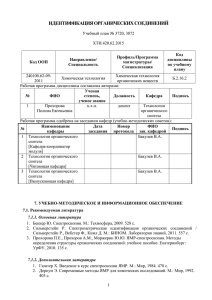
Диссертация - Институт Нефтехимии и катализа РАН
реклама

ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ УЧРЕЖДЕНИЕ НАУКИ ИНСТИТУТ НЕФТЕХИМИИ И КАТАЛИЗА РОССИЙСКОЙ АКАДЕМИИ НАУК На правах рукописи САМОЙЛОВА ЕЛЕНА ВЛАДИМИРОВНА НОВЫЕ РЕАКЦИИ Al- И Zn-ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ С 1АЛКЕНАМИ И НОРБОРНЕНАМИ, КАТАЛИЗИРУЕМЫЕ КОМПЛЕКСАМИ ТАНТАЛА 02.00.03- Органическая химия 02.00.15-Кинетика и катализ ДИССЕРТАЦИЯ на соискание ученой степени кандидата химических наук Научный руководитель: доктор химических наук, старший научный сотрудник СУЛТАНОВ Р.М. Научный консультант: доктор химических наук, член-корр. РАН ДЖЕМИЛЕВ У.М. Уфа-2015 2 ОГЛАВЛЕНИЕ стр. ВВЕДЕНИЕ.............................................................................................................4 ГЛАВА 1. ЛИТЕРАТУРНЫЙ ОБЗОР РЕАКЦИИ КАРБОАЛЮМИНИРОВАНИЯ И КАРБОЦИНКИРОВАНИЯ НЕПРЕДЕЛЬНЫХ СОЕДИНЕНИЙ......................................................................8 1.1. Карбоалюминирование алкенов и алкинов....................................................8 1.1.1.Межмолекулярное карбоалюминирование α-олефинов.............................8 1.1.2. Некатализированное карбоалюминирование алкилацетиленов.............10 1.1.3. Каталитическое карбоалюминирование олефинов..................................12 1.1.4. Каталитическое карбоалюминирование ацетиленов...............................19 1.2. Карбоцинкирование непредельных соединений.........................................24 1.2.1 Некатализируемое карбоцинкирование алкенов.......................................24 1.2.2. Некатализируемое карбоцинкирование алкинов.....................................26 1.2.3. Каталитическое карбоцинкирование алкенов..........................................27 1.2.4. Каталитическое карбоцинкирование алкинов..........................................32 ГЛАВА 2. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ……………………………......39 2.1. Карбоалюминирование и восстановительное β-алюминийэтилирование 1алкенов с помощью Et3Al.....................................................................................39 2.2. Восстановительное β-алюминийэтилирование норборнена и некоторых его производных с помощью Et3Al в присутствии TaCl5........................................................................................................................46 2.3. TaCl5 - катализируемая реакция α-олефинов с помощью высших алюминийорганических соединений...................................................................54 3 2.4. Восстановительное β-цинкэтилирование 1-алкенов с помощью Et2Zn в присутствии TaCl5.................................................................................................56 2.5. Реакция 1-алкенов с помощью R2Zn в присутствии TaCl5.........................63 ГЛАВА 3. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ..............................................68 ВЫВОДЫ............................................................................................................. 82 ЛИТЕРАТУРА.....................................................................................................84 4 ВВЕДЕНИЕ Актуальность работы. 1 Успешное внедрение современных методов металлокомплексного катализа в химию металлоорганических соединений непереходных металлов (Al, Mg, Zn, Sn, B, Hg) привело к открытию и развитию новых направлений исследований в области синтетической и металлоорганической высокоэффективные химии. В результате каталитические методы были гидро-, созданы карбо- и циклометаллирования непредельных соединений, которые открыли широкий путь к однореакторным методам получения сложных по своей структуре соединений, синтез которых классическими методами является многостадийным или основан на применении малодоступных реагентов и мономеров. Важное место среди этих пионерских достижений занимают работы Уфимской школы химиков под руководством члена-корреспондента РАН Джемилева У. М. К числу наиболее эффективных и широко применяемых катализаторов, разработанных для проведения реакций алюминийорганических, магнийорганических и цинкорганических соединений с непредельными субстратами, относятся комплексы Ti и Zr, в меньшей степени соединения Pd, Co, Ni, V и редкоземельных элементов. В то же время, до начала наших исследований в мировой литературе полностью отсутствовали исследования в области синтеза Mg-, Zn- и Alорганических соединений с участием соединений и комплексов Ta. Между тем,как мы полагали, применение комплексов Ta в металлоорганическом синтезе могло бы привести к разработке эффективных методов синтеза новых классов металлоорганических соединений. В связи с этим разработка новых реакций, эффективных металлоорганических реагентов на основе доступных органических соединений Al и Zn, перспективных для применения в органическом и металлоорганическом синтезе, развитие 1 Автор выражает глубокую благодарность члену-корреспонденту РАН У.М. Джемилеву за выбор направления исследования и постоянную помощь при обсуждении полученных результатов. 5 оригинальных направлений исследований в области применения танталсодержащих металлокомплексных катализаторов для осуществления реакций непредельных соединений с металлоорганическими реагентами является важной и актуальной научной задачей. Цель исследования. Исследование возможности применения Taсодержащих комплексных катализаторов для осуществления карбометаллирования α-олефинов и норборненов с помощью доступных Alи Zn-органических соединений с целью получения новых классов металлоорганических соединений непереходных металлов. В рамках диссертационной работы были определены следующие наиболее важные задачи: − исследование каталитической активности комплексов тантала в реакции алкенов с Et3Al; − изучение реакций высших АОС с 1-алкенами в присутствии TaCl5; − осуществление реакции восстановительного β-цинкэтилирования 1-алкенов с помощью Et2Zn под действием Та-содержащего катализатора; − карбометаллирование 1-алкенов с помощью R2Zn (где R= Prn, Bun, Penn, Hexn и т.д.) в присутствии катализатора TaCl5. Научная новизна. В рамках данной диссертационной работы выполнено целенаправленное исследование по применению металлокомплексных катализаторов на основе TaCl5 в реакциях триалкилаланов и диалкильных производных цинка с олефинами с получением новых классов АОС и ЦОС. Осуществлено карбоалюминирование алюминаэтилирование терминальных и алкенов восстановительное с помощью βEt3Al катализируемое комплексами тантала, приводящее к образованию двух региоизомерных АОС: диэтил-2-(R-замещенного)-н-бутилалюминия и диэтил-3-(R-замещенного)-н-бутилалюминия с общим выходом ~ 75-85%. Разработаны условия, позволяющие проводить данную реакцию с высокой региоселективностью. 6 − Проведено стереоселективное восстановительное β-алюминаэтилирование норборнена и его производных с помощью Et3Al, катализируемое TaCl5 с получением диэтил[2-экзо-(2'-норборнилэтил)]алюминия с высокими выходами. − Проведена оценка термодинамической вероятности элементарных стадий предложенного механизма на примере реакции норборнена с Et3Al катализируемого TaCl5, с применением метода функционала плотности PBE/3z с псевдопотенциалом Стивена-Баша-Краусса. − Впервые изучена реакция 1-алкенов катализируемая и TaCl5 бутилалюминиевым и приводящая с помощью высших АОС, к (2R',3R-дизамещенным)-н- (2R,3R-дизамещенным)-н-бутилалюминиевым производным. − Осуществлено строго региоселективное восстановительное β- цинкэтилирование α-олефинов с помощью Et2Zn, катализируемое TaCl5 с получением этил-3-(R-замещенным)-н-бутилцинкорганических соединениий с высокими выходами. − С применением DFT расчетов проведена оценка термодинамической вероятности элементарных стадий предложенного механизма реакции 1алкенов с Et2Zn под действием катализатора TaCl5. − Впервые изучено карбометаллирование 1-алкенов с помощью высших ЦОС, катализируемое TaCl5, приводящее к (2R',3R-дизамещенным)-нбутилцинкорганическим и (2R,3R-дизамещенным)-н- бутилцинкорганическим соединениям. − Предложены схемы каталитических циклов образования ациклических АОС и ЦОС в результате Та-катализируемых превращений 1-алкенов, норборнена и его производных с помощью триалкилаланов и диалкилпроизводных цинка. Практическая ценность работы. В результате проведенных исследований разработаны новые препаративные методы синтеза ранее неописанных Al- и Zn- органических соединений с помощью доступных 7 триалкилаланов и диалкилпроизводных цинка с участием Та-содержащих металлокомплексных катализаторов, основанные на применении реакции карбометаллирования и восстановительного β-металлэтилирования терминальных и циклических олефинов. С применением разработанной реакции региоселективного восстановительного β-цинкэтилирования 1-алкенов с помощью Et2Zn в присутствии TaCl5 предложен высокоэффективный оригинальный "однореакторный" метод синтеза 6-метилоктан-3-она – компонента феромона тревоги муравьев рода Crematogaster. Апробация работы и публикации. Материалы диссертационной работы были представлены на международном симпозиуме «FOC-2012 and 2nd Taiwan–Russian Symposium on Organometallic Chemistry, всероссийской конференции с международным участием «Современные достижения химии непредельных соединений: алкинов, алкенов, аренов и гетероаренов», посвященной научному наследию М.Г. Кучерова, XXI Молодежной конференции студентов, аспирантов и молодых ученых «Ломоносов». По материалам диссертации опубликовано 3 статьи в зарубежных журналах, индексируемых Web of Science, тезисы 3 докладов научных конференций. Структура и объем диссертации. Диссертация состоит из введения, литературного обзора, обсуждения результатов, экспериментальной части, выводов и списка цитируемой литературы, включающего 130 наименований. Работа изложена на 97 страницах машинописного текста, содержит 23 схемы, 6 таблиц, 4 рисунка. Работа выполнена в лаборатории химии высоких энергий и катализа Института нефтехимии и катализа РАН в соответствии с научным направлением Института по бюджетной теме «Металлокомплексный катализ в химии металлоорганических и кластерных соединений» (№ Госрегистрации 0120.0850048). 8 ЛИТЕРАТУРНЫЙ ОБЗОР РЕАКЦИИ КАРБОАЛЮМИНИРОВАНИЯ И КАРБОЦИНКИРОВАНИЯ НЕПРЕДЕЛЬНЫХ СОЕДИНЕНИЙ 1.1. Карбоалюминирование алкенов и алкинов Реакция карбоалюминирования представляет собой частный случай реакции карбометаллирования и заключается в присоединении связи Al-C алюминийорганического соединения (АОС) к кратной связи, приводящее к новому АОС: С Al C + С C С С Al Реакция карбоалюминирования широко изучена, так как имеет огромный синтетический потенциал для получения новых высших АОС, которые в свою очередь могут служить интермедиатами биологически активных соединений. 1.1.1.Межмолекулярное карбоалюминирование α-олефинов Согласно [1,2] присоединение Et3Al к α-олефинам приводит не только к продукту карбоалюминирования 1, но и продуктам дегидроалюминирования и димеризации. Et Et3Al R AlEt2 R 1 Триалкилалюминий с разветвленным радикалом, например, But3Al реагирует с 1-октеном при 60-80ºС за 65 ч с образованием АОС 2 и 3 [3]. C5H11 C6H13 But 3Al C6H13 t-Bu al -alH -alH t-Bu 2 C6H13 t-Bu 3 9 Стереоселективно реагирует бицикло[2.2.1]гептен с Et3Al, давая экзоэтилнорборнан 4 с выходом 49% [4]. Et3Al H2O al Et бензол, 20оС Et 4 Взаимодействие дициклопентадиена с Et3Al приводит к образованию АОС 5 и 6. Et3Al легко Et Et + Et 6 5 Алкены H2O al Et al + подвергаются арилированию с помощью трифенилалюминия [5-7]. C6H5 (C6H5)3Al C6H13 C6H5 al C6H13 C6H13 C6H5 -alH C6H13 Трифенилалюминий также реагирует с 1,1-дифенилэтиленом, давая АОС 7. (C6H5)2C CH2 (C6H5)3Al (C6H5)2 H2O C6H5 (C6H5)2 C6H5 al 7 Производные бицикло[2.2.1]гептена взаимодействуют с Ph3Al с хорошими выходами. трифенилалюминием в В результате кипящем фенилбицикло[2.2.1]-2-гептена дифенилбицикло[2.2.1]гептана 9 [5]. реакции бензоле 8 норборнадиена образуется и экзо, смесь с экзо-5экзо-2,5- 10 Ph3Al al Ph al + Ph Ph al H2O Ph Ph + Ph 8 9 Согласно [7] Ph3Al легко присоединяется к бензонорборнадиену 10 с образованием изомерных АОС 11 и 12. al Ph al Ph3Al Ph + 12 11 10 1.1.2. Некатализированное карбоалюминирование алкилацетиленов Некатализированное карбоалюминирование алкинов обычно проходит неселективно и сопровождается конкурирующими реакциями металлирования, гидро- и дегидроалюминирования. Было установлено [8], что реакция алкилацетиленов с триалкилаланами (Et3Al, Pr3Al, Bui3Al) приводит к образованию не только продуктов карбоалюминирования 13 и 14, но и продукта металлирования 15. 2 1 R 2 R1 R3Al 1 AlR2 H R + R2 H H + 2 R2 R2Al 13 1 2 R 14 AlR2 15 Фенилацетилен реагирует с Et3Al и Pr3Al с образованием АОС 16 и 17 [9,10]. Ar H R3Al 70oC Ar H Ar + R2Al R медленнее, чем ацетилены 17 подвергаются монозамещенные. AlR2 R 16 Дизамещенные H карбоалюминированию Особенностью реакции 11 карбоалюминирования дизамещенных ацетиленов является то, что реакция не останавливается на первой стадии присоединения, а продолжается за счет взаимодействия образовавшихся алкенилаланов с исходными ацетиленами. Так, согласно работе [11] взаимодействие 3-гексина с Et3Al происходит при 80-90 ºC с образованием после гидролиза 1,1,2,3,4-пентаэтил-1,3-бутадиена 18. Et Et Et Et3Al Et Et Et Et Et AlEt2 80-90oC Et AlEt2 H2O Et Et Et H Et Et Et Et Et 18 В отличие от 3-гексина, при взаимодействии дифенилацетилена с Et3Al происходит преимущественное образование продуктов карбоалюминирования 19 и гидроалюминирования 20. Ph Ph Et3Al 80-90oC Ph Ph Ph Ph + Et AlEt2 H 20 19 AlEt2 Реакция дифенилацетилена с Ph3Al приводит к образованию продукта карбоалюминирования который 21, при дальнейшем термическом превращении дает алюмогетероцикл 22 [12-18]. Ph Ph Ph Ph3Al Ph Ph Ph 200oC AlPh2 Al 21 22 Ph Ph Селективно проходит реакция 1-фенилпропина с Ph3Al с образованием единственного АОС 23 [16,17]. Ph Me Ph3Al Me Ph Ph AlPh2 23 Согласно [16,17] фенил-трет-бутилацетилен реагирует с Ph3Al при длительном кипячении в толуоле. 12 t Ph Bu Ph3Al Ph Bu Ph t AlPh2 Замещенные дифенилацетилены также реагируют с Ph3Al. Так, было установлено [12], что при 160ºC Ph3Al взаимодействует с соответствующими алкинами с образованием продуктов карбоалюминирования 24 и 25. R R R Ph3Al Ph + Ph Ph R= Me2N, MeO, Me, MeS, Cl Ph Ph2Al AlPh2 Ph 25 24 Авторы работы [18] осуществили взаимодействие дифенилацетилена с трибензилалюминием при длительном кипячении реагентов в ксилоле. Ph (PhCH2)3Al Ph Ph Ph PhCH2 Al(CH2Ph)2 1.1.3. Каталитическое карбоалюминирование олефинов Реакции каталитического карбоалюминирования алкенов и алкинов открывают новые возможности получения Al-C и C-C связей, что позволяет синтезировать новые типы высших АОС. Некаталитическое карбоалюминирование алкенов обычно проходит в жестких условиях с образованием побочных продуктов гидроалюминирования [1]. Первого упоминание реакции каталитического карбоалюминирования датируется 1979 г. [19]. Авторы [20-22] осуществили селективное алкилирование α-олефинов с помощью Et2AlCl в присутствии каталитических количеств Zr(OBu)4 с образованием алкенов 26 диалкилалюминийхлоридов: с этильной группой в β-положении и 13 Et 2R + R AlEtCl R 26 Et [Zr] Et2AlCl 4R + R 2R 2 AlCl R= C6H13, C8H17, C10H21, CH3CH CHCH2 Было установлено [23-25], что диэтилалюминийхлорид (Et2AlCl) селективно карбоалюминирует α-олефины в присутствии 0,3-3 мол% Ti(OBu)4 или TiCl4 в толуоле при 20ºС, давая соответствующие диалкилалюминийхлориды 27 с высокими выходами: Et2AlCl [Ti] R толуол, 3 ч Et Et D2O, DCl AlEtCl R R D 27 Реакция нашла свое применение в синтезе АОС и биологически полезных природных соединений [21,23,25]. В работе [26] описано взаимодействие триалкилаланов с 1-алкенами, в том числе функциональнозамещенными, в присутствии стехиометрических количеств Cp2TiCl2, приводящее к образованию 1,1-дизамещенных алкенов 28. AlR32 R2 Cp2TiCl2 (2экв) R' CH2Cl2, 23oC, 1-24 ч R' 28 R' = C8H17, MeCO2(CH2)4, Br(CH2)4, NC(CH2)4 ;R2 = Me, Et, Bun Карбометаллирование гомоаллильных спиртов с участием Et2AlCl в присутствии стехиометрических количеств Ti-содержащих соединений (Cp2TiCl2, TiCl4, Ti(acac)2Cl2) проходит неселективно с образованием смеси продуктов карбо - и гидроалюминирования, гидрирования, изомеризации и β- 14 гидридного элиминирования [26-29]. При этом образование целевого продукта 29 не превышало 30%. Et2AlCl [Ti] OH H2O + HO + HO HO 29 + HO HO Комплексы титана (Cp2TiCl2, Ti(acac)2Cl2, TiCl4) также проявили активность в реакции карбоалюминирования замещенных норборненов с помощью диалкилгалогеналанов (Et2AlCl, с Me2AlCl) образованием алкилированных норборнанов с высокой стереоселективностью [30,31]. , [Ti] 1. Et D 2. D3O+ Et2AlCl , [Ti] 1. Et D 2. D3 O+ Несмотря на успешное Zr-катализируемое карбоалюминирование алкинов [32-34] аналогичная реакция с алкенами давала только следовые количества целевого продукта. Так, карбоалюминирование 1-октена [35] в стандартных условиях (1 эквив. AlMe3, 8 мол% Cp2ZrCl2) приводило к распределению продуктов как показано на схеме: AlMe3 (1 экв) Cp2ZrCl2 (8 мол%) C6H13 Me C8H17 C6H13 + C6H13 Me + C6H13 H Предполагаемый механизм включает первоначальное формирование интермедиата 2-метилоктилметалла 30, который быстро подвергается β-H элиминированию с образованием 2-метил-1-октена 31 и гидрида металла 32. 