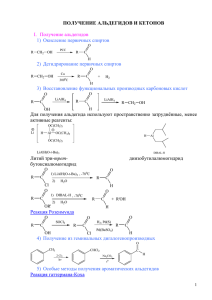
Диссертация Кошелевой А.М. - Институт химии и химической
реклама

ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ УЧРЕЖДЕНИЕ НАУКИ ИНСТИТУТ ХИМИИ И ХИМИЧЕСКОЙ ТЕХНОЛОГИИ СИБИРСКОГО ОТДЕЛЕНИЯ РОССИЙСКОЙ АКАДЕМИИ НАУК На правах рукописи Кошелева Александра Михайловна НЕПРЯМОЕ ЭЛЕКТРОКАТАЛИТИЧЕСКОЕ ОКИСЛЕНИЕ АЛИФАТИЧЕСКИХ СПИРТОВ ДО КАРБОНОВЫХ КИСЛОТ IN SITU ГЕНЕРИРОВАННЫМИ АКТИВНЫМИ ФОРМАМИ КИСЛОРОДА 02.00.04 — физическая химия Диссертация на соискание ученой степени кандидата химических наук Научный руководитель: д.х.н., профессор В.Л. Корниенко Красноярск — 2014 2 СОДЕРЖАНИЕ ВВЕДЕНИЕ................................................................................................................. 4 ГЛАВА 1. Литературный обзор .............................................................................. 9 1.1 . Физико-химические свойства спиртов и карбоновых кислот алифатического ряда. Применение карбоновых кислот. .......................................................... 9 1.1.1.Физико-химические свойства алифатических спиртов. ............................ 9 1.1.2. Физико-химические свойства алифатических карбоновых кислот. ...... 11 1.1.3. Применение карбоновых кислот ............................................................... 14 1.2. Общие лабораторные и промышленные методы получения карбоновых кислот ......................................................................................................................... 15 1.3. Электрохимические методы получения карбоновых кислот ........................ 18 1.3.1.Электрохимическое окисление спиртов .................................................... 19 1.3.2. Редокс-медиаторное и непрямое электрокаталитическое окисление органических веществ........................................................................................... 21 1.3.3. Химически связанные активные формы кислорода. ............................... 26 1.3.4. Электрокаталитическое окисление спиртов на основе редокс- медиаторных систем ............................................................................................. 28 1.4. Влияние условий электролиза на процесс электрокаталитического окисления ............................................................................................................................... 33 1.4.1. Материал анода, состав электролита и величина потенциала ................... 33 1.4.2. Строение спирта и температура .................................................................... 38 1.5. Постановка задачи исследования. .................................................................... 39 ГЛАВА 2. Экспериментальная часть.................................................................. 41 2.1. Условия эксперимента ....................................................................................... 41 2.1.1. Рабочие растворы и реактивы. ................................................................... 41 2.1.2. Электродные материалы, электроды и электролиты. ............................. 41 2.1.3. Методика проведения электрокаталитического окисления спиртов..... 44 2.2. Приборы, оборудование и методы исследования ........................................... 50 2.3. Обработка экспериментальных данных........................................................... 54 3 ГЛАВА 3. Экспериментальные результаты и их обсуждение ....................... 55 3.1. Кинетические закономерности непрямого электрокаталитического окисления алифатических спиртов ......................................................................... 55 Заключение ................................................................................................................ 68 3.2. Влияние основных параметров электролиза на эффективность электрокаталитического окисления алифатических спиртов............................... 70 Заключение ................................................................................................................ 76 3.3. Влияние химической природы электродного материала и способа генерации АФК .......................................................................................................... 77 Заключение ................................................................................................................ 81 3.4. Постановка опытов по наработке целевого продукта .................................... 81 ВЫВОДЫ .................................................................................................................. 87 СПИСОК ЛИТЕРАТУРЫ ..................................................................................... 89 4 ВВЕДЕНИЕ Актуальность работы. В последние годы в связи с ужесточившимися требованиями к экологической безопасности химико-технологических процессов, разработка экологически чистых и эффективных процессов, соответствующих принципам «Green Chemistry», привлекает большое внимание исследователей и продолжает быть главной тенденцией в развитии современных химических технологий. Алифатические спирты являются сравнительно дешевым исходным сырьѐм для получения карбоновых кислот, которые широко востребованы в различных отраслях современного промышленного производства: фармацевтической, витаминной, косметической и автомобильной. При этом окисление спиртов до соответствующих карбоновых кислот является одним из основных процессов в органическом синтезе, который протекает с использованием сравнительно дорогих и токсичных химических окислителей (перманганат калия, соединения хрома и т.п.). В то же время, согласно литературным данным, методы непрямого электрокаталитического окисления in situ генерированными активными формами кислорода (АФК), различающимися по окислительной способности на 5–6 порядков, являются экологически чистыми и перспективными для получения органических продуктов и разрушения органических субстратов в сточных водах. В связи с этим изучение возможности их применения для окисления спиртов до целевых продуктов — соответствующих карбоновых кислот является актуальным. Известно, что АФК сравнительно просто in situ генерировать из кислорода, пероксида водорода и воды на электрокаталитической поверхности электрода и в объеме электролита. В связи с этим появляется возможность проводить различной глубины окисление органических субстратов наряду с гетерогенными и в гомогенных эффективность процесса. условиях, что позволяет увеличить 5 Алифатические спирты в зависимости от длины углеродной цепи обладают различной растворимостью в воде и адсорбционной способностью на поверхности электродных материалов. Из литературы известно, что использование различных анодных материалов позволяет получать отличные по реакционной способности АФК. Таким образом, подбор и исследование эффективных электродных материалов для генерации АФК, установление кинетических закономерностей протекания процессов окисления алифатических спиртов с различной длиной углеродной цепи, выявление влияния основных параметров электролиза на эффективность их непрямого электрокаталитического окисления до карбоновых кислот является актуальной задачей. В связи с этим в качестве модельных электрокатализаторов нами выбраны следующие электродные материалы: оксидо-гидроксид-никеля (ОГН), диоксид свинца (Pb/PbO2), допированный бором алмаз (ДБА), в качестве реагентовсубстратов выбраны 1-бутанол, 1-гексанол, 1-нонанол, 1-деканол. Цель работы: Установление общих закономерностей протекания непрямого электрокаталитического окисления алифатических спиртов до соответствующих карбоновых кислот химически связанными активными формами кислорода, in situ генерированными из О2, Н2О2 и Н2О. Для достижения поставленной цели необходимо решить следующие задачи: ─ изучение кинетических закономерностей протекания непрямого электрокаталитического окисления алифатических спиртов до электролиза на соответствующих карбоновых кислот; ─ установление влияния основных параметров эффективность процесса электрокаталитического окисления алифатических спиртов: плотности температуры; тока, количества пропущенного электричества, 6 ─ установление влияния химической природы электродного материала и способа генерации химически связанных активных форм кислорода на эффективность процесса окисления алифатических спиртов; ─ постановка опытов по наработке и выделению целевого продукта. Научная новизна защищаемых в диссертации положений заключается в следующем: ─ впервые проведены систематические исследования непрямого электрокаталитического окисления алифатических спиртов до карбоновых кислот in situ генерированными активными формами кислорода; ─ установлены общие закономерности и найдены оптимальные условия электросинтеза масляной, капроновой, пеларгоновой и каприновой кислот; ─ впервые изучено применение допированного бором алмазного электрода для электросинтеза карбоновых кислот непрямым окислением соответствующих алифатических спиртов. Практическая значимость. Установленные нами закономерности непрямого электрокаталитического окисления алифатических спиртов могут быть востребованы при разработке и практической реализации экологически безопасных и высокоэффективных процессов получения карбоновых кислот. Связь темы с планами работы Института: Диссертационная работа выполнена в соответствии с планами научно-исследовательских работ ИХХТ СО РАН по проекту V 39.1.2. «Разработка новых методов получения востребованных химических веществ, основанных на целенаправленной трансформации растительных полимеров» и в рамках Федеральной целевой научно-технической программы «Исследование и разработка по приоритетным направлениям развития науки и техники гражданского назначения» (подпрограмма «Экологически безопасные и ресурсосберегающие процессы химии и химической технологии»). 7 Апробация работы. Основные результаты диссертационной работы доложены и обсуждены на VI школе-семинаре молодых ученых России «Проблемы устойчивого развития региона» (Улан-Удэ, 2011); на Всероссийской научно-практической конференции «Молодые ученые в решение актуальных проблем науки» (Красноярск, 2011); на Шестой всероссийской конференции молодых ученых, аспирантов и студентов с международным участием. Менделеев-2012 (Санкт-Петербург, 2012); на VII Всероссийской с международным участием школы по электрохимии органических соединений. Актуальные проблемы электрохимии органических соединений (Тамбов, 2012); на Всероссийской научно-практической конференции (с международным участием) «Молодые ученые в решении актуальных проблем науки» (Красноярск, 2012); на Кластере конференций по органической химии «ОргХим-2013» (Санкт-Петербург, 2013); на Научных конференциях молодых учѐных ИХХТ СО РАН (Красноярск, 2012, 2013, 2014). Публикации. По результатам исследования опубликовано 13 научных работ, в том числе 4 статьи в журналах, рекомендованных ВАК. На защиту выносятся следующие основные положения: ─ кинетические скорости) протекания закономерности непрямого (порядок реакции, константы электрокаталитического окисления алифатических спиртов (1-бутанола и 1-нонанола) до соответствующих карбоновых кислот на оксидо-гидроксидно-никелевом и диоксидно-свинцовом электродах; ─ процесса влияние основных параметров электролиза на эффективность электрокаталитического окисления алифатических спиртов: плотности тока, количества пропущенного электричества и температуры при использовании различных схем генерации АФК на оксидо-гидроксидноникелевом, диоксидно-свинцовом и допированном бором алмазном электродах; 8 ─ результаты исследования влияния химической природы электродного материала и способа генерации химически связанных активных форм кислорода на эффективность процесса окисления алифатических спиртов; ─ результаты опытов по наработке целевого продукта. Личный вклад автора. Все эксперименты, обработка и анализ результатов, подготовка и оформление публикаций выполнены лично или при непосредственном участии автора. Структура и объѐм работы. Диссертация состоит из введения, трѐх глав, выводов и списка литературы. Работа изложена на 103 страницах, включает в себя 19 рисунков, 17 таблиц и библиографический список из 137 наименований. Благодарности. Автор выражает благодарность за помощь в проведении исследований методами газожидкостной хроматографии — В.И. Власенко (Сибирский Государственный Технологический Университет, г. Красноярск), УФ- спектроскопии — Н.Г. Максимову (ИХХТ СО РАН, г. Красноярск), хроматомасс-спектроскопии — Г.С. Калачевой (Институт биофизики СО РАН), рентгеноспектрального анализа — В.Ф. Каргину; атомно-абсорбционной спектроскопии — Г.В. Колесниченко (ИХХТ СО РАН, г. Красноярск); рентгенофазового анализа — Г.Н. Бондаренко (ИХХТ СО РАН, г. Красноярск). 9 ГЛАВА 1. Литературный обзор 1.1. Физико-химические свойства спиртов и карбоновых кислот алифатического ряда. Применение карбоновых кислот 1.1.1. Физико-химические свойства алифатических спиртов Спирты представляют собой обширный и разнообразный класс соединений: они весьма распространены в природе и являются важными соединениями с точки зрения органического синтеза [1]. Спиртами, или алкоголями, называют гидроксильные производные углеводородов, имеющие общую формулу , или – [2]. Физические свойства некоторых спиртов приведены в таблице 1.1 [2–4]. Спирты до С10 при комнатной температуре — жидкости; начиная с С11 и выше — твердые тела. Спирты С1–С3 смешиваются с водой во всех соотношениях и имеют характерный запах [5]. По мере увеличения числа атомов углерода в молекуле – , растворимость жидких спиртов в воде уменьшается из- за гидрофобных свойств радикала R [1] и запах их становится неприятным [5]. Твердые спирты лишены запаха и почти не растворимы в воде, жидкие спирты являются хорошими растворителями многих органических веществ [2, 5]. Температура кипения спиртов возрастает по мере увеличения их молекулярной массы (табл. 1.1) [3, 4]. Таблица 1.1 Физико-химические характеристики спиртов [3, 4] МолеНазвание Формула кулярная масса 1 2 3 Растворимость Плотность, Tкип, °С г/см3 4 при 20°С, г/100мл В воде 5 6 В орг. раств. 7 10 Продолжение таблицы 1.1 1 2 3 4 5 6 7 74,12 0,810 117,7 12,5 сп., э. 102,18 0,819 157,2 0,59 сп., э. 144,26 0,828 213,5 н. 158,28 0,8292 231,0 н. Бутиловый спирт (1-бутанол) Гексиловый спирт (1-гексанол) Нониловый спирт (1-нонанол) Дециловый спирт (1-деканол) сп., э., хл. р. сп., ∞ э. Химические свойства спиртов обусловлены присутствием реакционноспособной гидроксильной группы –ОН [5]. Молекула спирта характеризуется тремя реакционными центрами [2]: 1) О–Н-связь: реакция с разрывом О–Н-связи определяет кислотность спирта; 2) неподеленная электронная пара атома кислорода определяет основность и нуклеофильность молекулы спирта; 3) С–О-связь: разрыв С–О-связи характерен для реакций нуклеофильного замещения и β-элиминирования. Под действием различных окислителей первичные спирты окисляются до альдегидов и далее — до карбоновых кислот (1.1), причѐм остановить реакцию на стадии образования альдегидов, предотвратив их дальнейшее окисление, удается, только за счѐт использования специальных реагентов (хлорхромата пиридиния PCC и дихромата пиридиния PDC) [2, 6]. 11 R CH2OH [O] первичный спирт R O C альдегид H [O] O R C OH карбоновая кислота (1.1) Спирты являются чрезвычайно слабыми кислотами (в водных растворах диссоциации с образованием карбониевого иона и OH- не происходит), однако они дают соли с металлами (алкоголяты) [5]: (1.2) Алкоголяты спиртов легко разлагаются водой: (1.3) Спирты взаимодействуют с минеральными и органическими кислотами с образованием соответствующих сложных эфиров и выделением воды [1, 5]: (1.4) Большинство реакций спиртов протекает с расщеплением связи O–H, и лишь при превращении в алкилгалогениды наблюдается разрыв связи C–O [2]. 1.1.2. Физико-химические свойства алифатических карбоновых кислот Карбоновые кислоты алифатического ряда представляют собой производные углеводородов, у которых один атом водорода замещен карбоксильной группой (-COOH), представляющей собой сочетание карбонильной и гидроксильной групп [1, 2, 5]. Общая формула одноосновных предельных карбоновых кислот или . Эту группу соединений можно также рассматривать как конечный продукт процесса окисления спиртов, не связанного с разрушением углеродной цепи (1.1) [2, 5]. Низшие кислоты с числом атомов углерода до 3 — легкоподвижные бесцветные жидкости с характерным острым запахом; они смешиваются с водой в 12 любых соотношениях. Большинство кислот С4–С9 — маслянистые жидкости с неприятным запахом [5]. Растворимость их в воде сильно уменьшается по мере возрастания молекулярных масс. Кислоты от С10 и выше — твердые вещества, нерастворимые в воде. Температура кипения кислот возрастает по мере увеличения молекулярной массы [1, 2, 5]. Физические свойства некоторых карбоновых кислот приведены в таблице 1.2 [2–4]. Таблица 1.2 Физико-химические характеристики карбоновых кислот [2–4] МолеНазвание куляр- Формула ная масса Масляная кислота (бутановая) C3 H7 O C Капроновая кислота C5H11 C Пеларгоновая кислота C8H17 ность, г/см3 Tкип, о С при 20°С, г/100мл В воде В орг. раств. 88,10 0,959 163,5 3,7 сп., э. 116,16 0,929 205,3 0,97 сп., э. 158,24 0,906 255,0 0,03 172,27 0,88640 268,4 0,02 OH OH O C (нонановая) Каприновая (декановая) Плот- O (гексановая) кислота Растворимость OH O C9H19 C OH сп., э., хл. сп., э., хл. Кислотный характер карбоновых кислот ярко выражен [1, 2, 5]. Это объясняется взаимным влиянием атомов в карбоксильной группе; в ней электронная 13 плотность смещена в сторону наиболее электрофильного (электроноакцепторного) атома — кислорода [5]: - O C O H + Тем самым ослаблена связь между кислородом и водородом, и облегчено отделение иона водорода, т.