы ионитов и ионов, участвующих в обменной адсорбции, их концентрации, размера зерна, температуры проведения процесса ионного обмена установленные равновесия колеблются от нескольких минут до многих суток. 4. Химическая стойкость и механическая прочность. Наиболее важной характеристикой прежде всего является стойкость к действию агрессивных сред (кислот, щелочей, солей, органических веществ, окислителей, восстановителей), под действием которых может произойти разрушение структуры ионита. Химическая стойкость определяется по потере обменной емкости после действия агрессивных сред и долговечности воздействия. Механическая прочность определяет многократность использования ионита в процессе обменной адсорбции. Снижение механической прочности вызывает изменение фракционного состава зерен ионита после определенного числа циклов адсорбции и десорбции, а также в результате встряхивания ионита в вибрационном аппарате. Наилучшими химическими и механическими характеристиками обладают иониты, полученные полимеризацией. 5. Селективность. Селективность ионита характеризуется константой обменной адсорбции, отражает сродство твердого тела к тому или иному нону и имеет следующий вид: k c2 c1 1 z2 1 z1 1 z1 c1 1 z2 c2 (69) аналогично уравнению Никольского (66) с учетом коэффициентов активности ионов, участвующих в ионном обмене. При одинаковой валентности ионов уравнение (69) выражает коэффициент разделения. Селективность ионита зависит от упругости сетки полимера, из которого состоит зерно. По мере сопротивления набуханию за счет упругости ионит поглощает менее гидратированный ион, поэтому меньшему размеру совместно с сольватной оболочкой в лиотропном ряду Li - Cs и отвечает адсорбционная способность Li+ < Na+ < K+ < Rb+ < Cs+. Если ионы имеют разные по величине заряды, то селективность адсорбции заключается в том, что ионит предпочитает адсорбировать или поглощать противоион с большим зарядом. В этом случае по адсорбционной способности катионы располагаются согласно возрастанию валентности Na+ < Ca2+ < Al3+ < Th4+. Селективность обменной адсорбции зависит от состояния ионов в растворе; чем меньше активность или сродство, тем хуже идет процесс ионного обмена. Ионный обмен можно представить в виде изотермы адсорбции одного или второго иона, и уравнение изотермы обменной адсорбции аналогично уравнению молекулярного 63 обмена (34) (рис. 14). Широкое применение ионный обмен нашел не только в умягчении к очистке воды, но и извлечении ценных компонентов - урана, золота, платины и т.д. Ионный обмен используется в производстве редкоземельных металлов с высокой степенью чистоты и для исследований методом ионообменной хроматографии. 9. Адсорбция полимеров на твердом теле. Адсорбция высокомолекулярных соединений (ВМС) - полимеров на твердых поверхностях существенно отличается от адсорбции низкомолекулярных веществ, что связанно не только с макромолекулярной природой адсорбирующихся молекул, но и с возникновением большого числа конформаций как в объеме раствора, так и на границе раздела. Каждой концентрации раствора в зависимости от природы растворителя твердой поверхности (кроме самых разбавленных) соответствует своя присущая только этой системе конформация макромолекулярного клубка степень взаимопроникновения ассоциатов и их агрегация как в объеме, так и на поверхности. Для гибких полимерных молекул в растворе происходит значительное уменьшение энтропии при адсорбции, по сравнению с низкомолекулярными или жесткими полимерными цепями. Уменьшение энтропии при адсорбции макромолекул полимера еще усугубляется и тем, что присоединение цепей к активным центрам поверхности происходит многими возможными способами. Это дает возможность конформации применения макромолекул представления об различных на статистических поверхности особенностях и адсорбции методов разработать макромолекул расчета теоретические и строения адсорбционного слоя. Процессы адсорбции полимеров на твердых поверхностях из растворов играют большую роль в формировании свойств многих композиционных материалов. Одним приходится учитывать