15 Гидрометаллирование исходного 1-октена приводит к интермедиату 33, который затем карбоалюминирует 1-октен, давая 34: C6H13 Me Me AlMe3 Cp2ZrCl2 (8 мол%) MLn C6H13 C6H13 H 30 MLn = Zr или Al 31 18% C8H17 M-H C6H13 C6H13 32 34 C6H13 MLn C6H13 33 MLn = Zr или Al Применение хиральных производных неоментилинденил)цирконийдихлорида 35, тетрагидроинденил)цирконийдихлорида и цирконоцена – бис(1- бис(1-нео-изо-ментил-4,5,6,7др. в данной реакции с последующим окислением позволило получить оптически активные спирты 36 [35]. 1. Me3Al [Zr](8мол%) R CH2Cl2, 0oC, 18-24ч 2. O2 Me R * OH 62-92% (65-85% ee) 36 [Zr]= ZrCl2 Pr i R= C6H13, PriCH2, Ph, PhCH2, HO(CH2)4, Et2N(CH2)3 2 35 16 Таблица 1. Zr-катализированное энантиоселективное метилалюминирование α-олефинов [35] Алкен №/№ Время Продукт реакции, Выход, ее, % % 88 72 92 74 80 65 77 70 30 85 81 74 79 75 68 71 ч 1 12 OH 5 5 2 12 OH 12 3 OH 4 24 5 528 OH OH 6 12 Si 7 Si OH 12 HO OH 3 HO 8 3 96 Et2N OH 2 Et2N 2 Согласно[36], введение воды или метилалюмоксана (МАО) приводит к ускорению реакции метилалюминирования олефинов. R Me3Al/35 H2O/МАО R AlMe2 17 При карбоалюминировании децена-1 с помощью Et3Al в присутствии каталитических количеств Cp2ZrCl2 наряду с получением целевого продукта 37 наблюдается образование продукта гидроалюминирования 38 и βэтилированного исходного α-олефина 39 (1) [37]. Использование объемных инденовых производных в циклопентадиеновом лиганде позволило в некоторой степени подавить βгидридное элиминирование и, следовательно, повысить селективность образования продуктов карбоалюминирования [38]. C8H17 Cp2ZrCl2 C8H17 AlR2 + Et CH2Cl2, 23oC 37 37% C8H17 ZrCl2 + AlEt3 C8H17 AlR2 CH2Cl2, 23oC AlR2 H 38 20% O2 [O] Al Et H Et 39 20% OH (1) (2) Et >85% C8H17 CH3CHCl2 + C8H17 Et [Zr] C8H17 C8H17 H OH (3) Et 64%, 92%ee Реакция не ограничивается этилалюминированием. Так, авторам [37] удалось получить R- и S-изомеры 2-(н-пропил)-1-деканола с помощью нпропилалюминирования 1-децена и н-октилалюминирования 1-пентена, соответственно, используя катализатор 35: (R) n-Oct 1. (Prn)3Al, [Zr] CH3CHCl2 2. [O] n-Oct n-Pr OH H 62%, 92%ee (S) n-Pr 1. (Octn) 3Al, CH3CHCl2 2. [O] [Zr] n-Oct n-Pr OH H 59%, 85%ee Применение двухкомпонентного катализатора – Cp2*ZrMe2 – B(C6F5)3 (Cp* = η5-C5Me5) в среде толуола за 3 ч позволило селективно провести 18 реакцию карбоалюминирования терминальных олефинов с помощью Me3Al [39]: Me3Al Cp2ZrMe2 , B(C6F5)3 R толуол, 0оС, 3ч Me Me O2 AlMe2 R R OH 71-82% R= H2C C(Me)(CH2)2 , Me(CH2)2 Согласно [36,40,41] добавление аллилфенилового эфира 40 к смеси AlMe3, катализатора 35 и воды в CH2Cl2 с последующим окислением дает продукт метилалюминирования-перегруппировки Кляйзена 41: 1. AlMe3 (4 экв), [Zr] (5 мол%) H2O (1 экв) O CH2Cl2, 0оС, 12ч HO 2. O2 40 OH 41 75%, 75%ee Реакция 2,5-дигидрофурана 42 с AlEt3 в присутствии 5 мол% комплексов Zr (45 и 46) приводит к энантиоселективному образованию диола 43 и моноспирта 44. Энантиоселективность 25 более 99% ее, 24 – 85-90% ее (Схема 1) [42-44]. AlEt3 H H 24 или 25 O 42 без раств-ля 6-20ч AlEt O Et + 1. O2 2. H2O H AlEt2 O H2O (или D2O) OH H OH OH 43 H(D) 44 19 O H Cl Zr Zr O Cl 45 46 Схема 1. Zr-катализируемое энантиоселективное 2-этилалюминирование алкенов. 1.1.4. Каталитическое карбоалюминирование ацетиленов Каталитическое карбоалюминирование ацетиленов достаточно широко обобщено[34, 45-49]. Наиболее оптимальные карбоалюминированию алкинов результаты были по получены каталитическому с использованием катализаторов на основе Ti [50-57] и Zr [28,34,58-60], хотя в литературе также имеются сведения о применении металлокомплексных катализаторов на основе Fe [61,62], Mn [62,63] и Ni [62,64,65] в данной реакции. Катализаторы на основе соединений Ti проявили активность в реакции Me3Al с гомопропаргильными спиртами [66,67]. В результате реакции образуются АОС 47 и 48. Аналогичная реакция с моно- и дизамещенными алкинами, не содержащими OH-группы, малоселективна, за исключением дифенилацетилена, карбоалюминирование которого проходит в мягких условиях с образованием 49 [68,69]. 20 al R Me3Al, TiCl4 CH2Cl2, HO Me R -78...-45oC H 47 alO R= H, Me, Et al R Me Me3Al, [Ti] R HO alO R= Me, Et, i-Pr, Pent,Ph 48 H2O Me3Al/Cp2TiCl2 Ph (CH2Cl)2, 20-25oC Me Ph Ph Ph Me H 84% PhC CPh AlMe2 J2 Ph Ph 49 Me Взаимодействие терминальных и интернальных 75% J алкинолов с диэтилалюминийхлоридом в присутствии Cp2TiCl2 проходит с разной региоселективностью [54]. Регио- и стереоселективность наблюдается только со спиртами с интернальной связью: (CH2)2OH Et2AlCl, [Ti] (CH2)2OH + (CH2)2OH Et2AlCl, [Ti] (CH2)2OH R R (CH2)2OH В 1978 г. Негиши с сотрудниками осуществили Zr-катализированное карбоалюминирование ацетиленов [69]. Так, реакция моно- и дизамещенных ацетиленов с Me3Al в присутсвии Cp2ZrCl2 стереоселективностью с получением АОС 50 и 51. проходит с высокой 21 Me3Al Cp2ZrCl2 R H RC CH Me 50 Me3Al Cp2ZrCl2 R AlMe2 R RC CR Me 51 AlMe2 Механизм и региоселективность реакции широко изучен [34,58-60,7073]. Авторы полагают, что механизм реакции включает прямое карбоалюминирование ацетиленов (Схема 2) [33,58]. Cl Me2Al Me3Al + Cp2ZrCl2 RC CR ZrCp2Me RC CR δδ+ Me2Al Cl ZrCp2Me Cl R R Cl R R Cl Me Me + Cp2ZrCl2 ZrCp2Me Al Me AlMe2 Cl Схема 2. Механизм метилалюминирования алкинов. Полученные винилаланы 51 являются синтетически полезными реагентами (Схема 3) [74]. В работах [75-77] приведены способы синтезов природных изопреноидов из функционализированных алкенилаланов, получаемых методом каталитического карбоалюминирования терминальных ацетиленов AlMe3. 22 R R' H Me RC CH PhSO2Cl O R' OH Me SO2Ph R' 1.BuLi 2.ClCOOEt H Me COOEt H R' H Me 51 AlR2 ClZn n R' R' R' O Me I2 H R H X O R' H H n Me Cat. PdLn Me R H Me R' Me Cat. PdLn (NiLn) I XMgCH2SiMe3 RZnX (MgX) Cat. PdLn (NiLn) Cat. NiLn R' H Me SiMe3 Схема 3. Превращения (Е)-2метил-1-алкенилаланов в различные практически важные соединения. Согласно [78,79] добавка метилалюмоксана или H2O ускоряет Zrкатализированное метилалюминирование алкинов. В стандартных условиях, 1-октин карбоалюминируется за 3ч при комнатной температуре, давая смесь региоизомеров 52 и 53 в соотношении При 95:5. добавлении стехиометрических количеств воды данная реакция проходит в течение нескольких минут при -70оС с образованием региоизомерной смеси в соотношении 97:3. 1. AlMe3 (2 экв) Cp2ZrCl2 (0.2 экв) H2O (1,5 экв)/МАО C6H13 C6H13 C6H13 + CH2Cl2, -70oC,10 мин Me Me 2. 3н HCl 53 52 Эта методология была использована для 3х-стадийной каскадной реакции, включающей добавлением воды, Zr-катализируемое перегруппировку карбоалюминирование Кляйзена и с нуклеофильное 23 карбонильное присоединение, превращающее терминальные алкины и аллилвиниловые эфиры в аллильные спирты 54, имеющие 3 близко расположенных ассиметрических центра [80]: AlMe3/Cp2ZrCl2 H2O R1 R AlMe2 O 2 R2 R3 O H R1 OH R2 * R3 * R3 R1 * 54 Взаимодействие других триалкилаланов с алкинами в отличие от метилалюминирования проходит с меньшей региоселективностью. Так, присоединение 1-октина к Pr3Al приводит к образованию смеси АОС 55 и 56 в соотношении 4:1 [32]. Pr3Al, [Zr] C6H13 C6H13 H C6H13 H R2Al Pr + AlR2 Pr 55 56 4:1 Реакция бутен-3-ола с Et3Al также приводит к изомерной смеси АОС 57 и 58 в соотношении 63:37 соответственно [54]. HO(CH2)2C CH al-O(CH2)2 Et3Al al-O(CH2)2 H H + Et Et 58 57 Ацетилены также способны карбоалюминироваться трибензилалюминием в присутствии Cp2ZrCl2 [81]. Bn3Al, Cp2ZrCl2 R H RC CH H + Bn В R работе [82] AlBn2 Bn2Al авторами получены Bn R Hex Ph H2C CH(CH2)4 Me3SiC CCH2 хорошие 70 >98 75 65 30 результаты при 25 35 использовании в качестве катализатора комплексов циркония с 5,10,15,20тетрафенилпорфирином (TPP) в реакции Et3Al с ацетиленами. 24 Et3Al, TPPZrX2 R H R H AlEt2 AlEt2 Et + RC CH Et R Pr i-Bu t-Bu Pent Ph >99 97 1 99 90 <1 3 99 1 10 1.2. Карбоцинкирование непредельных соединений Первые цинкорганические соединения (ЦОС) были открыты Э. Франклендом еще в 1849 году и широко использовались в органическом синтезе вплоть до введения в практику реактивов Гриньяра. В настоящее время их использование в различных синтезах весьма ограничено вследствие их высокой пожароопасности, а также замены ЦОС на менее чувствительные к воздуху и достаточно активные магнийорганические соединения. Энантиоселективные реакции вернули ЦОС из забвения, в котором они пребывали практически со времен открытия реагентов Гриньяра. 1.2.1 Некатализируемое карбоцинкирование алкенов Некатализируемое присоединение алкилцинка к алкенам обычно не происходит за исключением более реакционного ди(трет-бутил)цинка [83]. Карбоцинкирование этилена проводится под давлением при 50-75 ºС и дает преимущественно диалкилцинк соответствующего углеводорода. который 59, Реакция также гидролизуется проходит с до менее реакционноспособными монозамещенными олефинами, такими как пропен или 1-октен. Стерически объемная трет-бутильная группа ЦОС присоединяется к менее замещенному углероду алкена с образованием вторичных цинкдиалкилов 60 и 61 соответственно с высокой региоселективностью (≥ 98/≤ 2). Первичные и вторичные ЦОС 59 и 60 далее не присоединяются к исходному олефину, однако при повышенной температуре диалкилцинк 61 далее реагирует с 1-октеном с образованием 62 и небольших количеств бипродукта 63 (Схема 4) [83]. 25 R HCl t-Bu t-Bu2Zn + H2C CHR 2 t-BuCH2CH2R Zn 94% R=H 60 атм, C6H6, 50-75oC R=Me 22-40oC R=Hex 75oC 70% 41% 58% 59 60 61 75oC H2C CHHex Hex Hex t-Bu 2 Hex HCl Hex t-Bu Zn 62 63 13% Схема 4 1,3-бутадиен проявляет более высокую реакционноспособность по отношению к t-Bu2Zn. В результате реакции первоначально образуются два аллилцинкдиорганических изомера 64 и 65. При тщательном контроле условий реакция останавливается на стадии моноприсоединения и после гидролиза получают смесь регио- и стереоизомерных у.в.: t-Bu2Zn н-пентан, от -10оС до rt t-Bu 2 t-Bu Zn 2 65 64 Zn HCl t-Bu + t-Bu 70% Me + t-Bu 9% 6% Me Моноприсоединение t-Bu2Zn к стиролу происходит при 15ºС с получением бензилцинкдиорганического соединения 66 [83]. Ph t-Bu2Zn -15оС Ph HCl t-Bu 2 66 Zn t-BuCH2CH2Ph 46% 26 1.2.2. Некатализируемое карбоцинкирование алкинов Ди(трет-бутил)цинк реагирует с различными терминальными алкинами, имеющими спиртовые, эфирные, ацетатные или аминные функциональные группы [84]. Присоединение проходит региоселективно, так как объемная трет-бутильная группа присоединяется к менее замещенному углероду алкина. Однако реакция не стереоселективна и дает смесь геометрических изомеров: 1. ТГФ, поток t-Bu2Zn + H t-Bu R t-Bu + 2. H2O R R (E)/(Z) = 35/65 до 88/12 R= Bu, (CH2)nOH (n=1-3), CH2OBu, CH2OTHP, CH2NHEt, CH2NEt2 20-65% Фенилацетилен, в котором тройная связь соединена с ароматическим кольцом проявляет высокую реакционную способность и быстро подвергается стереоселективному анти-присоединению t-Bu2Zn в потоке кипящего эфира, давая (Z)-алкены 67 [85]. Различные терминальные енины также реагируют хемоселективно в этих условиях, давая 1,3-диены 68 [86,87]: t-Bu2Zn + H R t-Bu Et2O R= Ph R= (R1)C=CHR2 R1= H, R2= Bu, CH2OH, CH2OBu,NHEt, NEt2 R1 R2 = R 67 70% 68 30-55% (CH2)n Карбоцинкирование алкенов и алкинов, катализируемое переходными металлами. Углерод-углеродные ненасыщенные связи, как правило, инертны по отношению к Zn-органическим реагентам, поэтому для проведения реакции карбоцинкирования с участием различных субстратов и реагентов часто используются переходные металлы. 27 1.2.3. Каталитическое карбоцинкирование алкенов В 2000 г. авторам [88] удалось осуществить этилцинкирование монозамещенных алкенов в присутствии катализатора, образованного in situ из Cp2ZrCl2 с 2 молярными эквивалентами EtMgBr с получением диизоалкилцинкорганических соединений 69 с высокими выходами. Cp2ZrCl2 (10 мол%) EtMgBr (20 мол%) Et2Zn (0.5 экв) R ТГФ, 4 ч I2 R 2 Zn Et2O R I 69 96% Реакция является довольно общей по отношению к замещенным алкенам, хотя аллилбензиловый и аллилфениловый эфиры не реагируют. Предложенный авторами механизм (Схема 5) предполагает первоначальное образование диэтилцирконоцена из цирконоцендихлорида и 2х молярных эквивалентов EtMgBr. Диэтилцирконоцен подвергается βгидридному переносу с образованием цирконоцен-этиленового комплекса (циклопентадиенилцирконоциклопропан) [89], который реагирует с исходным алкеном давая β-замещенный цирконоциклопентан. Последний подвергается региоселективному трансметаллированию под действием Et2Zn, давая биметаллический интермедиат. Региоселективность, как предполагают авторы, наблюдается вследствие уменьшения стерических затруднений, поскольку цирконоценовая часть присоединяется к менее замещенной части. Диалкилцирконоценовый интермедиат подвергается хемоселективному β-гидридному переносу, включающему селективный перенос β-H атома от этильной группы, давая активный этилен-цирконоценовый комплекс и этилзамещенное цинкорганическое соединение, которое может вступать во второй каталитический и, в конечном счете, приводит к этилзамещенному диалкилцинку. 28 R Cp2ZrCl2 + 2EtMgBr R ZnR' Et Cp2ZrEt2 -EtH ZrCp2 R R EtZnR' ZnR' ZrCp2Et ZrCp2 1 цикл R' = Et 2 цикл R' = CH2CH(Et)R Схема 5. Механизм этилцинкирования монозамещенных алкенов Наряду с ациклическими олефинами в реакцию карбоцинкирования были вовлечены циклопропены [90,91]. Взаимодействие цинкдиорганических соединений с циклопропеновыми кеталями, содержащими напряженную двойную связь, происходит под действием катализатора с FeCl3 образованием алкилированных циклопропановых кеталей 70 с высокими выходами. Me O O Me Me Me 1. R2Zn FeCl3 O O ТГФ/толуол, -25оС 2. H+/H2O R (a) R= Et 73% (b) R= Pen 91% (c) R= (CH2)2CO2Pri 76% 70 Энантиоселективность была достигнута с помощью введения тройной каталитической системы, состоящей из FeCl3, хирального дифосфина, такого как p-TolBINAP и бидентатного ахирального диамина, например, TMEDA. Реакция проводится в смеси толуол/ТГФ. 29 Me O Me O Et2Zn (2 экв) FeCl3 (5 мол%) (R)-p-TolBINAP (7,5 мол%) Me Me P(C6H4Me-p)2 O TMEDA (5 экв) толуол/ТГФ, 10 оС, 24 ч O P(C6H4Me-p)2 Et (R)-p-TolBINAP Позднее было карбоцинкирование осуществлено циклопропенов с энантиоселективное [91] использованием палладиевого катализатора и хирального лиганда. Взаимодействие 71 с диэтилцинком в присутствии каталитических количеств палладиевой соли, (R)-TolBINAP и трифлата цинка с последующей обработкой бензоил хлоридом приводит к продукту 72 с выходом 75%. 