е. облегчена диссоциация кислоты: R O C O R C OH O + H+ - (1.5) Степень диссоциации, а, следовательно, и сила кислот зависят также от величины и характера радикала, связанного с карбоксильной группой: константа диссоциации уменьшается с увеличением радикала. Таким образом, органические кислоты являются слабыми кислотами [5]. В общей оценке реакционной способности следует отметить, что карбоновая кислота имеет, по крайней мере, три реакционных центра [2]: R CH2 + O C - O H + 1. Связь O–H, разрыв этой связи наблюдается при кислотной диссоциации; 2. Карбонильная группа С=О, эта группа способна присоединять нуклеофильные реагенты; 3. С–Н-связи при -углеродном атоме, эти связи подвержены ионизации с образованием енолятов. Электролиз солей карбоновых кислот протекает с отщеплением диоксида углерода и известен как реакция Кольбе (1849 г.). В этой реакции наблюдается димеризация углеводородного фрагмента [1, 2]. O 2R C O Na H2O электролиз R R + CO2 + NaOH + H2 (1.6) 14 Характерной особенностью карбоновых кислот является их способность образовывать со спиртами сложные эфиры. Эта реакция катализируется сильными кислотами (H2SO4 и другие) и известна как реакция этерификации (реакция Фишера-Шпайера, 1895 г.) [2, 5]: O R C O + R' H O H2SO4 H O R C O R' сложный эфир карбоновая кислота + H2O (1.7) Также органические кислоты способны образовывать соли с металлами, с их оксидами и гидрооксидами [2, 5]: – – – – (1.8) 1.1.3. Применение карбоновых кислот Карбоновые кислоты, их соли и эфиры имеют большое биологическое значение и находят широкое практическое применение в различных отраслях промышленности: фармацевтической, витаминной, косметической, парфюмерной, пищевой, автомобильной и других отраслях [6–8]. Масляная кислота применяется для получения ароматизирующих добавок (эфиры метилбутират и изоамилбутират — ароматизаторы в промышленности), пластификаторов и флотореагентов, как экстрагент щелочно-земельных металлов [10]. Капроновая кислота и производные капроновой кислоты используются для получения полиамидных синтетических волокон [9]. Высшие жирные карбоновые кислоты и их соли широко применяют в качестве сырья для производства мыла, лаков и красок, поверхностно-активных веществ, как эмульгаторы в производстве каучуков, как пластификаторы в производстве резин и др. Эфиры кислот — пищевые добавки и растворители [12]. 15 Пеларгоновая кислота — применяют в производстве полиэфирных алкидных смол, красителей, стабилизаторов; эфиры пеларгоновой кислоты используют в качестве душистых веществ [12, 13]. Каприновая — находит широкое применение в производствах фармацевтической и косметической продукции. Также используется в производстве пластичных смазок, лакокрасочных материалов, латексов, каучуков, неионогенных ПАВ и др. [13]. В медицинской практике широко используют соли карбоновых кислот: калия ацетат, кальция лактат, натрия цитрат для инъекций, кальция глюконат, натрия вальпроат и др. [13, 14]. 1.2. Общие лабораторные и промышленные методы получения карбоновых кислот В последние годы исследованиям процесса окисления спиртов посвящается значительное количество работ, из которых можно выделить три следующих основных направлений: синтез целевых продуктов — карбоновых кислот [6–8, 15–18], окислительная деструкция спиртовых отходов, содержащихся в промышленных сточных водах [19] и разработка топливных элементов путем низкотемпературного окисления низших спиртов [20–22]. Широкий спектр работ [6–9, 15–17] посвящен синтетическому окислению спиртов до карбоновых кислот, которые являются, как упоминалось в разделе 1.1, значимыми продуктами и промежуточными веществами в различных промышленных производствах. В органическом синтезе окислительные процессы протекают с использованием значительного ряда окислительных агентов. На практике применяют неорганические кислоты, перманганат калия, оксид марганца (IV), соединения хрома, соли и оксиды металлов переменной валентности и др. [1, 2, 5, 7]. Известно, что в лабораторной практике спирты окисляют кислородом воздуха в присутствии медных и других катализаторов при 300–500 °С, а также такими окислителями как хромовая смесь, хромовый ангидрид, твердый 16 комплекс CrO3 с пиридином, KMnO4, специально приготовленный диоксид марганца, RO4+NaIO4, Ag2CO3 (осажденный на цеолите) и др. [23]: R R O CrO3,H2SO4 CH2OH H2O, 15-20°C CH2OH + KMnO4/H R C OH (1.9) O R C OH (1.10) Также спирты могут быть дегидрированы при 100–180 °С над Cu, Ag, Ni, Co, Pt и Pd. Во всех этих случаях первичные спирты окисляются до альдегидов, которые при дальнейшем окислении дают кислоты с тем же числом углеродных атомов [1, 2, 5, 23]. Из литературы известно [23], что карбоновые кислоты также получают окислительной деструкцией алкенов смесью перманганата калия и периодата натрия в водном ацетоне в нейтральной среде. Реакция идѐт в две стадии — на первой перманганат окисляет алкен до диола, на второй периодат окисляет диол до кислоты, избыток периодата окисляет Mn+4 до Mn+7 (перманганата требуется каталитическое количество): R1 CH CH R2 KMnO4, NaIO4 R1 COOH +R2 COOH (1.11) Помимо окислительных методов, в лабораторной практике карбоновые кислоты также получают гидролизом нитрилов при нагревании (как правило, при кипячении) с водными растворами минеральных кислот (реже щелочей) [2, 5]: R C H2O N H SO , t° 2 4 R COOH (1.12) Либо гидролизом сложных эфиров. Эта реакция наиболее гладко протекает в разбавленных щелочах при нагревании [2]. R NaOH, t° COOR' H O 2 H3O+ R COOH + R' OH (1.13) 17 Также карбоновые кислоты получают карбоксилированием металлорганических реагентов (в основном реактивов Гриньяра и литийорганических соединений) [2, 5]: R Mg Br абс.эфир R MgBr CO2 R HCl COOMgBr H O R 2 COOH (1.14) Одним из основных методов получения карбоновых кислот в промышленном масштабе является окисление парафиновых углеводородов воздухом или техническим кислородом при высокой температуре в присутствии или в отсутствие катализаторов [3, 23–25]. Низшие углеводороды (с числом атомов углерода до 8) окисляются главным образом в паровой фазе при повышенном давлении, а высшие (от 16 до 30 атомов карбона для получения кислот от 10 до 20 атомов карбона) — преимущественно в жидкой фазе. Окисление проводят при температуре около 500 °C и атмосферном давлении или при 400 °C под давлением 10-20 МПа (130-200 атмосфер). Катализаторами служат металлы, их оксиды и соли (KMnO4, MnO2, соли марганца). При получении высших жирных кислот в присутствии катализаторов температуру снижают до 130–150 °C. Следует отметить, что при окислении углеводородов обычно образуется смесь кислот с различным количеством углеродных атомов [24], что значительно осложняет процесс получения целевого продукта. В некоторых случаях в промышленности для получения карбоновых кислот применяют оксосинтез, который возможен в двух вариантах [5]: а) с его помощью получают альдегиды и далее окисляют их до соответствующих кислот; б) взаимодействием олефинов с оксидом углерода (II) и водяным паром в присутствии тетракарбонила никеля или фосфорной кислоты при температуре 300–400 °C и давлении 200–500 атмосфер получают смесь кислот нормального и изостроения: 18 главное направление R CH CH2 + CO + H2O побочное направление R COOH R COOH CH3 (1.15) Обобщая, следует отметить, что все традиционные лабораторные и промышленные методы получения карбоновых кислот, несмотря на их разнообразие, имеют ряд существенных недостатков. К этим недостаткам относятся: использование дорогих и дефицитных окислителей (таких как перманганат калия), большое количество отходов, применение в качестве катализаторов токсичных соединений тяжелых металлов (соединения хрома), использование высоких температур и давлений (специальное аппаратурное оформление), сложных технологических схем, невысокая селективность и малая эффективность. Этим объясняется постоянный интерес исследователей к изучению процессов окислительных превращений спиртов, поиску новых, экологически чистых высокоэффективных окислителей и усовершенствованию технологий. 1.3. Электрохимические методы получения карбоновых кислот С точки зрения промышленной реализации известно, что электрохимические процессы имеют ряд преимуществ [26–28]: – мягкие условия, исключающие использование высоких температур и давлений; – экологичность и селективность процесса (электрон как реагент не загрязняет окружающую среду); – дешевые исходные реагенты (сравнительно низкая себестоимость электроэнергии как реагента и соответственно синтезированного продукта); – получение продуктов с высокой степенью чистоты; – возможность проводить различной глубины окислительные процессы органических субстратов; 19 – удобный операционный контроль с использованием таких параметров, как плотность тока и потенциал, контроль над протеканием электрохимических процессов легко автоматизировать; – упрощение в целом технологической схемы процесса. Исходя из вышеперечисленных достоинств, электрохимические реакции окисления различных субстратов, и в частности спиртов, в течение длительного времени привлекают внимание исследователей [27, 28, 15–18]. 1.3.1. Электрохимическое окисление спиртов Условия электрохимического окисления спиртов до соответствующих карбоновых кислот весьма разнообразны. Анод может быть изготовлен из платины [22, 29–31], никеля [32–41], свинца [42–43], на основе сажевых гидрофобизированных электродов с формированием на его высокоразвитой поверхности высших оксидов никеля [44, 45]. Направление процесса и выход продуктов окисления зависит не только от материала анода, но и от природы растворителя и электролита. Также значительное влияние оказывают плотность тока, строение субстрата, температура и подготовка поверхности электродов [15, 16, 32, 44]. Из литературы известно, что алифатические спирты окисляют в водных и ацетонитрильных растворах, а также в безводных спиртах, содержащих в качестве электролитов алкоголяты щелочных металлов, перхлорат натрия или тетрафторбораттетрабутиламмония. Последний, наряду с перхлоратом лития, служит электролитом и в ацетонитрильных растворах. Также известно, что в водных растворах окисление спиртов может происходить в присутствии как щелочей, так и минеральных кислот, главным образом серной и хлорной [15, 16]. Электрохимическое окисление спиртов протекает через ряд промежуточных стадий. Основную схему реакции можно представить следующим образом [15]: 20 – (1.16) Таким образом, первой стадией окисления спирта является образование альдегида, который окисляется до кислоты. При надлежащих условиях кислота вступает в реакцию Кольбе (1.6). Первая стадия окисления спирта до альдегида протекает по электронно-радикальному механизму [15]: (1.17) (1.18) (1.19) (1.20) Электрохимическое окисление алифатических спиртов до соответствующих кислот можно проводить с использованием свинцового анода, который в процессе электролиза покрывается слоем диоксида свинца, несколько предохраняющем его от коррозии. Однако, слой диоксида свинца, образующийся при электролизе на поверхности свинца, является неплотным и незначительно предохраняет свинцовую основу от разрушения в процессе электролиза, особенно в присутствии органических кислот, образующих растворимые соли свинца [42]. Для уменьшения износа анода в сернокислых растворах рекомендуется использовать аноды из диоксида свинца, электроосажденной на инертную основу [46]. Также стоит отметить, что окисление спиртов на свинцовом аноде в водном растворе серной кислоты сопровождается побочной реакцией этерификации, осложняющей процесс [21, 43]. Образование эфиров обусловлено частичным взаимодействием образовавшейся кислоты с исходным реагентом – спиртом, где катализатором этого процесса выступает серная кислота (1.7) [43]. Важно отметить, что отдельные процессы электрохимического окисления спиртов получили практическое применение, например, окисление изобутило- 21 вого спирта в изомасляную кислоту на аноде из электроосажденного диоксида свинца при плотности тока ~ 70 мА/см2 с выходом 40 % [43]. Однако свинец и его соединения токсичны и проблема загрязнения ими окружающей среды в настоящее время является острой. Предельно допустимые концентрации (ПДК) соединений свинца следующие: в атмосферном воздухе 0,003 мг/м³ [47], в воде 0,03 мг/л [48], почве 20,0 мг/кг [49]. Таким образом, известный способ окисления спиртов до соответствующих кислот с использованием анода из диоксида свинца требует дальнейшего исследования с целью усовершенствования, устранения недостатков и оптимизации. 1.3.2. Редокс-медиаторное и непрямое электрокаталитическое окисление органических веществ В последнее время большое распространение среди современных методов электрохимического органического синтеза приобретает «медиаторное» или непрямое окисление органических соединений, при котором на электроде происходит генерация редокс-реагентов медиаторов [26, 50–55]. Интерес к ним во многом обусловлен широкими, а в ряде случаев, уникальными возможностями для осуществления разнообразных превращений органических соединений, создания экономичных и экологически чистых процессов [26–28]. Непрямое электрокаталитическое окисление позволяет осуществлять превращение органических соединений при более низких окислительных потенциалах, чем их стандартные потенциалы окисления, приводит к повышению скорости, селективности и выхода целевых продуктов по сравнению с прямым электроокислением, при котором происходит непосредственный перенос электрона от субстрата на поверхность электрода [18, 50, 55]. На рисунке 1.1 показана схема процесса окисления, где сравнивается простая редокс-реакция (схема 1а) с непрямым электрохимическим процессом, при котором тот же редокс-реагент генерируется электрохимически in situ (схема 1б), и с прямым электрохимическим процессом (схема 1в) [26]. 22 Окислитель (стехиометрически) Cубстратred а Переработка отходов Израсходованный окислитель Продуктox Cубстратred Окислитель (каталитически) Продуктox Израсходованный окислитель б eАнод в Cубстратred e- Продуктox Анод Рисунок 1.1. Схема редокс-реакций окисления: 1а – простая редокс реакция окисления, 1б – редокс-реагент генерируется электрохимически in situ, 1в – прямой электрохимический процесс [26]. В классической редокс-реакции реагенты, а часто это токсичные соли тяжелых металлов, используют, как правило, в стехиометрических количествах, что является дорогостоящим и критичным для окружающей среды. Медиатор, по своей сути, является окислителем, регенерируемым электрохимически, что позволяет применять его не в стехиометрическом, а в каталитическом количестве, отработанных и тем самым реагентов, избавиться от представляющих необходимости серьезную утилизации проблему для окружающей среды. Прямой или непрямой электрохимический процесс используют в зависимости от типа целевой реакции. Селективность в непрямом электрохимическом процессе можно достичь подходящим выбором редокскатализатора, тогда как в прямых электрохимических процессах селективность 23 обычно зависит от потенциалов электрода и других параметров электролиза [26, 56]. В условиях непрямого анодного окисления медиаторы (М) играют роль либо посредников при переносе электронов от органического субстрата (RH) на анод без образования интермедиатов с компонентами реакционной среды (внешнесферное окисление, редокс-катализ), либо реагентов, превращающихся при переносе электрона в окислителе (внутрисферное окисление, химический катализ). В последнем случае конечные продукты образуются, например, в результате отрыва электрогенерированными химическими окислителями атомов водорода от субстратов [50, 57–60]. В обзорной работе [50] представлен общий механизм промотирования медиаторами процесса анодного окисления органических соединений, который включает следующие стадии. 1. Электрогенерирование окисленной (активной) формы медиатора (Mox). (1.21) 2. Внешнесферное одноэлектронное окисление субстрата активной формой медиатора (т.е. связь между комплексом металла и субстратом не образуется) с регенерированием исходной формы последнего или внутрисферное одноэлектронное окисление субстрата (т.е. через стадию образования промежуточного активированного комплекса, распад которого и последующие превращения приводят к продукту) с отрывом атома H: (1.22) или (1.23) 3. Депротонирование продукта внешнесферного окисления субстрата (В – основание): 24 (1.24) 4. Регенерирование медиатора. (1.25) 5. Образование продуктов окисления: . (1.26) Ионы переходных металлов в высоких степенях окисления обычно реагируют с субстратами как внешнесферные окислители. Важно отметить, что в настоящее время особое место в ряду медиаторов непрямого электрохимического окисления занимают системы на основе пероксида водорода, генерированного из молекулярного кислорода, и ионов переходных металлов [50, 55, 57, 61]. В этом случае в окислении принимает участие катодный ток, который в большинстве процессов прямого и непрямого окисления органических соединений нерационально расходуется на образование водорода за счет восстановления протонов [50]. Это дает возможность экономить потребление ресурсов путем генерирования продуктов в больших объемах одновременно как на аноде, так и на катоде. Такие процессы называют парными (paired) электросинтезами [26, 27]. Некоторые исследователи [50, 62], электроокисление с участием катодного тока называют «катодным окислением». В обзоре [50], авторы указывают возможные реакции промотирования непрямого катодного окисления органических субстратов, реализующиеся в процессе парного электросинтеза: 1. Генерирование пероксида водорода катодным восстановлением молекулярного кислорода: (1.27) 25 2. Гомолитическое разложение электрогенерированного H2O2 под действием медиатора (иона металла в низшем валентном состоянии Mn+1): (1.28) 3. Окисление субстрата образующимися гидроксильными радикалами (например, или в результате отрыва атома водорода от субстрата, или в результате присоединения к субстрату): (1.29) 4. Превращение радикальных интермедиатов под действием окисленной формы медиатора (M(n+1)+) в продукты с регенерированием исходной формы последнего: (1.30) Таким образом, непрямое окисление органических субстратов и электрогенерирование медиаторов могут протекать как в гомогенных, так и в гетерогенных условиях (при иммобилизации медиатора на поверхности анода). В обоих случаях использование медиатора приводит к уменьшению перенапряжения электрохимического процесса и повышению его селективности и эффективности [50, 63]. Проведение электролиза с участием медиаторов осуществляется либо «ex situ», либо «in situ». В первом случае окисление субстрата электрогенерированной активной формой медиатора осуществляют в две стадии с использованием отдельного реактора, а во втором – в одну стадию непосредственно в электролизере, что является важным преимуществом «in situ»-процесса, существенно упрощающим технологическое оформление [50]. Для обеспечения эффективного протекания электрохимического окисления медиаторы и медиаторные системы должны удовлетворять следующим требованиям [50]: 26 1) химическая стабильность окисленной формы медиатора в условиях электролиза; 2) высокая скорость и (по возможности) необратимость переноса электрона от медиатора на электрод и реакции активированной формы медиатора с субстратом; 3) хорошая растворимость в электролите при проведении электролиза в однофазной системе. В большей степени этим требованиям отвечают медиаторы неорганической природы. Активированные формы органических медиаторов, которыми обычно являются радикалы или катион-радикалы, часто вступают в необратимые побочные реакции. Самую многочисленную группу неорганических медиаторов электроокисления органических соединений составляют электрохимически регенерируемые соединения и комплексы переходных металлов [50, 57–60]. 