испарение растворителя при создании подобных материалов, а, это в свою очередь, требует необходимости детально рассматривать адсорбцию полимеров из разбавленных и концентрированных растворов отдельно. Если имеющиеся теории адсорбции полимеров на твердых поверхностях из разбавленных растворов в значительной мере применимы, то теории адсорбции полимеров из концентрированных растворов практически отсутствуют. Это связанно с отсутствием количественной теории концентрированных растворов, что затрудняет интерпретацию полученных результатов. Термодинамическое качество растворителя по отношению к полимерной или другой молекуле раствора в настоящее время характеризуется параметром Хаггинса-Флори χ. Исходя из теоретических расчетов параметр χ меньше нуля для “очень хороших” растворителей, проходя через нуль для “плохих” растворителей и 64 достигает величины 0,5 в квазиидеальном случае (Ө-растворитель). Экспериментальные значения χ не превышают (0,3-0,5), для водных растворов(0,45-0,5). Физический смысл параметра χ становится ясен при рассмотрении процесса обмена сегментов из объема чистого молекулярного полимера с молекулой чистого растворителя, с учетом всех межвалентных энергий взаимодействия. Примем следующие обозначения: Z-координационное число 1растворитель 2-полимер. Тогда процесс обмена происходит при разных Z контактов 1-1 или 2-2 с образованием 2Z контактов 1-2. Энтальпия этого процесса (h) в расчете на единичный контакт определяется как Z(2h12-h11-h22).Поскольку в процессе обмена образуются 2Z контактов 1-2, параметр χ определяется в расчете на одну молекулу (в единицах KT). χ =Z(h12-h11-h22)/KT. (70) При анализе этого уравнения видно, что при χ >0 растворитель “плохой”, в нем достаточно сильны взаимодействия 1-1 и 2-2, и не образуются 1-2. Однако, и в этом случае, растворы полимеров термодинамически устойчивы, при условии, что их энтропия смешения превосходит эту высокую энтальпию. С появлением третьего компонента – твердого тела – в теории адсорбции полимеров вводится параметр χ s. Физический смысл χ s состоит в том, что молекула растворителя, находящаяся на поверхности, обменивается с частью макромолекулой полимера (сегментом), находящимся в растворе, при этом значительно изменяется взаимодействие между полимером и растворителем – χ. С учетом происходящего обмена параметр энтальпии адсорбции χ s имеет такой вид: χ s=Z' ( hs1 – hs2+1/2h22 – 1/2h11)*(kT) (71) Z'- доля контактов растворителя, приходящихся на поверхность (S). χs имеет положительный знак, при условии предпочтительного взаимодействия: «твердое тело – сегмент», и имеет величину (0,1 – 0,2kT) на сегмент. В адсорбированной линейной макромолекуле различают три вида расположения сегментов на поверхности твердого тела. Согласно Каргину под сегментом понимают значения молекулярной массы, которое должны были бы иметь молекулы полимера для того, что бы полимер или его раствор подчинялись закономерностям низкомолекулярных веществ. Для гибких макромолекул сегмент представляет собой отрезок цепи, состоящий из 20 – 30 атомов элемента, образующего главную цепь. С появлением полярных групп, двойных и тройных связей, разветвление и его частота в значительной мере уменьшается длина сегмента за счет дополнительных взаимодействий внутри самой макромолекулы и молекула становится жесткой (менее гибкой). На гибкость макромолекулы оказывает влияние температура и 65 время воздействия внешней среды. Известно, что релаксационные переходы от одной конформации макромолекулы к другой требуют определенного времени. Быстрое высыхание, например, способствует значительному объединению конформационного набора, что приводит к увеличению гибкости молекулы и возрастанию длины сегмента. Рис 33. Макромолекула на твердой поверхности 1) хвост, 2) петли, 3) сегменты, контактирующие с поверхностью твердого тела, 4) эшелон. При расчете χs учитываются аддитивные составляющие энтропии адсорбции сегмента, а также ориентационное действие растворителя, которое приводит к гидрофобному связыванию, однако, энтропия конформационного перехода « петли