1. PdCl2(MeCN)2 (5 мол%) (R)-p-TolBINAP (5 мол%) Zn(OTf)2 (10 мол%) Et2Zn CH2Cl2, -78oC O Et Ph 2. CuCN*2LiCl, THF 3. PhCOCl 71 72 75% В 2009 г., Fox с сотрудниками [92] осуществили Cu-катализированную реакцию карбометаллирования циклопропенов. Обработка циклопропена 73 диметилцинком в цинкорганический присутствии каталитических интермедиат 74, количеств который после CuI дает протонолиза превращается в конечный продукт 75. Me CO2Et 1. Me2Zn (4 экв) CuI (20 мол%) толуол, 0-18оС 2. H+ Hex OEt Me Hex Zn X Me 73 Позднее O [93] было CO2Et Me 75 74 авторами Me Hex сообщено 70% стереоселективное арилцинкирование циклопропенов. Цинкорганические реагенты в данном 30 случае были приготовлены с помощью йод/магниевого обмена с последующим трансметаллированием хлоридом цинка, и дальнейшим превращением в одну стадию: ArI 4 экв Ph 1. PriMgX ТГФ (Et2O) CO2Me Ph , CuCN (20 мол%) Ar2Zn 2. ZnCl2 3. диоксан Интересным классом 0оС-18оС, толуол соединений, CO2Me Ar показывающим высокую реактивную способность по отношению к различным металлоорганическим реагентам, являются оксабициклические алкены. Алкилирование с последующим раскрытием кольца таких субстратов дает интересную стратегию для синтеза циклических и ациклических структур, имеющих смежные стереоцентры. Согласно данным авторов [94], оксабензонорборнен 76 подвергается реакции алкилирования с раскрытием кольца с помощью Et2Zn в присутствии палладиевого катализатора и хирального лиганда с образованием син-дигидронафтола 77. O OH Et2Zn PdCl2 (MeCN)2 (5 мол%) (R)-p-TolBINAP (5 мол%) 76 CH2Cl2 Et 77 В реакцию также вступают цинкорганические галогениды [95]. По сравнению с оксабензонорборненами, оксабицикло[2.2.1]гептены и оксабицикло[3.2.1]октены менее реакционноспособные субстраты и для проведения данной реакции обычно требуется нагревание в потоке 1,2дихлорэтана. Добавление Zn(OTf)2 приводит к конверсии 78 в циклогептенол 79, которая проходит уже при комнатной температуре в CH2Cl2 [96]. В отсутствии Zn(OTf)2 реакция дает 78 и продукт энантиоселективного этилцинкирования 79 [97]: 31 OH Me OH Et2Zn Me OH Me Me Me cat. ((R)-p-TolBINAP)PdCl2 O + OH CH2Cl2 Me O Et 77 Et 78 79 В случае, когда спиртовая часть защищена силиловым эфиром образующиеся субстраты, такие как становятся 80 менее реакционноспособны и Pd-каталитический процесс останавливается на стадии карбоцинкирования даже в присутствии Zn(OTf)2. Образующийся конфигурационно реагент устойчивый может 81 оксабициклический подвергаться цинкорганический протонолизу, иодонолизу или трансметаллированию с получением Zn-Cu органического реагента, который реагирует с аллилбромидом давая оксабицикло[3.2.1]октен 82. ЦОС 81 подвергается раскрытию кольца при нагревании в потоке 1,2дихлорэтана или при добавлении тяжелых электрофилов, таких как бензоилхлорида или Me3SiCl, вероятно, вследствие активации мостикового кислорода. Добавление EtMgBr индуцирует раскрытие кольца 81 предположительно путем формирования цинкатов [97]. Me OTIPS Me O Et2Zn, Zn(OTf)2 OTIPS Me Me cat. ((R)-p-TolBINAP)PdCl2 O EtZn 80 81 Et TIPS=Si(Pr-i)3 Me3SiCl, BzCl EtMgBr R= SiMe3 65% R=Bz 80% R=H 84% E+ H+ 83% I2 58% 1.CuCN*LiCl 64% 2.AllBr Me E+ OTIPS Me OTIPS Me Me O OR Et E Et 82 32 Примеры Pd-катализированного карбоцинкирования до сих пор распространены только на замещенные оксабициклические алкены. Использование лиганда 1,2-бис(дифенилфосфино)бензола (dppbz) [98] в реакции оксабензонорборнена с дифенилцинком в присутствии FeCl3 приводит к соответствующему продукту карбоцинкирования 83 с высокими выходами. O FeCl3 (1 мол%) F-dppbz (2 мол%) Ph2Zn (1.5 экв) ТГФ/толуол, 0оС, 2ч OH Ph Ph H+ O O ZnX Ph + 83 95% H 5% 1.2.4. Каталитическое карбоцинкирование алкинов Первый пример карбоцинкирования алкинов датируется 1983 г., и включает взаимодействие диалкилцинка с алкинами в присутствии X2ZrCp2 (X= I, Br, Cl) [99]. Так, при взаимодействии Et2Zn с 1-октином в присутствии Cp2ZrI2 образуется смесь региоизомеров 84 и 85 в соотношении 75:25 соответственно. Реакция стереоселективна, и протонолиз или иодонолиз дает региоизомерную смесь соответственно алкенов или алкенилиодидов. Авторы утверждают, что Cp2ZrI2 более эффективен, чем Cp2ZrBr2 и Cp2ZrCl2, с последним скорость реакции заметно ниже, хотя региоселективность значительно выше (84/85=96/4). C6H13 Et2Zn (экв) Cp2ZrI2 (1 экв) C6H13 C6H13 + Et EtZn ZnEt 84 Et 85 7/8 = 75/25 E+ = H+ или I2 C6H13 + Et E C6H13 E Et 33 Авторы работы [100] осуществили превращение 5-иодо-1-пентина в алкинилэтилцинк с последующим этилцинкированием в присутствии Cp2ZrI2 с получением гем-цинкорганического соединения 86. Et Et2Zn Cp2ZrI2 ZnEt 1.n-BuLi гексан ZnEt ZnEt I 2. EtZnCl CH2Cl2 I Интернальные I 86 алкины также способны подвергаться карбоцинкированию в присутствии каталитических количеств Cp2ZrI2 [99].Так, этилцинкирование 5-децина с последующим иодонолизом приводит к тетразамещенному алкенилиодиду 87. 1. Et2Zn (2 экв) Cp2ZrI2 (1 экв) C4H9 C4H9 C4H9 2. I2 C4H9 Et I 87 Дизамещенные арилацетилены вступают в реакцию карбоцинкирования с цинкдиорганическими соединениями в присутствии катализатора Ni(acac)2 в смеси ТГФ/NMP (NMP – N-метилпирролидинон). В этих условиях реакция между и Et2Zn дифенилацетиленом дает тетразамещенный алкенилцинк 88, образующийся путем син-присоединения с высокой стереоселективностью [101,102]. Ph Et2Zn Ni(acac)2 (25 мол%) Ph Ph THF/NMP (3/1), -35oC Et ZnEt Ph E+= H+/I2 Ph Ph Et E 73-79% 88 Предполагаемый механизм рассматриваемой реакции (Схема 6) включает первоначальное трансметаллирование диалкил- (или дифенил)цинка с помощью Ni(acac)2 с образованием никельорганического соединения. Последний затем подвергается карбоникилированию алкинов с 34 получением комплекса алкенилникеля (II). Трансметаллирование с помощью RZn(acac) и последующая регенерация катализатора NiII дает соединение αарилалкенилцинк [101,102]. R' R 2 Zn + R' Ar Ar ZnR R Ni(acac)2 R 2Zn RZn(acac) RZn(acac) R' Ar RNi(acac) Ni(acac) R R' Ar Схема 6. Механизм карбоцинкирования дизамещенных арилацетиленов Таким образом, каталитическая активность никеля объясняется заменой неактивного карбоцинкирования на более благоприятное карбоникилирование, в результате чего каталитический цикл имеет две реакции трансметаллирования. В случае силил-замещенных арилацетиленов региоселективность обратная, в условиях реакции преимущественно образуется конфигурационно стабильный α-силилалкенилцинк 89. Et2Zn Ni(acac)2 Me3Si Ph ТГФ/NMP, -30oC Me3Si Ph EtZn Et I2 89 Me3Si Ph I Et 61% Дифенилцинк также может региоселективно присоединяться к алкину, давая после иодонолиза соответствующие (Z)-алкенилиодиды, которые нашли свое применение в синтезе (Z)-тамоксифена гидрохлорида 90 антиэстрогенного противоракового препарата [101-103]: 35 Me2N 1. Ni(acac)2 (25 мол%) Et Ph + Ph2Zn TГФ/NMP,-35oC 2. I2 Et Ph BrZn Ph I 88% O [Pd(dba)2] (4 мол%) PPh3 (16 мол%) ТГФ, 55оС, 10ч, 2. HCl Et Ph Ph NMe2*HCl O 75% (Z/E>99:1) 90 На основе производственного данной реакции также был разработан синтеза (Z)-1-бромо-2-этилстильбена – метод ключевого интермедиата модулятора рецепторов эстрогена [104]: N O Et Ph Ph2Zn (0.7 экв)/Et2Zn (0.7 экв) Ni(acac)2 (25 мол%) ТГФ/NMP, 20oC Et Ph Ph Zn O N Br Et Ph Ph Br 58% Авторы [105] осуществили арилметаллирование диалкилацетиленов с помощью арилцинка в присутствии кобальтового катализатора. Обработка диалкилацетиленов с помощью арилцинка в ацетонитриле в присутствии каталитических количеств бромида кобальта приводит к соответствующему арилированному интермедиату 91. При дальнейшем изучении было показано [106,107], что при использовании Xantphos в качестве лиганда полностью изменяется конечный продукт реакции. 36 R PhZnX CoBr2 (5 мол%) R R R R R Co MeCN, 60oC, 1,5-6 ч ZnX 91 [CoCl2(Xantphos)] (5 мол%) ТГФ, 60оС, 4-12 ч R R R ZnX Co Позже авторам [108] R удалось осуществить Co-катализируемое арилцинкирование алкинов, приводящее к три- или тетразамещенным алкенам с высокой стереоселективностью. Было установлено, что данная каталитическая система имеет двойное действие: комплекс CoBr2(bpy) работает в качестве катализатора не только для арилцинкирования, но и для формирования арилцинка: R CoBr2(bpy) (10 мол%) Zn (2 экв) ZnBr H2C CH CH2Cl FG MeCN 0,5-1,5 ч Цирконоценйодид R R' (0,33 экв) R' H 0,5-5 ч FG может FG способствовать присоединению диалкилцинка к α,β-дизамещенным неактивированным алкинам [109]. Так, в случае 5-децина после иодонолиза была получена смесь двух изомерных алкенилиодидов 92 и 93, образованных соответственно при син- и антиприсоединении к тройной связи, в соотношении 94/6. 37 Bu 1. Cp2ZrI2 (1экв) Zn Bu + 2 Bu Bu Bu I + 2. I2 Bu I 93 92 94 : 6 84% Zr-катализируемое кротилцинкирование 5-децина с высоким выходом дает продукт 94 [107]. Bu Bu 1. Cp2ZrI2 Bu + Me Bu Zn 2. H+ 2 92% 94 Me Было установлено, что CoCl2 катализирует аллилцинкирование различных 1-арил-1-алкинов [110]: Cl Cl ZnBr Hex Cl Hex CoCl2 (5 мол%) Hex I2 ТГФ ZnBr I 95 57% Как показано для 95, син-присоединение к тройной связи происходит с высокой региоселективностью, и может быть в дальнейшем функционализирован. Этот метод может быть использован для получения некоторых три- и тетразамещенных стиролов. Авторы [111] провели бензилцинкирование простых алкинов с получением производных аллилбензола с высокими выходами: Pr Pr + ZnBr CoBr2 (5 мол%) P(p-MeOC6H4)3 (10 мол%) EtCN, 25oC, 1,5 ч Pr Pr Pr ZnBr Pr I2 I 81% (E/Z<1:99) 38 Анализ литературных данных в области синтеза АОС и ЦОС с использованием гомогенных показывает, реакция что карбоцинкирования металлокомплексных каталитического являются катализаторов карбоалюминирования эффективными способами и превращения олефинов, ацетиленов и других ненасыщенных соединений в ациклические металлоорганические соединения различных структур, которые в свою очередь могут быть успешно использованы в синтезе различных функциональнозамещенных соединений. Рассмотренные выше реакции обладают исключительно богатым синтетическим потенциалом и находят широкое применение в органической и металлоорганической химии. Несмотря на достигнутые успехи в области каталитического синтеза АОС и ЦОС до наших исследований в литературе совершенно отсутствовали работы по применению комплексов металлов V группы (V, Nb, Ta) Периодической системы элементов в реакциях простейших АОС и ЦОС с непредельными соединениями. Настоящая диссертационная работа является одной из попыток восполнения этого пробела, т.к. осуществление вышеперечисленных реакций, а также обнаружение новых направлений реакций открывают путь к получению новых ранее труднодоступных АОС и ЦОС. 39 2. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ 2.1. Карбоалюминирование и восстановительное βалюминаэтилирование 1-алкенов с помощью Et3Al Известные примеры каталитического карбоалюминирования простейших α-олефинов позволяют получать различные по строению АОС. Среди переходных металлов в данной реакции наиболее эффективными являются каталитические системы на основе Ti и Zr. Состав образующихся продуктов зависит не только от применяемого катализатора, но и от условий проведения реакции. С целью расширения ассортимента эффективных катализаторов реакции карбоалюминирования разработки новых непредельных направлений реакций соединений, АОС с а также ненасыщенными углеводородами мы изучили взаимодействие α-олефинов с Et3Al в присутствии TaCl5 в качестве катализатора. Из литературных источников известно, что комплексы тантала используют в качестве катализаторов в реакциях димеризации, содимеризации и олигомеризации 1-алкенов [112-120], метатезиса [121-124] и полимеризации этилена [125]. В то же время данные о каталитическом поведении танталовых комплексов в реакциях непредельных углеводородов с металлоорганическими соединениями практически отсутствуют. В результате проведенных исследований установили, что при взаимодействии 1-октена, взятого в качестве модельного 1-алкена, с Et3Al в соотношении 1:1 в присутствии 5 мол% TaCl5 в гексане при 20ºС за 7ч образуются продукты реакций карбоалюминирования 1б и восстановительного β-алюминаэтилирования 2б с общим выходом 75%, рассчитанным на исходный 1-октен. Необходимо отметить, что наряду с 1б и 2б в минорных количествах (до 10-15%) образуется гидроалюминирования исходного 1-октена – 3б (Схема 1). продукт 40 Схема 1 al [Ta] + Et3Al R гексан, 20оС 7ч R al + + R 1а,б,в al R 3а,б,в 2а,б,в R = Bun (а) O2 D3O+ H3O+ Hexn (б) Octn (в) R R R D 9а,б,в 6а,б,в 4а,б,в + + + OH D OH R 5а,б,в R R 7а,б,в 10а,б,в + + R D R 8а,б,в OH 11а,б,в Структура полученных соединений доказывалась на основании спектров ЯМР 1Н и 13С продуктов дейтеролиза и окисления. При разложении продуктов реакции 1б и 2б 8%-ным раствором HCl образуется единственный продукт – 3-метилнонан, а дейтеролиз реакционной массы приводит к образованию двух монодейтерированных углеводородов 6б и 7б, соотношение которых определено измерением площадей хроматографических пиков, соответствующих региоизомерам 6б и 7б (1:1). В спектре ЯМР 13С региоизомера 6б характерно наличие сигнала δ (С10) 18.85 м.д. с характерной константой спин-спинового взаимодействия (КССВ) 1 JC-D = 19.0 Гц и диагностического сигнала δ (С1) 11.34 м.д. Изотопный эффект атома дейтерия при С10 метильной группы приводит к ее 41 экранированию по сравнению с аналогичной группой соединения 7б. Величина этого эффекта составляет 0.31 м.д. В спектре ЯМР 13 С региоизомерного продукта 7б также наблюдается характерный триплетный сигнал δ (С1) 11.04 м.д. (1JC-D = 19.0 Гц). Аналогично изомеру 6б, изотопный эффект в соединении 7б приводит к экранированию метильной группы (С1), а его величина составляет ~ 0.30 м.д. Следует также отметить присутствие β-изотопных эффектов в соединениях 4б (∆δβ-изотоп С3 = 0.06 м.д.) и 7б (∆δβ-изотоп С2 = 0.07 м.д.). Окислением реакционной массы получены региоизомерные спирты 9б и 10б, у которых гидроксильные группы расположены при разных атомах углерода. В спектре 13 ЯМР С региоизомера наблюдается 10б сигнал карбинольного углеродного атома δ С1 61.07 м.д., локализующийся в более сильном поле по сравнению с сигналом карбинольного атома углерода региоизомера 9б (δ С10 65.18 м.д.). Также существенно отличается положение слабопольной метильной группы δ С10 19.65 м.д. спирта 10б от положения экранированной группы 9б (δ С1 11.08 м.д.). Интегрированием однозначно отличающихся протонов гидроксиметиленовых групп (δ С1Н2 3.51 м.д. – для спирта 9б и δ С1Н2 3.64 м.д. – для спирта 10б) установлено соотношение региоизомеров, составляющее 9б:10б = 1:1. С целью разработки оптимальных условий проведения реакций карбоалюминирования и восстановительного β-алюминаэтилирования олефинов триэтилалюминием в присутствии TaCl5 нами исследовано влияние различных факторов: природа растворителя, соотношение исходных реагентов, концентрация катализатора, продолжительность реакции на выход и состав целевых продуктов реакции 1б и 2б (табл.1). 42 В результате проведенных экспериментов взаимодействие 1-октена с помощью установили, что Et3Al в присутствии TaCl5 удается осуществить в углеводородных растворителях: гексан, бензол, толуол за 7 часов с хорошими выходами целевых продуктов 1б и 2б (соотношение 1б:2б = 1:1). В 1,2- дихлорэтане эта же реакция приводит, в основном, к образованию АОС 1б. Использование эфирных растворителей (Et2O, ТГФ, МТБЭ) не приводит к образованию АОС 1 и 2. Использование в реакции в качестве катализатора 5 мол.% TaCl5 является оптимальным, снижение количества катализатора до 3 мол.