1.3.3. Химически связанные активные формы кислорода В настоящее время значительное внимание исследователей привлекают процессы с использованием химически связанных активных форм кислорода (reactive oxygen species), к которым относят ионы кислорода, свободные радикалы и пероксиды как неорганического, так и органического происхождения ( – , , – , ). Это, как правило, небольшие молекулы с исключитель- ной реактивностью благодаря наличию неспаренного электрона на внешнем электронном уровне [61, 63–65]. Из-за своей высокой активности АФК быстро вступают в реакции с растворенными субстратами и друг с другом, поэтому стационарная концентрация их в растворах очень мала, а исследование реакций с их участием затруднительно [63, 66]. Поэтому все известные методы доказательства присутствия АФК в реальных редокс-системах и определения их концентрации носят косвенный характер и, как правило, основаны на кинетических проявлениях АФК [66]. 27 В таблице 1.3 представлена относительная окислительная способность некоторых окислителей [67, 68]. Необходимо отметить, что АФК различаются по окислительной способности на 5–6 порядков, наиболее активными из которых являются НО•-радикалы [55, 61], обладающие высоким редокс-потенциалом (2,8 В относительно стандартного водородного электрода) [69]. В связи с этим, непрямое электрохимическое окисление органических веществ АФК в основном осуществляется с целью получения новых продуктов (непрямой электросинтез) и разрушения субстратов в сточных водах (минерализация) [55, 70]. Таблица 1.3 Относительная окислительная способность некоторых окислителей [67, 68] Окислитель Относительная окислительная способность Cl2 1,00 1,24 H2O2 1,31 O3 1,52 O 1,78 HO• 2,05 Известно, что АФК могут быть сравнительно легко in situ электрохимически генерированы из О2, Н2О2, и Н2О на поверхности электрода и в объеме электролита путем катодного восстановления кислорода на углеграфитовых электродах в зависимости от pH среды по следующим реакциям [55, 61]: pH ≥ 7 pH < 7 а так же анодным окислением Н2О2 и Н2О: (1.31) (1.32) 28 (1.33) (1.34) (1.35) В работах [33, 71] была показана возможность повышения эффективности процесса окисления алифатических спиртов при использовании химически связанных АФК и в значительной мере преодолеть ряд существенных недостатков, присущих известным химическим способам окисления. Также следует отметить, что возрастающее количество работ, посвященных использованию АФК, как экологически чистых окислителей, в различных процессах окисления, свидетельствует о дальнейшей перспективе создания на их основе высокоэффективных и высокоэкологичных процессов, отвечающих принципам «Green Chemistry» [27]. 1.3.4. Электрокаталитическое окисление спиртов на основе редоксмедиаторных систем В настоящее время прогресс в развитии электрохимических превращений спиртов, гликолей, углеводов и их производных, а также алкенов в карбонильные соединения и карбоновые кислоты достигнут благодаря эффективному катализу реакций медиаторными редокс-системами на основе ионов переходных металлов, например, рутения [72–74], марганца [75], церия [76–78], хрома [79], кобальта [80, 81] и оксидо-гидроксида никеля (Ni(O)OH) [32–41, 45, 82–86]. В работах [72–74] исследовано непрямое электроокисление спиртов, углеводов и их производных в условиях бездиафрагменного электролиза с медиаторной системой RuVIII/RuIV–[Cl+]/Cl– в двухфазной системе, состоящей из водного раствора NaCl в фосфатном буфере (pH 4) и несмешивающегося с ним органического растворителя (например, CCl4). Таким образом, процесс проводят в разных фазах, что позволяет исключить контакт образующихся продуктов 29 с электродами и тем самым предотвратить нежелательные побочные реакции. Такие условия обеспечивают высокий выход целевого продукта (80–90 %) [50]. Аналогичным образом и с сопоставимой эффективностью катализируют непрямое электроокисление спиртов и другие рутениевые медиаторы — RuCl3 (H2O)x, Ru3(CO)12, RuCl2(Ph3P)3 и [Ru3O(OAc)6(H2O)3]OAc [72]. В качестве электродов с закрепленными на их поверхности редокс-медиаторами, способными непрерывно регенерироваться в процессе электролиза и катализировать окисление спиртов, наибольшее использование нашел оксидогидроксидный никелевый анод [32–41, 45, 71, 82–93]. Первые работы, посвященные окислению органических веществ на Ni(O)OH электроде, в частности и спиртов, были выполнены в начале 70-х годов Фиошиным М.Я. и Авруцкой И.А. с сотрудниками [32, 39, 40, 45, 87–93]. Также параллельно активные исследования в этой области были проведены Vertes G., Horanyi G. с сотрудниками [36, 37, 82–84], Pletcher D. с сотрудниками [38, 80, 82] и Schafer H.J. с сотрудниками [34, 41, 85]. В обзорной статье [35] Б.В. Лялиным и В.А. Петросяном обобщены литературные данные по окислению органических соединений с использованием Ni(O)OH электрода. Авторы [35] описывают реализацию такого процесса следующей модельной схемой на примере окисления спиртов (рисунок 1.2): + - 12 OH - 4Ni Ni 2+ -8e +12e, -6H2 4Ni(OH)2адс 12 H 2O + H2 O +4OH-, -4H2 O, -4e RCH2-OH k RCOOH 4NiOOHадс Рисунок 1.2. Схема процесса электроокисления спиртов на Ni(O)OHэлектроде [35]. 30 Согласно этой схеме, в основу которой положены данные [34, 41, 85], первая стадия окисления спиртов на оксидо-гидроксидном никелевом электроде включает электрохимическую реакцию образования высшего оксида никеля — Ni(O)OH (1.36). Далее происходит адсорбция спирта на поверхности Ni(O)OH-электрода (1.37), причем адсорбция спирта уменьшается с увеличением длины углеродной цепи, это является причиной снижения скорости окисления. Лимитирующей стадией является отщепление водорода от адсорбированного спирта с образованием β-гидроксиметильного радикала на поверхности Ni(O)OH-электрода (1.38). Образовавшийся радикал далее окисляется до карбоновой кислоты в результате реакций (1.39–1.40): (1.36) (1.37) (1.38) (1.39) (1.40) Авторами [35] сообщается, что, поскольку окисление субстрата протекает на поверхности электрода, процесс в целом квалифицируется как гетерогенный электрокатализ с постоянно регенерируемым катализатором. В гетерогенном катализе важную роль играет количество каталитических центров на поверхности катализатора. То же следует из схемы 5, согласно которой эффективность процесса (при заданном токе I) зависит от величины k, характеризующей скорость регенерации катализатора и в пределе сохраняющей количество таких центров (Ni(O)OH) на поверхности анода в единицу времени. Таким образом, авторы [35] делают заключение, что в целом эффективная реализация процессов окисления в щелочном электролите на Ni(O)OH-электроде зависит от ве- 31 личины k и, в тех случаях, когда k велика, достигается при использовании Niанода, в других – необходимы добавки в электролит небольших количеств солей Ni, и в третьих – требуется специальная обработка (активация) Ni-анода. В двух последних случаях величина k невысокая и эффективность процесса удается поднять уже не за счет быстрой регенерации Ni(O)OH, а за счет специальной обработки электрода, приводящей к механическому увеличению количества каталитических центров на поверхности анода и, как следствие, к увеличению токовой нагрузки. Непрямое окисление спиртов на Ni(O)OH-электроде проводят в условиях бездиафрагменного амперостатического электролиза в щелочной среде при pH 10–14. Используют либо одно-, либо двухфазную систему и ток низкой плотности (0,5–2,5 мА см-2) [34, 50]. Отмечено, что при более высокой плотности тока резко снижается выход целевых продуктов по току из-за конкурентного процесса электролиза воды [50]. Как уже отмечено в разделе 1.3.1, эффективность электрохимического превращения органических веществ до целевых продуктов можно существенно повысить благодаря использованию катодного тока. Так авторами [71] было показано, что при окислении n-бутанола с использованием парного электролиза при плотности тока ~ 2 мА∙см-2, где Ni(O)OH и H2O2 электрогенерируются в щелочной среде в присутствии кислорода, выход масляной кислоты по току повышается от 80 % до 170 %. На рисунке 1.3 представлены реакции, протекающие в такой электрохимической системе [50]. Таким образом, при парном электролизе наряду с реакциями (1.36) – (1.40) дополнительно протекают реакции (1.31), (1.33)–(1.35). 32 Катод Анод 2 HO- O О2 + 2 Н2О R OH +2e- 2 Ni(OH)2 -2e- Ni0 2 Ni(O)OH Н2О2 + 2 HO- 2 НО• R OH 2 Н2О О2 Рисунок 1.3 Схема реакций, протекающих при окислении спиртов до карбоновых кислот на Ni(O)OH при парном электролизе. Использование непрямого редокс-медиаторного окисления спиртов до соответствующих кислот в щелочной среде на оксидо-гидроксидно-никелевых электродах позволяет избежать реакции этерификации, осложняющей процесс в кислых растворах на анодах из платины и диоксида свинца [87]. Однако недостатком в проведении процесса является использование малых плотностей тока (0,5–2,5 мА∙см-2) [34, 50]. В литературе также известны работы [94, 95], направленные на получение карбоновых кислот в укрупненном масштабе путем окисления алифатических спиртов на Ni(O)OH-электроде в щелочной среде в электролизере непрерывного действия с токовой нагрузкой 20 А [35]. Таким образом, достигнутые селективность и эффективность процесса окисления алифатических спиртов до соответствующих карбоновых кислот в условиях непрямого электрокатализа позволяют рассматривать оксидогидроксидный никелевый электрод, как весьма перспективный анодный материал [35, 50]. Следует отметить, что влияние дополнительной генерации активных форм кислорода на эффективность в процессах электрокатализа до настоящего времени изучено недостаточно глубоко и требует дальнейшего исследования с целью более глубокого понимания их влияния и создания новых высокоэффек- 33 тивных и высокоэкологичных процессов электрокаталитического окисления спиртов до соответствующих карбоновых кислот. 1.4. Влияние условий электролиза на процесс электрокаталитического окисления 1.4.1. Материал анода, состав электролита и величина потенциала Одной из наиболее важных проблем электрохимии является правильный выбор материала и конструкция электродов. От решения этих задач зависят механизм, селективность и в целом эффективность процесса электрохимического окисления [96, 97]. Специфическое электрокаталитическое воздействие на получение определенного продукта состоит в том, что электрод фактически играет роль гетерогенного катализатора. Это связано как с его каталитической активностью, так и с адсорбцией различных частиц из раствора, точнее, с адсорбцией реагентов и их соадсорбцией с компонентами раствора [98]. Как упоминалось в разделе 1.3, электроокисление органических веществ можно проводить двумя различными путями: прямое окисление, где в качестве электрокаталиаторов, как правило, применяют металлы платиновой группы [99]. Среди них наиболее активны для окисления спиртов палладий и платина, на иридии и родии селективное окисление протекает в значительно меньшей степени [15]. Либо непрямое электрокаталитическое окисление, которое происходит с участием медиаторов постоянно регенерируемых на поверхности анода (раздел 1.3.2) [99]. В работах органических было [100–103] соединений в установлено, водной среде что электроокисление протекает без потери электрокаталитической активности анодов только при высоких значениях потенциалов, соответствующих области выделения О2. В этих условиях становится возможным образование на поверхности реакционноспособных радикалов, которые участвуют в последующих химических превращениях. 34 Значения потенциалов выделения О2, наиболее активно используемых анодных материалов, представлены в таблице 1.4 [70]. Таблица 1.4 Значения потенциалов выделения кислорода, отн. н.в.э. [70] Материал анода E, В Условия Pt 1,3 0,5 M H2SO4 Pt 1,6 0,5 M H2SO4 IrO2 1,6 0,5 M H2SO4 Графит 1,7 0,5 M H2SO4 PbO2 1,9 1M HClO4 SnO2 1,9 0,5 M H2SO4 Pb-Sn (93:7) 2,5 0,5 M H2SO4 Оксид титана 2,2 1 M H2SO4 Si/ДБАЭ 2,3 0,5 M H2SO4 Ti/ДБАЭ 2,7 0,5 M H2SO4 DiaChem 2,8 0,5 M H2SO4 Comninellis и сотрудниками был предложен механизм [99, 100, 104, 105], позволяющий классифицировать электродные материалы по способности адсорбировать НО -радикалы. Схематическое изображение реакций, протекающих на электродных материалах, отличающихся по этому признаку, представлено на рисунке 1.4. 35 H2O + - H +e 1/2 O2 MOx e a RO d R MOx(HO ) b 1/2 O2+ MOx+1 +H++ec H++eРисунок 1.4 Схема реакций электрокаталитического окисления органических веществ: a – реакция разряда H2O; b – реакция выделения O2, в отсутствии субстрата на активных центрах анода; с – образование высшего оксида металла; d – окисление органического соединения; e – выделение O2 в отсутствии субстрата на активных центрах. Согласно модели Comninellis, на поверхности анода присутствуют два вида активных центров. На одних происходит физическая адсорбция НО радикалов (1.41), а на других – внедрение адсорбированных HO -радикалов в кристаллическую решетку оксида, что приводит (1.42) к образованию высших оксидов MOx+1 (хемосорбированный активный кислород ). Процесс окисления инициируется разрядом воды в кислом электролите или OH–-иона в щелочном с образованием HO• (1.33). (1.41) (1.42) В отсутствие субстрата на активных центрах выделяется О2: (1.44) 36 (1.45) Окисление органического субстрата на центрах, осуществляющих физическую адсорбцию НО - радикалов, происходит преимущественно с минерализацией и образованием СО2 и Н2О (1.46), а хемосорбированный «активный кислород» участвует с образованием частично окисленных продуктов (1.47): (1.46) (1.47) В таблице 1.5 представлены электродные материалы со значениями их потенциалов окисления и зависимость вида адсорбции HO•-радикалов на дальнейший маршрут процесса окисления органических субстратов. Таблица 1.5 Влияние материала электрода и вида адсорбции HO•-радикалов на механизм процесса окисления органических субстратов [45, 99, 105, 106] Материал анода Ni(O)OHэлектрод (ОГНЭ) Потенциал Вид адсорбции окисления органики, OH•-радикалов В (отн. н.в.э.) 0,8–1,1 Pb/PbO2 1,3–2,0 ДБАЭ 2,2–2,7 хемосорбция — образование целевого продукта Физ. адсорбция — минерализация до CO2 и H2O Таким образом, из всего вышесказанного следует, что электрокаталитические превращения субстрата до целевого продукта преимущественно 37 протекают на анодах, создающих концентрацию MOx(HO ) близкой к нулю. К таким электродам можно отнести оксидо-гидроксидно-никелевый электрод [99, 104, 107]. Электроокисление с минерализацией происходит на анодах с высокой концентрацией HO -радикалов на поверхности, скорость реакции (1.42) в этом случае минимальна. Эффективность по току для этих типов анодов зависит от соотношения скоростей протекания реакций (1.46) и (1.47), которые в свою очередь зависят от соответствующих реакций выделения О2 по реакциям (1.44) и (1.45) [99, 100, 104]. Для допированных бором алмазных электродов основными реакциями являются (1.41) и (1.46), где происходит физическая адсорбция НО - радикалов, что преимущественно и приводит к полной минерализации органических соединений до СО2 и Н2О [99, 105–109]. На электроде из диоксида свинца может происходить как физическая адсорбция НО - радикалов, так и хемосорбция, в зависимости от модификации анода, pH раствора и потенциала электрода [99, 105, 107]. В работах [105, 106], также упоминается, что в отсутствии органического субстрата, электрогененированные свободные HO•-радикалы могут реагировать друг с другом с образованием H2O2: (1.48) Далее H2O2 может окисляться до O2 либо путем его прямого разряда на поверхности электрода (1.49), либо при участии HO•-радикалов (1.50): (1.49) (1.50) В последние годы активно ведутся исследования процессов окисления органических субстратов с использованием новых высокоэффективных электродов — допированных бором алмазных электродов [105, 106, 109–112]. Из 38 литературы известно [105, 109, 113–115], что ДБАЭ являются электродными материалами, обладающими высокой механической прочностью, широкой областью рабочих потенциалов, устойчивостью к коррозии и высоким перенапряжением радикалов. кислорода Вследствие с высокоселективным этого, ДБАЭ образованием используются в НО•- качестве высокоэффективного анода в основном для разрушения органических субстратов в сточных водах [101, 106, 108–112], а также для электроаналитических определений [116]. Однако, известен незначительный ряд работ по исследованию возможности использования ДБА электродов в препаративных целях [115, 117–119]. Проанализировав существующую в настоящее время литературу, следует отметить, что работ по исследованию окисления алифатических спиртов до соответствующих карбоновых кислот с использованием ДБА электродов на данный момент не известно. 