сегмент», контактирующий с поверхностью - « хвост » в расчете χs не выполняется из-за сложности определения. Существует множество теорий адсорбции полимеров на твердых поверхностях, но наиболее принятые в последнее время теории ЛалаСтепто и Сильберберга, Рое, Шетьенса и Флира, основанные на анализе конформационной статистики адсорбированной молекулы, состоящей из сегментов, контактирующих с поверхностью, петель, хвостов и учитывающих взаимодействие между отдельными сегментами и между сегментом и растворителем. Модель Лала-Степто предполагает переход от одной конформации к другой происходит как последовательность изменений вращательных положений небольшого числа последовательно соединенных связей. При этом принимается число связей минимальное (Z = 4), решетка – тетраэдрическая и остальные цепи молекулы не претерпевают изменений. Используя метод Монте-Карло учитывали только те конформации, в которых макромолекула взаимодействовала с поверхностью, и описывается тремя энергетическими переходами от транс и гош – конформациям с резкой энергией ∆E взаимодействия. При этом учитывается только энергия, отвечающая углам между группами 0, 120, -120 градусов. ∆E12=E12 - 1/2 (E11+E22). E12 - энергия взаимодействия между сегментом и растворителем. 66 (72) E11 – энергия взаимодействия между молекулами растворителя. E22 – то же между сегментами макромолекул полимера. Проведенные расчеты позволили определить среднюю долю адсорбированных сегментов « P », как функцию энергии адсорбции Es/kT цепи, состоящую из 100 сегментов и толщиной δм . При этом учитывается, что в результате увеличения доли связанных сегментов и уменьшение толщины слоя, вызывающее изменение конформации цепей полимера энергии связи сегмента с в адсорбционном слое поверхностью. С и приводяott ее к росту учетом размера и формы адсорбированной цепи и цепи со свободным вращением через среднеквадратичный размер молекулы (R2) с радиусом вращения (r2), стало возможным описать плотность расположения сегментов по толщине слоя для разных энергий адсорбции. Расчет показал, что при E=0,5 kТ плотность сегментов мало меняется в первых слоях вблизи поверхности. Затем медленно падает, а молекулы находятся на поверхности в растянутом состоянии. Средняя доля адсорбционных сегментов возрастает по мере ухудшения взаимодействия между полимером и растворителем, а толщина адсорбционного слоя уменьшается. На рис.34 показано изменение конформаций сегментов, рассчитанных методом Монте-Карло при ΔE1=0,5; 0,00 и -0,5kТ. Конформация выступает в виде высоты каждого сегмента над поверхностью в одномерном изображении, встречающихся с высокой частотой. ΔЕ=0,5 kT ΔЕ=0,0 67 Е=-0,5 kT R2=100-6000 R2=1500-2000 R2=100-300 V=20-40 V=20-60 V=30-40 S2=290-700 S2=170-250 S2=60-100 τ=10-21 τ=5-15 τ=6-7 Рис.34. Схема изменения конформации при адсорбции. Как видно из рисунка, в «хорошем» растворителе отталкивание сегментов вместе с положительным значением и приводит к растянутому жесткому положению цепи. В «плохом» растворителе наибольшая вероятность взаимодействия сегментов друг с другом и с поверхностью, а молекулы в этом случае находятся в состоянии плотно упакованного клубка с большим числом сегментов, связанных с поверхностью. В теориях Хуве и состоящая из набора Сильберберга рассматривается изолированная цепь, петель, хвостов и сегментов, адсорбированных на поверхности. Сильберберг нашёл выражение суммы по состояниям, исходя из предположения об узком распределение петель по размерам, с учётом самопересечения изолированных полимерных молекул, в том числе и для θ растворителя (∆ε=0,5kT). Хуве вывел приближенные аналитические выражения для адсорбции полимеров при любых величинах параметра χ, определяющего взаимодействие «полимер-растворитель». Эти теории адсорбции полимеров на твёрдом теле показали неправомерность подразделения адсорбционного слоя на поверхностный и диффузионный. На практике плотность слоя как функция распределения в основном зависит от конкретной конформации макромолекул. При расчете профиля плотностей и средней толщины слоя учитывается и ислюченный обмен, который всегда присутствует в реальной системе как результат межцепного взаимодействия. 