% снижает общий выход до 53%. Оптимальным соотношением исходных реагентов является 1:1. Таблица 1. Влияние соотношения исходных реагентов, природы растворителя и концентрации катализатора на выход и состав продуктов карбоалюминирования и восстановительного β-алюминаэтилирования 1октена № Молярное Молярное п/п соотноше- Растворитель Общий Молярное соотношение выход соотношение ние 1-октен: 1б+2б, % 1б/2б Et3Al: TaCl5 Выход 3б 1-октен 1 1: 1 100: 5 гексан 75 1: 1 11 2 1: 1 100: 3 гексан 53 1: 1 10 3 2: 1 100: 5 гексан 85 1: 1 15 4 1: 1 100: 10 гексан 81 1: 1 15 5 1: 1 100: 5 дихлорэтан 74 2: 1 10 6 1: 1 100: 5 бензол 70 1: 1 12 7 1: 1 100: 5 толуол 71 1: 1 13 Условия реакции: 20 ºС, 7ч, CEt3Al = 2 ммоль/мл. С целью повышения региоселективности реакции мы изучили влияние лигандного окружения центрального атома катализатора. Так, экспериментально было установлено, что среди испытанных P- и N- 43 содержащих соединений двухкомпонентная Ta-содержащая система TaCl5 + (OPri)3P в соотношении 1:1 позволяет проводить реакцию с образованием АОС 2б с селективностью 85% (табл. 2). Модифицирование катализатора другими лигандами (Ph3P, [(CH3)2N]3P, Et3N, C5H5N) снижает общий выход продуктов реакции с преимущественным образованием продукта 1б. В случаях использования в каталитической системе бидентатных лигандов, например 1,2-бис(дифенилфосфино)этана или других лигандов, указанных в табл.1, в соотношении Ta:L = 1:2 реакция не катализируется и продукты 1б и 2б не образуются. Таблица 2. Влияние лигандного окружения катализатора на выход и состав продуктов карбоалюминирования и восстановительного β- алюминаэтилирования 1-октена № Лиганд, L п/п Общий Молярное выход соотношение 1б+2б, % 1б/2б Выход 3б 1 Ph3P 70 5: 4 11 2 (OPri)3P 72 1: 6 10 ― ― 59 1: 1 14 66 3: 2 13 65 5: 1 11 3 PPh2 Ph2P CH3 4 P( N CH3 5 Et3N 6 )3 N Условия реакции: 1-октен: Et3Al: [Та] = 10:10:0.5, CEt3Al = 2 ммоль/мл, гексан, 20 ºС, 7ч, Ta:L = 1:1. Заметное влияние на выход и соотношение продуктов 1б и 2б оказывает температура. Проведение реакции при повышенной температуре (60 ºС) снижает выход АОС 2б (общий выход 1б+2б = 78%, 5ч, 1b:2b = 44 70:30). Общий выход продуктов 1б и 2б при проведении реакции при более низкой температуре (~0 ºC) не превышает 20% (1б:2б = 4:5). По аналогии с 1-октеном в реакцию карбоалюминирования с помощью Et3Al были вовлечены 1-гексен и 1-децен. Мы установили, что реакции с их участием проходят с сохранением описанных выше закономерностей. В каждом опыте наряду с целевыми АОС 1а-в и 2а-в образуются продукты гидроалюминирования, общее количество которых колеблется в пределах 10 – 15%. Исходя из экспериментальных результатов, а также имеющихся в литературе данных мы предположили возможные пути образования АОС через танталациклопентановые интермедиаты. Согласно предложенной схемы (Схема 2) взаимодействие TaCl5 с исходным Et3Al дает диэтилтанталовый комплекс 12, в котором за счет реакции β-гидридного переноса происходит элиминирование молекулы этана с получением алкенового комплекса 13. Последний превращается в комплекс 14, содержащий в своей координационной сфере молекулу этилена и исходного 1-алкена. Внутримолекулярная окислительная циклизация в комплексе 14 приводит к β-алкилзамещенному танталоциклопентану 15. Реакция лабильного осуществляется по танталоциклопентана двум практически 15 с избытком равнозначным Et3Al параллельным маршрутам. Первый маршрут предполагает образование биметаллического комплекса 16 в результате присоединения исходного Et3Al к танталациклопентану 15 с последующим раскрытием по связи C1 – Ta, βгидридный перенос в котором (16) приводит к АОС 1. Второй маршрут реализуется поэтапным осуществлением реакции присоединения Et3Al к интермедиату 15 с последующим раскрытием по связи C4 – Ta, приводящей к биметаллическому комплексу 17, который в результате β-гидридного переноса превращается в продукт 2. Как уже отмечалось выше, в ходе эксперимента в реакционной массе были также обнаружены продукты гидроалюминирования исходного 1- 45 алкена. Их частичное образование возможно объясняется тем, что алкильные производные тантала довольно легко переходят в гидриды, которые могут являться промоторами процесса гидрометаллирования. Схема 2 TaCl5 2Et3Al [Ta] = TaCl3 2Et2AlCl [Ta] 12 C2H6 [Ta] 13 R R [Ta] R 14 R [Ta] 15 R R Et3Al AlEt2 [Ta] маршрут 2 маршрут 1 Et2Al [Ta] H H 16 17 R R AlEt2 1 Et2Al 2 46 Таким образом, реакция 1-алкенов с помощью Et3Al в присутствии TaCl5 приводит к образованию смеси двух АОС, а именно диэтил-3-(Rзамещенных)-н-бутилалюминиевых и диэтил-2-(R-замещенных)-н бутилалюминиевых производных. 2.2. Восстановительное β-алюминаэтилирование норборнена и некоторых его производных с помощью Et3Al в присутствии TaCl5 Положительные результаты по катализируемому TaCl5 карбометаллированию и восстановительному β-алюминаэтилированию алкенов с помощью Et3Al стимулировали проведение 1- дальнейших исследований в данном направлении с целью расширения в области приложения открытой нами реакции, а также изучения возможности осуществления реакций карбоалюминирования и восстановительного βалюминаэтилирования циклических олефинов и получению новых типов АОС. В качестве объектов исследования выбрали бицикло[2.2.1]гепт-2-ен, эндо-трицикло[5.2.1.02.6]дека-3,8-диен, экзо- и эндо-5- метилбицикло[2.2.1]гепт-2-ены (экзо: эндо=2:1). Так, бицикло[2.2.1]гепт-2-ен в присутствии каталитических количеств TaCl5 взаимодействует с эквивалентным количеством Et3Al, образуя с выходом ~ 85% единственный продукт восстановительного β- алюминийэтилирования 18 (Схема 3). Экспериментально было установлено, что оптимальные результаты в исследуемой реакции восстановительного β-алюминаэтилирования циклических олефинов достигаются в течение 6 часов при температуре ~ 20ºC, концентрации катализатора 5 мол% и с использованием в качестве растворителя гексана. При разложении продукта реакции 18 8%-ным водным раствором HCl образуется 2-экзо-этилбицикло[2.2.1]гептан 19, а его дейтеролиз приводит к образованию монодейтеросодержащего этилнорборнана 20. Окислением 18 молекулярным кислородом получен соответствующий спирт 21. 47 Схема 3 + Et3Al TaCl5 (5 мол%) AlEt2 гексан, 6 ч 18 20 oC H3O+ O2 D3O+ OH D 20 19 21 Для выяснения влияния структуры норборнена на направление и стереохимию восстановительного β-алюминаэтилирования исследовали реакции Et3Al с эндо-трицикло[5.2.1.02.6]дека-3,8-диеном и смесью экзо-5метилбицикло[2.2.1]гепт-2-ена и эндо-5-метилбицикло[2.2.1]гепт-2-ена (2: 1) в разработанных оптимальных условиях. Эндо-дициклопентадиен реагирует с давая Et3Al, смесь двух региоизомерных АОС 22 и 23 в эквимолярных количествах, различающихся положением алюминазамещенной этильной группы по отношению к двойной связи пятичленного цикла в молекуле эндо-дициклопентена (Схема 4). Общий выход 22 и 23 составил 88%. Схема 4 + Et3Al TaCl5 (5 мол%) AlEt2 + AlEt2 гексан, 20 oC 22 23 D3O+ D + 24 1 : D 1 25 48 Дейтеролизом и 22 трицикло[5.2.1.02.6]дец-3-ен получены 23 экзо-8-(1-дейтероэтил)-эндо- и 24 экзо-9-(1-дейтероэтил)-эндо- трицикло[5.2.1.02.6]дец-3-ен 25 соответственно. Для смеси эндо- и экзо-метилзамещенных норборненов характерно образование 4 изомерных АОС 26, 27, 28, 29, которые относятся друг к другу не только как стереоизомеры, но и характеризуются различным положением алюминийорганического заместителя (Схема 5). Схема 5 Et3Al + 20 oC TaCl5 (5 мол%) 6 ч гексан AlEt2 + 26 27 7 7 1 6 2 4 28 29 7 31 1 6 5 30 AlEt2 7 4 3 5 + AlEt2 D3O+ 1 2 + D 3 + AlEt2 + D 1 2 6 + 4 3 D 5 D 4 3 32 6 2 5 33 (30+31) : (32+33) = 2 :1 Дейтеролиз реакционной массы в данном случае приводит к монодейтерозамещенным 2-метил-6-этил-, а также 2-метил-5- этилнорборнанам 30, 31, 32, 33 с суммарным выходом 86%. Выходы целевых АОС определяли по продуктам их гидролиза и дейтеролиза. Структура полученных АОС доказывалась на основании спектров ЯМР 13 С, 1 Н, данных ИК, хромато-масс спектроскопии их продуктов гидролиза, дейтеролиза и окисления. 49 В спектрах ЯМР 13С производных 20 и 21 наблюдается по 9 сигналов магнитно неэквивалентных углеродных атомов норборнанового каркаса. Это указывает на замещение атома водорода по С-2 метиленовой группе. Дополнительным тому подтверждением также служат кросс-пики кросс за счет дальних спин-спиновых спиновых взаимодействий между сигналом С-2 метиленовой группы с сигналами протонов заместителя (для производного 20 – это кросспики δС 44.37 с δН 1.35 и 0.80-0.88 м.д., для 21 – кросс-пики пики между δС 38.35 и δН 1.38, 1.59 и 3.64 м м.д.). Заместители в молекулах обоих производных находятся в экзо-ориентации ориентации, на что указывают значения химических сдвигов сигналов характеристичных С-2, С-5 и С-7 углеродных атомов (δС 44.37, 28.90 и 35.22 для соединения 20 и δС 38.35, 30.06 и 35.30 – для 21). Рис.1.Спектр ЯМР 13С соединения 20 Наличие в спектре спект ЯМР 13С соединений 24 и 25 двух сигналов δС 11.94 и 12.32 монодейтеро--этильных заместителей в соотношении 1:1указывает на образование только двух (24 и 25) из четырех возможных изомеров. изомеров Сигнал интенсивностью 2С δС 38.37, отнесенный к С-10 мостиковому углеродному атому обоих изомеров изомеров, свидетельствует об экзо-расположении монодейтерированных фрагментов фрагментов. Кросс-пик между сигналом протонов С12 группы заместителя δН 0.82 и сигналом δС 38.64 в спектре ЯМР НМВС изомера 25 указывает на замещение в каркасе по С-99 метиленовой группе. Тогда как для изомера 24 характерно замещение по С-88 углеродному атому, что подтверждается кросс кросс-пиком между δН 0.82 и δС 35.55 в спектре НМВС. Более сложный случай был характерен для бицикло[2.2.1]гептанов гептанов 30–33. Согласно спектрам ЯМР дизамещенных 13 С, в смеси 50 присутствовало четыре изомера, которые относятся друг по отношению к другу не только как стереоизомеры (экзо- и эндо-изомеры), но и характеризуются различным положением монодейтероэтильной группы (при С-5 и С-6) относительно метильного заместителя. Четыре характеристичных сигнала в спектре DEPT135 соответствовали метиновым углеродным атомам, два из которых δС 44.77 и 44.90 отнесены к С-5 и С-6 экзо-изомерам соответственно а, δС 46.42 и 47.64 – С-2 и С-3 к эндо-изомерам соответственно. Мостиковые С-7 углеродные атомы в экзо-изомерах являются случайно изохронными и имеют химический сдвиг δС 36.97 м.д., в то время как для эндо-изомеров сигналы данных углеродных атомов разрешились, и имеют значения δС 39.12 и 40.18 м.д. Таким образом, замещение в эндо- и экзо-изомерах произошло по С-5 и С-6 положениям бициклического каркаса. Строение и состав образующихся АОС, а также имеющиеся в литературе данные дают основание предположить возможный путь осуществления обсуждаемой реакции. На схеме 6 на примере бицикло[2.2.1]гепт-2-ена представлен возможный каталитический цикл по формированию АОС 18, согласно которой на начальном этапе образуется диалкилтанталовый комплекс 12, в котором за счет β-гидридного переноса происходит элиминирование молекулы этана с получением этиленового комплекса 13. Последний в результате координации исходного норборнена превращается в бис-алкеновый комплекс 34, внутримолекулярная окислительная циклизация в котором приводит к танталациклопентановому интермедиату 35, взаимодействие которого с исходным Et3Al приводит к раскрытию 35 с образованием Al - и Ta-содержащего биметаллического комплекса 36. Внутримолекулярный β-гидридный перенос в 36 сопровождается образованием целевого продукта 18. Региоспецифичность реакции возможно определяется стадией переметаллирования танталациклопентана, так как норборнановый фрагмент в 35 препятствует переметаллированию с его стороны. 51 Схема 6 TaCl5 2Et3Al 2Et2AlCl [Ta] = TaCl3 H [Ta] Et 12 C2H6 Et2Al [Ta] 18 13 [Ta] Et2Al [Ta] 34 H 36 Et3Al [Ta] 35 С целью объяснения вышеуказанной закономерности взаимодействия норборнена и его производных на примере реакции норборнена и Et3Al в присутствии TaCl5 была проведена оценка термодинамической вероятности элементарных стадий предложенного каталитического цикла (Схема 7) с применением метода функционала плотности PBE [126] с псевдопотенциалом Стивена-Баша-Краусса [127] (программа ПРИРОДА-06 [128]). Строение всех участников реакций было оптимизировано без 52 ограничений на симметрию. Рассчитанные частоты гармонических колебаний характеризуют оптимизированные структуры как минимумы на поверхности потенциальной энергии. Изменение энергии Гиббса ∆G для реакций рассчитывали как разницу полных энергий продуктов и исходных веществ с учетом энергий нулевых колебаний и термических поправок (Т= 293К, ∆G-в кДж/моль). Схема 7 G, кДж/мол TaCl5 + 2Et3Al (1) Cl3TaEt2 -543.4 + 2Et2AlCl 12 Cl3TaEt2 (2) TaCl3 + C2H6 +13.9 13 (3) 13 -41.0 + TaCl3 35 (4) Et2Al -113.0 Cl3EtTa 35 + 36 Et3Al (5) Cl3EtTa -100.0 Et2Al (6) 36 37 Et2Al H (7) + 13 -10.2 13 + 8.1 18 Et 37 + Et2Al 38 53 Согласно схемы 7, представляющей последовательный набор элементарных стадий каталитического цикла, реакция Et3Al с TaCl5 (реакция (1)) завершается образованием танталоорганического соединения Cl3TaEt2 12, которое затем превращается в π-комплекс этилена с хлоридом тантала 13 (реакция(2)). Реакция (2) эндотермична и происходит, скорее всего, за счет предыдущей стадии, характеризующейся высоким экзотермическим тепловым эффектом. Образование ключевого интермедиата 35 (реакция (3)) происходит также с экзотермическим эффектом. Как уже нами показано, реакция норборнена с Et3Al в присутствии TaCl5 приводит к образованию только одного продукта 18 с экзорасположением алюминаэтильной группы. Для выяснения причин такого поведения норборнена в данной реакции нами рассмотрены два варианта дальнейшего взаимодействия интермедиата 35 с Et3Al: 1) с сохранением химической связи между атомом тантала с норборнильным фрагментом (реакция (4)), приводящим к продукту 36 и 2) без сохранения упомянутой связи Та-С с образованием соединения 37 (реакция (5)). Квантовахимический расчет показал, что эти альтернативные каналы реакции экзотермичны и характеризуются близкими значениями ∆G. В пользу равной вероятности взаимодействия свидетельствует очень танталоциклопентана небольшая разница с Et3Al рассчитанных длин 35 неэквивалентных связей Та-С в 35, которые равны 2.152 и 2.122 Å (Рис.2). Рис. 2. Рассчитанные длины связей танталоциклопентанового кольца интермедиата 35. 54 Более существенная разница термодинамических параметров элементарных стадий может быть обнаружена для дальнейших превращений соединений 36 и 37. Согласно предложенной схеме, на заключительных этапах каталитического цикла происходит регенерация активного πкомплекса 13 с образованием продуктов 18 и 38 соответственно. Согласно расчетам PBE/SBK, эти продукты характеризуются разной устойчивостью, в связи с чем эти реакции имеют значения ∆G разного знака: образование 38 эндотермично (реакция (7)), тогда как образование 18 характеризуется экзотермическим обнаружение тепловым эффектом единственного исследованиях обусловлено продукта его Таким (реакция(6)). в 18 большей образом, экспериментальных термодинамической устойчивостью. 