1.4.2. Строение спирта и температура В литературе отмечается [98], что температура играет двоякую роль в процессах электролитического превращения органических веществ: с одной стороны, она снижает диффузионные ограничения и увеличивает эффективную концентрацию вещества у поверхности электрода. С другой стороны, при повышении температуры снижается величина активационного перенапряжения, что может неблагоприятно сказаться на величине выхода продукта по току, хотя скорость процесса возрастет. В связи с этим целесообразно проводить электроокисление при комнатной температуре. Однако, из литературы известно [33-35, 41, 54], что для спиртов нормального строения с увеличением длины углеводородной цепи, ростом молекулярной массы и снижением их адсорбируемости на аноде процесс окисления до соответствующих карбоновых кислот замедляется в связи с низкой растворимостью в водных растворах. Так же известно [34, 41, 54], что повышение температуры электролиза (> 60 °С) позволяет повысить конверсию при окислении 39 спиртов с 6 и более атомами углерода в цепи и как следствие этого – увеличить выход целевого продукта. В работе [120] для повышения эффективности наработки карбоновых кислот при окислении плохо растворимых спиртов электросинтез на оксидогидроксидно никелевом электроде проводили в двухфазной системе «органический растворитель – водный раствор щелочи». В качестве растворителей использовали третбутиловый спирт и четырѐххлористый углерод, которые являются устойчивыми в процессе электрокаталитического окисления, облегчают процессы перемешивания, предохраняют от дальнейшего окисления наработанный целевой продукт. Авторами Фиошиным М.Я., Авруцкой И.А. и др. [32, 39, 40] было отмечено, что в процессе окисления спиртов до соответствующих карбоновых кислот основным побочным продуктом окисления спиртов является низший гомолог образовавшейся целевой карбоновой кислоты. 1.5 Постановка задачи исследования Анализ литературы по традиционным методам окисления алифатических спиртов до соответствующих карбоновых кислот показал перспективность применения непрямого электрокаталитического окисления с использованием в качестве окислителей экологически чистые реагенты, а именно химически связанные активные формы кислорода, in situ генерированные из O2, H2O2 и H2O. Ряд недостатков, существующих в настоящее время промышленных технологий окисления алифатических спиртов, ограничивает высокоэффективное, экологически чистое получение карбоновых кислот. Определение оптимальных условий осуществления процессов непрямого электрокаталитического окисления с применением эффективных окислителей и высокоэффективных электродных материалов создает определенные перспективы для создания экономичных и экологически уравновешенных технологий, соответствующих принципам «Green Chemistry». 40 Повышение эффективности окисления алифатических спиртов до соответствующих карбоновых кислот с применением экологически безопасных реагентов-окислителей, in situ электрокаталитически генерированных активных форм кислорода, представляет большой научный и практический интерес. В связи с этим основной целью исследования являлось установление общих закономерностей протекания процессов непрямого электрокаталитического окисления алифатических спиртов с различной длиной углеродной цепи (1-бутанол, 1-гексанол, 1-нонанол, 1-деканол) до целевых продуктов — соответствующих карбоновых кислот активными формами кислорода, генерированными in situ из О2, Н2О2 и Н2О на оксидо-гидроксидном никелевом, диоксидно-свинцовом и допированном бором алмазном электродах. На основании проведенного литературного обзора были поставлены следующие исследовательские задачи: ─ выявление общих кинетических закономерностей протекания непрямого электрокаталитического окисления алифатических спиртов до соответствующих карбоновых кислот; ─ установление влияния основных параметров электролиза на эффективность процесса электрокаталитического окисления алифатических спиртов: плотности тока, количества пропущенного электричества, температуры; ─ установление влияния химической природы электродного материала и способа генерации химически связанных активных форм кислорода на эффективность процесса окисления алифатических спиртов; ─ постановка опытов по наработке и выделению целевого продукта. 41 ГЛАВА 2. Экспериментальная часть 2.1. Условия эксперимента 2.1.1. Рабочие растворы и реактивы В работе в качестве объектов исследования использовали модельные растворы алифатических спиртов, различающиеся по длине углеродной цепи, растворимости и адсорбционной способности на анодных материалах: 1бутанол, 1-гексанол, 1-нонанол и 1-деканол марок х.ч. Растворы использовали без дополнительной очистки, чистоту реагентов проверяли методом газожидкостной хроматографии. Концентрации модельных растворов были выбраны исходя из растворимости реагента в водных средах и составляли: 1-бутанол и 1гексанол — 0,036 использовании моль/л, 1-нонанол и 1-деканол — 0,05 оксидо-гидроксидно-никелевого и моль/л при диоксидно-свинцового электродов. При использовании допированного бором алмазного электрода концентрация 1-бутанола составляла 0,55 моль/л, 1-нонанола — 0,05 моль/л. В экспериментах и аналитических исследованиях использовали реактивы квалификации не ниже «ч.д.а.», без дополнительной очистки. Для приготовления растворов использовали дистиллированную воду. 2.1.2. Электродные материалы, электроды и электролиты Окисление алифатических спиртов химически связанными активными формами кислорода (HO2–, HO2•, HO•), in situ генерированными из O2, H2O2 и H2O, проводили с использованием следующих электродных материалов: оксидо-гидроксидно-никелевый электрод (ОГНЭ), анод из диоксида свинца (Pb/PbO2), допированный бором алмазный электрод (ДБАЭ). Электродные материалы были выбраны исходя из их химической стойкости, каталитической активности и способности адсорбировать НО радикалы на поверхности электрода (раздел 1.5.1.). 42 Оксидо-гидроксидно-никелевый электрод (ОГНЭ) ОГНЭ изготавливали по следующей методике: пористую углеграфитовую матрицу (композитный материал, состоящий из технического углерода А437-Э (80 масс. %) и гидрофобного связующего фторопласта-4Д (20 масс. % )) катодно поляризовали в растворе 1 М NiSO4 ∙7 H2O при плотности тока 10 мА∙см-2 в течение 1 часа, затем анодной поляризацией в растворе 1,2 М NaOH формировали высшие оксиды никеля при Е=1,1 В относительно х.с.э. в течение 1 часа. Для улучшения воспроизводимости характеристик рабочего слоя ОГНЭ проводили периодическое изменение направления его поляризации (4-5 циклов с интервалом 10 минут) в растворе 0,1 N NiSO4 + 0,1 N CH3COONa + 0,005 N NaOH при плотности тока 1 мА∙см-2 [34]. Состояние поверхности ОГНЭ и его химический состав анализировали рентгеноспектральным методом с помощью сканирующего электронного микроскопа ТМ-1000 со спектрометром EDS. Сканы снимали в 5–9 точках на поверхности электрода. Содержание никеля в составе химических элементов, определенных в поверхностном слое электрода, составляло 90,1–97,3% (рисунок 2.1). Рисунок 2.1. Спектральная характеристика, определяющая химический состав поверхностного слоя композитного электрода с нанесенными высшими оксидами никеля. 43 В экспериментах электролизы на ОГНЭ проводили в растворе 1,2 М NaOH. Площадь видимой поверхности электрода составляла 8,0 см2.. Анод из диоксида свинца (Pb/PbO2) На поверхности свинцового анода слой PbO2 формировали по методике, описанной в работах [43, 121, 122]. Свинцовую пластину, с площадью видимой поверхности 8,0 см2, погружали в раствор 0,7 М H2SO4 и анодно окисляли при силе тока 500 мА в течение 1 часа при температуре 20–25 °С. Химический состав анода из диоксида свинца определяли с помощью рентгенофазового анализа на приборе ДРОН-3: PbO– -PbO2 в соотношении 3:1 [122]. При проведении электрокаталитического окисления алифатических спиртов с использованием анода из диоксида свинца использовали электролит следующего состава: 0,1 М H2SO4 + 0,5 M Na2SO4 (в соотношении 1:3). Допированный бором алмазный электрод (ДБАЭ) Допированный бором алмазный электрод был сформирован на подложке из монокристаллического кремния методом горячей нити, разработанным в ИФХ АН СССР [123, 124]. Суть метода заключается в следующем. Смесь метана (или другого углеродсодержащего газа) и водорода активируется вольфрамовой (Ta, Re) проволокой, нагретой до температуры ~ 20000C, и разлагается на поверхности подложки, поддерживаемой при Т≈900–700 0C, размещаемой в непосредственной близости (в 5-10 мм) от W-активатора. Образующиеся при активации углеводородные радикалы обеспечивают высокую скорость осаждения, а высокая концентрация атомарного водорода — селективность осаждения углерода в форме алмаза, газифицируя sp2-углерод. Для придания осаждаемым поликристаллическим слоям алмаза высокой электрической проводимости в газовую смесь добавляют борсодержащие соединения — в нашем случае пары триметилбората. Детали эксперимента описаны в работе [125]. Плѐнки толщиной в несколько 44 мкм осаждали на поверхность пластин кремния и в экспериментах использовали центральную часть плѐнки площадью 1 и 0,15 см2, соответственно. В экспериментах при использовании ДБАЭ электролитом служил раствор 0,1 М H2SO4.. Площадь видимой поверхности электрода составляла 4,16 см2. 2.1.3. Методика проведения электрокаталитического окисления спиртов В разделе 1.5.2 сообщалось, что электроокисление спиртов, содержащих 6 и более атомов углерода, проводят, как правило, при нагревании с целью повышения растворимости спирта и улучшения адсорбируемости его на аноде. В связи с этим, электрокаталитическое окисление 1-гексанола, 1-нонанола и 1-деканола на ОГНЭ и на аноде из диоксида свинца проводили при 70 ºC. Исходя из высокой селективности генерации HO•-радикалов из воды на ДБАЭ, электролизы с его использованием проводили при температуре 20–25 °С. Такой температурный интервал должен способствовать более мягкому окислению использованных субстратов. Окисление алифатических спиртов проводили в гальваностатическом режиме в трехэлектродной бездиафрагменной ячейкеэлектролизере, объемом 150 мл с перемешиванием (при использовании ОГНЭ и Pb/PbO2) и 50 мл без перемешивания (при использовании ДБАЭ). Принципиальная схема ячейки-электролизера представлена на рисунок 2.2. Перемешивание электролита осуществляли с помощью магнитной мешалки. Во всех экспериментах вспомогательным электродом-катодом служил графитовый стержень (спектрально чистый графит) общей площадью 8 см 2. В качестве электрода сравнения во всех экспериментах применяли хлорид-серебрянный электрод (х.с.э). В работе использовали потенциостат марки IPC-Pro MF, соединенный с компьютером. 45 Катод Анод гебер электрода сравнения электролит Рисунок 2.2. Принципиальная схема ячейки-электролизера. Электрокаталитическое окисление алифатических спиртов проводили с реализацией 3-х схем генерации АФК: 1-я схема — окисление на аноде (Анодно): известно, что на ДБАЭ и на Pb/PbO2-электроде в диапазоне потенциалов выделения O2 окисление органических субстратов может протекать при участии образованных АФК [124]. На ОГНЭ окисление спиртов протекает при участии высших оксидов никеля в области потенциалов регенерации последних [45]. Таким образом, процессы при использовании анодной схемы могут протекать как в гомогенных, так и в гетерогенных условиях (раздел 1.3.2) [50, 63]. 2-я схема — окисление на аноде с добавлением Н2О2 в электролит, где дополнительно происходит генерация АФК согласно уравнениям (1.33)–(1.35). Раствор Н2О2 с концентрацией 0,025 М порционно добавляли в электролит каждый час (Анодно + H2O2); 3-я схема — парный электролиз: окисление на аноде с параллельной генерацией HO2– на катоде при пропускании кислорода через раствор электролита (реакции (1.31), (1.32) в зависимости от pH раствора). Скорость пропускания кислорода составляла 30 мл сек-1 (Анодно + O2). Плотность тока рассчитывали на единицу видимой поверхности электрода (габаритная плотность тока). Электролизы проводили в интервале 46 плотностей тока от 5 до 50 мА∙см-2, соответствующих областям рабочих потенциалов для выбранных электродных материалов. Выбор области рабочих значений потенциалов электродов для проведения электрокаталитического окисления алифатических спиртов был сделан на основании литературных данных [45, 99, 104–106] и анодных ветвей снятых циклических вольтамперных (ЦВА) кривых на исследованных электродных материалах с участием спиртов. Все значения потенциалов приведены относительно х.с.э. Для демонстрации вышеизложенного подхода приводим анодные ветви снятых ЦВА кривых с участием 1-бутанола (рисунки 2.3–2.5). Все измерения проводили с помощью потенциостата IPC Pro-MF в трехэлектродной бездиафрагменной ячейке, описанной выше (рисунок 2.2). Скорость развертки потенциала для ОГНЭ и Pb/PbO2-электрода составляла 50 мВ/сек, для ДБАЭ — 100 мВ/сек. 6 1 2 5 i, мА см-2 4 3 2 1 0 0,2 0,4 0,6 0,8 1 1,2 E, В (отн. х.с.э.) Рисунок 2.3. Поляризационные кривые электроокисления 1-бутанола в 1,2 М NaOH на ОГНЭ: 1 – электролит; 2 – реагент-бутанол. Скорость развертки – 50 мВ/сек. 47 Из рисунка 2.3 видно, что добавление регента в электролит приводит к смещению бестокового электродного потенциала в анодную область. Это может быть объяснено адсорбцией данного реагента-спирта на поверхности электродного материала. Согласно литературным данным оксидо-гидроксидноникелевый электрод эффективно работает в области потенциалов регенерации высших оксидов никеля [39, 45, 126]. Таким образом, электрокаталитическое окисление спиртов проводили в диапазоне потенциалов 0,6–1,1 В. 9 8 7 i, мА см-2 6 5 4 3 1 2 2 1 0 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 E, В (отн. х.с.э.) Рисунок 2.4. Поляризационные кривые электроокисления 1-бутанола в электролите состава: 0,1 М H2SO4 + 0,5 M Na2SO4 на Pb/PbO2-электроде: 1 – электролит; 2 – реагент-бутанол. Скорость развертки – 50 мВ/сек. Из рисунка 2.4 видно, что анодные ветви ЦВА для Pb/PbO2-электрода при потенциале 1,1 В имеют явный перегиб с резким увеличением плотности тока в электролите и при добавлении регента-спирта. Это может свидетельствовать о 48 начале генерации АФК через выделение O2 в области потенциалов 1,1 В и выше. Таким образом, основываясь на литературных данных [43] и полученных нами поляризационных кривых, электрокаталитическое окисление спиртов генерированными АФК на диоксидно-свинцовом электроде проводили в области рабочих потенциалов 1,1–1,6 В. 45 1 40 35 i, мА см-2 30 25 2 20 15 10 5 0 0 1 2 3 4 5 6 E, В (отн. х.с.э.) Рисунок 2.5 Поляризационные кривые электроокисления 1-бутанола в 0,1 М H2SO4 на ДБАЭ: 1 – электролит; 2 – реагент-бутанол. Скорость развертки – 100 мВ/сек. На рисунке 2.5 приведены анодные ветви ЦВА кривых электроокисления 1-бутанола в 0,1 М H2SO4 на ДБАЭ. На поляризационных кривых при потенциале 2,9 В наблюдается явный перегиб с ростом плотности тока, что свидетельствует о начале генерации АФК. Также видно, что при потенциале 3,2 В при добавлении реагента на кривой 2 имеется перегиб и смещение в более положительную область потенциалов, в отличие от кривой 1. Это может 49 свидетельствовать об адсорбции субстрата и о начале взаимодействия субстрата с АФК. Из литературы известно, что на ДБАЭ образование АФК происходит при потенциалах положительнее 2,58 В (относительно х.с.э.) [105]. Таким образом, на основании полученных нами результатов и имеющихся литературных данных, для электрокаталитического окисления спиртов на ДБАЭ была выбрана область рабочих потенциалов 2,6–3,5 В. Расчѐт количества электричества, необходимое для проведения электрокаталитического окисления алифатических спиртов (Qтеор) проводили согласно уравнению реакции [127]: (2.1) Из литературы известно [127], что для получения 1 г-экв вещества необходимо определѐнное количество электричества равное числу Фарадея, F, 26,8 А ч. Из уравнения (2.1) следует, что на окисление одного моля спирта требуется затратить 4F, или . Исходя из этого, теоретически необходимое количество электричества на окисление определѐнного количества спирта-субстрата рассчитывали по формуле (2.2), согласно [127]: (2.2) где mисх – количество исходного вещества, г; Mr – грамм-молекулярный вес спирта (таблица 1.1). Результаты расчѐтов требуемого количества электричества (Qтеор) на окисление алифатических спиртов (1-бутанола, 1-гексанола, 1-нонанола и 1деканола) приведены в таблицах 2.1–2.2. 