4 доля заполнения, θ 1 2 3 2 4 0 68 r длина цепи Рис.37. Зависимость степени заполнения (θ) от длины цепи при χ=0,5 для различных теорий адсорбции полимеров. 1.Хуве; 2.Сильберберг; 3.Шетьенс-Флир; 4.Роэ -2 плотность lg 10 -4 10 -6 10 1 2 3 4 20 40 60 количество адсорбированных слоев Рис.38 .Профиль плотности в адсорбционном слое от количества полимерных слоёв при χ=0,5 для различных теорий адсорбции полимеров (обозначения аналогичны рис.37).. В теориях Роэ и Шетьенса-Флира также применялась решеточная модель адсорбции полимерных сегментов в растворе любой природы на твёрдых поверхностях. При этом предполагалось, что пространственное распределение любого сегмента цепи не зависит от его порядкого номера в цепи и конформация характеризуется указанием порядкового номера слоя, в котором находится каждый из последующих сегментов цепи. 69 Из рис.38 видна существенная разница в моделях расчёта адсорбции полимеров различными методами, хотя в разбавленных растворах это не очень влияет на толщину адсорбционного слоя. Теорию адсорбции полимеров, основанную на соотношениях скейлинга для полуразбавленных растворов в хороших (атермичных) растворителях, предложил Де Женн, как чисто аналитическую без использования численных методов. Рассматривалась модельная цепь, состоящая из сегментов, для которых реальный объём и исключенный объём равны между собой, хотя это предположение не является очень жестким. Следующее ограничение состоит в том, что для обеспечения отсутствия эффектов, связанных с концентрированием молекул вблизи поверхности (взаимодействие отталкивания высшего порядка), энергия адсорбции мала (χs<<1). Для этого Де Жен использует параметр γi, который связан с χs следующим выражением: γi =kT /a² (73) где a² - площадь узла на поверхности. При этом предполагается, что любые вклады в "энергоприлипание" (γi), связанные с растворителем, обращаются в ноль в результате сродства полимера и растворителя. В своих предположениях Де Жен выделяет три области (режима) в адсорбционном слое: 1. Ближайшая область, в которой осуществляется взаимодействие “сегментповерхность” имеет плотность сегментов, зависящую от энергии адсорбции и убывает, т.к. толщина этого режима соизмерима с размерами сегмента. Образуется некий домен контактируемый с поверхностью. 2. Центральная область, в которой адсорбированные молекулы образуют нестационарную сетку ,соединенных между собой цепей также как в полуразбавленном растворе. Характеристический сегментов, размер аналогично ячейки сетки зависимости зависит от координационной локальной длины плотности сегмента от концентрации его в объёме полуразбавленного раствора. Де Женн считает, что в адсорбционном слое расстояние от межфазной поверхности играет роль корреляционной длины для плотности сегментов по мере удаления от границы раздела. При этом образуется так называемая “самоподобная” структура центрального режима. 3. Периферийная область, в которой концентрация резко падает до уровня концентрации сегментов в объёме. При этом в периферийном режиме в основном 70 доминирует «хвосты», и нет никаких предсказаний относительно величины плотности числа сегментов. Несмотря на то, что поверхностный избыток поддаётся расчету, но в этом случае не устанавливается зависимость этой величины от концентрации в объёме, так как предполагается, что свободная энергия сегментов адсорбированного полимера значительно (или несколько) превышает свободную энергию системы и не зависит от молекулярной массы полимера, аналогично результатам Роэ или Шетьенса и Флира. Изотерма адсорбции макромолекул полимера на твёрдых поверхностях, выведена Сильбербергом для плотноупакованных самоагрегированных глобул в растворе. При этом предполагалось, что электростатические эффекты, специфические для данной системы, носят второстепенное значение по сравнению с большим размером макромолекулы. Рассматривается компактная кубическая макромолекула, объём которой в p раз больше объёма молекулы растворителя, то есть