2.3. TaCl5 - катализируемая реакция α-олефинов с помощью высших алюминийорганических соединений В продолжение карбоалюминированию и исследования восстановительному по каталитическому β-алюминаэтилированию терминальных алкенов нами была изучена возможность осуществления данных реакцийс участием высших АОС. Проведенные эксперименты показали, что TaCl5 также проявляет высокую каталитическую активность в реакции н-алкильных производных алюминия ((н-C6H13)3Al, (н-C5H11)3Al) с 1-алкенами. Так при взаимодействии (н-C6H13)3Al с 1-октеном (20°С, толуол, 8ч., (нC6H13)3Al : 1-алкен : TaCl5 = 15 : 10 : 0,5) образуются АОС 39 и 40 в соотношении ~40 : 60 с суммарным выходом ~75% для 1-октена (Схема 8). При переходе от (н-C6H13)3Al к (н-C8H17)3Al все закономерности рассматриваемой реакции сохраняются. Образующиеся при этом АОС можно разделить на две группы в зависимости от состава углеводородного радикала (Схема 9): А – АОС, углеводородный радикал которых образован из молекулы исходного 1-алкена и радикала исходного АОС; Б – АОС, 55 углеводородный радикал которого образован из двух молекул исходного 1алкена. Схема 8 Bun [Ta] R + Hex3Al R R R + 20 oC, 7 ч al al 40 39 R =Hexn H3O+ Bu n D3O+ n Bu R R D R 41 + 43 + R R D 42 R 44 Схема 9 R' [Ta] R 3 Al + R' 20 oC, 8 ч R R R + al al А Б R =Bun, R' = Hexn R = Hexn, R' = Bun D3O+ R' R R R + D D 44 45 Если углеводородный радикал исходного АОС содержит то же число атомов углерода, что и исходный 1-алкен, как, например, при взаимодействии (н-C8H17)3Al с 1-октеном или (н-C6H13)3Al с 1-гексеном, то после дейтеролиза реакционной массы образуется соответственно по единственному дейтероалкану с выходами 70–75% при соотношении АОС: 1алкен : TaCl5 = 150:100 :5 (Схема 10). 56 Схема 10 R [Ta] + Oct3Al R R толуол, 20оС al R=Hexn R H3O+ ( D3O+ ) R X 40 X= H 42 X= D 44 Выход продуктов реакции определяли методом ГЖХ по продуктам гидролиза 41 и 42. Структура полученных АОС доказывалась на основании спектров ЯМР 13 С, 1Н, данных хромато-масс спектрометрии продуктов их гидролиза, дейтеролиза и окисления. 13 В спектре ЯМР С соединения 42 сильнопольные значения химических сдвигов метильных групп δС15 = δС16 = 14.36 м.д. указывают на их эритро-конфигурацию. Для дейтерированного продукта характерна 44 частичная дисимметризация молекулы, которая приводит к изотопному экранированию δС15 = 14.10 м.д. в отличие от аналогичной группы δС15 = 14.40 м.д. для незамещенной метильной группы. Сигнал δС16 = 14.10 м.д. также расщепляется в триплет с константой спин-спинового взаимодействия КССВ 1 JC–D = 19.0 Гц (Рис.3). 15 5 3 1 16 10 7 2 4 12 14 8 6 9 42 11 5 3 1 10 7 2 D 16 15 13 4 12 14 8 9 6 11 13 44 Рис.3. Структуры 7,8-диметилтетрадекана 42 и 1-дейтеро-2-гексил-3метилнонана 44. 2.4. Восстановительное β-цинкэтилирование 1-алкенов с помощью Et2Zn в присутствии TaCl5 Приведенные выше результаты по изучению каталитической активности TaCl5 в реакции карбоалюминирования и восстановительного β- 57 алюминийэтилирования терминальных олефинов стимулировали проведение дальнейших исследований в области синтеза ЦОС. В развитие этих исследований мы попытались использовать TaCl5 для осуществления реакции терминальных олефинов с помощью R2Zn (R= C2H5; C3H7; C4H9; C5H11). Согласно [88], каталитическое карбоцинкирование α-олефинов с помощью Et2Zn проходит региоселективно под действием катализатора Cp2ZrCl2 в сочетании с 20 мол% EtMgBr (см. главу 1.2.3). Экспериментально установлено, что взаимодействие 1-октена и Et2Zn в соотношении 1:1 в присутствии TaCl5 (октен: [Та]= 100:5) в гексане (20ºС, 4ч) приводит к образованию ЦОС 46 с общим выходом 90%, рассчитанным на исходный олефин (Схема 11). Стоит отметить, что аналогичная реакция с Et3Al приводит к образованию двух региоизомеров (см. главу 2.1). Схема 11 R TaCl5 R + Et2Zn гексан, 20 oC, 4ч zn 46a,б,в R = Bun (а) Hexn (б) Octn (в) zn = 1/2 Zn H3 O+ D3O+ R R D 4a,б,в 7a,б,в При разложении продукта реакции 46б 8%-ным водным раствором HCl образуется 3-метилнонан 4б, а дейтеролиз реакционной массы приводит к образованию монодейтерированного углеводорода 7б. По аналогии с 1октеном в реакцию с Et2Zn были вовлечены 1-гексен и 1-децен. Выходы продуктов реакции определяли методом ГЖХ по продуктам гидролиза 46 а,б,в (табл. 3). Структура полученных ЦОС доказывалась на основании спектров ЯМР 1Н, 13 С, данных хромато-масс спектрометрии продуктов их гидролиза и дейтеролиза. 58 Независимо от структуры исходных α-олефинов в каждом опыте образуется исключительно один из региоизомеров, причем наиболее высокие выходы (> 90%) последних удается получить в углеводородных растворителях. В ТГФ и диэтиловом эфире выходы целевых ЦОС не превышают 35%. Таблица 3. Влияние соотношения исходных реагентов, времени реакции, растворителя на выход продукта восстановительного β-цинкэтилирования 1алкенов с помощью Et2Zn в присутствии TaCl5 №№ 1-алкен п/п Молярное Растворитель соотношение Время Выход 46, % реакции, ч Et2Zn:1-алкен 1 1-гексен 1:1 гексан 4 89 2 1-октен 1:1 гексан 4 90 3 1-децен 1:1 гексан 4 87 4 1-октен 1:1,5 гексан 4 92 5 1-октен 1:1 ТГФ 4 35 6 1-октен 1:1 толуол 4 86 7 1-октен 1:1 бензол 4 89 8 1-октен 1:1 диэтиловый 4 15 эфир 9 1-децен 1:1 гексан 8 90 10 1-октен 1:1 гексан 12 92 Возможный каталитический цикл восстановительного β- цинкэтилирования 1-алкенов с помощью Et2Zn в присутствии TaCl5 предполагает первоначальное образование диэтилтанталового комплекса 12, в котором за счет β-гидридного переноса происходит элиминирование молекулы этана с получением этиленового комплекса 13. Координационноненасыщенный олефиновый комплекс 13 взаимодействуя с исходным олефином дает диолефиновый комплекс 14. Внутримолекулярная 59 окислительная циклизация в 14 приводит к танталоциклопентану 15, реакция которого с исходным Et2Zn приводит к образованию Zn- и Ta-содержащего биметаллического комплекса 47, последующая трансформация которого в результате β-гидридного переноса сопровождается образованием 46 (Схема 12). Схема 12 TaCl5 2Et2Zn [Ta] = TaCl3 2EtZnCl 12 [Ta] C2H6 R EtZn R [Ta] 46 13 R EtZn [Ta] [Ta] H R 47 14 R Et2Zn [Ta] 15 С целью объяснения наблюдаемых закономерностей взаимодействия алкенов с диэтилцинком в присутствии солей тантала (V) была предложена следующая схема (Схема 13) и рассчитаны энергии Гиббса ∆G возможных элементарных стадий реакции 1-алкенов с Et2Zn в присутствии TaCl5 (табл. 60 4). Согласно схемы 13, на начальном этапе происходит взаимодействие Et2Zn с TaCl5, завершающееся образованием танталорганического соединения Cl3TaEt2 (реакция (1)), которое затем превращается в π-комплекс этилена с хлоридом тантала 13 (реакция (2)). Реакция (2) эндотермична и происходит, скорее всего, за счет предыдущей стадии, характеризующейся высоким экзотермическим тепловым эффектом. Далее происходит образование ключевого интермедиата реакции рассматриваемой реакции – замещенного танталациклопентана 15 (реакция (3)). Экспериментально показано, что в реакции 1-алкенов с Et2Zn в присутствии хлорида тантала(V) образуется только один продукт 46. Для выяснения причин протекания реакции в одном из направлений нами были изучены два варианта дальнейшего взаимодействия интермедиата 15 с Et2Zn – 1) с сохранением химической связи между атомом тантала и алкильным фрагментом R (реакция (4)), приводящим к продукту 47, и 2) без сохранения упомянутой связи Ta–C с образованием соединения 48 (реакция (5)). Квантово-химический расчет показал, что эти альтернативные каналы реакции экзотермичны и характеризуются близкими значениями ∆G (различия – в пределах 6–10 кДж/моль). В пользу равной вероятности взаимодействия танталациклопентана 15 с цинкорганическим соединением свидетельствует очень небольшая разница рассчитанных длин неэквивалентных связей Ta–C, которые равны 2.150 и 2.130 Å (Рис. 4). Рисунок 4. Рассчитанные длины связей танталациклопентанового кольца интермедиата 15 (R = nBu; Å). 61 Более существенной разницей термодинамических параметров характеризуются дальнейшие превращения соединений 47 и 48. Согласно предложенной схеме, на заключительных этапах каталитического цикла происходит регенерация активного π-комплекса 13 с образованием конечных продуктов 46 и 49 соответственно. Согласно расчетам PBE/SBK, эти продукты характеризуются разной устойчивостью. Так, в случае R = nBu для обеих реакций ∆G < 0, и разница в изменении энергии Гиббса составляет ~18.0 кДж/моль в пользу продукта 46. При увеличении длины алкильного фрагмента R эти реакции приобретают значения ∆G разного знака: образование 49 эндотермично (реакция (7)), тогда как образование 46 характеризуется экзотермическим тепловым эффектом (реакция (6)). Таким образом, обнаружение экспериментальных единственного исследованиях продукта реакции обусловлено его (46) в большей термодинамической устойчивостью. Таблица 4. Рассчитанные изменения энергии Гиббса ключевых стадий взаимодействия алкенов с диэтилцинком в присутствии хлорида тантала(V), кДж/моль Реакция (3) (4) (5) (6) (7) ∆G (кДж/мол) n Bu -38.2 -88.6 -82.0 -18.0 -0.2 n Hex -37.7 -90.0 -82.2 -16.2 +0.6 n Oct -39.9 -92.0 -83.0 -17.6 +2.9 62 Схема 13 (1) TaCl5 + Cl3TaEt2 Et2Zn Cl3TaEt2 (2) + ZnCl2 + C2H6 TaCl3 13 R (3) 13 + R TaCl3 15 R (4) EtZn Cl3EtTa 47 15 + Et2Zn R (5) ZnEt TaEtCl3 48 R 47 (6) EtZn + 13 + 13 46 R (7) 48 ZnEt 49 Одним из отличительных свойств ЦОС является их взаимодействие с хлорангидридами одноосновных карбоновых кислот с образованием кетонов. С целью получения биологически активных кетонов мы попытались вовлечь образующийся в результате реакции карбоцинкирования олефинов с помощью Et2Zn продукт 46 в реакцию с хлорангидридом карбоновой кислоты. Так, взаимодействие бут-1-ена и Et2Zn в присутствии TaCl5 (Et2Zn: бут-1-ен: TaCl5 = 110:100:5, концентрация Et2Zn 1,0 ммол/мл в гексане, 20ºC, 5 ч) приводит к образованию ЦОС 50 с выходом 92%. К последнему при -5÷0 63 ºC медленно прикапывали хлорангидрид пропионовой кислоты (1:1) и получили рацемический 6-метилоктан-3-он 51– компонент феромона тревоги муравьев рода Crematogaster с выходом 95%, считая на взятый хлорангидрид (Схема 14). Схема 14 + Et2Zn 1. C2H5COCl TaCl5 гексан, 5ч 2. H3O+ EtZn O 51 50 В результате проведенных исследований нами установлено, что реакция Et2Zn с 1-алкенами в присутствии TaCl5 приводит к образованию изоалкильных производных цинка с высокой региоселективностью и высокими выходами. Проведенная с помощью DFT расчетов оценка термодинамической вероятности элементарных стадий предложенного механизма взаимодействия Et2Zn с 1-алкенами хорошо согласуется с экспериментальными данными. Показана возможность эффективного применения разработанной реакции региоселективного восстановительного β-цинкэтилирования 1алкенов в однореакторном синтезе 6-метилоктан-3-она – аналога феромона тревоги муравьев рода Crematogaster. 2.5. Реакция 1-алкенов с R2Zn в присутствии TaCl5 Положительные результаты по восстановительному β- цинкэтилированию α-олефинов с помощью Et2Zn побудили нас испытать реакционную способность других ди-н-алкильных производных цинка в вышеуказанной реакции. Мы исследовали взаимодействие Prn2Zn, Bun2Zn, Pentn2Zn и Hexn2Zn с 1-алкенами в присутствии TaCl5. Так, при взаимодействии Bun2Zn, выбранного нами в качестве модельного ЦОС, с 1-гексеном или 1-октеном (20ºС, 7ч, Bun2Zn:1-алкен: TaCl5= 100:100:5) образуются ЦОС 52а,б и 53а,б в соотношении ~ 40:60 с суммарным выходом 84% для 1-гексена и 85% для 1-октена (Схема 15). 64 Выходы продуктов определяли методом ГЖХ по продуктам гидролиза 54а,б и 42a,б. Схема 15 + Bun2Zn R R TaCl5 гексан, 20 oC,7ч R R + Bun Zn ZnBu 52a,б R = Bun (42a,44a,52a-55a) Hexn (42б,44б,52б-55б) 53a,б H3O+ D3O+ R R 54a,б 55a,б D + R n + R R 42a,б R 44a,б D Структура полученных ЦОС доказывалась на основании спектров ЯМР 1 H, 13C, данных хромато-масс спектрометрии, элементного анализа продуктов их гидролиза и дейтеролиза. На примере взаимодействия 1-октена с Bun2Zn исследовано влияние соотношения исходных реагентов, температуры, продолжительности процесса, концентрации катализатора и природы растворителя на выход и соотношение продуктов 52б и 53б. Таблица 5. № п/п 1 2 3 4 5 6 7 8 9 Молярное соотношение Растворитель 1-октен: Bun2Zn 1:1 2:1 3:1 1:1.5 1:3 1:1 1:1 1:1 1:1 гексан гексан гексан гексан гексан ТГФ Et2O 1,4-диоксан толуол Общий выход 52б+53б, % 85 49 30 90 93 40 30 45 84 Селективность продуктов, % 52б 53б 40 20 8 45 50 43 90 80 42 60 80 92 55 50 67 10 20 58 65 Условия реакции: TaCl5: Bun2Zn= 5:100, 20ºC, 7ч, [Bun2Zn] = 1.0 ммол/мл. Проведение реакции 1-октена с Bun2Zn (1-октен: Bun2Zn:[Ta]=100:100:5, гексан, 5ч) при повышенных температурах (65-70ºC) приводит к преимущественному образованию ЦОС 53б (54б:55б=10:90, суммарный выход 52б+53б= 89%). При пониженных температурах (1-октен: Bun2Zn: [Ta]=100:100:5, 5ч, 0ºC) выход образующихся ЦОС составляет не более 20%. Однако в этом случае в реакционной смеси преобладает ЦОС 52б (52б:53б=60:40). При проведении данной реакции в эфирных растворителях суммарный выход целевых ЦОС составляет не более 45%. При переходе к другим членам гомологического ряда н-алкилов (например, Prn2Zn, Pentn2Zn и Hexn2Zn) все закономерности рассматриваемой реакции сохраняются. В результате реакции, также как и в случае с Bun2Zn образуются 2 типа ЦОС (Схема 16). Суммарный выход целевых продуктов (56+53) при этом остается также на уровне 81-85%. Схема 16 + R 20 oC 7ч R' R' 2 Zn [Ta] гексан R R R + Zn R' Zn 56a,б,в R' 53б D3O+ R' R R R + D D 45a,б,в 44б R = Hexn, R' = CH3 (56a,45a) R = Hexn, R' = Prn (56б,45б) R = Hexn, R' = Bun (56в, 45в) Активность различных 1-алкенов в данной реакции практически одинакова. 66 На основании строения и состава синтезированных ЦОС, а также литературных данных по синтезу и превращениям комплексов тантала можно предположить каталитический цикл обсуждаемой реакции через танталоциклопентановые интермедиаты. На схеме 17 представлены возможные пути формирования ЦОС, полученных реакцией н-алкильных производных цинка с 1-алкенами в присутствии TaCl5. Согласно схемы, взаимодействие TaCl5 с исходным ЦОС дает диалкилтанталовый комплекс 57, который за счет реакции β-гидридного переноса элиминирует молекулу алкана, соответствующего радикалу исходного ЦОС, с образованием алкенового комплекса 58. Комплекс 58 трансформируется в бис-алкеновый комплекс 59 в результате внедрения исходного 1-алкена в координационную сферу тантала. Удаление элиминированного алкена из координационной сферы металла приводит к координационно-ненасыщенному комплексу 60. Дополнительное внедрение молекулы исходного 1-алкена в комплекс 60 завершается образованием комплекса 61 с двумя молекулами исходного олефина. Внутримолекулярная окислительная циклизация в комплексах 59 и 61 приводит соответственно к β,β'-диалкилзамещенным танталоциклопентанам 62и 63. Последние в результате взаимодействия их с исходным ЦОС превращаются в соответствующие Zn- и Ta-содержащие биметаллические комплексы 64 и 65, внутримолекулярный β-гидридный перенос в которых приводит к образованию конечных ЦОС 56 и 53. Таким образом, нами было установлено, что катализируемая TaCl5 реакция 1-алкенов с н-диалкильными производными цинка приводит к образованию изоалкилцинкорганических соединений двух типов. Предложены предполагаемые каталитические циклы реакций, которые предусматривают образование ключевых интермедиатов. замещенных танталоциклопентанов как 67 Схема 17 TaCl5 2 R' Zn 2 ZnCl 2 R' R' [Ta] = TaCl3 [Ta] 57 R' R' 58 [Ta] R' R R R R' R R [Ta] [Ta] [Ta] R' R R' 59 R 60 61 R R' путь 2 путь 1 62 R' R' [Ta] 63 Zn 2 R R Zn R' R Zn [Ta] [Ta] 64 65 H H R' R' R' R R [Ta] R' R R R R' Zn 56 H H R' R Zn 53 68 ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ Продукты гидролиза и дейтеролиза АОС и ЦОС анализировали на хроматографе SHIMADZU GC– 2014 в токе He, колонка 2000*3 мм, 5% SE30 на Chromaton N-AW-HMDS (0.125-0.160 мм), рабочая температура 50 – 300ºС. Одномерные (1H, 13 С) и двумерные (COSY, HSQC, HMBC) спектры ЯМР записаны в CDCl3 на спектрометре “Bruker Avance-400” (100 МГц для 13 С и 400 МГц для 1H) при 298 К, химические сдвиги даны относительно TMS. Масс-спектры соединений регистрировали на приборе SHIMADZU GCMS– QP 2010 (SUPELCO SLB™- 5 ms, стеклянная капиллярная колонка 60000*0.25мм*0.25µм, газ-носитель – гелий, программирование температуры от 50 до 260ºС со скоростью 5 град/мин, температура источника ионов 260 ºС, 70эВ). Элементный анализ образцов определяли на элементном анализаторе фирмы Karlo Erba, модель №1106. Расчет хроматограмм реакционных смесей производили с использованием внутреннего стандарта (декан, додекан, гексадекан). ИК спектры зарегистрировали на спектрометре VERTEX 70V фирмы “Bruker”. Выходы и соотношение продуктов определяли с помощью ГЖХ-анализа. Очистка исходных реагентов и растворителей В работе использованы мономеры чистотой не менее 99%. Эфирные растворители выдерживали над KOH, затем кипятили с натриевой стружкой и перегоняли в токе аргона над LiAlH4. Ароматические растворители перед применением промывали концентрированной H2SO4 для удаления тиофена, водой до нейтральной реакции, кипятили с натриевыми стружками и перегоняли в токе аргона. Гексан перегоняли над натриевой стружкой. Галогенсодержащие соединения перегоняли над P2O5. Исходные олефины обезвоживались добавлением триизобутилалюминия (0,5 % от массы углеводородов) с последующей перегонкой в токе аргона. Для создания инертной атмосферы использовали аргон марки “чистый” (ГОСТ 10157-73), дополнительно очищенный от следов влаги и кислорода путем пропускания 69 через систему, состоящую из последовательно соединенных колонн, содержащих силикагель, молекулярные сита (4Å) и 30% раствор триизобутилалюминия в декалине. Использовали коммерчески доступные 1алкены (гекс-1-ен, окт-1-ен, дец-1-ен), норборнен, дициклопентадиен (чистота 99,5%), экзо-, эндо-метилнорборнен (экзо/эндо= 2:1), Et3Al (98%) (ОАО «Редкинский опытный завод»), TaCl5 (99,8%, Aldrich). Получение исходных Al- и Zn- органических соединений Высшие алюминийорганические соединения синтезировали согласно методике [129]. Цинкорганические реагенты (Et2Zn, Prn2Zn, Bun2Zn, Amn2Zn) синтезировали по методике Ноллера [130]. Общая методика карбоалюминирования и восстановительного βалюминийэтилирования 1-алкенов с помощью Et3Al в присутствии TaCl5 В стеклянный реактор, установленный на магнитной мешалке, в атмосфере сухого аргона при 0ºC загружали 2.5 ммоль TaCl5, 50 ммол 1алкена и 50 ммоль Et3Al (2М раствор в гексане) и 20 мл сухого гексана. Температуру повышали до комнатной (~20ºC) и перемешивали 7 часов. Для идентификации полученных АОС реакционную смесь обрабатывали 8%-ной HCl в H2O или DCl в D2O. Органический слой отделяли, промывали NAHCO3 до нейтральной реакции и сушили MgSO4. Смесь продуктов выделяли фракционной перегонкой. 