50 Таблица 2.1 Требуемое количество электричества на окисление алифатических спиртов. Электроды: ОГНЭ и Pb/PbO2. Исходный Сспирта, V, P, Qтеор, реагент моль/л мл г Ач 1-бутанол 0,36 5,0 4,05 5,8 1-гексанол 0,36 6,7 5,50 5,9 1-нонанол 0,05 1,34 1,10 0,8 1-деканол 0,05 1,5 1,25 0,9 Таблица 2.2 Требуемое количество электричества на окисление алифатических спиртов. Электроды: ДБАЭ. Исходный Сспирта, V, P, Qтеор, реагент моль/л мл г Ач 1-бутанол 0,55 2,5 2,03 2,9 1-нонанол 0,05 0,5 0,36 0,3 2.2. Методы исследования По окончании процесса электрокаталитического окисления выделение карбоновых кислот проводили методом дискретной экстракции [128]. В качестве экстрагента использовали хлороформ. В экспериментах с использованием щелочного электролита конечным продуктом электролиза являются натриевые соли карбоновых кислот. В этом случае, с целью перевода натриевой соли в карбоновую кислоту, перед экстракцией раствор электролита предварительно подкисляли до pH ~ 4 концентрированной H2SO4. Для всех экспериментов экстракцию повторяли три раза для полного извлечения продукта. Далее экстракт очищали от посторонних веществ промыванием 51 дистиллированной водой. После чего проводили аналитические исследования с использованием химических и физико-химических методов: газожидкостной хроматографии, хроматомасспектроскопии, потенциометрического титрования, метода УФ-спектроскопии, определения химического потребления кислорода (ХПК). При использовании Pb/PbO2-электрода и ОГНЭ электролит проверяли на содержание свинца абсорбционной или никеля, спектроскопии с соответственно, помощью методом атомно- спектрофотометра AAS-30. Содержание никеля в электролите после окончания электролизов не превышало 0,2 мг/л, свинца не более 0,3 мг/л. В поставленных балансовых опытах по наработке целевых продуктов электролит подвергали регенерации. Во всех экспериментах оставшуюся часть водного раствора после экстракции (рафинат) качественно проверяли на содержание спиртов по реакции с реактивом Несслера [129]. Отсутствие карбоновых кислот хроматографии. в рафинате Далее проверяли концентрацию методом газожидкостной электролита определяли титриметрическими методами анализа [130], после чего раствор электролита доводили до требуемой концентрации. Количественное определение карбоновых кислот проводили методом газожидкостной хроматогрфии (ГЖХ) на хроматроне-N-AW-HMDS. Условия хроматографирования были следующие: колонка длиной 1,5 м, диаметром 4 мм заполнена смешанной фазой: полиэтиленгликольадипинат и ФФАП, температура термостата колонок 180 °С, газ-носитель гелий, скорость протока 30 мл/мин. Количественное содержание компонентов вычисляли по площадям газохроматографических пиков. В некоторых количественное опытах определение (где отсутствовали карбоновых кислот побочные проводили потенциометрического титрования параллельно с методом ГЖХ. продукты) методом 52 Состав продуктов электролиза анализировали с помощью методов УФспектроскопии. Электронные спектры поглощения регистрировали на спектрофотометре Shimadzu UV-300 в диапазоне длинны волны 150–350 нм. Известно, что карбоновые кислоты обладают слабой интенсивностью поглощения света со значением максимума поглощения λmax 200 nm и практически не пригодны для количественного определения. Однако следует отметить, что в этом случае по данным УФ-спектра может быть достаточно надежно установлено наличие или отсутствие карбоксильной группы в исследуемом веществе [131]. В данной работе для всех экспериментов наблюдали идентичные спектры поглощения (рисунок 2.6), которые качественно доказывают наличие карбоновых кислот в исследуемом растворе. Это свидетельствует об отсутствии процессов более глубокого окисления с формированием новых сопряженных связей. 1,6 1,4 Absorbance (AU) 1,2 1 0,8 0,6 0,4 0,2 0 150 200 250 300 350 Wavelength (nm) Рисунок 2.6. Спектры поглощения 1-нонанола. Также продукты электрокаталитического окисления анализировали методом хромато-масс-спектроскопии на газовом хроматографе Agilent Technologies 7890 GC System с квадрупольным масс-спектрометром 5975 С. 53 Качественный анализ основан на сравнении времен удерживания и полных масс-спектров с соответствующими данными библиотеки масс спектрометра. Степень минерализации контролировали с помощью показателя химического потребления кислорода (ХПК) по методике [132]. Реактивы для определения ХПК были следующие: Бихромат калия, 0,25 N титрованный раствор. Соль Мора, приблизительно 0,25 N раствор. N-фенилантраниловая кислота, индикатор. Расчет ХПК, выраженный числом миллиграммов кислорода на 1 л анализируемой пробы, проводили по формуле: (2.3) где a – объем раствора соли Мора, израсходованного на титрование в холостом опыте, мл; b – объем того же раствора, израсходованного на титрование пробы, мл; N – нормальность титрованного раствора соли Мора; V – объем анализируемой пробы, мл; 8 – эквивалент кислорода. Присутствие альдегидов качественно проверяли параллельно по трѐм методикам, описанным в [129, 133]: реакция с реактивом Феллинга, реакция с аммиачным раствором оксида серебра и реакция с реактивом Несслера. Количественное определение альдегидов в растворе проводили по прямому методу Бенетта [129]. Выход по току (Вт) выраженный в % рассчитывали по следующей формуле [127]: где mпрод – количество полученного продукта, г; 54 Qфакт – фактически затраченное количество электричества в процессе, А ч; Mrпрод – грамм-молекулярный вес полученного продукта (таблица 1.2). В настоящей работе под эффективностью процесса мы понимаем получение целевого продукта электрокаталитического окисления спиртов с наиболее высоким выходом по току, то есть эффективное количественное использование окислительного реагента – электрического тока, прошедшего через электролизер на образование целевого продукта. В связи с этим, особое внимание мы уделяем выходу целевого продукта по току. 2.3 Обработка экспериментальных данных Статистическую обработку экспериментальных результатов проводили согласно методике [134], пользуясь методом вычисления средних величин, исключением выскакивающих вариант. Доверительные интервалы для генерального среднего измеряемых величин при доверительной вероятности, равной 0,95, рассчитывали, воспользовавшись функцией распределения Стьюдента. С целью контроля воспроизводимости результатов, эксперименты повторяли 3–5 раз. Анализ эмпирических зависимостей проводился с использованием программных средств Microsoft Excel2007, Visio2007, Mathcad15. 55 ГЛАВА 3. Экспериментальные результаты и их обсуждение 3.1. Кинетические закономерности непрямого электрокаталитического окисления алифатических спиртов Кинетику накопления карбоновых кислот в процессе окисления алифатических спиртов исследовали с использованием в качестве модельных субстратов-реагентов: 1-бутанол, как представитель широкого ряда алифатических спиртов, обладающий высокой растворимостью в водных электролитах и хорошей адсорбционной способностью на анодных материалах, и 1-нонанол, обладающий низкой растворимость и адсорбируемостью (раздел 1.1.1, 1.5.2). В качестве электродных материалов использовали оксидогидроксидно никелевый и диоксидно-свинцовый электроды, отличающиеся по своей способности генерировать и адсорбировать HO•-радикалы (раздел 1.4.1). Кинетические исследования по накоплению продукта электролиза проводили при плотности тока 10 мА см-2 с различными схемами генерации АФК (раздел 2.1.3). При окислении 1-бутанола и 1-нонанола за первые 4 часа электролиза наблюдали изменения потенциала электрода от 0,91 до 0,96 В на ОГНЭ, и от 1,51 до 1,57 В на Pb/PbO2-электроде. Концентрацию образующегося продукта во время электролиза контролировали методом ГЖХ (раздел 2.2). В таких условиях можно ожидать наличия различных порядков реакции окисления спиртов до соответствующих карбоновых кислот. Для определения порядка реакции использовали известное формальное уравнение кинетики химических реакций: (3.1) где n – порядок реакции; k – эффективная константа скорости окисления, зависящая от силы тока, активности анода и других параметров электрохимической ячейки; C – концентрация спирта в активной зоне ячейки. 56 Следует подчеркнуть, что уравнение (3.1) не представляет истинную кинетику реакции, а n и k скорее кажущиеся порядок реакции и константа скорости [135], характеризующие изменение концентрации вещества не только непосредственно на поверхности электрода, но и в объеме электролита [136]. Эти параметры уравнения могут быть определены по экспериментально измеренным данным как функции от времени. Для определения параметров, входящих в кинетическое уравнение, использовали интегральный метод расчета, при котором уравнение (3.1) интегрировали, и получили вид функции, отражающей изменение количества нарабатываемого продукта во времени. Таким образом, решение уравнения (3.1) можно представить в общем виде: где ; C0, C – концентрации спирта-субстрата на входе в электрохимическую ячейку и на выходе из ячейки, соответственно; t – среднее время пребывания раствора электролита в электрохимической ячейке. При решении уравнения (3.2) значение n варьировали в пределах 0,3–2 с целью установления порядка реакции. Далее строили графики в координатах . При построении кривых хорошие линейные зависимости с минимальным отклонением были получены в большинстве случаев при n = 0,8 0,5, что является близким к псевдо-первому порядку реакции. Таким образом, далее кинетику превращения органического компонента рассматривали согласно реакции псевдо-первого порядка, для которого кинетическое уравнение (3.2) принимает вид: (3.3) 57 Результаты решения уравнения (3.3) для 1-бутанола и 1-нонанола на ОГНЭ и на Pb/PbO2-электроде графически представлены на рисунках 3.1–3.4. Согласно полученным данным можно сделать заключение, что порядок реакции окисления спиртов до соответствующих кислот мало зависит от концентрации реагента, из чего можно предположить, что лимитирующим фактором процесса окисления является концентрация химически связанных активных форм кислорода. Таким образом, скорость накопления продукта будет пропорциональна концентрации АФК. 0,14 3 0,12 lnC0/C 0,10 2 0,08 1 0,06 0,04 0,02 0,00 0 1 2 3 4 5 6 7 Время, ч Рисунок 3.1. Кинетика накопления масляной кислоты на ОГНЭ при плотности тока 10 мА см-2 с использованием схем окисления: 1 – Анодно; 2 – Анодно+H2O2; 3 – Анодно+O2. Исходная концентрация 1-бутанола составляла 0,36 моль/л. 58 0,3 1 0,25 3 lnC0/C 0,2 0,15 0,1 2 0,05 0 0 0,5 1 1,5 2 2,5 3 3,5 Время, ч Рисунок 3.2. Кинетика накопления пеларгоновой кислоты на ОГНЭ при плотности тока 10 мА см-2 с использованием схем окисления: 1 – Анодно; 2 – Анодно+H2O2; 3 – Анодно+O2. Исходная концентрация 1-нонанола 0,05 моль/л. 59 0,04 3 0,035 0,03 lnC0/C 0,025 1 0,02 0,015 0,01 2 0,005 0 0 0,5 1 1,5 2 2,5 3 3,5 4 4,5 Время, ч Рисунок 3.3. Кинетика накопления масляной кислоты на Pb/PbO2 электроде при плотности тока 10 мА см-2 с использованием схем окисления: 1 – Анодно; 2 – Анодно+H2O2; 3 – Анодно+O2. Исходная концентрация 1-бутанола составляла 0,36 моль/л. 60 0,09 0,08 3 0,07 lnC0/C 0,06 0,05 1 0,04 0,03 0,02 2 0,01 0 0 0,5 1 1,5 2 2,5 3 3,5 Время, ч Рисунок 3.4. Кинетика накопления пеларгоновой кислоты на Pb/PbO2 электроде при плотности тока 10 мА см-2 с использованием схем окисления: 1 – Анодно; 2 – Анодно+H2O2; 3 – Анодно+O2. Исходная концентрация 1-нонанола составляла 0,05 моль/л. Из данных, представленных на рисунках 3.1–3.2, видно, что на ОГНЭ в первоначальный момент процесс окисления 1-бутанола протекает с большей эффективностью при дополнительном введении окислителей в электролит (O2 или H2O2). Однако при окислении 1-нонанола дополнительная генерация АФК снижает эффективность процесса в первые часы электролиза. Это объясняется плохой адсорбционной способностью на электроде 1-нонанола в следствие стерических затруднений и плохой растворимостью спирта, что приводит к снижению скорости подвода субстрата к поверхности электрода. В таком случае, в отсутствии субстрата на активных центрах электрода выделяется кислород согласно уравнению (1.45), а дополнительно генерированные АФК 61 рекомбинируют между собой с образованием H2O2 по реакции (1.48), который далее окисляется до O2 (1.49–1.50). Как видно из рисунков 3.3–3.4, на аноде из диоксида свинца эффективность процесса накопления целевых продуктов увеличивается с использованием схемы парного электролиза за счет вклада непрямого окисления, где АФК дополнительно катодно генерируются согласно уравнению (1.32). Необходимо отметить, что в первоначальный момент добавление пероксида водорода в электролит снижает эффективность окисления. Это может быть объяснено разложением Н2О2 с образованием различных АФК по реакциям (1.34)–(1.35), что приводит к дезактивации поверхности электрода при наработке карбоновых кислот в первые часы электролиза. Найденные графически значения констант скорости окисления спиртов в начальный момент при различных схемах генерации АФК представлены в таблице 3.1. Таблица 3.1 Константы скорости окисления (k (ч-1)) 1-бутанола и 1-нонанола до соответствующих кислот на Pb/PbO2 и ОГНЭ с различными схемами электролиза при плотности тока 10 мА см-2. Спирт 1-бутанол 1-нонанол Электрод Константа скорости, k, ч-1, при схеме электролиза Анодно Анодно+Н2О2 Анодго+О2 Pb/PbO2 0,006±0,001 0,002±0,001 0,009±0,001 ОГНЭ 0,12±0,001 0,13±0,001 0,22±0,002 Pb/PbO2 0,012±0,001 0,003±0,001 0,028±0,001 ОГНЭ 0,085±0,002 0,022±0,001 0,067±0,001 Исходные концентрации 1-бутанола – 0,36 моль/л, 1-нонанола – 0,05 моль/л. Константы скорости реакции показывают, что в начале процесса электролиза окисление спиртов происходит с небольшой скоростью. Это, 62 вероятно, может быть связано с наработкой достаточно высокой концентрации активных форм кислорода, которые далее взаимодействуют с субстратом. Из таблицы видно, что в начальный период времени лучшие условия для окисления 1-бутанола до масляной кислоты реализуются на оксидо- гидроксидно никелевом электроде при парном электролизе. При окислении 1нонанола до пеларгоновой кислоты лучшими условиями являются использование Ni(O)OH электрода при анодной схеме окисления. Анализ полученных экспериментальных результатов и литературных данных (раздел 1.3) позволил предположить возможные маршруты непрямого электрокаталитического соответствующих окисления карбоновых кислот алифатических in situ спиртов генерированными до АФК. Рассмотрим возможные варианты непрямого окисления. Например, начальным этапом процесса окисления является образование АФК по уравнениям (1.33), (1.34), которые могут адсорбироваться на поверхности анода согласно уравнениям (1.41), (1.42). Далее происходит взаимодействие спирта с генерированными АФК в объѐме электролита или на поверхности электрода. Можно предположить, что при взаимодействии спирта с -радикалом процесс пойдѐт по следующему пути (3.1): H R C OH + HO2 H OH R C + H2O + H+ + e O (3.1) Формально эта реакция возможна, но в силу сложности процесса может протекать только на поверхности электрода – гетерогенный вариант. При взаимодействии спирта с водорода от спирта с образованием -радикалом возможно отщепление -гидроксиметильного радикала по уравнению (3.2), который далее может взаимодействовать с различными типами АФК по уравнениям (3.3)–(3.5) в гетерогенных и гомогенных условиях: 63 H H R C OH + HO H H R C (3.2) + HO2 OH (3.3) + H2O2 OH OH + H2O + H+ + e R C O (3.4) H R C + H2O OH OH + H2O R C O H R C R C H + HO OH + H2O R C O (3.5) Анализ уравнений (3.4)–(3.5) позволяет сказать, что взаимодействие гидроксиметильного радикала спирта с - формально возможно, но является маловероятным как и при реализации уравнения (3.1). При взаимодействии радикала спирта с HO -радикалом образуется альдегид (3.5), который далее может окисляться до соответствующей карбоновой кислоты. Протекание реакции (3.3), где взаимодействие -гидроксиметильного радикала спирта с -радикалом приводит к образованию карбоновой кислоты является возможным по причине простоты реализации. Таким образом, из вышесказанного видно, что возможные пути реакции зависят от соотношения и концентрации той или иной формы химически связанного активного кислорода, а, следовательно, и от используемых электродных материалов, различающихся по способности адсорбировать HO радикалы (раздел 1.4.1). Следует отметить, что образование -радикала происходит в процессе рекомбинации HO -радикалов до пероксида водорода по реакции (1.48) с 64 последующим его разложением (1.34), либо при введении H2O2 в электролит. Также -радикал может образовываться по уравнению (1.50). Генерация HO -радикалов происходит по маршруту, как описано в разделе 1.4.1, в зависимости от материала используемого электрода и pH электролита. Из литературы известно, что промежуточными продуктами окисления спиртов до кислот являются соответствующие им альдегиды (раздел 1.3.1). Поскольку скорость окисления альдегидов значительно выше по сравнению со спиртами, соответствующих альдегидов в продуктах окисления 1-бутанола, 1гексанола, 1-нонанола и 1-деканола на всех используемых электродных материалах практически обнаружить не удалось. На рисунках 3.5 и 3.6 представлены хроматограммы продуктов окисления 1-нонанола до пеларгоновой кислоты и 1-деканола до каприновой кислоты на ОГНЭ с использованием различных схем генерации АФК при пропущенном теоретически необходимом количестве электричества, при плотности тока 10 мА∙см-2. Из данных рисунков 3.5 и 3.6 видно, что в растворах электролитов обнаружен ряд алифатических кислот в следовых количествах: гептановая, гексановая при окислении 1-нонанола; октановая при окислении 1-деканола. Эти кислоты образуются в ходе дальнейшего последовательного окисления побочных продуктов. 65 c Анодное окисление a g Показание детектора Чувств. 1:32 f Время, мин 50 g 40 e 30 20 10 Анодное окисление + H2O2 a Показание детектора c Чувств. 1:32 f Время, мин b d 50 40 g e 30 d 20 b 10 a Чувств. 1:32 c f e Время, мин 50 40 30 d 20 Показание детектора Анодное окисление + O2 b 10 Рисунок 3.5. Хроматограммы продуктов окисления 1-нонанола на ОГНЭ при различных способах генерации АФК: (а) хлороформ, (b) бутановая кислота, (c) 1-нонанол, (d) гексановая кислота, (e) гептановая кислота, (f) октановая кислота, (g) пеларгоновая кислота. Q=Qтеор, i=10 мА∙см-2. 66 a Анодное окисление Показание детектора b Чувств. 1:32 d c Время, мин 50 40 30 20 10 a Показание детектора Анодное окисление + H2O2 Чувств. 1:32 b d c e Время, мин 50 40 30 20 10 a Анодное окисление + O2 Показание детектора Чувств. 1:32 d b c Время, мин 50 e 40 30 20 10 Рисунок 3.6. Хроматограммы продуктов окисления 1-деканолана на ОГНЭ при различных способах генерации АФК: (а) хлороформ, (b) 1-деканол, (c) пеларгоновая кислота, (d) каприновая кислота, (e) октановая кислота. Q=Qтеор, i=10 мА∙см-2. 