в трёхмерной решётке такая макромолекула имеет p мест. Каждое место в решетке имеет Zв соседей объёме и Zs соседей на поверхности. При этом число эквивалентных слоёв Ө - система заполнения поверхности. ρ-доля связанных сегментов. зависит как от параметра взаимодействия полимер с растворителем χ, так и полимер-твердое тело χs при условии, что при адсорбции изменяется доля сегментов в равновесном растворе Ф*, изотерма адсорбции имеет следующий вид: ln(Ө/ Ф*)=Рln(1- Ө)/ln(1- Ф*)+Pln ρ +(1- ρ)ln(1- ρ) + ρ χ/Zв[Zp(1-2 Ф*)-(Zp-1)(1-2Ө)]+ + ρ χs/Zs, (74) где Zp-координационные числа системы., Итак, сложный математический вид изотермы адсорбции с учётом процессов, происходящих при агрегации макромолекул полимера на твёрдой поверхности с образованием нового равновесия «агрегат-поверхность» позволяет однозначно интерпретировать полученные экспериментальные данные. При адсорбции длинноцепочечных полимеров в растворе не все активные центры будут заняты частями цепей макромолекул. Согласно современным представлениям об адсорбции полимеров на твёрдых поверхностях адсорбционные центры делятся на занятые (1), свободные (2), экранируемые (3) и недоступно экранируемые (4),представленные на схеме рис.35. петля хвост 71 3 1 1 4 12 Рис.39. Активные центры твердой поверхности. В процессе адсорбции все меньше становится свободных активных центров, по истечении времени при достижении равновесия осуществляется взаимодействие цепей макромолекул и с экранированными центрами, а недоступные центры зачастую остаются и после высыхания полимера на поверхности твёрдого тела, являясь дефектами, сказывающимися на прочности связи между полимером и подложкой. Решение ряда дифференциальных уравнений с помощью численных методов на ЭВМ позволяет раскрыть временную картину изменения числа макромолекул, частиц твёрдой фазы и их агрегатов, а также адсорбированных макромолекул в виде функции распределения макрообъектов по размерам. Типичная изотерма адсорбции для широкого интервала концентраций полимера при χ=0,5представлена на рис.25 доля заполнения поверхности, lg 10-2 III * 10-4 II 10-6 I 72 10-30 10-20 10-10 lg , концентрация Рис.40. Изотерма адсорбции полимера χ=0,5 по расчету Сильберберга. I - область Генри, адсорбированные молекулы ведут себя как изолированные цепи. II-область псевдоплато, где молекулы начинают собираться в агрегаты и адсорбция слабо зависит от концентрации. III-область концентрирования, где θ постепенно увеличивается, стремясь достичь величины, характерной для объёма. Таким образом, изотерма адсорбции носит сложный характер. При малых концентрациях полимера, который ведет себя как отдельная несвязанная молекула, адсорбция соответствует изотерме Н-типа (рис.11). Это связано с тем, что согласно мультиконтактной теории макромолекула считается адсорбированной, если адсорбировался хотя бы один её сегмент. Это происходит при самых малых концентрациях даже в θ-растворителе. По мере увеличения количества полимера в растворе преобладает межмолекулярное взаимодействие между адсорбированными молекулами(поверхностная агрегация) и его влияние на конформацию адсорбированных цепей и толщину слоя определяет специфическое отличие адсорбции полимеров от адсорбции низкомолекулярных соединений. При этом для адсорбции полимеров из умеренных растворов в отличие от адсорбции из разбавленных растворов наблюдается инверсия термодинамического характера растворителя на адсорбцию. В основном, среднеконцентрированные растворы предпочтительнее адсорбируются из растворов в “хорошем” растворителе. В концентрированных растворах (в зависимости от природы растворителя, молекулярной массы полимера, температуры) взаимодействие агрегатов молекул приводит к образованию плотноупакованных клубков размером от 50 до 200нм с продолжительным временем жизни. Эти вновь образовавшиеся клубки (кластеры, домены, рои) преимущественно адсорбируются на поверхности твёрдого тела, особенно в первый промежуток времени, до наступления равновесия, на что требуется несколько суток. Этим и объясняется переходы агрегатов на поверхность, что