1-дейтеро-2-этилоктан (6б) D Т.кип. 75 oC (30 mm Hg). ИК-спектр, см–1: 2175 (C– D). Спектр ЯМР 1H (δ, м.д.): 1.12–1.45 (м, 13Н, CH2, СН), 0.86–0.97 (м, 8Н, 70 CH3, СН2D). Спектр ЯМР 13C (δ, м.д.): 36.8 (C4), 34.5 (C3), 32.1 (C7), 29.8 (C6), 29.6 (C2), 27.2 (C5), 22.8 (C8), 18.9 (C10, т, 1JC–D = 19 Hz), 14.1 (C9), 11.3 (C1). [M]+143. Найдено (%): C, 84.20; H+D, 15.49. Вычислено (%): C, 83.92; H, 14.68; D, 1.40. 1-дейтеро-3-метилгептан (7а) D Т.кип. 56 oC (80 mm Hg). ИК-спектр, см–1: 2180 (C–D). Спектр ЯМР 1H (δ, м.д.): 1.45 (м, 1Н, СН), 1.29 (м, 2Н, СН2), 1.20 (м, 2Н, СН2), 1.16 (м, 2Н, CН2), 1.12 (м, 2H, СН2), 0.87 (т, 3H, CH3, 3J = 7.2 Hz), 0.86 (м, 2Н, СН2D), 0.83 (ч, 3H, CH3, 3J = 6.4 Hz). Спектр ЯМР 13C (δ, м.д.): 36.6 (C4), 34.7 (C3), 29.4 (C2), 28.6 (C5), 22.8 (C6), 18.6 (C8), 14.1 (C9), 10.8 (C1, t, 1JC–D = 19.0 Hz). [M]+115. Найдено (%): C, 83.41; H+D, 16.49. Вычислено (%): C, 83.48; H, 14.78; D, 1.74. 1-дейтеро-3-метилнонан (7б) D Т.кип. 75 oC (30 mm Hg). ИК-спектр, см–1 : 2175 (C–D). Спектр ЯМР 1H (δ, м.д.): 1.12–1.45 (м, 13Н, CH2, СН), 0.86–0.97 (м, 8Н, CH3, СН2D). Спектр ЯМР 13C (δ, м.д.): 36.8 (C4), 34.5 (C3), 32.1 (C7), 29.8 (C6), 29.5 (C2), 27.2 (C5), 22.8 (C8), 19.2 (C10), 14.1 (C9), 11.0 (C1, t, 1JC–D = 19.0 Hz). [M]+143. Найдено (%): C, 83.79; H+D, 15.97. Вычислено (%): C, 83.92; H, 14.68; D, 1.40. 1-дейтеро-3-метилундекан (7в) D Т.кип. 110 oC (30 mm Hg). ИК-спектр, см–1 : 2175 (C–D). Спектр ЯМР 1H (δ, м.д.): 1.43 (м, 1Н, СН), 1.35 (м, 16Н, СН2), 0.93 (м, 8H, СН3, CH2D). Спектр ЯМР 13 C (δ, м.д.): 34.4 (C2), 34.4 (C4), 32.0 (C5), 30.2 (C6), 29.8 (C7), 29.5 (C3), 29.5 (C8), 27.2 (C9), 22.7 (C10), 19.1 (C12), 18,8 (C1, т, 1JC–D = 19.0 Hz), 14.0 (C11). [M]+171. Найдено (%): C, 84.18; H+D, 15.71. Вычислено (%): C, 84.21; H, 14.62; D, 1.17. 71 Общая методика окисления алюминийорганических соединений Окисление алюминийорганических соединений проводили в стеклянных термостатируемых реакторах пропусканием пузырьков сухого чистого кислорода через раствор, температура которого поддерживалась на уровне 5-8 ºC. По истечении 120 минут реакционную смесь выливали в 5%ный раствор HCl в H2O, и образовавшийся спирт экстрагировали тремя MgSO4 и после удаления эфира порциями эфира (3*50 мл), сушили анализировали методом ГЖХ. Выделение спиртов осуществляли методом вакуумной перегонки. 2-этилгексан-1-ол (9а) HO Т.кип. 101 oC (30 mm Hg). ИК-спектр, см–1: 1375, 1465, 1045 (C–O), 3430 (O–H). Спектр ЯМР 1H (δ, м.д.): 3.52 (ч, 3JH–H = 4.8 Hz, 2Н, СН2O), 1.39 (м, 2Н, СН2), 1.37 (м, 1H, СН), 1.32 (м, 2H, CH2), 1.28 (м, 4H, CH2), 0.89 (м, 6H, CH3). Спектр ЯМР 13 C (δ, м.д.): 65.1 (C1), 41.9 (C2), 30.1 (C3), 29.2 (C4), 23.3 (C7), 23.1 (C5), 14.1 (C6), 11.1 (C8). [M]+130. Вычислено (%): C, 73.85; H, 13.84; O, 12.31. Найдено (%): C, 73.63; H, 13.75. 2-этилоктан-1-ол (9б) HO Т.кип. 76–77 ºC (3 mm Hg). ИК-спектр, см–1: 3420 (O–H), 1045 (C–O), 1465, 1375. Спектр ЯМР 1H (δ, м.д.): 3.51 (д, 3JH–H = 5 Hz, 2Н, СН2O), 1.12–1.41 (м, 13H, CH2, CH), 0.87–0.89 (м, 6H, CH3). Спектр ЯМР 13 C (δ, м.д.): 65.2 (C1), 31.9 (C6), 30.5 (C2), 29.8 (C3), 29.7 (C5), 30.0 (C4), 23.4 (C9), 22.7 (C7), 14.1 (C8), 11.1 (C10). [M]+158. Найдено (%): C, 75.91; H, 13.69. Вычислено (%): C, 75.95; H, 13.92; O, 10.13. 72 2-этилдекан-1-ол (9в) HO Т.кип. 151 ºC (30 mm Hg). ИК-спектр, см–1: 3425 (O–H), 1045 (C–O), 1465, 1375. Спектр ЯМР 1H (δ, м.д.): 3.35 (м, 2Н, СН2O), 1.76 (м, 3H, CH3), 1.45 (м, 1H, CH), 1.17 (м, 16H, CH2), 0.76 (м, 3H, CH3). Спектр ЯМР 13C (δ, м.д.): 64.3 (C1), 41.8 (C2), 37.2 (C3), 31.9 (C4), 30.4 (C5), 29.6 (C6), 29.3 (C7), 26.9 (C8), 23.2 (C11), 22.3 (C9), 13.8 (C10), 10.8 (C12). [M]+186. Найдено (%): C, 77.39; H, 13.89. Вычислено (%): C, 77.42; H, 13.98; O, 8.60. 3-метилгептан-1-ол (10а) HO Т.кип. 97 oC (30 mm Hg). ИК-спектр, см–1 : 1375, 1465, 1045 (C–O), 3425 (O–H). Спектр ЯМР 1H (δ, м.д.): 3.65 (м, 2Н, СН2O), 1.59 (м, 2H, CH2), 1.52 (м, 1H, CH), 1.28 (м, 6H, CH2), 0.89 (м, 6H, CH3). Спектр ЯМР 13 C (δ, м.д.): 61.1 (C1), 39.9 (C2), 36.8 (C4), 29.5 (C3), 29.1 (C5), 22.9 (C6), 19.6 (C8), 14.0 (C7). [M]+130. Найдено (%): C, 73.71; H, 13.80. Вычислено (%): C, 73.85; H, 13.84; O, 12.31. 3-метилнонан-1-ол (10б) HO Т.кип. 76–77 ºC (3 mm Hg). ИК-спектр, см–1: 3420 (O–H), 1045 (C–O), 1465, 1375 . Спектр ЯМР 1H (δ, м.д.): 3.64 (м, 2Н, СН2O), 1.59 и 1.35 (м, 2H, H2C2), 1.57 (м, 1H, HC3), 1.18–1.31 (м, 10H, CH2), 0.87–0.89 (м, 6H, CH3). Спектр ЯМР 13C (δ, м.д.): 61.1 (C1), 40.0 (C2), 37.2 (C4), 31.9 (C7), 29.8 (C6), 29.6 (C3), 26.9 (C5), 22.7 (C8), 19.7 (C10), 14.1 (C9). [M]+158. Найдено (%): C, 75.91; H, 13.69. Вычислено (%): C, 75.95; H, 13.92; O, 10.13. 73 3-метилундекан-1-ол (10в) HO Т.кип. 156 ºC (30 mm Hg). ИК-спектр,см–1 : 3425 (O–H), 1045 (C–O), 1465, 1375. Спектр ЯМР 1H (δ, м.д.): 3.47 (м, 2Н, СН2O), 1.45 (м, 1H, CH), 1.17 (м, 16H, CH2), 0.76 (м, 6H, CH3). Спектр ЯМР 13 C (δ, м.д.): 60.2 (C1), 39.7 (C2), 31.9 (C4), 30.1 (C5), 30.0 (C6), 29.6 (C7), 29.4 (C3), 29.3 (C8), 26.9 (C9), 22.3 (C10), 19.4 (C12), 13.8 (C11). [M]+186. Вычислено (%): C, 77.42; H, 13.98; O, 8.60. Найдено (%): C, 77.43; H, 13.85. 2-экзо-(2-гидроксиэтил)бицикло[2.2.1]гептан (21) OH Т.кип.= 135 ºС (30 mm Hg). ИК-спектр, см−1: 3435 (О−Н), 1045 (С−О). Спектр ЯМР 1Н (δ, м.д.): 3.64 (т, 3J = 7.2 Hz, 2H, CH2O), 2.21 (м, 1Н, СН), 1.98 (м, 1Н, СН), 1.59 (м, 1Н, СН), 1.50 (м, 1Н, СН), 1.49 (м, 1Н, СН), 1.47 (м, 1Н, СН), 1.45 (м, 1Н, СН), 1.38 (м, 1Н, СН), 1.31 (м, 1Н, СН), 1.15 (м, 1Н, СН), 1.10 (м, 3Н, СН). Спектр ЯМР 13С (δ, м.д.): 61.7 (С9), 41.1 (С1), 40.0 (С8), 38.4 (С2), 38.1 (С3), 36.6 (С4), 35.3 (С7), 30.1 (С5), 28.8 (С6). [M+−H2O]122. Найдено (%): С, 77.21; Н, 11.39. Вычислено (%): С, 77.14; Н, 11.43; O, 11.43. Общая методика восстановительного β-алюминаэтилирования норборнена и его производных с помощью Et3Al в присутствии TaCl5 В стеклянный реактор в атмосфере сухого аргона при 0°С и перемешивании последовательно загружали 20 мл гексана, 30 ммоль норборнена или его производного, 30 ммоль Et3Al и 1,5 ммоль TaCl5. Температуру реакционной массы повышали до 20°С и перемешивали 6 часов. Реакционную массу обрабатывали 8%-ной DCl в D2O. Продукт реакции экстрагировали эфиром (3*50 мл), сушили вакуумной перегонкой. MgSO4. Продукты выделяли 74 2-экзо-(2-дейтероэтил)бицикло[2.2.1]гептан (20) D Т.кип. = 64 ºС (30 mm Hg.). ИК-спектр, см−1: 2180 (C−D). Спектр ЯМР 1Н (δ, м.д.): 2.19 (м, 1Н, СН), 1.98 (м, 1Н, СН), 1.48 (м, 1Н, СН), 1.43 (м, 1Н, СН), 1.41 (м, 1Н, СН), 1.33 (м, 2Н, СН2), 1.31−1.33 (м, 1Н, СН), 1.28 (м, 1Н, СН), 1.24 (м, 1Н, СН), 1.16 (м, 1Н, СН), 1.10 (м, 1Н, СН), 1.04 (м, 1Н, СН), 0.83−0.88 (м, 2Н, CH2D). Спектр ЯМР 13 С (δ, м.д.): 44.4 (С2), 40.8 (С1), 38.0 (С3), 36.5 (С4), 35.2 (С7), 30.2 (С6), 29.5 (С8), 28.9 (С5), 12.1 (С9, 1JC−D = 19 Hz). [M+]125. Найдено (%): С, 86.35; Н+D, 13.61. Вычислено (%): С, 86.4; Н, 12.00; D, 1.60. 8-экзо-(1-дейтероэтил)-эндо-трицикло[5.2.1.02.6]дец-3-ен (24) D Т.кип. = 118 ºС (30 mm Hg). ИК-спектр, см−1 : 1370, 1480, 1620, 2190 (C-D), 2860, 2960, 3060. Спектр ЯМР 1Н (δ, м.д.): 5.66 (м, 1Н, СН), 5.54 (м, 1Н, СН), 2.52 (м, 1Н, СН), 2.21 (м, 2Н, СН2), 2.11 (м, 1Н, СН), 1.88 (м, 1Н, СН), 1.54 (м, 1Н, СН), 1.49 (м, 1Н, СН), 1.48 (м, 1Н, СН), 1.29 (м, 2Н, 2СН), 1.23 (м, 2Н, СН2), 0.82 (т, 2Н, СН2D), 0.77 (м, 1Н, CH). Спектр ЯМР 13С (δ, м.д.): 132.6 (С4), 130.8 (С3), 53.7 (С2), 45.7 (С6), 42.5 (С7), 41.1 (С1), 38.4 (С10), 35.6 (С8), 33.6 (С9), 32.0 (С5), 29.6 (С11), 12.3 (С12, т, 1JC−D = 19 Hz). [M+]163. Найдено (%): С, 88.41; Н+ D, 11.60. Вычислено (%): С, 88.34; Н, 10.43; D, 1.23. 9-экзо(1-дейтероэтил)-эндо-трицикло[5.2.1.02.6]дец-3-ен (25) D Т.кип. = 118 ºС (30 mm Hg). ИК-спектр, см−1: 1370, 1480, 1620, 2190 (C-D), 2860, 2960, 3060. Спектр ЯМР 1Н (δ, м.д.): 5.66 (м, 1Н, СН), 5.54 (м, 1Н, СН), 2.52 (м, 1Н, СН), 2.27 (м, 1Н, СН), 2.21 (м, 1Н, СН), 2.07 (м, 75 1Н, СН), 1.54 (м, 1Н, СН), 1.52 (м, 1Н, СН), 1.48 (м, 1Н, СН), 1.46 (м, 1Н, СН), 1.29 (м, 1Н, СН), 1.12 (т, 2Н, СН2), 0.83 (м, 1Н, CH), 0.82 (т, 2Н, СН2D). Спектр ЯМР 13С (δ, м.д.): 132.6 (С4), 130.4 (С3), 52.4 (С2), 44.1 (С6), 41.8 (С7), 40.0 (С1), 38.6 (С9), 38.4 (С10), 32.4 (С5), 30.3 (С8), 29.2 (С11), 11.9 (С12, т, 1JC−D = 19 Hz). [M+]163. Найдено (%): С, 88.41; Н+ D, 11.60. Вычислено (%): С, 88.34; Н, 10.43; D, 1.23. 2-экзо-метил-6-экзо-(1-дейтероэтил)бицикло[2.2.1]гептан (30) D Т.кип. = 78 ºС (30 mm Hg). ИК-спектр, см−1: 2180 (C−D). Спектр ЯМР 1Н (δ, м.д.): 2.15 (м, 1Н, СН), 1.90 (м, 1Н, СН), 1.64 (м, 1Н, СН), 1.46 (м, 1Н, СН), 1.41 (м, 1Н, СН), 1.25 (м, 1Н, СН), 1.22−1.25 (м, 1Н, СН), 1.20 (м, 2Н, СН2), 1.15 (м, 1Н, CH), 0.97 (д, 3Н, СН3, 3J = 7.2 Hz), 0.95 (м, 1Н, СН), 0.93 (м, 1Н, СН), 0.86 (д, 2Н, CH2D, 3J = 7.2 Hz). Спектр ЯМР 13 С (δ, м.д.): 44.8 (С6), 38.9 (С3), 37.0 (С7), 36.9 (С1), 35.3 (С2), 31.7 (С5), 22.3 (С9), 16.7 (С8), 12.3 (С4), 12.1 (С10, т, 1JC−D = 19 Hz). [M+]139. Найдено (%): С, 86.21; Н+D, 13.61. Вычислено (%): С, 86.33; Н, 12.23; D, 1.44. 2-экзо-метил-5-экзо-(1-дейтероэтил)бицикло[2.2.1]гептан (31) D Т.кип. = 78 ºС (30 mm Hg). ИК-спектр, см−1: 2180 (C−D). Спектр ЯМР 1Н (δ, м.д.): 2.15 (м, 1Н, СН), 1.81 (м, 1Н, СН), 1.48 (м, 1Н, СН), 1.46 (м, 1Н, СН), 1.37 (м, 1Н, СН), 1.29 (м, 2Н, 2СН), 1.27 (м, 1Н, СН), 1.20 (м, 1Н, СН), 1.13 (м, 1Н, СН), 1.11 (м, 1Н, СН), 1.02 (м, 1Н, CH), 0.95 (м, 1Н, СН), 0.93 (д, 3Н, СН3, 3J = 6.8 Hz), 0.86 (д, 2Н, CH2D). Спектр ЯМР 13 С (δ, м.д.): 44.9 (С5), 43.3 (С4), 38.7 (С3), 37.0 (С1), 37.0 (С7), 35.7 (С2), 31.7 (С6), 29.4 (С9), 17.4 (С8), 12.1 (С10). [M+]139. Найдено (%): С, 86.21; Н+D, 13.61. Вычислено (%): С, 86.33; Н, 12.23; D, 1.44. 76 1-эндо-метил-6-экзо-(1-дейтероэтил)бицикло[2.2.1]гептан (32) D Т.кип. = 78 ºС (30 mm Hg). ИК-спектр, см−1 : 2180 (C−D). Спектр ЯМР 1Н (δ, м.д.): 2.14 (м, 1Н, СН), 1.94 (м, 1Н, СН), 1.90 (м, 1Н, СН), 1.80 (м, 2Н, 2СН), 1.78 (м, 1Н, СН), 1.65 (м, 1Н, СН), 1.48 (м, 1Н, СН), 1.41 (м, 1Н, СН), 0.90 (д, 3Н, СН3, 3J = 7.2 Hz), 0.86 (д, 2Н, СН2D), 0.84 (м, 1Н, CH), 0.57 (м, 1Н, СН), 0.55 (м, 1Н, СН). Спектр ЯМР 13 С (δ, м.д.): 47.6 (С5), 41.9 (С4), 39.1 (С7), 37.9 (С1), 37.6 (С3), 34.1 (С2), 30.7 (С6), 29.6 (С9), 22.2 (С8), 12.1 (С10). [M+]139. Найдено (%): С, 86.21; Н+D, 13.61. Вычислено (%): С, 86.33; Н, 12.23; D, 1.44. 1-эндо-метил-5-экзо-(1-дейтероэтил)бицикло[2.2.1]гептан (33) D Т.кип. = 78 ºС (30 mm Hg). ИК-спектр, см−1: 2180 (C−D). Спектр ЯМР 1Н (δ, м.д.): 1.90 (м, 1Н, СН), 1.80 (м, 1Н, СН), 1.46 (м, 1Н, СН), 1.41 (м, 1Н, СН), 1.40 (м, 1Н, СН), 1.25 (м, 1Н, СН), 1.15 (м, 1Н, СН), 0.97 (м, 3Н, СН2, СН), 0.90 (д, 3Н, СН3, 3J = 7.2 Hz), 0.86 (м, 2Н, СН2D), 0.84 (м, 1Н, CH). Спектр ЯМР 13 С (δ, м.д.): 46.4 (С6), 41.8 (С4), 40.2 (С7), 38.2 (С3), 37.9 (С1), 33.3 (С2), 30.7 (С5), 29.3 (С9), 22.0 (С8), 12.1 (С10). [M+]139. Вычислено (%): С, 86.33; Н, 12.23; D, 1.44. Найдено (%): С, 86.21; Н+D, 13.61%. Общая методика реакции 1-алкенов с высшими АОС, катализируемой TaCl5 В стеклянный реактор, установленный на магнитной мешалке, в атмосфере сухого аргона при 0ºC загружали 2.5 ммоль TaCl5, 50 ммол 1гексена или 1-октена и 50 ммоль Hex3Al или Oct3Al (2М раствор в толуоле). Температуру повышали до комнатной (~20ºC) и перемешивали 7 часов. Для идентификации полученных АОС реакционную смесь обрабатывали 8%-ной HCl в H2O или DCl в D2O. Органический слой отделяли, промывали NAHCO3 77 до нейтральной реакции и сушили MgSO4. Смесь продуктов выделяли фракционной перегонкой. 7,8-диметилтетрадекан (42) Т.кип. 158 ºC (30 mm Hg). Спектр ЯМР 1H (δ, м.д.): 1.17-1.44 (м, 22H, CH, CH2), 0.86 (т, 6H, CH3), 0.73 (д, 1J= 6.6 Hz, 6H, CH3). Спектр ЯМР 13 C (δ, м.д.): 36.54, 34.93, 31.96, 29.70, 27.67, 27.70, 14.36, 14.11. [M+] 226. Найдено (%): C, 84.92; H, 15.01%. Вычислено (%): C, 84.96; H, 15.04. 1-дейтеро-2-гексил-3-метилнонан (44) D Т.кип. 159 ºC (30 mm Hg). ИК-спектр, cм-1: 2175 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.11-1.44 (м, 22H, CH, CH2), 0.740.91 (м, 11H, CH3, CH2D). Спектр ЯМР 13C (δ, м.д.): 36.58, 36.50, 34.95, 34.94, 31.98, 29.68, 27.68, 22.71, 14.40, 14.11, 14.10 (т, 1JC-D=19Hz). [M+]227. Найдено (%): C, 84.56; H, 15.39.Вычислено (%): C, 84.60; H+D, 15.40. 1-дейтеро-2-бутил-3-метилнонан (45) D Т.кип. 132 ºC (30 mm Hg). ИК-спектр, cм-1: 2175 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.09-1.46 (м, 18H, CH, CH2), 0.75-0.89 (м, 11H, CH3, CH2D). Спектр ЯМР 13 C (δ, м.д.): 36.81, 36.25, 34.71, 34.12, 31.68, 29.12, 27.86, 27.71, 22.61, 22.12, 14.26, 13.95, 13.68, 13.34 (т, 1JC- 78 D=19Hz). [M+]199. Найдено (%): C, 83.87; H, 16.05. Вычислено (%): C, 83.90; H+D, 16.10. Методика получения 6-метилоктан-3-он – компонента феромона тревоги муравьев рода Crematogaster В стеклянный реактор в атмосфере сухого аргона при -15 °С помещали 20 мл гексана, 20 ммоль охлажденного до –30°С бутена-1, 22 ммоль Et2Zn и 1 ммоль TaCl5. Температуру доводили до комнатной и перемешивали 5ч. При –5°С прикапывали 20 ммоль (1,74мл) хлорангидрида пропионовой кислоты в гексане и перемешивали 2 ч. Реакционную массу обрабатывали 10%-ным водным раствором HCl, продукт реакции экстрагировали диэтиловым эфиром (3x50 мл), сушили безводным MgSO4. После отгонки растворителей остаток хроматографировали (SiO2, гексан). Выход продукта 2,69 г. 6-метилоктан-3-он (51) O ИК-спектр, см−1: 2980, 2860, 1720, 1460, 1375. Спектр ЯМР 1Н (δ, м.д.): 0.84 (д, 3Н, СН3), 0.92 (м, 6Н, 2СН3), 1.00-1.80 (бр, 5Н, СН2СНСН2), 1.93-2.4 (м, 4Н, СН2СОСН2). Спектр ЯМР 13 С (δ, м.