67 В таблице 3.2 приведены количественные данные о побочных продуктах, которые образуются при окислении 1-нонанола и 1-деканола. Данные получены по результатам хроматографии и хромато-масс-спектроскопии. Установлено, что основным побочным продуктом окисления 1-нонанола является октановая кислота в количестве 4,2 % при анодном окислении и 6,0–9,0 % при дополнительном использовании АФК. При окислении 1-деканола основным побочным продуктом является пеларгоновая кислота в количествах 4,7 % при анодном окислении и ~12,0 % при дополнительной генерации АФК. Это подтверждает известный факт, что и при наших условиях электролиза основными побочными продуктами окисления спиртов являются низшие гомологи образовавшихся целевых карбоновых кислот (раздел 1.5.2). Таблица 3.2 Содержание побочных продуктов при окислении 1-нонанола и 1-деканола до пеларгоновой и каприновой кислот, соответственно, на ОГНЭ. Плотность тока i=10 мА∙см-2, количество электричества Q=Qтеор. Целевой продукт Пеларгоновая кислота Каприновая кислота Методами Основной побочный продукт, % Общее количество побочных продуктов, % Анодно 4,2 1 7,3 2 Анодно+H2O2 6,5 1 10,8 2 Анодно+O2 8,9 1 11,1 1 Анодно 4,7 2 8,1 2 Анодно+H2O2 12,3 1 19,3 1 Анодно+O2 12,0 1 19,7 1 Схема окисления хромато-масс-спектроскопии и УФ-спектроскопии установлено, что на ОГНЭ, Pb/PbO2-электроде и ДБАЭ при окислении бутилового, гексилового, нонилового и децилового спиртов при использовании 68 2-ой и 3-ей схем электролиза (раздел 2.1.3) с дополнительным введением активных форм кислорода состав продуктов электрокаталитического окисления идентичен схеме с анодным окислением. Однако стоит отметить, что при использовании анода из диоксида свинца в состав продуктов входят кроме исходного спирта, эфиры и соответствующие карбоновые кислоты. Так с использованием схемы с катодной генерацией H2O2 из O2 при окислении 1-бутанола в составе продуктов окисления доля масляной кислоты составила 80,6 %, эфиров – 17,9 %, в составе продуктов окисления 1гексанола капроновая кислота составила 17,5 % и эфиры – 31,5 %. Причиной образования эфиров является реакция этерификации, которая обусловлена характерной особенностью карбоновых кислот образовывать со спиртами сложные эфиры в присутствии сильных кислот (раздел 1.1.2). При окислении спиртов на аноде из диоксида свинца катализатором процесса является серная кислота (раздел 1.3.1). При электрокаталитическом окислении спиртов на оксидо-гидроксидноникелевом и допированном бором алмазном электродах основными продуктами являются соответственные карбоновые кислоты. Заключение В результате проведенных исследований установлено, что превращение органических субстратов в процессе непрямого электрокаталитического окисления алифатических спиртов протекает по псевдо-первому порядку реакции. Сделано предположение, что лимитирующим фактором процесса окисления спиртов является концентрация химически связанных активных форм кислорода. Таким образом, скорость накопления продукта будет пропорциональна концентрации АФК. Установлено, что при окислении плохо растворимых спиртов (1нонанол), вследствие снижения скорости подвода субстрата к поверхности электрода, в первые часы электролиза дополнительная генерация АФК снижает 69 эффективность процесса. В этом случае в отсутствии субстрата на активных центрах электрода выделяется кислород, а генерированные АФК рекомбинируют между собой с образованием H2O2, который далее окисляется до O2. Согласно рассчитанным константам скорости окисления сделано заключение о том, что в начальный период электролиза для окисления хорошо растворимых спиртов (1-бутанол) лучшие условия реализуются на ОГНЭ при парном электролизе. При окислении плохо растворимых спиртов (1-нонанол) лучшими условиями являются использование ОГНЭ при анодной схеме окисления. В начале процесса электролиза окисление спиртов происходит с небольшой скоростью. Это, вероятно, связано с наработкой достаточно высокой концентрации активных форм кислорода, которые далее взаимодействуют с субстратом. На основании полученных нами результатов и известных из литературы данных установлено, электрокаталитического что возможные окисления маршруты алифатических непрямого спиртов до соответствующих карбоновых кислот зависят от соотношения и концентрации той или иной формы химически связанного активного кислорода и, как следствие, от используемых электродных материалов. Установлено, что при электрокаталитическом окислении спиртов на ОГНЭ и ДБАЭ основными продуктами являются соответствующие карбоновые кислоты, при использовании Pb/PbO2-электрода в состав продуктов входят кроме исходного спирта, эфиры и соответствующие карбоновые кислоты. Установлено, что при окислении 1-бутанола, 1-гексанола, 1-нонанола и 1деканола основными побочными продуктами электролиза являются низшие гомологи образовавшихся целевых карбоновых кислот при использовании всех схем генерации АФК на ОГНЭ, ДБАЭ и на Pb/PbO2-электроде. 70 3.2. Влияние основных параметров электролиза на эффективность электрокаталитического окисления алифатических спиртов Исследование влияния плотности тока, количества пропущенного электричества и температуры на эффективность процесса окисления алифатических спиртов до соответствующих карбоновых кислот проводили на ОГНЭ, Pb/PbO2-элетроде и на ДБАЭ. В качестве модельных реагентов были использованы 1-бутанол, 1-гексанол и 1-нонанол. Выбор был обусловлен различием в длине углеродной цепи молекул спиртов и способности адсорбироваться на материале поверхности электрода, что позволяет выявить закономерности влияния основных параметров электролиза на эффективность окисления выбранных представителей широкого гомологического ряда нормальных алифатических спиртов. Экспериментальные результаты исследования влияния плотности тока на эффективность процесса электрокаталитического окисления 1-бутанола и 1-нонанола до масляной и пеларгоновой кислот, соответственно, представлены в таблицах 3.3–3.4. Из таблиц 3.3–3.4 видно, что окисление алифатических спиртов до соответствующих карбоновых кислот на ОГНЭ, ДБАЭ и Pb/PbO2-электроде эффективно протекают при плотностях тока 5–10 мА∙см-2 с использованием различных схем генерации АФК. Так, например, с использованием анодной схемы окисления на ОГНЭ при 10 мА∙см-2 выход по току масляной кислоты составляет 91,5 %, выход по току пеларгоновой кислоты составляет 56,4 %; на Pb/PbO2-элетроде выход по току масляной кислоты — 69,5 %, пеларгоновой — 30,5 %. С использованием ДБАЭ при плотности тока 5 мА∙см-2 выход по току масляной кислоты составляет 91,6 %, пеларгоновой кислоты –– 98,0 %. Дальнейшее повышение плотности тока в интервале 15–60 мА∙см-2 резко снижает выход целевого продукта (таблицы 3.3–3.4). Вероятно, это связано с тем, что с увеличением плотности тока значительно возрастает скорость генерации HO•-радикалов и это смещает процесс в сторону дальнейшего 71 неселективного окисления образовавшейся карбоновой кислоты с минерализацией до CO2 и H2O. Такой вариант протекания процесса подтверждают данные УФ-спктроскопии и снижение показателя химического потребления кислорода (ХПК). На примере окисления 1-нонанола при анодном окислении на ДБАЭ снижение показателя ХПК в зависимости от плотности тока составило: при i = 15 мА∙см-2 — 79,9 %; при i = 50 мА∙см-2 — 89,4 %. Таблица 3.3 Результаты окисления 1-бутанола до масляной кислоты на ОГНЭ, Pb/PbO2-электроде (Сбутанола=0,36 моль/л) и ДБАЭ (Сбутанола=0,55 моль/л) при разных значениях плотности тока. Материал электрода Схема электролиза Анодно ОГНЭ Анодно+H2O2 Анодно+O2 Масляная кислота i, мА∙см-2 Q, А ч Вт, % 10 0,8 91,5±2,1 15* 0,8 36,2±0,7 30 0,8 21,6±1,2 50* 3,5 6,2±0,6 10 0,8 87,6±1,4 * 0,8 16,9±0,6 10 0,8 167,5±2,1 * 3,5 12,4±1,1 0,8 0,8 0,3 0,3 0,7 69,5±1,1 42,2±0,7 91,6±0,5 66,4±1,0 11,0±1,2 50 35 10 60 5 ДБАЭ Анодно 15 25 * – электролизы проводили при температуре 40 ºC; Pb/PbO2 Анодно 72 Таблица 3.4 Результаты окисления 1-нонанола до пеларгоновой кислоты на ОГНЭ, Pb/PbO2, ДБАЭ при разных значениях плотности тока (Снонанола = 0,05 моль/л), количество пропущенного электричества Q=Qтеор. Материал электрода Схема электролиза Анодно ОГНЭ Анодно+H2O2 Анодно+O2 Анодно Pb/PbO2 Анодно ДБАЭ Анодно+H2O2 Анодно+O2 Пеларгоновая кислота i, мА∙см-2 Вт, % 5 10 15 35 10 15 10 20 5 10 15 5 10 15 5 10 15 5 10 15 60,0±1,5 56,4±2,1 22,6±1,6 9,4±0,5 79,8±0,7 3,8±0,6 83,7±0,6 15,9±1,1 29,0±1,3 30,5±1,7 22,3±1,5 98,0±0,6 68,2±1,2 0,1±0,01 76,0±1,0 57,8±2,1 26,0±1,6 33,0±1,5 60,5±1,2 30,1±1,6 При высоких плотностях тока снижение выхода по току целевых продуктов также связано с увеличением вклада конкурентного процесса электролиза воды (п. 1.3.4). Также из таблиц 3.3–3.4 видно, что на ДБАЭ при малых плотностях тока (5 мА см-2) выход целевого продукта высок и составляет более 90 %, что связано, по-видимому, с присутствием высоко 73 реакционноспособных НО•-радикалов на поверхности электрода в концентрациях больших, чем при использовании ОГНЭ и Pb/PbO2-электрода. В разделе 1.5.2 отмечено, что с ростом температуры скорость окисления плохо растворимых спиртов возрастает за счѐт повышения их растворимости и, как следствие, снижения диффузионных ограничений по подводу субстрата к поверхности электрода. Так при окислении 1-гексанола на ОГНЭ с использованием 2-ой схемы электролиза (Анодно+H2O2) при плотности тока 10 мА см-2 и пропущенном количестве электричества 0,8 А ч выходы по току и веществу капроновой кислоты при 20 °С составили 93,7 % и 13,0 %, соответственно, а при 60 °С — 108,9 % и 15,1 %, соответственно. Однако результаты исследования показали, что при окислении спиртов с короткой углеродной цепью повышение температуры приводит к увеличению степени минерализации образовавшейся кислоты до CO2 и H2O. Контроль за изменением ХПК на примере 1-бутанола показал, что при температуре 20 °C степень минерализации кислоты составила 21,3 %, при температуре 40 °С — 65,0 %. В разделе 1.5.2 указано, что применение органического растворителя (трет-бутилового спирта или четырѐххлористого углерода), добавленного в электролит, позволяет проводить процесс окисления плохо растворимых спиртов без нагревания. На основании этого были проведены эксперименты по окислению 1-нонанола на ОГНЭ без нагревания при использовании анодной схемы, при плотности тока 10 мА∙см-2 и Q=Qтеор, где выход по току пеларгоновой кислоты с добавлением в электролит CCl4 увеличивается с 56,5 % до 69,0 %. Результаты исследования, показывающие влияние количества пропущенного электричества на эффективность процесса на примере окисления 1-бутанола до масляной кислоты, 1-гексанола до каприновой кислоты и 1-нонанола до пеларгоновой кислоты, представлены в таблицах 3.5–3.7, соответственно. 74 Таблица 3.5 Влияние количества пропущенного электричества Q на выход масляной кислоты при окислении 1-бутанола на ОГНЭ и Pb/PbO2-электроде. Концентрация 1-бутанола 0,36 моль/л. Qтеор = 5,9 А ч. Материал электрода Схема электролиза i, мА см-2 Q, Ач Вт, % ОГНЭ Анодно, 20°С 10 10 60 60 60 60 0,8 5,9 0,8 3,5 0,8 3,5 91,5±2,1 7,4±1,4 79,8±1,6 58,2±1,9 133,7±1,2 74,8±1,0 Pb/PbO2 Анодно +H2O2, 40°С Анодно +О2, 40°С Таблица 3.6 Влияние количества пропущенного электричества Q на выход капроновой кислоты при окислении 1-гексанола на ОГНЭ и Pb/PbO2-электроде. Концентрация 1-гексанола 0,36 моль/л. Qтеор = 5,8 А ч. Материал электрода Схема электролиза i, мА×см-2 Q, Ач Вт, % ОГНЭ Анодно, 20°С 30 30 0,8 3,5 69,7±1,5 22,2±1,6 60 0,8 28,8±1,7 60 3,5 25,0±1,0 60 60 60 60 0,8 3,5 0,8 3,5 51,6±0,9 16,7±0,7 98,0±1,0 30,6±1,7 Анодно Pb/PbO2 Анодно +H2O2, 40°С Анодно +О2, 40°С 75 Таблица 3.7 Влияние количества пропущенного электричества Q на выход пеларгоновой кислоты при окислении 1-нонанола на ОГНЭ, i = 10 мА∙см-2, Т = 70 °С. Концентрация 1-нонанола — 0,05 моль/л. Qтеор = 0,8 А ч. Схема окисления Q, А∙ч 0,8 Выход по веществу, % 56,5±2,0 Выход по току, % 56,4±2,1 Анодно 1,1 93,1±1,0 41,0±1,3 Анодно 0,8 79,9±1,0 79,8±0,7 + Н2О2 1,1 77,7±1,2 59,7±1,2 Анодно 0,8 83,8±0,8 83,7±0,6 + О2 1,1 82,5±0,8 63,4±0,8 Известно [127], что полная конверсия исходного вещества всегда приводит к непропорциональному удлинению электролиза и снижению выхода целевого вещества по току. Так из таблиц 3.5–3.7 видно, что превращение исходного спирта до соответствующей карбоновой кислоты протекает с высокой токовой эффективностью при пропускании определенного количества электричества (Qэффект), зависящее от концентрации реагента-спирта. С увеличением количества пропущенного электричества свыше Qэффект выход по току карбоновых кислот снижается при использовании всех трѐх схем окисления. Это связано с дальнейшим деструктивным окислением целевого продукта. Результаты подтверждены УФ-спектроскопией и снижением показателя ХПК. Так, на примере окисления 1-бутанола на ОГНЭ при плотности тока 10 мА см-2 с использованием анодной схемы окисления, выход по току масляной кислоты при пропускании 0,8 А ч составил 91,5 %, а при 5,9 А ч — 7,4 %. Из таблицы 3.7 видно, что при небольшой концентрации исходного спирта (1-нонанола) при пропускании теоретически необходимого количества 76 электричества наработка пеларгоновой кислоты происходит с высокой токовой эффективностью, при увеличении количества электричества свыше теоретически необходимого выход по току снижается. При высокой концентрации реагента-спирта (таблица 3.5–36) целесообразно выделять из реакционной среды продукт (карбоновую кислоту) и с целью сохранения токовой эффективности использовать не вступивший в реакцию спирт для повторного синтеза [127]. Заключение Установлено, что наиболее эффективное окисление алифатических спиртов до карбоновых кислот протекает при малых плотностях тока 5–10 мА∙см-2 при использовании всех трех схем генерации АФК (раздел 2.1.3) на ОГНЭ, ДБАЭ и на Pb/PbO2-электроде. Дальнейшее повышение плотности тока в интервале 15–60 мА см-2, как и увеличение количества пропущенного электричества свыше Qэффект резко снижают выход целевого продукта. Повидимому, это связано с возрастанием скорости и селективности генерации HO -радикалов, что смещает процесс в сторону дальнейшего неселективного окисления образовавшейся карбоновой кислоты с минерализацией до CO2 и H2O. Согласно литературным данным и нашим экспериментальным результатам, установлено, что при окислении плохо растворимых спиртов повышение температуры электролиза ведѐт к увеличению скорости их окисления вследствие увеличения их растворимости и снижения диффузионных ограничений по подводу субстрата к поверхности электрода. Также установлено, что при окислении спиртов с короткой углеродной цепью повышение температуры приводит к увеличению степени минерализации образовавшейся кислоты до CO2 и H2O. 77 3.3 Влияние химической природы электродного материала и способа генерации АФК Исследование влияния способа генерации химически связанных активных форм кислорода и химической природы электродного материала на эффективность процесса непрямого электрокаталитического окисления спиртов до соответствующих карбоновых кислот проводили с использованием в качестве субстратов 1-бутанола, 1-нонанола и 1-деканола. В экспериментах были реализованы 3 схемы генерации АФК (раздел 2.1.3). Результаты исследования представлены в виде диаграмм на рисунках 3.7– 3.9. 180 3 Выход по току, % 150 3 3 2 120 1 2 2 90 1 1 60 30 0 Схема 1 Схема 2 Схема 3 ОГНЭ 91,5 87,1 167,5 ДБАЭ 66,4 114,8 137,0 Pb/PbO2 69,5 86,3 136,0 Рисунок 3.7. Диаграмма влияния схемы электролиза на выход по току масляной кислоты при окислении 1-бутанола на ОГНЭ, ДБАЭ, Pb/PbO2электроде. Плотность тока i=10 мА см-2; электричества Q/Qтеор=0,14; температура Т=20 °С. количество пропущенного 78 1 100 2 3 2 Выход по току, % 80 1 60 40 1 3 3 2 20 0 Схема 1 Схема 2 Схема 3 ОГНЭ 56,4 79,8 83,7 Pb/PbO2 30,4 22,5 29,2 ДБАЭ* 98,0 76,0 30,6 Рисунок 3.8. Диаграмма влияния схемы электролиза на выход по току пеларгоновой кислоты при окислении 1-нонанола на ОГНЭ, ДБАЭ, Pb/PbO2электроде. Плотность тока i=10 мА см-2; количество пропущенного электричества Q/Qтеор= 1; температура Т=70 °С, * – электролиз при температуре Т=20 °С. 79 70 3 3 2 60 ВЫход по току, % 50 40 30 2 20 1 1 10 0 Схема 1 Схема 2 Схема 3 ОГНЭ 10,7 56,4 63,6 Pb/PbO2 8,9 17,4 56,9 Рисунок 3.9. Диаграмма влияния схемы электролиза на выход по току каприновой кислоты при окислении 1-деканола на ОГНЭ, Pb/PbO2-электроде. Плотность тока i=10 мА см-2; количество пропущенного электричества Q/Qтеор=1; температура Т=70 °С. Из сравнения диаграмм, представленных на рисунках 3.7–3.9, видно, что на оксидо-гидроксидно-никелевом электроде процесс окисления спиртов до соответствующих карбоновых кислот протекает более эффективно с использованием схемы парного электролиза (схема 3), чем с анодным окислением и окислением с добавлением H2O2 в электролит. Так, на примере окисления 1-бутанола (рисунок 3.7) выход по току масляной кислоты с 80 использованием схемы анодного окисления составляет 91,5 %, при парном электролизе — 167,5 %; при окислении 1-нонанола (рисунок 3.8) выход по току пеларгоновой кислоты при анодном окислении — 56,4 %, а при парном электролизе 83,7 %; при окислении 1-деканола (рисунок 3.9) выход по току каприновой кислоты с использованием схемы анодного окисления составляет 10,6 %, а при парном электролизе 63,6 %. Увеличение эффективности процесса при использовании парного электролиза происходит за счет вклада непрямого окисления при целевом использовании катодного тока. В результате этого вклада происходит дополнительная генерация АФК (раздел 1.3.2) и, как следствие, увеличение их концентрации в объеме электролита. Также следует отметить, что на ОГНЭ происходит хемосорбция HO -радикалов (раздел 1.4.1) и электролиз проводили в интервале потенциалов регенерации высших оксидов никеля (раздел 2.1.3, рисунок 2.3). Указанные выше факторы являются благоприятными для протекания мягкого окисления и образования целевой карбоновой кислоты по реакциям (1.39)–(1.40), (1.47). Схема реакций, протекающих в такой системе, представлена на рисунке 1.3. При переходе к допированному бором алмазному электроду, эффективное электрокаталитическое окисление спиртов до карбоновых кислот наблюдается при использовании 1-ой схемы (Анодно) электролиза (рисунок 3.8), дополнительная генерация АФК приводит к минерализации целевого продукта до СО2 и Н2О. Так при окислении 1-нонанола выход по току пеларгоновой кислоты при анодной схеме окисления составляет 98,0 %, при дополнительной генерации АФК при использовании 2-ой и 3-ей схем окисления — 76,0 и 33,9 %, соответственно. Следует отметить, что использование ДБАЭ позволило проводить процесс электроокисления алифатических спиртов с длиной углеродной цепи > 6 без нагревания, в отличие от процессов с использованием ОГНЭ или Pb/PbO2электрода. Это значительно упрощает аппаратурное оформление процесса электролиза. 