вызывает большие величины адсорбции, инверсию влияния природы растворителя и наличие максимумов на изотермах адсорбции. От выяснения механизма адсорбции полимеров, находящихся в виде отдельных молекул, ассоциатов, агрегатов, клубков целесообразно рассматривать процессы, протекающие при адсорбции макромолекул; адсорбцию, ассоциацию макромолекул и агрегацию частиц адсорбента. Кинетика адсорбции описывается следующим уравнением: 73 dA 4nF * DRC1C2 , dt (75) где A-число адсорбированных молекул, t-время; n-отношение числа адсорбированных молекул к общему числу молекул в растворе. F*-коэффициент эффективности взаимодействия макромолекул с поверхностью. D- коэффициент диффузии макромолекул полимера. C1-концентрация частиц адсорбента. С2-общее число макромолекул и их ассоциатов в единице объёма. R = (r1+r2)-радиус взаимодействия частиц адсорбента и макромолекулы. Вид кривой, полученной при расчете уравнения адсорбции, соответствует типичной зависимости с насыщением, которая тем медленнее, чем выше молекулярная масса полимера. В случае полидисперсной системы известно, что обладающие большой низкомолекулярные скоростью фракции диффузии сначала высокомолекулярного адсорбируются соединения, которые постепенно вытесняются, обладающими большей адсорбционной способностью высокомолекулярными цепями. Это и приводит к смещению установления равновесия. Увеличение температуры при адсорбции сказывается на изотерме адсорбции, так как возрастание теплового движения молекул вызывает разрушение агрегатов в разбавленных растворах и уменьшение размера клубков считать тот в концентрированных растворах. Следовательно, адсорбционным слоем можно слой макромолекулы, который образуется на поверхности вследствие адсорбции и в котором часть сегментов макромолекул и молекулы растворителя находятся в поле действия поверхности твёрдого тела. Толщина такого слоя в основном зависит от концентрации полимера и определяется структурой агрегата Ниже приведена таблица, где показано изменение толщины адсорбционного слоя, определённого по изотерме адсорбции пентафталевой смолы ПФС на TiO2. Таблица2. Изменение толщины адсорбционного слоя ПФС на TiO2 в А° от концентрации смолы ,масс.%. Концентрация смолы, масс.% 5 20 40 45 50 55 60 65 Толщина слоя, А 26 100 150 300 460 590 800 1020 74 При этом изотерма адсорбции, а следовательно и толщина адсорбционного слоя зависит от природы растворителя и адсорбента, а также метода оценки. 4 2 Адсорбция, г/г 1 0,1 0,2 3 концентрация, масс % Рис.41. Изотерма адсорбции эпоксидной смолы ЭД-5 на твёрдом теле. 1 и 2-стеклосферы; 3 и 4-сажа ПЖ-50;1 и 3-из раствора в толуоле; 2 и 4-из раствора в ацетоне Рис.42. иллюстрирует сложность процессов адсорбции макромолекул полимера в широких концентрациях и показывает многообразие видов кривых, которые при расчете дают искаженные значения толщины адсорбционного слоя, определенную динамическими методами. Однако, с использованием неразрушающих методов установлено, что толщина адсорбционного слоя возрастает с увеличением молекулярной массы полимера и зависит от природы подложки и растворителя. 75 Рис.42. Эллипсометрическое измерение толщины адсорбционных слоёв полистирола, адсорбированного из θ-растворителя, ( χ=0,5), от молекулярной массы. 1-из метанола на золоте(Au); 2-из метанола на хроме(Cr); 3-из циклогексана на платине(Pt); 4-из циклогексана на хроме(Cr). Резюмируя вышесказанное о процессе адсорбции полимеров на твёрдых поверхностях можно считать, что большинство полимеров адсорбируются на поверхностях, так как при адсорбции молекула полимера теряет часть конформационной энтропии. При этом требуетя минимальная свободная энергия 76 адсорбции, а учитывая многочисленность сегментов в макромолекуле, адсорбированных на поверхности, количество связанного с твёрдым телом молекул полимера значительно увеличивается, если свободная энергия адсорбции превышает некоторую величину. Характер изотерм адсорбции полимеров отражает высокое сродство к поверхности, поэтому в начальной стадии, в случае разбавленных растворов, величина адсорбции резко увеличивается, а