д.): 7.78, 11.45, 20.35, 30.24, 30.89, 34.58, 36.95, 39.56, 210.65. Общая методика взаимодействия 1-алкенов с помощью R2Zn В стеклянный реактор в атмосфере сухого аргона при 0°С и перемешивании последовательно загружали 2.5 ммоль TaCl5, 50 ммол 1алкена, 50 ммоль Bu2Zn (Prn2Zn, Amn2Zn, Hexn2Zn) и 50 мл сухого гексана. Температуру повышали до комнатной (~20ºC) и перемешивали 7часов. Для идентификации полученных АОС реакционную смесь обрабатывали 8%ной HCl в H2O или DCl в D2O. Органический слой отделяли, промывали NAHCO3 до нейтральной реакции и сушили MgSO4. Смесь продуктов выделяли фракционной перегонкой. 79 3,4-диметилоктан(54а) Т.кип. 68ºC (30 mm Hg). Спектр ЯМР 1H (δ, м.д.): 0.931.31 (м, 8H, 4CH2), 0.83-0.92 (м, 12H, 4CH3), 0.82-0.88 (м, 2H, 2CH). Спектр ЯМР 13C (δ, м.д.): 38.13, 37.82, 36.75, 35.24, 27.06, 22.81, 17.43, 16.65, 14.12, 12.33. [M+]142. Найдено (%): C, 84.47; H, 15.46. Вычислено (%): C, 84.51; H, 15.49. 3,4-диметилдекан (54б) Т.кип. 105 ºC (30 mm Hg). Спектр ЯМР 1H (δ, м.д.): 1.08-1.44 (м, 14H, CH, CH2), 0.73-0.96 (м, 12H, CH3). Спектр ЯМР 13C (δ, м.д.): δ= 38.55, 36.32, 35.00, 32.00, 29.73, 27.68, 27.52, 22.71, 14.29, 14.03, 13.88, 11.31. [M+] 170. Найдено (%): C, 84.68; H, 15.27. Вычислено (%): C, 84.71; H, 15.29. 1-дейтеро-2-этил-3-метилгептан (55а) D Т.кип. 69 ºC (30 mm Hg). ИК-спектр, cм-1: 2185 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.08-1.44 (м, 10H, CH, CH2), 0.77-0.88 (м, 11H, CH3, CH2D). Спектр ЯМР 13C (δ, м.д.): 37.66, 36.41, 35.25, 34.95, 25.82, 21.71, 13.58, 12.65 (т, 1JC-D=19Hz), 11.10. [M+]143. Найдено (%): C, 83.82; H, 16.05. Вычислено (%): C, 83.90; H+D, 16.10. 80 1-дейтеро-2-этил-3-метилнонан (55б) D Т.кип. 105 ºC (30 mm Hg). ИК-спектр, cм-1: 2175 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.08-1.44 (м, 14H, CH, CH2), 0.88 (т, JH-H= 7 Hz, 3H, CH3), 0.86 (т, JH-H= 7 Hz, 3H, CH3), 0.74 (д, JH-H= 7 Hz, 2H, CH2D), 0.73 (д, JH-H= 7 Hz, 3H, CH3). Спектр ЯМР 13C (δ, м.д.): 38.41, 36.25, 34.95, 31.98, 29.71, 27.66, 27.50, 22.71, 14.12, 13.94, 13.65 (т, 1JC-D=19Hz), 12.21. [M+]171. Найдено (%): C, 84.16; H, 15.77. Вычислено (%): C, 84.20; H+D, 15.80. 1-дейтеро-2,3-диметилнонан (45а) D Т.кип. 104 ºC (30 mm Hg). ИК-спектр, cм-1: 2175 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.08-1.44 (м, 12H, CH, CH2), 0.75-0.87 (м, 11H, CH3, CH2D). Спектр ЯМР 13C (δ, м.д.): 39.20, 33.92, 32.27, 32.08, 29.01, 27.65, 22.89, 21.78, 19.44 (т, 1JC-D=19Hz), 16.61, 14.11. [M+]157. Вычислено (%): C, 84.08; H+D, 15.92. Найдено (%): C, 84.05; H, 15.90. 1-дейтеро-2-пропил-3-метилнонан (45б) D Т.кип. 118 ºC (30 mm Hg). ИК-спектр, cм-1: 2175 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.08-1.44 (м, 16H, CH, CH2), 0.74-0.88 (м, 11H, CH3, CH2D). Спектр ЯМР 13 C (δ, м.д.): 37.95, 35.91, 35.51, 34.15, 31.58, 28.80, 27.26, 22.21, 19.90, 14.25, 14.12, 13.65 (т, 1JC-D=19Hz). [M+]185. Вычислено (%): C, 84.32; H+D, 15.68. Найдено (%): C, 84.30; H, 15.65. 81 5,6-диметилдекан (42а) Т.кип. 62 ºC (2.5 mm Hg). Спектр ЯМР 1H (δ, м.д.): 0.98-1.31 (м, 12H, 4CH3), 0.82-0.94 (м, 14H, 6CH2, 2CH). Спектр ЯМР 13 C (δ, м.д.): 36.89, 35.64, 28.21, 22.72, 16.91, 15.49. [M+] 170. Найдено (%): C, 84.47; H, 15.48. Вычислено (%): C, 84.51; H, 15.49. 1-дейтеро-2-бутил-3-метилгептан (44а) D Т.кип. 102 ºC (30 mm Hg). ИК-спектр, cм-1: 2185 (C-D). Спектр ЯМР 1H (δ, м.д.): 1.09-1.44 (м, 14H, CH, CH2), 0.75-0.95 (м, 11H, CH3, CH2D). Спектр ЯМР 13 C (δ, м.д.): 36.51, 36.24, 36.11, 34.21, 27.92, 22.63, 14.01, 12.41 (т, 1JC- D=19Hz), 12.40. [M+]171. Найдено (%): C, 84.17; H, 15.74. Вычислено (%): C, 84.21; H+D, 15.79. 82 ВЫВОДЫ Выполнена 1. разработке программа методов синтеза карбометаллированием и ориентированных исследований новых АОС классов восстановительным и по ЦОС β-металлэтилированием терминальных алкенов и норборненов с помощью триалкилаланов и диалкильных производных цинка с участием катализатора ТаCl5. 2. Впервые изучено каталитическое действие Та-содержащих металлокомплексных катализаторов в реакции Et3Al с 1-алкенами. В результате было обнаружено, что наряду с реакцией карбоалюминирования параллельно происходит восстановительное β-алюминаэтилирование α- олефинов. Разработаны оптимальные условия, позволяющие проводить реакцию с высокой региоселективностью. 3. Разработано принципиально новое направление реакции норборнена и его производных с помощью Et3Al в присутствии TaCl5, позволяющее проводить реакцию стереоселективно с высокими выходами целевых диэтил[2-экзо-(2'-норборнилэтил)]алюминиевых оценка термодинамической вероятности производных. Проведена элементарных стадий предложенного каталитического цикла на примере реакции норборнена с Et3Al в присутствии катализатора TaCl5 с применением метода функционала плотности PBE/3z с псевдопотенциалом Стивена-Баша-Краусса. 4. Впервые изучена реакция α-олефинов с помощью высших АОС под действием катализатора TaCl5. Установлено, что в углеводородных растворителях реакция проходит с образованием (2R',3R-дизамещенных)-нбутилалюминиевых и (2R,3R-дизамещенных)-н-бутилалюминиевых производных. 5. Осуществлено строго региоселективное восстановительное βцинкэтилирование α-олефинов с помощью Et2Zn, катализируемое TaCl5, с получением соответствующих 3-(R-замещенных)-н-бутилпроизводных цинка с выходами ~ 92%. Предложенный каталитический цикл реакции 83 предполагает образование β-замещенных танталациклопентанов. Проведенные квантово-химические расчеты по оценке термодинамической вероятности предложенного механизма реакции восстановительного βцинкэтилирования α-олефинов подтверждают полученные экспериментальные данные. 6. Впервые осуществлена реакция 1-алкенов с помощью высших ЦОС (R2Zn, где R= Prn, Bun, Amn, Hexn) в присутствии TaCl5, позволяющая синтезировать (2R',3R-дизамещенные)-н-бутилцинкорганические и (2R,3Rдизамещенные)-н-бутилцинкорганические соединения. 7. С применением разработанной реакции региоселективного восстановительного β-цинкэтилирования 1-алкенов с помощью Et2Zn предложен оригинальный метод синтеза аналога феромона тревоги муравьев рода Crematogaster. 84 ЛИТЕРАТУРА 1. Hay J. N., Hooper P. G., Robb J. C. Kinetics of reaction of metal alkyl compounds with alkenes. Part 5.−Aluminium triethyl and substituted alkenes // Trans. Faraday Soc.− 1970.− 66.− р. 2045-2054. 2. Allen P.E. M. , Lough R. M. Kinetics of the addition of triethylaluminium to alkenes in hydrocarbon solution // J. Chem. Soc., Faraday Trans 1.− 1973.− 69.− р. 2087-2095. 3. Lehmkuhl H., Olbrysch O., Reinehr D., Schomburg G., Henneberg D. Vergleich der Additionsrichtungen von tert.-Butyl- und Isopropyl-verbindungen des Magnesiums, Aluminiums und Lithiums an 1-Alkene // Liebigs Ann. Chem. −1975.− р.145-159. 4. Schimpf R., Heimbach P. Reaktionen von AlH- und AlC-Bindungen mit gespannten Olefinen und 1.5-Dienen // Chem.Ber.−1970 .−103.−р. 2122-2137. 5. Eisch J.J., Liu Sh. J. Y. Selective phenylation of olefins with triphenylaluminum // J.Organomet.Chem.− 1970.− 21.− p. 285. 6. Eisch J. J., Burlinson N. E., Boleslawski M. Organometallic compounds of group III : XXXIV. Steric factors in the carbalumination of olefins. The question of the anomalous, reduced reactivity of olefins versus acetylenes // J.Organomet.Chem.− 1976.− 111.− p.137-152. 7. Eisch J. J., Burlinson N. E. Organometallic compounds of Group III. XXXIII. Polar and stereochemical factors in the carbalumination of olefins. Addition of triphenylaluminum to 6-substituted benzonorbornadienes // J. Am. Chem. Soc.−1976.− 98.− p. 753–761. 8. Westmijze H., Kleijn H., Vermeer P. A New, Stereospecific Synthesis of trans2-Alkenenitriles from 2-Alkynenitriles and Lithium Tetrahydridoaluminate // Synthesis.− 1979.− p.430-431. 9. Mole T., Surtess J. R. // Chem. Ind. (London).− 1963.− p. 1727. 10. Mole T., Surtees J. R. Organoaluminium compounds. VII. The reactions of organoaluminium compounds with phenylacetylene // Austral. J.Chem.− 1964.− 17.− p.1229- 1235. 85 11. Eisch J. J., Kaska W. C. Stereochemistry and Orientation in the Reactions of 1Phenylpropyne with Diisobutylaluminum Hydride // J. Am. Chem. Soc.−1966.− 88.− p. 2213–2220. 12. Eisch J. J., Kaska W. C. The Synthesis of Aluminoles via the Addition and Cyclization Reactions of Arylaluminum Compounds // J. Am. Chem. Soc.−1966.− 88.− p. 2976–2983. 13. Несмеянов А.Н., Борисов А.Е., Савельев И.С. Присоединение триэтилалюминия к толану // Изв. Ан СССР. ОХН. 1959.− с. 1034-1036. 14. Eisch J. J., Kaska W. C. The Novel Synthesis of Aluminoles by the Metalative Cyclization of Unsaturated Organoaluminum Compounds // J. Am. Chem. Soc.−1962.− 84.− p. 1501–1502. 15. Eisch J. J., Hordis Ch. K. Organometallic compounds of Group III. XVI. Polar and stereochemical effects in the addition of triphenylaluminum to para-substituted diphenylacetylenes // J. Am. Chem. Soc.−1971.− 93.− p. 2974–2981. 16. Johncock P. // J.Organomet. Chem. 1969.− 19.− p. 257-265. 17. King R.B., Eavenson C.W. A novel transannular cyclization of a macrocyclic diacetylene to form a substituted π-cyclopentadienylmetal derivative // J.Organomet. Chem. −1969.− 16.− p. 55-77. 18. Eisch J. J., Amtmann R. Organometallic compounds of group III. XXII. Steric effects in the hydralumination, carbalumination, and oligomerization of tertbutyl(phenyl)acetylene // J. Org. Chem.− 1972.− 37.− p. 3410-3415. 19. Голубев В.К., Смагин В.М., Румянцева М.Р., Гавриленко В.В., Захаркин Л.Л. А.С.687076 СССР. Бюл. изобрет. – 1979. – 35. − С.108. 20. Джемилев У.М., Вострикова О.С., Толстиков Г.А., Ибрагимов А.Г. Новый метод введения этильной группы в β-положение высших α-олефинов с помощью диэтилалюминийхлорида // Изв. АН СССР.Сер.хим.− 1979.−№ 11.− c.2626-2627. 21. Джемилев У.М., Ибрагимов А.Г., Вострикова О.С., Толстиков Г.А., Зеленова Л.М. Новый метод β-алкилирования α-олефинов с помощью 86 диалкилалюминийхлоридов в присутствии каталитических количеств Ti, Zr, Hf // Изв. АН СССР.Сер.хим.−1981.−№2.− c.361-364. 22. Ибрагимов А. Г. // Дис. …канд.хим.наук. Ин-т химии БФ АН СССР, Уфа, 1982. 23. Джемилев У.М., Ибрагимов А.Г., Вострикова О.С., Толстиков Г.А. Катализированное комплексами титана и циркония карбалюминирование высших α-олефинов // Изв. АН СССР. Сер.хим.− 1985.−№1.−с.207-209. 24. Ибрагимов А.Г., Джемилев У.М. Способ получения 2-этилгексанола // А.С. 1051054 СССР. Б.И. – 1983. –№40– С.89. 25. Dzhemilev U. M., Vostrikova O. S. Some novelties in olefin carbometallation assisted by alkyl-magnesium and -aluminium derivatives and catalyzed by zirconium and titanium complexes // J.Organomet.Chem.−1985.−285.− p.43-51. 26. Barber J. J., Willis C., Whitesides G. M. Conversion of monoalkyl olefins to 1,1-dialkyl olefins by reaction with bis(cyclopentadienyl)titanium dichloridetrialkylaluminum // J.Org.Chem.− 1979.−44.− p.3603–3604. 27. Tweedy H. E., Hahn P. E., Smedley L. C., Youngblood A.V., Coleman R. A., Thompson D. W. The insertion of 3-buten-1-ol into metal−alkyl bonds utilizing typical titanium−aluminum ziegler−natta catalysts // J.Mol.Catal.− 1978.−3.− 239243. 28. Youngblood A.V., Nichols S.A., Coleman R.A., Thompson D.W. Transition metal promoted alkylations of unsaturated alcohols: the methylation and ethylation of 3-buten-1-ol using titanium tetrachloride-organoaluminum systems // J.Organomet.Chem.− 1978.−146.−p. 221-228. 29. Harris T. V., Coleman R.A., Dickson R. B., Thompson D.W. Transition metal promoted alkylations of alkenols // J.Organomet.Chem.− 1974.−69.− p. C27-C30. 30. Richey Jr. H. G., Moses L. M., Hangeland J. J. Stereochemistry of titaniumassisted additions of organoaluminum compounds to hydroxybicyclo[2.2.1]hept-2enes // Organometallics.− 1983.−2.−p.1545-1549. 31. Джемилев У.М., Ибрагимов А.Г., Минскер Д.Л., Толстиков Г.А. Карбоалюминирование норборненов с помощью Et2AlCl катализируемое 87 TiCl4 // В кн. Тез. докл. V Всесоюзн. конф. по металлоорганической химии. Рига.− 1991.−С.100. 32. Van Horn D. E., Negishi E. Selective carbon-carbon bond formation via transition metal catalysts. 8. Controlled carbometalation. Reaction of acetylenes with organoalane-zirconocene dichloride complexes as a route to stereo- and regiodefined trisubstituted olefins // J.Am.Chem.Soc.− 1978.−100.−p.2252-2254. 33. Negishi E., Van Horn D. E., Yoshida T. Controlled carbometalation. 20. Carbometalation reaction of alkynes with organoalene-zirconocene derivatives as a route to stereo-and regiodefined trisubstituted alkenes // J.Am.Chem.Soc.− 1985.−107.− p. 6639-6647. 34. Negishi E. Bimetallic catalytic systems containing Ti, Zr, Ni, and Pd. Their applications to selective organic syntheses // Pure Appl.Chem.−1981.− 53.− p. 2333-2356. 35. Kondakov D. Y., Negishi E. Zirconium-catalyzed enantioselective methylalumination of monosubstituted alkenes // J.Am.Chem.Soc.− 1995.−117.− p.10771–10772. 36. Wipf P., Ribe S. Water/MAO Acceleration of the Zirconocene-Catalyzed Asymmetric Methylalumination of α-Olefins // Organic Lett.− 2000.−2.− p.17131716. 37. Kondakov D. Y., Negishi E. Zirconium-Catalyzed Enantioselective Alkylalumination of Monosubstituted Alkenes Proceeding via Noncyclic Mechanism // J.Am.Chem.Soc.− 1996.− 118.− p.1577-1578. 38. Negishi E., Ojima I. Catalytic Asymmetric Synthesis. In Asymmetric carboaluminations.− New York.− 2000.−165. 39. Shaughnessy K. H., Waymouth R.M. Carbometalation of .alpha.,.omega.Dienes and Olefins Catalyzed by Zirconocenes // J.Am.Chem.Soc.− 1995.−117.−5873-5874. 40. Wipf P., Ribe S. Water-Accelerated Tandem Claisen Rearrangement−Catalytic Asymmetric Carboalumination // Org.Lett.−2001.− 3.− p.1503-1505. 88 41. Ribe S.,Wipf P. Water-accelerated organic transformations // Chem. Commun.− 2001.−4.− p. 299-307. 42. Bell L., Brookings D. C., Dawson G. J., Whitby R. J., Jones R. V. H., Standen M.C.H. Asymmetric ethylmagnesiation of alkenes using a novel zirconium catalyst // Tetrahedron.−1998.− 54.− p.14617–14634. 43. Dawson G., Durrant Ch.A., Kirk G. G., Whitby R. J., Jones R. V.H., Standen M. C.H. Zirconium-catalysed enantioselective 2-aluminoethylalumination of alkenes // Tetrahedron Lett.− 1997.− 38.− p. 2335-2338. 44. Bell L., Whitby R. J., Jones R. V. H., Standen M.C.H. Catalytic asymmetric carbomagnesiation of unactivated alkenes. A new, effective, active, cheap and recoverable chiral zirconocene // Tetrahedron Lett.− 1996.−37.− p. 7139-7142. 45. Джемилев У.М., Вострикова О.С., Толстиков Г.А. Металлокомплексный катализ в алюминийорганическом синтезе // Усп. химии.− 1990.−59.−С. 19722002. 46. Dzhemilev U. M., Vostrikova O. S., Tolstikov G. A. Homogeneous zirconium based catalysts in organic synthesis // J.Organomet.Chem.− 1986.−304.− p.17-39. 47. Normant J. F. , Alexakis A. Carbometallation (C-Metallation) of Alkynes: Stereospecific Synthesis of Alkenyl Derivatives // Synthesis.− 1981.− p.841-870. 48. Negishi E., Takahashi T. Organozirconium Compounds as New Reagents and Intermediates // Aldrichim.Acta. −1985.− 18.− p. 26-44. 49. Negishi E. Controlled carbometalation as a new tool for carbon-carbon bond formation and its application to cyclization // Acc.Chem.Res.− 1987.−20.− p.6572. 50. Coleman R.A., O'doherty C.M., Tweedy H.E., Harris T.V., Thompson D.W. Transition metal promoted alkylations of alkynols // J.Organomet.Chem.− 1976.− 107.− p.C15-C17. 51. Smedley L.C., Tweedy H. E., Coleman R. A., Thompson D. W. Alkylations of alkynols with organoaluminum reagents promoted by bis(η5- cyclopentadienyl)titanium dichloride // J.Org.Chem.−1977.− 42.− p.4147-4148. 89 52. Tweedy H.E., Coleman R.A., Thompson D.W. Transition metal promoted alkylations of unsaturated alcohols: The ethylation of alkynols via the reactions of cis-alkynoxy(chloro)bis(2,4-pentanedionato)titanium(IV) complexes with diethylaluminium chloride // J.Organomet.Chem.−1977.− 129.− p.69-78. 53. Snider B. B., Karras M. Stereocontrolled carbotitanation of alkynylsilanes // J.Organomet.Chem.−1979.− 179.− p.C37-C41. 