81 Из данных рисунка 3.8 видно, что при использовании ДБАЭ дополнительная генерация АФК (2-ая и 3-ья схемы окисления) снижает выход по току пеларгоновой кислоты за счѐт еѐ дальнейшей деструкции. Это обусловлено, по-видимому, увеличением концентрации НО -радикалов как на поверхности электрода, так и в объѐме электролита. При окислении бутанола-1 на ДБАЭ (рисунок 3.7) увеличение выхода по току масляной кислоты при парном электролизе объясняется условиями проведения процесса и справедливо для первоначального момента окисления (при доле количества пропущенного электричества равной 0,14 от требуемого). Как упоминалось выше (раздел 3.2), с ростом количества пропущенного электричества селективность генерации АФК возрастает и также увеличивается возможность их контакта с субстратом. Это ведѐт к дальнейшему неселективному окислению и минерализации образующихся кислот до CO2 и H2O. Экспериментальные данные, представленные выше в настоящей работе (раздел 3.1), подтверждают тот факт, что процесс окисления алифатических спиртов на аноде из диоксида свинца осложняется побочной реакцией этерификации (уравнение (1.7), разделы 1.1.2, 1.3.1). Это свидетельствует о более низкой эффективности этого анода в процессах электросинтеза карбоновых кислот в сравнении с ОГНЭ и ДБАЭ. Как видно из рисунка 3.8, при окислении 1-нонанола выходы по току пеларгоновой кислоты не превышают 31 % с использованием всех трѐх схем генерации АФК. Из данных рисунков 3.7–3.9 также видно, что при электросинтезе каприновой и пеларгоновой кислот наблюдается снижение токовой эффективности в сравнении с масляной. Так, например, при использовании ОГНЭ и схемы анодного окисления выходы по току целевых продуктов составили: масляной кислоты 91,5 %, пеларгоновой кислоты 56,4 %, каприновой кислоты 10,7 %. Это, связано с тем, что с удлинением углеродной 82 цепи, снижается адсорбируемость спирта на материале анода и уменьшается растворимость субстрата в воде, что затрудняет процесс их окисления. Заключение Установлено, что окисление спиртов до соответствующих карбоновых кислот протекает более эффективно с использованием схемы парного электролиза на ОГНЭ, за счет вклада непрямого окисления при целевом использовании катодного тока, где происходит дополнительная генерация АФК. Установлено, что при использовании ДБАЭ, где реализуется высокая селективность генерации АФК в объѐме электролита, эффективное электрокаталитическое окисление спиртов до карбоновых кислот наблюдается при использовании 1-ой схемы электролиза (Анодно). 3.4 Постановка опытов по наработке целевого продукта В ходе экспериментальных исследований были установлены наиболее благоприятные условия электрокаталитического для эффективного окисления проведения алифатических процесса спиртов до соответствующих карбоновых кислот (таблица 3.8). На основании этих данных поставлены опыты по наработке и выделению целевых продуктов. Таблица 3.8 Условия проведения наиболее эффективного электрокаталитического окисления алифатических спиртов до соответствующих карбоновых кислот. Исходное Материал Схема вещество электрода электролиза Спирты с углеродной ОГНЭ Анодно+O2 цепью > 6 ДБАЭ Спирты с углеродной цепью < 6 i, Q, мА×см-2 А×ч 70 10 0,8 Анодно 20 5 0,3 ОГНЭ Анодно+O2 20 10 0,8 ДБАЭ Анодно 20 5 0,7 Т, °С 83 Препаративные опыты проводили на лабораторном электролизере, устройство которого было аналогично схеме описанной в разделе 2.1.3 (рисунок 2.2), согласно условиям, представленным в таблице 3.8. Для препаративного электролиза использовали оксидо-гидроксидно-никелевый и допированный бором алмазный электрод, подготовленные как описано в разделе 2.1.2. Катодом служил графитовый электрод. Площадь рабочей поверхности ОГНЭ составляла 8 см2, ДБАЭ – 4,16 см2. На ОГНЭ электросинтез проводили с использованием 1-деканола, 1гесканола и 1-бутанола, на ДБАЭ с использованием 1-нонанола. По окончании электролиза проводили количественное определение кислот. Результаты опытов по наработке и выделению целевых карбоновых кислот представлены в таблице 3.9. Для выделения целевого продукта использовали следующие операции: 1) экстракция продукта из электролита хлороформом; 2) выпаривание части хлороформа на водяной бане; 3) хроматографический анализ продукта. Необходимо отметить, что хроматограммы полученных продуктов соответствуют, описанным в разделе 3.1 (рисунки 3.5–3.6). Это свидетельствует о высокой воспроизводимости экспериментальных результатов, взятых за основу для постановки препаративных опытов по наработке целевых продуктов. Выход по веществу рассчитывали по загруженному субстрату-спирту согласно [127]: (3.6) где mпрод, mисх — массы полученного продукта–кислоты и исходного субстрата– спирта, соответственно, г; 84 Mrисх, Mrпрод — грамм-молекулярный вес исходного субстрата–спирта и полученного продукта–кислоты. Таблица 3.9 Результаты препаративных опытов по наработке и выделению целевых кислот. Продукт Каприновая кислота Капроновая кислота Масляная кислота Пеларгоновая Материал mисх, г mпрод, г Вв, % Вт, % ОГНЭ 1,2 0,94±0,2 72,0 2 64,0±2 ОГНЭ 5,5 1,3±0,1 21,0 2 148,0±2 ОГНЭ 4,0 1,8±0,1 38,0 1 168,0±2 ДБАЭ 0,4 0,38±0,05 98,0 1 89,0±1 электрода кислота При наработке масляной и капроновой кислот доля пропущенного количества электричества к теоретически необходимому составляла Q/Qтеор = 0,14, соответственно происходила неполная конверсия исходного субстратаспирта. В связи с этим, выходы по веществу масляной и капроновой кислот значительно отличаются от выхода каприновой и пеларгоновой, где Q/Qтеор= 1. Таким образом, с целью сохранения токовой эффективности целесообразно использовать не вступивший в реакцию спирт для повторного синтеза (раздел 3.2). Далее рассчитывали затраченную электроэнергию на окисление спиртов до соответствующих кислот. Приближенный расчѐт электроэнергии, затраченной на количество произведѐнного продукта, проводили по уравнению (3.7) согласно [137]: 85 (3.7) где W – расход электроэнергии, кВт ч/т Э – электрохимический эквивалент, г/(А ч); U – напряжение на электролизѐре, В, U=4 В; Вт – выход по току, доли единицы. Электрохимический эквивалент рассчитывали по уравнению: , (3.8) где Mrисх – грамм-молекулярный вес исходного субстрата; z – число электронов в реакции, которое соответствует количеству фарадеев электричества, участвующему в электрохимическом преобразовании одного моля исходного вещества, равное 4; F – число Фарадея, равное 96500 кул. = 26,8 А ч/г экв. Результаты приближѐнных расчѐтов расхода электроэнергии на 1 т произведенного продукта приведены в таблице 3.9. Таблица 3.9 Приближѐнный расход электроэнергии на 1 т произведенного продукта. Целевой продукт Исходное вещество Mr спирта Вт, доли единицы Э, г/(А ч) W, кВт ч/т масляная кислота 1-бутанол 74,12 1,675 0,69 3454,0 капроновая кислота 1-гексанол 102,18 1,480 0,95 2836,0 пеларгоновая кислота 1-нонанол 144,26 0,894 1,35 3325,0 каприновая кислота 1-деканол 158,28 0,636 1,48 4260,0 86 На основании результатов препаративных опытов и приближенных расчѐтов расхода электроэнергии на производство продукта можно сделать заключение, что непрямое электрокаталитическое окисление алифатических спиртов является перспективным и альтернативным способом для получения соответствующих карбоновых кислот. Согласно полученным данным (таблица 3.9), приближѐнный расход электроэнергии на окисление спиртов до соответствующих карбоновых кислот с использованием непрямого электрокаталитического метода составляет 2,84–4,26 кВт ч/кг. Различие величин расхода электроэнергии зависит от значений выхода по току целевой карбоновой кислоты. Такой расход электроэнергии справедлив для принятых условий электролиза (напряжение на ячейке 4 В), оптимизация электролитической ячейки по напряжению не проводилась. Однако при проведении должной оптимизации расход электроэнергии может быть значительно снижен при сохранении выходов по току. Также согласно литературным данным [26–28] и результатам настоящего исследования можно предположить, что метод непрямого электрокаталитического окисления алифатических спиртов до карбоновых кислот in situ активными формами кислорода открывают электросинтеза. новые возможности в области органического 87 ВЫВОДЫ 1. Установлены общие закономерности непрямого электрокаталитического окисления алифатических спиртов до карбоновых кислот. Определены начальные скорости и установлено, что реакция окисления спиртов протекает по псевдо-первому порядку при использовании исследованных схем генерации активных форм кислорода. 2. Найдены оптимальные условия электроокисления алифатических спиртов до соответствующих карбоновых кислот in situ активными формами кислорода. Установлено, что при малых плотностях тока 5–10 мА см-2 и при пропускании концентрации определенного количества реагента-спирта, наработка электричества, целевых зависящее карбоновых от кислот происходит с высокой эффективностью. Увеличение плотности тока, как и увеличение количества пропущенного электричества, снижают выход целевого продукта за счет его дальнейшего деструктивного окисления. 3. Установлено, что при использовании оксидо-гидроксидно-никелевого электрода наиболее эффективной схемой окисления алифатических спиртов до соответствующих карбоновых кислот является парный электролиз. На допированном бором алмазном электроде дополнительная генерация активных форм кислорода ведет к снижению выхода целевого продукта за счет его дальнейшего окисления. 4. Доказана электрокаталитического формами кислорода перспективность окисления для использования алифатических получения карбоновых непрямого спиртов активными кислот. Результаты препаративных опытов по наработке целевых продуктов показали: на ОГНЭ при парном электролизе и пропущенном количестве электричества Qтеор/Q = 0,14 выход по току кислот составил: масляной – 168,0 %, капроновой – 148,0 %, каприновой при Qтеор/Q = 1,0 составил 64,0 %, на ДБАЭ при анодной схеме окисления и Qтеор/Q = 1,0 выход по току пеларгоновой кислоты составил 89,0 %. 88 5. Разработана экспериментальная методика получения карбоновых кислот методом непрямого электрокаталитического окисления алифатических спиртов активными формами кислорода, in situ генерированными из кислорода, пероксида водорода и воды на различных электродных материалах. 89 СПИСОК ЛИТЕРАТУРЫ 1. Roberts J. D., Caserio M. C. Basic Principles of Organic Chemistry. – Second edition. – CA.: W. A. Benjamin, Inc. , Menlo Park, 1977. – 1596 p. 2. Травень В.Ф. Органическая химия: Учебник для вузов – Том 2. – М.: ИКЦ «Академкнига», 2006. – 582 с. 3. Рабинович В.А., Хавин З.Я. Краткий химический справочник. – 2-е изд. перераб. и доп. – Л.: «Химия», 1978. – 392 с. 4. Гороновский И.Т. и др. Краткий справочник по химии / И.Т. Гороновский, Ю.П. Назаренко, Е.Ф. Некряч. – 4-е изд. перераб. и доп. – Киев: Наукова думка, 1974. – 992 с. 5. Петров А.А. и др. Органическая химия: Учебник для вузов / А.А. Петров, Х.В. Бальян, А.Т. Трощенко. – 5-е изд. перераб. и доп. – СПб.: Иван Федоров, 2002. – 624 с. 6. Pillai U.R., Sahle-Demessie. Oxidation of alcohols over Fe3+/montmorillonite-K10 using hydrogen peroxide // Appl. Catal. A: Gen. – 2003. – V. 245. – P. 103–109. 7. Sheldon R. A., Kochi J. K. Metal-Catalyzed Oxidations of Organic Compounds. – New York: Academic Press, 1981. – 350 p. 8. Hudlicky M. Oxidation in Organic Chemistry. – Washington, DC: American Chemical Society, 1994. – 433p. 9. Patai S. The Chemistry of carboxylic acid and esters. – NY.: John Wiley and Sons Itd., 1969. – 1155 p. 10. Химическая энциклопедия: В 5 томах. Том 3 / Под ред. Кнунянц И.Л. и др. – М.: Большая Российская энциклопедия, 1992. – 639 с. 11. Химическая энциклопедия: В 5 томах. Том 5 / Под ред. Зефиров Н.С. и др. – М.: Большая Российская энциклопедия, 1998. – 783 с. 12. Болотин И.М. и др. Синтетические жирные кислоты и продукты на их основе. Темат. обзор. Сер. "Поверхностно-активные вещества" / И.М. Болотин, П.Н. Милосердов, Е.И. Суржа. – М.: ЦНИИТЭнефтехим, 1970. – 78 с. 90 13. Солодунова Е.А., Лысенко К.Н. Лекарственные препараты карбоновых кислот алифатического ряда и их производных / Е.А. Солодунова, К.Н. Лысенко. – Волгоград: ВолГМУ, 2011. – 77 с. 14. Беликов В.Г. Фармацевтическая химия: Учебник для вузов. – Ч. 1-2. – Пятигорск: Пятигорская фармацевтическая государственная академия, 2003. – 720 с. 15. Томилов А.П. и др. Электрохимия органических соединений / А.П. Томилов, С.Г. Майрановский, М.Я. Фиошин, В.А. Смирнов. – Л.: Химия, 1968. – 592 с. 16. Томилов А.П. и др. Электрохимический синтез органических веществ / А.П. Томилов, М.Я. Фиошин, В.А. Смирнов. – Л.: Химия, 1976. – 424 с. 17. Томилов А.П. Развитие отечественной электрохимии органических соединений (1940–2002 гг.) // Российский химический журнал (Ж. Рос. хим. обва им. Д.И. Менделеева). 2005. – Т. XLIX. – № 5. – С. 4–16. 18. Томилов А.П. Развитие исследований в области электрохимии органических соединений в 2000–2006 гг. / А.П. Томилов, В.В. Турыгин, Л.В. Каабак // Электрохимия. 2007. – Т. 43. – № 10. – С. 1164–1181. 19. Canizares P. Electrochemical oxidation of alcohols and carboxylic acids with diamond anodes. A comparison with other advanced oxidation processes / P. Canizares, R. Paz, C. Saez, M.A. Rodrigo // Electrochimica Acta, 2008. – V.53. – P. 2144–2153. 20. Brandon Nigel N.P., Thompsett D. Fuel Cells Compendium. – London: Elsiver, 2005. – 632 p. 21. Vigier F. On the mechanism of ethanol electro-oxidation on Pt and PtSn catalysts: electrochemical and in situ IR reflectance spectroscopy studies / F. Vigier, C. Coutanceau, F. Hahn, E.M. Belgsir, C. Lamy // Journal of Electroanalytical Chemistry, 2004. – V. 563. – № 1. – P. 81–89. 91 22. Brown O.R. The anodic oxidation of benzyl alcohol to benzaldehyde / O.R. Brown, S. Chandra, J.A. Harrison // Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. – 1972. – V. 38. – № 1. – P. 185–190. 23. Реутов О.А. и др. Органическая химия Ч.2: Учебник. / О.А. Реутов, Курц А.Л., Бутин К.П. – М.: МГУ, 1999. – 624 с. 24. Юкельсон И.И. Технология основного органического синтеза. – М.: Химия, 1968. – 848 с. 25. Лебедев Н.Н. Химия и технология основного органического и нефтехимического синтеза: Учебник для вузов. – 4-е изд. перераб. и доп. – М.: Химия, 1988. – 592 с. 26. Будникова Ю.Г. Электросинтез органических соединений. Экологически чистые процессы и дизайн новых синтетических методов // Российский химический журнал. 2005. – Т. XLIX. – № 5. – С. 81–92. 27. Shafer H.J. Contributions of organic electrosynthesis to green chemistry // Comptes Rendus Chimie. 2011. – V. 14. – P. 745–765. 28. Frontana-Uribe B.A. Organic electrosynthesis: a promising green methodology in organic chemistry / B.A. Frontana-Uribe, R.D. Little, J.G. Ibanez, A. Palma, R. Vasquez-Medrano // Green Chemistry. 2010. – V. 12. – P. 2099–2119. 29. Lamy C. Electrocataltytic oxidation of aliphatic alcohols: Application to the direct alcohol fuel cell (DAFC) / C. Lamy, E.M. Belgsir, J-M. Leger // Joutnal of Applied Electrochemistry. 2001. – V. 31. – P. 799–809. 30. Horanyi G. Oxidation of secondary alcohols on a platinum electrode, I. The oxidation products / G.Horanyi, P.Konig, I.Telcs // Acta Chimica Scientiarum Hungaricae, Tomus. 1972. – V. 72. – № 2. – P. 165–178. 31. Horanyi G. Oxidation of secondary alcohols on a platinum electrode, II. Study of the connection between oxygen adsorption and the reaction / G.Horanyi, Vrtes G., P.Konig // Acta Chimica Scientiarum Hungaricae, Tomus. 1972. – V. 72. № 2. – P. 179–187. 92 32. Реморов Б.С. Влияние материала электрода на электроокисление изобутилового спирта в щелочной среде / Б.С. Реморов, И.А. Авруцкая, М.Я. Фиошин // Электрохимия. 1981. – Т. 17. – С. 1547-1551. 33. Чаенко Н.В. Непрямое электрохимическое окисление алифатических спиртов до карбоновых кислот активными формами кислорода в водных средах / Н.В. Чаенко, Г.В. Корниенко, А.М. Кошелева, Н.Г. Максимов, В.Л. Корниенко // Электрохимия. 2011. – Т.47. – № 10. – С.1228–1233. 34. Kaulen I. Oxidation of alcohols by electrochemically regenerated nickel oxide hydroxide. Selective oxidation of hydroxysteroids / I. Kaulen, H. Schafer // Tetrahedron. 1982. – V. 38. – № 22. – P. 3299–3308. 35. Лялин Б.В. Окисление органических соединений на NiOOH-электроде / Б.В. Лялин, В.А. Петросян // Электрохимия. 2010. – Т. 46 – № 11. – С. 1283– 1298. 36. Vertes G. Oxidation of the nickel hydroxide electrode. I. Principles of the study / G. Vertes, G. Horanyi, F. Nagy // Acta Chimica Academiae Scientiaricae Hungaricae. 1971. – V. 67. – P. 145-156. 37. Vertes G. Oxidation on the nickel hydroxide electrode / G. Vertes, G. Horanyi, F. Nagy // Croatica Chemica Acta. 1972. – V. 44. –Р. 21-29. 38. Fleischman M. The oxidation of organic compounds at a nickel anode in alkaline solution / M. Fleischman, K. Korinek, D. Pletcher // Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. 1971. – V. 31. – № 1. – P. 39–49. 39. Реморов Б.С. Поведение алифатических спиртов и продуктов их окисления на оксидно-никелевом электроде / Б.С. Реморов, И.А. Авруцкая, М.Я. Фиошин // Электрохимия. 1981. – Т. 17 – № 5. – С. 743–747. 40. Реморов Б.С. О продуктах окисления изобутанола регенерируемыми электрохимически высшими оксидами никеля / Б.С. Реморов, И.А. Авруцкая, М.Я. Фиошин // Электрохимия. 1980. – Т. 16. – № 5. – С. 723–727. 93 41. Schafer H.J. Oxidation of organic compounds at the nickel hydroxide electrode / H.J. Schafer // Electrochemistry I. Topics in Current Chemistry. 1987. – V.142. – P. 101–129. 42. Патент 3352768 (США). Acetylene dicarboxylic acid process / Chiddix Max E., Leon Katz // Опубл. 14.11.1967. 43. Джафаров Э.А. Электроосаждение, свойства и применение двуокиси свинца. – Баку: Аз. ССР. – 1967. – 100с. 44. Корниенко В.Л. и др. Электросинтез в гидрофобизированных электродах / В.Л. Корниенко, Г.А. Колягин, Ю.В. Салтыков. – Новосибирск: СО РАН, 2011. – 170 с. 45. Чаенко Н.В. Электроокисление алифатических спиртов на оксидноникелевых гидрофобизированных электродах / Н.В. Чаенко, В.Л. Корниенко, И.А. Авруцкая, М.Я. Фиошин // ЖПХ. – 1987. – № 6. – С. 1339–1343. 46. Фиошин М.Я., Смирнова М.