при концентрировании растворов наблюдается некоторое псевдонасыщение. При этом толщина адсорбционного слоя достигает 2-5 монослоёв и величина превышает 10 мг/м². В большинстве случаев адсорбция увеличивается по мере ухудшения качества растворителя, возрастает с повышением молекулярной массы полимера в «плохом» растворителе и практически не зависит от температуры. Произвести десорбцию макромолекул полимера разбавлением раствора практически невозможно из-за высокого сродства с твёрдой поверхностью, но макромолекулы могут в течение определённого времени обмениваться на другие, находящиеся в растворе молекулы. Адсорбция полимеров более длительный процесс по сравнению с адсорбцией низкомолекулярных веществ, что объясняется как медленной диффузией громоздких цепей макромолекул, так и необходимостью протекания конформационных изменений вблизи поверхности. 10. Методы исследования адсорбции на границе раздела раствор твердое тело 10.1. Термодинамические методы К ним относятся методы определения концентрации вещества в растворе до и после адсорбция и изменение энергии системы по данным калориметрии. Концентрацию компонента измеряют методом титрования, постоянного взвешивания в растворе, изменение окраски раствора оптическими методами, методом меченых атомов, с применением ИК, УФ, ЭПР и ЯМР спектроскопии. В настоящее время широко применяются современные методы, такие как термогравиометрия, элементный анализа (рентгено-флюоресценция) и другие. 10.2. Методы, связанные со спектроскопией и рассеянием К ним относится метод ИК-спектроскопии, основанный на наблюдении определенной колебательной моды (являющейся характеристикой адсорбата), которая сдвинута по частоте за счет специфического взаимодействия с подложкой. В настоящее время широкое применение находит метод усиленного поверхностью комбинированного рассеяния света (УПКРС), смысл которого состоит в том, что электронные колебательные моды в металле создают сильное электромагнитное поле в примыкающем к поверхности растворе, усиливая тем самым спектр 77 колебательного рассеяния молекул на подложке. Другими широко применяемыми методами безусловно являются резонансные методы ЯМР (ядерно-магнитный) и ЭПР (электронно-пара-магнитный), которые используют изменение подвижности молекулы у плоскости границы контакта по сравнению с сольватирующими молекулами и контактирующими непосредственно с молекулами. Недостатком этого метода является обязательное наличие элементных меток, обладающих собственными адсорбционными характеристиками. Плотность упаковки по толщине адсорбированных молекул, особенно макромолекул полимеров, на плоских оптически отражающих поверхностях хорошо контролируется методом эллипсометрии. Сущность метода состоит в том, что в результате взаимодействия молекул с твердой поверхностью в растворе образуется слой некоторой толщины, имеющий отличный показатель преломления по глубине в результате поляризации отраженного света. В последнее время широкое применение находит метод малоуглового рассеяния нейтронов, основанный на рассеянии нейтронов за счет взаимодействий отталкивания, происходящих на очень малых расстояниях. При этом интенсивность рассеяния на определенном ядре зависит от его спина и, в меньшей степени, от массы, интенсивность и фаза рассеянных нейтронов зависит от положения, динамики и структуры ядра. 10.3. Гидродинамические методы Методы вязкого течения и истечения через капилляр, являющиеся наиболее простыми и ранними по разработке, позволяют рассчитать толщину адсорбционного слоя за счет увеличения вязкости разбавленной суспензии или уменьшения вязкости раствора. Увеличение эффективной объемной фракции частицы рассчитывается по уравнению Стокса-Эйнштейна. 