54. Brown D. C., Nichols S. A., Gilpin A. B., Thompson D. W. Transition metal promoted alkylations of unsaturated alcohols. Alkylation of alkynols with organoalanes promoted by Group IVA metal-cyclopentadienyl compounds // J.Org.Chem.−1979.− 44.− p.3457-3461. 55. Schiavelli M. D., Plunkett J. J., Thompson D. W. Synthesis of (Z)-4methylhex-3-en-1-ol via the reaction of 3-hexyn-1-ol with trimethylaluminumtitanium tetrachloride // J.Org.Chem.− 1981.− 46.− p.807-808. 56. Ewing J.C., Ferguson G. S., Moore D. W., Schultz F. W., Thompson D.W. Transition metal promoted alkylations of unsaturated alcohols: the selective methylation of homopropargyl alcohols via titanium tetrachloride- trimethylaluminum // J.Org.Chem.− 1985.−50.− p.2124-2128. 57. Zitzelberger T. J., Schiavelli M. D., Thompson D. W. A facile and selective methylation of 5-en-3-yn-1-ols with titanium tetrachloride-trimethylaluminum yielding (3Z)-4-methylalka-3,5-dien-1-ols // J.Org.Chem.− 1983.− 48.− p.47814783. 58. Yoshida T., Negishi E. Mechanism of the zirconium-catalyzed carboalumination of alkynes. Evidence for direct carboalumination // J. Am. Chem. Soc.− 1981.−103.−p. 4985-4987. 59. Yoshida T., Negishi E. 1,1-Dimetalloalkenes containing aluminum as well as titanium or zirconium. Their structures and use as novel alkenylidene and alkenyl transfer agents // J. Am. Chem. Soc.− 1981.− 103.−p. 1276-1277. 60. Negishi E., Kondakov D. Y., Choueiry D., Kasai K., Takahashi T. Multiple Mechanistic Pathways for Zirconium-Catalyzed Carboalumination of Alkynes. 90 Requirements for Cyclic Carbometalation Processes Involving C−H Activation // J. Am. Chem. Soc.−1996.−118 .−p. 9577-9588. 61. Caporusso A. M., Giacomelli G., Lardicci L. Metal catalysis in organic reactions. Part 9. Iron-induced reaction of organoaluminium compounds with aliphatic alk-1-ynes // J. Chem. Soc., Perkin Trans. 1.−1979.− p. 3139-3145. 62. Caporusso A. M., Lardicci L., Giacomelli G. Metal catalysis in organic reactions. V. Selective alkylataion and oligomerization of aliphatic 1-alkynes catalyzed by transition metal salts // Tetrahedron Lett.−1977.− 18.− p.4351-4354. 63. Caporusso A. M., Giacomelli G., Lardicci L. Metal catalysis in organic reactions. 8. Alkylative dimerization of 1-alkynes induced by tris(acetylacetonato)manganese/trialkylalane systems // J.Org.Chem.−1979.−44.− p.1496-1501. 64. Caporusso A. M., Giacomelli G., Lardicci L. Metal catalysis in organic reactions. 3. Nickel-promoted reaction of triisobutylaluminum with terminal acetylenes as a synthetic route to (E)-2,4-dialkyl-1,3-butadienes and/or trialkylbenzenes // J.Org.Chem.− 1977.−42.− p.914-916. 65. Giacomelli G., Caporusso A. M., Lardicci L. Metal catalysis in organic reactions. 7. The role of nickel complexes in the reaction of triisobutylaluminum with terminal acetylenes // J.Org.Chem.−1979.−44.− p.231-237. 66. Schiavelli M. D., Plunkett J. J., Thompson D. W. Synthesis of (Z)-4methylhex-3-en-1-ol via the reaction of 3-hexyn-1-ol with trimethylaluminumtitanium tetrachloride // J. Org. Chem.− 1981.− 46.− p. 807–808. 67. Ewing J. C., Ferguson G. S., Moore D. W., Schultz F.W., Thompson D. W. Transition metal promoted alkylations of unsaturated alcohols: the selective methylation of homopropargyl alcohols via titanium tetrachloride- trimethylaluminum // J.Org.Chem. −1985.− 50.− p.2124-2128. 68. Van Horn D. E., Valente L. F., Idacavage M. J., Negishi E. Controlled carbometallation : II. The addition reaction of trimethylalane-titanocene dichloride with acetylenes // J.Organomet.Chem.− 1978.−156.− p.C20-C24. 91 69. Youngblood A.V., Nichols S.A., Coleman R.A., Thompson D.W. Transition metal promoted alkylations of unsaturated alcohols: the methylation and ethylation of 3-buten-1-ol using titanium tetrachloride-organoaluminum systems // J.Organomet.Chem. − 1978.− 146.− p.221-228. 70. Negishi E., Takahashi T. Patterns of Stoichiometric and Catalytic Reactions of Organozirconium and Related Complexes of Synthetic Interest // Acc.Chem.Rev. −1994.− 27.− p.124-130. 71. Negishi E., Kondakov D. Y. An odyssey from stoichiometric carbotitanation of alkynes to zirconium-catalysed enantioselective carboalumination of alkenes // Chem. Soc. Rev. −1996.−25.− p.417-426. 72. Negishi E., Kondakov D. Y., Van Horn D. E. Carbometalation Reactions of Diphenylacetylene and Other Alkynes with Methylalanes and Titanocene Derivatives // Organometallics.−1997.−16.− p.951-957. 73. Negishi E. Principle of Activation of Electrophiles by Electrophiles through Dimeric Association—Two Are Better than One // Chem.Eur.J.−1999.− 5.− p.411420. 74. Negishi E. A quarter of a century of explorations in organozirconium chemistry //Dalton Trans.− 2005.− p.827-848 75. Okukado N., Negishi E. One-step conversion of terminal acetylenes into terminally functionalized (E)-3-methyl-2-alkenes via zircomium-catalyzed carboalumination. A simple and selective route to terpenoids // Tetrahedron Lett. – 1978.−19.− p.2357-2360. 76. Negishi E., King A. O., Klima W. L., Patterson W., Silveira Jr. A. Conversion of methyl ketones into terminal acetylenes and (E)-trisubstituted olefins of terpenoid origin // J.Org.Chem.− 1980.−45.− 2526-2528. 77. Negishi E., Valente L. F., Kobayashi M. Palladium-catalyzed cross-coupling reaction of homoallylic or homopropargylic organozincs with alkenyl halides as a new selective route to 1,5-dienes and 1,5-enynes // J. Am. Chem. Soc.− 1980.− 102.− p. 3298–3299. 92 78. Deng H., Hoffmann R. How N2 Might Be Activated by the FeMo-Cofactor in Nitrogenase // Angew.Chem.,Int.Ed.Engl. −1993.−32.− p.1062–1065. 79. Caronna T., Gabbiadini S., Mele A., Recupero F. Approaches to the Azahelicene System: Synthesis and Spectroscopic Characterization of Some Diazapentahelicenes // Helv.Chim.Acta.− 2002.−85.− p. 1-8. 80. Wipf P., Waller D.L., Reeves J. T. Transition-Metal-Mediated Cascade Reactions: The Water-Accelerated Carboalumination-Claisen RearrangementCarbonyl Addition Reaction // J.Org.Chem.− 2005.−70.− 8096-8102. 81. Miller J.A., Negishi E. Zirconium-catalyzed allylalumination and benzylalumination of alkynes // Tetrahedron Lett.− 1984.−25.− p. 5863-5866. 82. Shibata K., Aida T., Inoue Sh. Zirconium porphyrins as novel active catalysts for highly regio- and stereo-selective ethylalumination of alkynes // Tetrahedron Lett.− 1992.− 33.−p.1077-1080. 83. Lehmkuhl H., Olbrysch O. Additionen von Di-tert.-butylzink an Olefine // Justus Liebigs Ann.Chem.− 1975.− p.1162-1175. 84. Courtois G., Miginiac L. C.R. Acad. Sci.Paris, Ser.C.− 1977.−285.− p.207. 85. Auger J., Courtois G., Miginiac L. Addition du di-t-butyl-zinc a la triple liaison des enynes conjugues, simples et fonctionnels // J. Organomet. Chem.− 1977.−133.− p. 285-291. 86. Courtois G., Miginiac L. Addition du di-t-butylzinc a la triple liaison des enynes conjugues, simples et fonctionnels: influence de divers facteurs sur la stereochimie de la reaction // J. Organomet. Chem.− 1980.−195.− p. 13-24. 87. Miginiac L. The synthetic possibilities of addition reactions between common organometallic compounds and conjugated enynes // J. Organomet. Chem.− 1982.−238.− p.235-266. 88. Gagneur S., Montchamp J.-L., Negishi E. Ethylzincation of Monosubstituted Alkenes Catalyzed by EtMgBr – Cl2ZrCp2 and Palladium-Catalyzed Cross Coupling of the Resultant Diisoalkylzinc Derivatives // Organometallics.−2000.− 19 .− p. 2417–2419. 93 89. Owczarczyk Z., Lamaty F., Vawter E. J., Negishi E. Apparent Endo-Mode Cyclic Carbopalladation with Inversion of Alkene Configuration via Exo-Mode Cyclization-Cyclopropanation-Rearrangement // J. Am. Chem. Soc.− 1992.− 114.− p.10091-10092. 90. Nakamura M., Hirai A., Nakamura E. Iron-Catalyzed Olefin Carbometalation // J.Am.Chem.Soc.−2000.−122.− p. 978-979. 91. Krämer K., Leong P., Lautens M. Enantioselective Palladium-Catalyzed Carbozincation of Cyclopropenes // Org. Lett.− 2011.− 13.− p. 819–821. 92. Tarwade V., Liu X., Yan N., Fox J. M. Directed Carbozincation Reactions of Cyclopropene Derivatives // J. Am. Chem. Soc.− 2009.− 131.− p. 5382–5383. 93. Tarwade V., Selvaraj R., Fox J. M. Facially Selective Cu-Catalyzed Carbozincation of Cyclopropenes Using Arylzinc Reagents Formed by Sequential I/Mg/Zn Exchange // J. Org. Chem.−2012.−77 .−p. 9900–9904. 94. Lautens M., Renaud J.-L., Hiebert Sh. Palladium-Catalyzed Enantioselective Alkylative Ring Opening // J. Am. Chem. Soc.−2000.−122 .− p. 1804–1805. 95. Bertozzi F., Pineschi M., Macchia F., Arnold L.A., Minnaard A.J., Feringa B.L. Copper Phosphoramidite Catalyzed Enantioselective Ring-Opening of Oxabicyclic Alkenes: Remarkable Reversal of Stereocontrol // Org. Lett.−2002.− 4.− p. 2703–2705. 96. Purohit V., Basu A. K. Synthesis and Characterization of Oligodeoxynucleotides Containing the Major DNA Adducts Formed by 1,6- and 1,8-Dinitropyrene // Org. Lett.−2000.− 2 .− p. 1871-1874. 97. Lautens M., Hiebert Sh. Scope of Palladium-Catalyzed Alkylative Ring Opening // J. Am. Chem. Soc.−2004.−126.− p.1437–1447. 98. Ito S., Itoh T., Nakamura M. Diastereoselective Carbometalation of Oxa- and Azabicyclic Alkenes under Iron Catalysis // Angew.Chem. −2011.− 123.− p. 474477. 99. Negishi E., Van Horn D. E., Yoshida T., Rand C. L. Selective carbon-carbon bond formation via transition metal catalysis. 31. Controlled carbometalation. 15. 94 Zirconium-promoted carbozincation of alkynes // Organometallics.−1983.− 2.− p. 563–565. 100. Negishi E., Sawada H., Tour J. M., Wei Y. Metal-promoted cyclization. 17. A novel bicyclization methodology via cyclialkylation of ω-halo-1-metallo-1-alkynes containing aluminum and zinc // J. Org. Chem.− 1988.−53.− p.913–915. 101. Stüdemann T., Knochel P. New Nickel-Catalyzed Carbozincation of Alkynes: A Short Synthesis of (Z)-Tamoxifen // Angew. Chem., Int. Ed. Engl.− 1997.− 36.− p. 93-95. 102. Stüdemann T., Ibrahim-Ouali M., Knochel P. A nickel-catalyzed carbozincation of aryl-substituted alkynes // Tetrahedron.− 1998.− 54.− p.12991316. 103. Stüdemann T., Knochel P. Eine neue, nickelkatalysierte Carbozinkierung von Alkinen – eine kurze Synthese von (Z)-Tamoxifen // Angew. Chem.− 1997.− 109.− p.132–134. 104. Cann R. O., Waltermire, R. E., Chung J., Oberholzer M., Kasparec J., Ye Y. K., Wethman R. Process Development for a Large Scale Stereoselective Synthesis of (Z)-(1-Bromobut-1-ene-1,2-diyl)dibenzene, a Key Intermediate of a Selective Estrogen Receptor Modulator // Org. Process Res. Dev.− 2010.− 14.− p.1147– 1152. 105. Murakami K., Yorimitsu H., Oshima K. Cobalt-Catalyzed Arylzincation of Alkynes // Org. Lett.− 2009.− 11.− p. 2373–2375. 106. Tan B.-H., Dong J., Yoshikai N. Cobalt-Catalyzed Addition of Arylzinc Reagents to Alkynes to Form ortho-Alkenylarylzinc Species through 1,4-Cobalt Migration // Angew. Chem.− 2012.−124.− p.9748-9752. 107. Tan B.-H., Dong J., Yoshikai N. Cobalt-Catalyzed Addition of Arylzinc Reagents to Alkynes to Form ortho-Alkenylarylzinc Species through 1,4-Cobalt Migration // Angew. Chem. Int. Ed.− 2012.− 51.− p. 9610–9614. 108. Corpet M., Gosmini C. Cobalt-catalysed synthesis of highly substituted styrene derivatives via arylzincation of alkynes // Chem. Commun.−2012.− 48.− p.11561-11563. 95 109. Negishi E., Miller J. A. Selective carbon-carbon bond formation via transition metal catalysis. 37. Controlled carbometalation. 16. Novel syntheses of α, βunsaturated cyclopentenones via allylzincation of alkynes // J. Am. Chem. Soc.− 1983.−105.− p. 6761–6763. 110. Nishikawa T., Yorimitsu H., Oshima K. Cobalt-Catalyzed Regio- and Stereoselective Allylzincation of 1-Phenyl-1-alkynes // Synlett.−2004.− p.15731574. 111. MurakamiK., Yorimitsu H., Oshima K. Cobalt-Catalyzed Benzylzincation of Alkynes // Chem.-Eur. J. −2010.− 16.− p. 7688–7691. 112. McLain S. J., Sancho J., Schrock R. R. Selective dimerization of monosubstituted .alpha.- olefins by tantalacyclopentane catalysts // J. Am. Chem. Soc.− 1980.− 102.− p. 5610- 5618. 113. McLain S. J., Sancho J., Schrock R. R. Metallacyclopentane to metallacyclobutane ring contraction // J. Am. Chem. Soc.− 1979.− 101.− p. 54515453. 114. McLain S .J.,Schrock R. R., Wood C. D. Multiple metal-carbon bonds. 6. The reaction of niobium and tantalum neopentylidene complexes with simple olefins: a route to metallocyclopentanes // J. Am. Chem. Soc. −1977.− 99.− p.3519-3520. 115. Schrock R. R., McLain S. J., Sancho J. Tantalacyclopentane complexes and their role in the catalytic dimerization of olefins // Pure Appl. Chem.− 1980.− 52.− p. 729-732. 116. McLain S. J., Schrock R. R. Selective olefin dimerization via tantallocyclopentane complexes // J. Am. Chem. Soc. −1978.− 100.− p. 1315-1317. 117. Fellmann J. D., Rupprecht G. A., Schrock R. R. Rapid selective dimerization of ethylene to 1-butene by a tantalum catalyst and a new mechanism for ethylene oligomerization // J. Am. Chem. Soc.− 1979.− 101.− p.5099-5101. 118. Andes C., Harkins S. B., Murtuza S., Oyler K., Sen A. New Tantalum-Based Catalyst System for the Selective Trimerization of Ethene to 1-Hexene // J. Am. Chem. Soc.−2001.−123.− p.7423-7424. 96 119. Mueller R. A., Tsurugi H., Saito T., Yanagawa M., Oda S., Mashima K. New Tantalum Ligand-Free Catalyst System for Highly Selective Trimerization of Ethylene Affording 1-Hexene: New Evidence of a Metallacycle Mechanism // J. Am. Chem. Soc.− 2009.− 131.− p. 5370-5371. 120. Yu Z. X., Houk K. N. Why Trimerization? Computational Elucidation of the Origin of Selective Trimerization of Ethene Catalyzed by [TaCl3(CH3)2] and An Agostic-Assisted Hydride Transfer Mechanism // Angew. Chem. Int. Ed.− 2003.− 42.− p. 808-811. 121. Rocklage S. M., Fellmann J. D., Messerle L. W., Schrock R. R. Multiple metal-carbon bonds. 19. How niobium and tantalum complexes of the type M(CHCMe3)(PR3)2Cl3 can be modified to give olefin metathesis catalysts // J. Am. Chem. Soc.− 1981.− 103.− p. 1440-1447. 122. Mauri O., Lefort L., Vidal V., Thivolle-Cazat J., Basset J.-M. Revisiting the Metathesis of 13C-Monolabeled Ethane // Organometallics.− 2010.−29.− p. 66126614. 123. Wallance K. C., Liu A. H., Dewan J. C., Schrock R. R. Preparation and reactions of tantalum alkylidene complexes containing bulky phenoxide or thiolate ligands. Controlling ring-opening metathesis polymerization activity and mechanism through choice of anionic ligand // J. Am. Chem. Soc.− 1988.− 110.− p. 4964-4977. 124. Roux E. L., Chabanas M., Baundonin A., de Mallman A., Coperet C., Quadrelli E. A., Thivolle-Cazat J., Basset J.-M., Lukens W., Lesage A., Emsley L., Sunley G. J. Detailed Structural Investigation of the Grafting of [Ta(=CHtBu)(CH2tBu)3] and [Cp*TaMe4] on Silica Partially Dehydroxylated at 700 °C and the Activity of the Grafted Complexes toward Alkane Metathesis // J. Am. Chem. Soc.− 2004.−126.− p. 13391- 13399. 125. Mashima K., Fujikama S., Tanaka Y., Urata H., Oshiki T., Tanaka E., Nakamura A. Living Polymerization of Ethylene Catalyzed by Diene Complexes of Niobium and Tantalum, M(η5-C5Me5)(η4-diene)X2 and M(η5-C5Me5)(η4-diene)2 97 (M = Nb and Ta), in the Presence of Methylaluminoxane // Organometallics.− 1995.− 14.− p. 2633-2640. 126. Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett.− 1996.− 77.− p. 3865-3869. 127. Giordan M., Custodio R., Morgon N.H. A universal basis set to be used along with pseudopotentials // Chemical Physics Letters.− 1997.−279.− p. 396-402. 128. Laikov D.N., Ustynyuk Yu.A. PRIRODA-04: a quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing // Russ. Chem. Bull.− 2005.− 54.− p.820-826. 129. Алюминийорганические соединения / Под ред. Жигача А.Ф. Москва.: Изд-во иностранной лит-ры.− 1962.−320 с. 130. Шевердина Н.И., Кочешков К.А. Методы элементоорганической химии (Цинк, Кадмий). М.: Наука.− 1964. − 235 с.