Г. Электрохимические системы в синтезе химических продуктов. – М.: Химия, 1985. – 256 с. 47. Гигиенические нормативы ГН 2.1.6.1338-03. Предельно допустимые концентрации (ПДК) загрязняющих веществ в атмосферном воздухе населенных мест, Российский регистр потенциально опасных химических и биологических веществ. – М.: Департамент госсанэпиднадзора Минздрава России, 2003. 48. Гигиенические нормативы ГН 2.1.5.1315-03. Предельно допустимые концентрации (ПДК) химических веществ в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования, Российский регистр потенциально опасных химических и биологических веществ. – М.: Департамент госсанэпиднадзора Минздрава России, 2003. 49. Гигиенические нормативы ГН 2.1.7.2041-06. Предельно допустимые концентрации (ПДК) химических веществ в почве. – М.: Роспотребнадзор, 2006. 94 50. Огибин Ю.Н. Органический электросинтез с использованием медиаторных систем окисления / Ю.Н. Огибин, М.Н. Элинсон, Г.И. Никишин // Успехи химии. 2009. – Т. 78. – Вып. 2. – С. 99–150. 51. Pletcher D. Indirect oxidation using electrogenerated hydrogen peroxide / D. Pletcher // Acta Chemica Scandinavica. 1999.– V. 53. – P. 745–750. 52. Scialdone O. Electrochemical oxidation of organic pollutants in water at metal oxide electrodes: A simple theoretical model including direct and indirect oxidation processes at the anode surface / O. Schaldone // Electrochimica Acta. 2009. – V. 54. – P. 6140–6147. 53. Torii S. The new role of electroreductive mediators in electroorganic synthesis / S. Torii // Synthesis.1986. – V. 11. – P. 873–886. 54. Bersier P.M. Electrochemistry for a better environment / P.M. Bersier, L. Carlsson, J. Bersier. Ed. E. Steckhan // Electrochemistry V: Topics in Current Chemistry. Berlin Heidelberg: Springer-Verlag. 1994. – V. 170. – P. 113–229. 55. Корниенко В.Л. Непрямое окисление органических веществ пероксидом водорода, электрохимически генерированным in situ из кислорода (обзор) / В.Л. Корниенко // Химия в интересах устойчивого развития. 2002. – Т. 10. – С. 391–399. 56. Nielsen M.F., Utley J.H.P. Organic electrochemistry. – 4-th ed.–New York: Marcel Dekker, 2001. – 795 p. 57. Органическая электрохимия: В двух книгах / Под ред. Бейзер М., Лунд Х. – Пер. с англ. / Под ред. В.А. Петросян, Л.Г. Феоктистова.– М.: Химия, 1988. – Кн. 2. – С. 470–1024. 58. Ёсида К. Электроокисление в органической химии. Роль катионрадикалов как интермедиатов в синтезе. – М.: Мир, 1987. – 336 с. 59. Магдесиева Т.В. Электрохимическая активация реакций с участием металлоорганических соединений / Т.В. Магдесиева, К.П. Бутин // Успехи химии. 2002. – Т. 71. – № 3. – С. 255-272. 95 60. Organic Electrochemistry / Eds H.Lund, O. Hammerich. – New York: Marcel Dekker, 2001. – 1416 p. 61. Колягин Г.А. Кинетика накопления пероксида водорода в кислых и нейтральном водном растворах при электровосстановлении кислорода в сажевом гиброфобизированном электроде / Г.А. Колягин, В.Л. Корниенко // Химия в интересах устойчивого развития. 2000. – Т. 8. – С. 803–807. 62. Li W. Development of a cathodic oxidation system and its application to paired electrosynthesis of sulfones and nitrones / W. Li, T. Nonaka // Journal of Electrochemical Society. 1999. – V. 146. – P. 592–599. 63. Высоцкая Н.А. Реакционная способность радикалов и атомов кислорода в водных растворах ароматических соединений / Н.А. Высоцкая // Успехи химии. 1973. – Т. 47. – Вып. 10. – С. 1843–1853. 64. Halliwell B. Free Radicals in Biology and Medicine / B.Halliwell, J.Gutteridge. – UK: Oxford University Press (Verlag), 2007. – 888 p. 65. Нагиев Т.М. Сопряженные реакции окисления перекисью водорода / Т.М. Нагиев // Успехи химии. 1985. – Т. 54. – Вып. 10. – С. 1654–1673. 66. Сычев А.Я. Каталитические реакции и охрана окружающей среды / А.Я. Сычев, С.О. Травин, Г.Г. Дука, Ю.И. Скурлатов. – Кишинев: Штиинца, 1983. – 272 с. 67. TECHCOMMENTARY: Advanced Oxidation Processes for Treatment of Industrial Wastewater // An EPRI Community Environmental Center Publ. – 1996. – V.1. 68. Carey J.H. An introduction to AOP for destruction of organics in wastewater / J.H. Carey // Water Pollut. Res. J. Can. 1992. – V.27. – P. 1–21. 69. Canizares P. Electrochemical oxidation of alcohols and carboxylic acids with diamond anodes. A comparison with other advanced oxidation processes / P. Canizares, R. Paz, C. Saez, M.A. Rodrigo // Electrochimica Acta. 2008. – V. 53. – P. 2144–2153. 96 70. Martínez-Huitle C.A. Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes / C.A. Martínez-Huitle, S.Ferro // Chemical Society Reviews. 2006. – V. 35. – № 5 – P. 1324–1340. 71. Chen Y.-L. Paired electrooxidation. Part III: Production of n-butyric acid from n-bytanol / Y.-L. Chen, T.-C. Chou // Journal of Applied Electrochemisrty. 1996. – V. 26. – P. 543–545. 72. Torii S. Indirect electrooxidation of alcohols and aldehydes by using a double mediatory system consisting of ruthenium tetraoxide/ruthenium dioxide and chlorine cation/chlorine anion redoxes in an aqueous-organic two-phase system / S. Torii , T. Inokuchi , T. Sugiura // J. Org. Chem. 1986. – V. 51. – P. – 155–161. 73. Rajendran S. Ruthenium tetroxide as a phase transfer catalyst in biphasic system and its in situ electrochemical regeneration: Oxidation of aromatic primary alcohol and aldehydes / S. Rajendran, D.C. Trivedi // Synthesis. 1995. – V. 1995 – № 2. – P. – 153–154. 74. Torii S. Indirect electrooxidation by using ruthenium tetraoxide and chloride ion as recycling mediators. Optimization for the oxidation of diisopropylidene-D-glucose to the ulose / S. Torii, T. Inokuchi, S. Matsumoto, T. Saeki, T. Oki // Bulletin of the Chemical Society Japan. 1989. – V. 62. – № 6. – P. – 2108–2110. 75. Vaudano P. The industrial electrolytic regeneration of Mn2(SO4)3 for the oxidation of substituted toluene to the corresponding benzaldehyde / P. Vaudano, E. Plattner, C. Comninellis // CHIMIA. 1995. – V. 49. – P. 12–16. 76. Kreh R.P. Lundquist Mediated electrochemical synthesis of aromatic aldehydes, ketones, and quinones using ceric methanesulfonate / R.P. Kreh, R.M. Spotnitz , J.T. Lundquist // J. Org. Chem. 1989. – V. 54 – № 7. – P. – 1526–1531. 77. Spotnitz R.M. Mediated electrosynthesis with cerium (IV) in methanesulphonic acid / R. M. Spotnitz, R. P. Kreh, J. T. Lundquist, P. J. Press // Journal of Applied Electrochemistry. 1990. – V.20. – № 2. – P. 209-215. 97 78. Патент 4639298 (США). Oxidation of organic compounds using ceric ions in aqueous methanesulfonic acid // Опубл. 27.01.1987. 79. Beck F. Kinetics of the anodic oxidation of isopropanol on chromium/titanium—oxide electrodes / F. Beck, H. Schulz // Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. 1987. – V. 229. – P. – 339–352. 80. Fleischmann M. The kinetics and mechanism of the oxidation of amines and alcohols at oxide-covered nickel, silver, copper, and cobalt electrodes / M. Fleischmann, K. Korinek, D. Pletcher // Journal of the Chemical Society, Perkin Transactions 2. 1972. – V. 10. – P.1396-1403. 81. Jafarian M. A study of the electrocatalytic oxidation of methanol on a cobalt hydroxide modified glassy carbon electrode / M. Jafarian, M.G. Mahjani, H. Heli, F. Gobal, H. Khajehsharifi, M.H. Hamedi // Electrochimica Acta. 2003. – V. 48. – № 23. – P. – 3423–3429. 82. Horanyi G. Oxidation of the nickel hydroxide electrode, II. Some problems of the practical application of the electrode with special reference to the oxidation of di-o-isopropylidenesorbose / G. Horanyi, G. Vertes, F. Nagy // Acta Chimica Academiae Scientiaricae Hungaricae. 1971. – V. 68. – № 3. – P. 217–228. 83. Vertes G. Some problems of the kinetics of the oxidation of organic compounds at oxide-covered nickel electrodes / G. Vertes, G. Horanyi // Electroanalytical Chemistry and Interfacial Electrochemistry. 1974. – V. 52. – P. 47– 53. 84. Vertes G. Oxidation on the nickel hydroxide electrode / G. Vertes, G. Horanyi, F. Nagy // Croatica chemical acta. – 1972. – V. 44. – P. 21–29. 85. Kaulen I. Oxidation of primary alcohols to carboxylic acids at the nickel hydroxide electrode / Kaulen I., Shafer H.J. // Synthesis. 1979. – V.7. – P. 481–560. 86. Amjad M. The oxidation of alcohols at a nickel anode in alkaline tbutanol/water mixtures / M. Amjad, D. Pletcher, C. Smith // Joutnal of Electrochemical Society. 1977. – V. 124. – P. 203–206. 98 87. Фиошин М.Я. Электроокисление органических соединений на анодах из окислов некоторых переходных металлов / М.Я. Фиошин, И.А. Авруцкая // Успехи химии. 1975. – Т. 44. – С. 2067–2077. 88. Фиошин М.Я. Влияние некоторых добавок на электролиз растворов бромида натрия / М.Я. Фиошин, И.А. Авруцкая, А.И. Борисов, Л.А. Чупина // Электрохимия. 1970. – Т. 6. – № 9. С.– 1393–1396. 89. Борисов А.И. Электросинтез диацетон-2-кето-L-гулоновой кислоты / А.И. Борисов, И.А. Авруцкая, М.Я. Фиошин // Электрохимия. 1970. – Т. 6. – № 9. – С.– 1397–1401. 90. Авруцкая И.А. Электросинтез диацетон-2-кето-L-гулоновой кислоты на окисноникелевых электродах / И.А. Авруцкая, М.Я. Фиошин, Т.Е. Мулина // Электрохимия. 1973. – Т. 9. – № 6. – С.– 897–901. 91. Мулина Т.Е. Некоторые закономерности электроокисления диацетон-2-кето-L-гулоновой кислоты на окисноникелевом электроде / Т.Е. Мулина, И.А. Авруцкая, М.Я. Фиошин, Т.А. Малахова // Электрохимия. 1974. – Т. 10. – №3. – С.– 481–484. 92. Фиошин М.Я. Влияние материала электрода на электроокисление некоторых моносахаридов / М.Я. Фиошин, И.А. Авруцкая, Т.А. Малахова, Т.Е. Мулина // Электрохимия. 1974. – Т. 10. – № 5. – С.– 796–800. 93. Реморов Б.С. Электросинтез изовалериановой кислоты в щелочной среде / Б.С. Реморов, И.А. Авруцкая, М.Я. Фиошин // Электрохимия. 1982. – № 5. – С.– 668–671. 94. Radoi, I. Contribution to the n-propanol electrochemical oxidation in basicmedium / I. Radoi, I. Taranu // Revista de Chimie. 1993. – V. 44. – P. 1034–1041. 95. Vaze A. S. Electrochemical oxidation of isobutanol to isobutyric acid in a flow cell / A. S. Vaze, S. B. Sawant, V. G. Pangarkar // Journal of Applied Electrochemistry. 1995. – V. 25. – P. 279–283. 96. Якименко Л.М. Электродные материалы в прикладной электрохимии. – М.: Химия, 1977. – 264 с. 99 97. Comninellis Ch. Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment / Comninellis Ch. // Electrochimica Acta. 1994. – V. 39. – P. 1857–1862. 98. Лукомский Ю.Я., Гамбург Ю.Д. Физико-Химические основы электрохимии: Учебник. – Долгопрудный: Издательский Дом «Интеллект», 2008. – 424 с. 99. Simond O. Theoretical model for the anodic oxidation of organics on metal oxide electrodes / O. Simond, V. Schaller, Ch. Comninellis // Electrochimica Acta. 1997. – V. 42. – P. 2009–2012. 100. Comninellis Ch. Electrochemical oxidation of phenol for wastewater treatment using SnO2, anodes / Ch. Comninellis, C. Pulgarin // Journal of Applied Electrochemistry. 1993. – V. 23. – I.2. – P. 108-112. 101. Swain G.M. The electrochemical activity of boron-doped polycrystalline diamond thin film electrodes / G.M. Swain, R. Ramesham // Analytical Chemistry. 1993. – V. 65. – P. 345–351. 102. Comninellis Ch. Anodic oxidation of phenol in the presence of NaCl for wastewater treatment / Ch. Comninellis, A. Nerini // Journal of Applied Electrochemistry. 1995. – V. 25. – P. 23–28. 103. Varkaraki E. Electrochemical promotion of IrO2 catalyst for the gas phase combustion of ethylene / E. Varkaraki, J. Nicole, E. Plattner, Ch. Comninellis, C. G. Vayenas // Journal of Applied Electrochemistry. 1995. – V. 25. – I. 10. – P. 978–981. 104. Simond O. Anodic oxidation of organics on Ti/IrO2 anodes using Nafion as electrolyte / O. Simond, Ch. Comninellis // Electrochimica Acta. 1997. – V. 42. – P. 2013–2018. 105. Brillias E, Martinez-Huitle C.A. Synthetic Diamond Films: Preparation, Chemistry, Characterization and Applications, 2011. – 619 p. 106. Kapalka A. The importance of electrode material in environmental electrochemistry. Formation and reactivity of free hydroxyl radicals on boron-doped 100 diamond electrodes / A. Kapalka, G. Foti, Ch. Comninellis // Electrochimica Acta. 2009. – V. 54. – P. 2018–2023. 107. Кошелева А.М. Влияние природы электродного материала на эффективность процесса окисления спиртов с использованием in situ активных форм кислорода // Актуальные проблемы электрохимии органических соединений (ЭХОС–2012): Тезисы докладов VII Всероссийской с международным участием школы по электрохимии органических соединений 20–25 октября 2012 г. – Тамбов, 2012. – С. 102–105. 108. Alfaro M.A.Q. Boron doped diamond electrode for the wastewater treatment / M.A.Q. Alfaro, S. Ferro, C.A. Martínez-Huitle, Y.M. Vong // Journal of the Brazilian Chemical Society. 2006. – V. 17. – P. 227–236. 109. Marselli B. Electrogeneration of hydroxyl radicals on boron-doped diamond electrodes / B. Marselli, J. Garcia-Gomezb, P.-A. Michauda, M. A. Rodrigob, Ch. Comninellisa // J. Electrochem. Soc. 2003. – V.150. – I. 3. – P. D79– D83. 110. Weiss E. A kinetic study of the electrochemical oxidation of maleic acid on boron doped diamond / E. Weiss, K. Groenen-Serrano, A. Savall, Ch. Comninellis // Journal of Applied Electrochemistry. 2007. – V. 37. – P. 41–47. 111. Scialdone O. Electrochemical incineration of oxalic acid at boron doped diamond anodes: Role of operative parameters / O. Scialdone, A. Galia, Ch. Guarisco, S. Randazzo, G. Filardo // Electrochimica Acta. 2008. – V. 53. – P. 2095– 2108. 112. Janssen L.J.J Electrochemical oxidation of iodate to periodate / L.J.J Janssen, M.H.A Blijlevens // Electrochimica Acta. 2003. – V. 48. – P. 3959–3964. 113. Плесков Ю.В. Электрохимия алмаза. – Москва: Едиториал УРСС, 2003. – 101 с. 114. Einaga Y., Fujishima A. Diamond electrochemistry. – Amsterdam: Elsevier Tokyo BKC, 2005. – 586 p. 101 115. Panizza M. Application of diamond electrodes to electrochemical processes / M. Panizza, G. Cerisola // Electrochimica Acta. 2005. – V. 51. – № 2. – P. 191–199. 116. Денисов А.Е. Электроокисление этилендиаминтетрауксусной кислоты на аноде из легированного бором поликристаллического алмаза / А.Е. Денисов, Ю.В. Плесков // Электрохимия. 2008. – Т.44. – №. 9. – С. 1166-1168. 117. Griesbach U. Evaluation of boron doped diamond electrodes for organic electrosynthesis on a preparative scale / U. Griesbach, D. Zollinger, H. Putter, Ch. Comninellis // Journal of Applied Electrochemistry. 2005. – V. 35. – P. 1265–1270. 118. Zollingera D. Methoxylation of p-tert-butyltoluene on boron-doped diamond electrodes / D. Zollingera, U. Griesbachb, H. Pütterb, Ch. Comninellisa // Electrochemistry Communications. 2004. – V.6. – № 6. – P. 600–604. 119. Zollingera D. Electrochemical cleavage of 1,2-diphenylethanes at borondoped diamond electrodes / D. Zollingera, U. Griesbachb, H. Pütterb, Ch. Comninellisa // Electrochemistry Communications. 2004. – V.6. – № 6. – P. 605– 608. 120. Морозова А.П. Электрохимическое окисление изобутанола на оксидноникелевом электроде с основой из никелированного углеродного волокна // Новости электрохимии органических соединений 2010. XVII Всероссийское совещание по электрохимии органических соединений с международным участием: тезисы докладов. – Тамбов, 2010. – С. 90–91. 121. Carr J.P. The Lead dioxide electrode / J.P. Carr, N.A. Hampson // Chemical Reviews. 1972. – V. 72. – № 6. – P. 679–703. 122. Корниенко Г.В. Непрямое электрохимическое окисление фенола пероксидом водорода, генерированном in situ из кислорода в газодиффузионном электроде в кислой и нейтральной средах / Г.В. Корниенко, Н.В. Чаенко, В.Л. Корниенко // Химия в интересах устойчивого развития. 2006. – Т. 14. – С. 23–26. 102 123. Корниенко Г.В. Электрохимическое окисление фенола на допированном бором алмазном электроде / Г.В. Корниенко, Н.В. Чаенко, Н.Г. Максимов, В.Л. Корниенко, В.П. Варнин // Электрохимия. 2011. – Т. 47. – № 2. – С. 225–229. 124. Сахарова А.Я. Электроды из синтетического полупроводникового алмаза: фотоэлектрохимическое поведение легированных бором и нелегированных поликристаллических тонких пленок, осажденных из газовой фазы / А.Я. Сахарова, Ю.В. Плесков, Ф. Ди Кварто, С. Пьяцца, К. Сунсери, И.Г. Теремецкая, В.П. Варнин // Электрохимия. 1995. – Т. 31. – № 2. – С. 188–193. 125. Федосеев Д.В. Синтез алмаза в области его термодинамической метастабильности / Д.В. Федосеев, В.П. Варнин, Б.В. Дерягин // Успехи химии. 1984. – Т. 53. – С. 753–771. 126. Чаенко Н.В. Редокс-медиаторное окисление алифатических спиртов до карбоновых кислот на гидрофобизированных оксидно-никелевых и оксиднокобальтовых электродах / Н.В. Чаенко, В.Л. Корниенко, Г.В. Корниенко // Журнал Сибирского федерального университета. Серия: Химия. 2009. – Т. 2 . – С. 64–70. 127. Томилов А.П. и др. Препаративная органическая электрохимия: Учеб. пособие / А.П.Томилов, Е.Ш. Каган, В.А.Смирнов, И.Ю. Жукова. – 2-е изд. перераб. и доп. – Новочеркасск: ЮРГТУ, 2002. – 153 с. 128. Органикум. Практикум по органической химии / Г. Беккер, В. Бергер, Г. Домшке и др. – М.: «Мир», 1979. – Том 1. – 456 с. 129. Полюдек-Фабини Р. Органический анализ: Пер. с нем. –Л.: Химия, 1981. – 624 с. 130. Алексеев В.Н. Количественный анализ. – изд. 4-е, перераб. – М.: «Химия», 1972. – 504 с. 131. Миронов В.А., Янковский С.А. Спектроскопия в органической химии. Сборник задач: Учеб. пособие для ВУЗов.–М.: «Химия», 1985. – 232 с. 103 132. Губен-Вейль Методы органической химии. Методы анализа. – 4-е изд. полностью перераб. – М.: Госхимиздат, 1963. – Том 2. – 1032 с. 133. Лурье Ю.Ю., Рыбникова А.И. Химический анализ производственных сточных вод. – 4-е изд. перераб. и доп. – М.: Химия, 1974. – 336 с. 134. Саутин С.Н. Планирование эксперимента в химии и химической технологии. – Л: «Химия», 1975. – 48 с. 135. Севодина К.В. О влиянии содержания сахаров на интенсивность изменения окраски облепиховых вин в процессе хранения // Техника и технология пищевых производств. 2013. – № 2. – С. 1 – 5. 136. Герасимов Я.И. Курс Физической химии. – 2-е изд., перераб. – М.: «Химия», 1973. – Том 2. – 624 с. 137. Прикладная электрохимия. Учеб. для вузов. / Под ред. А.П. Томилова. – 3-е изд., перераб. – М.: «Химия», 1984. – 520 с.