10.4. Электро-химические методы Эти методы основаны на изменении состояния двойного электрического слоя ДЭС в результате взаимодействия молекул и ионов с твердой поверхностью и различаются на статические и динамические. В результате адсорбции молекул и ионов на поверхности твердого тела образуется диффузионный слой, обращенный к объему раствора, противозарядов, в котором и потенциал и плотность зарядов монотонно убывает с расстоянием. В отличии от потока растворителя составляющие потока расположены исключительно в ДЭС (вблизи поверхности, 100-400 А), где отклонение профиля потока от "нормального" является весьма большим. В результате электрокинетическая "плоскость скольжения" не фиксируется и сильно зависит от 78 толщины двойного слоя, ионной силы, скорости движения частицы или потока, вязкости среды и длины адсорбированных молекул и, тем более, макромолекул полимеров. 10.5. Методы разъединяющего давления Эти методы основаны на определении пространственного взаимодействия между двумя сближающимися поверхностями. Расстояние между поверхностями в момент появления силы пространственного взаимодействия между адсорбированными слоями дает двойную толщину каждого адсорбционного слоя. К этим методам диспергированными относятся измерение частицами, взаимного запакованными в разнесения некоторую между геометрически правильную решетку и подвергающихся воздействию внешней силы, приводящей к сжатию (механическое, ультразвуковое, центрифугирование и т.д.). Широкое применение находит метод тонких пленок, которые находятся на микроскопически малых поверхностях, и под воздействием внешнего давления вызывается деформация адсорбционного слоя, в результате чего образуется микроскопическая плоскопараллельная пленка. Независимо от того, что используется в качестве твердой поверхности, частицы размером в несколько мкм, цилиндры, кварцевые нити и плоскости, тонкие пленки в месте контакта позволяют оценить взаимодействие слоев с ошибкой в 5-10%, что для данного метода является весьма эффективным. 79 11. Список литературы 1. Воюцкий С.С. Курс коллоидной химии. М.: Химия, 1964 и 1975. 2. Фролов Ю.Г. Курс коллоидной химии. М.: Химия, 1982. 3. Фридрихсберг Д.А. Курс коллоидной химии. Л.: Химия, 1984. 4. Адамсон А. Физическая химия поверхностей. М.: Химия, 1979. 5. Липатов Ю.С. Коллоидная химия полимеров. Киев: Наукова думка, 1984. 6. Де Бур Л. Динамический характер адсорбции. М.: И.Л., 1962. 7. Липатов Ю.С. Межфазные явления в полимерах. Киев: Наукова думка, 1980. 8. Адсорбция из растворов на поверхности твердых тел / Под ред. Г.Партита и К.Рочерста. М.: Мир, 1986. 9. Лопаткин А.А. Теоретические основы физической адсорбции. М.: МГУ, 1983. 10. Абрамзон А.П. Поверхностно-активные вещества. Л.: Химия, 1981. 11. Сумм Б.Д., Горюнов Ю.В. Физико-химические основы смачивания и растекания. М.: Химия, 1976. 12. Грег С., Синг А. Адсорбция, удельная поверхность, пористость. М.: Мир, 1984. 13. Твердохлебова И.И. Конформация макромолекул. М.: Химия, 1980. 14. Липатов Ю.З., Сергеева Л.С. Адсорбция полимеров. Киев: Наукова думка, 1972. 15. Липатов Ю.С. Физико-химия наполненных полимеров. Киев: Наукова думка, 1977. 16. Русанов А.И. Фазовые равновесия и поверхностные явления. Л.: Химия, 1967. 17. Измайлов И.А. Электрохимия растворов. М.: Химия, 1966. 18. Фиалков Ю.Я., Житомерский А. Я., Тарасенко Ю.А. Физическая химия неводных растворов. М.: Химия, 1972. 19. Курс физической химии / Под ред. чл. корр, АН СССР Я.Л. Герасимова. М.: Химия, 1970, 1973. 20. Щукин Е.Д., Перцев А.В., Амелина Е.А. Коллоидная химия. М.: МГУ, 1982. 21. Джейкоп М., Парфит Дж. Химия поверхностей раздела фаз. М.: Мир, 1984. 22. Неппер Д, Стабилизация коллоидных дисперсий полимерами. М.: Мир, 1986. 23. Kawaguchi M., Tekahashi A. The structure of macromolecules adsorbed on interfaces.// Advances in Polymer Science, Behavier of Macromolecules. Springer. New York. 1982.v.42.P.1-65 – перевод 84/12006 80 Издание учебное Киселев Владимир Яковлевич, Комаров Владимир Михайлович Адсорбция на границе раздела «твердое тело – раствор» Учебное пособие Подписано в печать ……………. …..2005. Формат 60x84x16. Бумага писчая. Отпечатано на ризографе. Уч. изд. листов …… Тираж 100 экз. Заказ № ………. Лицензия на издательскую деятельность ИД №03507 (рег. №003792) код221 Московская Государственная Академия тонкой химической технологии им. М.В.Ломоносова Издательско-полиграфический центр 119574, Москва, пр. Вернадского, 86. 81 .