Б.М. Искаков, А.Е. Рахимбекова МЕТОД ВЫЧИСЛЕНИЯ ЭНЕРГИИ ВЗАИМОДЕЙСТВИЯ АТОМОВ В КУБИЧЕСКОМ КРИСТАЛЛЕ
реклама

Б.М. Искаков, А.Е. Рахимбекова
МЕТОД ВЫЧИСЛЕНИЯ ЭНЕРГИИ ВЗАИМОДЕЙСТВИЯ АТОМОВ
В КУБИЧЕСКОМ КРИСТАЛЛЕ
Основой метода погруженного атома (МПА) является идея зависимости энергии системы от
электронной плотности. Поэтому в рамках теории функционала плотности полная электронная
энергия системы может быть записана как функционал полной электронной плотности.
В работе [1] была предложена модель межатомных взаимодействий, в которой каждый атом
рассматривается как примесь в системе остальных атомов, кроме выделенного атома. Другой
отправной точкой МПА был наблюдаемый факт, что полная электронная плотность в металле
достаточно хорошо апроксимируется линейной суперпозицией вкладов от отдельных атомов.
Теоретическое обоснование МПА в рамках теории функционала локальной плотности было
проведено в [2].
В МПА, описанном в работе [3], полная энергия металла состоит из двух частей. Первая
определяет энергию погружения выделенного атома в электронную плотность, создаваемую
суперпозицией всех остальных атомов в области расположения выделенного атома и отражает
многочастичные эффекты. Вторая часть – это сумма парных потенциалов, описывающих отталкивание
двух экранированных ионов. В итоге, полная энергия металла представляется выражением:
= ∑ ( ) + 1 2 ∑ Ф( ).
(1)
Здесь
( ) − энергия погружения атома i, как функция локальной плотности ρ, а
Ф( ) −парный потенциал взаимодействия атома i и центрального атома, расположенных на
расстоянии друг от друга.
Локальная электронная плотность системы атомов в точке погружения выделенного атома,
например, центрального атома – определяется в виде суперпозиции плотностей ( ) – вкладов
от электронов атомов i .
Так как
= ∑ ( ) ( ),
т.е. является функцией расстояния между атомами, то и ( ) является функцией расстояния
между атомами и может быть апроксимирована формулой
( ) = −2
− 1 exp −
−1 ,
(2)
где R0 – расстояние между атомами, соответствующее минимальному значению энергии
взаимодействия атомов в металле, а A и β являются параметрами также, как и R0 , определяемыми
через экспериментальные характеристики металла.
В работе [3] при выборе формы парного потенциала предпочтение было отдано парному
потенциалу взаимодействия экранированных ионов в среде с металлической проводимостью в
виде
zi ( R ) z j ( R )
(3)
Фij ( R ) =
R
где эффективный заряд имеет следующую форму:
( ) = (1 +
) exp(− ).
(4)
Здесь
- число внешних электронов; =10 для Ni, Pd, Pt;
= 11 для Cu, Ag, Au; =3 для Al.
Величина параметра γ выбиралась равной 1 для Ni, Pd, Pt и γ=2 для Cu, Ag, Au и Al [3]. Параметры ε и λ
являются подгоночными. Таким образом, при использовании потенциала парного взаимодействия
экранированных ионов (3) и (4) приходится использовать 3 подгоночных параметра: ε, λ и γ.
В принципе, здесь можно применить потенциал парного взаимодействия Морзе, также
имеющий 3 параметра, которые хорошо определяются через экспериментальные значения
постоянной кристаллической решетки, энергии сублимации и модуль упругости металла [4]
Z(R)=Z0(1+εRγ)exp(-λR)
(4)
Здесь D, α и R0 являются параметрами, причем R0 – это расстояние между атомами,
соответствующее минимальному значению энергии взаимодействия атомов в металле D.
Учитывая то, что при
= , полная энергия взаимодействия двух атомов металла должна
быть минимальной и равной –D, а также то, что по (1) полная энергия является суммой энергии
погружения атомов в электронную плотность и энергии парного взаимодействия и, проведя
некоторое преобразование формул (2) и (4), получим следующее выражение:
{exp[−2 ( − )] − 2 exp[− ( − )]} − (1 − )
=
[− ( − ).
(5)
) и η = exp (βR ), то выражение (5) можно представить
Если ввести постоянные ζ = exp(
в виде
) − 2 ζ exp(−
) − (1 − ) exp (−
=
ζ exp(−2
)
(6)
Для кубических кристаллов расстояние между атомами можно выразить, используя
кристаллографические индексы и параметр решетки. Тогда
=
где
,
,
+
+
=
,
−длина ребра элементарной ячейки кубического кристалла (параметр решетки),
− кристаллографические индексы положения i-го атома относительно центрального
атома в решетке кристалла. С учетом этого соотношения выражение (6) можно представить
удобной для расчетов формулой
) − 2 ζ exp(−
) − (1 − ) exp (−
( )=
ζ exp (−2
)
Энергия взаимодействия атомов в металле в соответствии с (1) будет определяться
выражением
ζ ∑ exp(−2
( )=
)−
ζ ∑ exp(−
)−
(7)
∑ exp (−
− (1 − )
).
Суммирование производится по всем атомам один раз, а так как атомы металла идентичны,
то полученную сумму умножают на число атомов N . Коэффициент ? появляется из-за того, что в
качестве центрального может быть любой атом решетки, а энергия взаимодействия центрального
атома с i-м атомом равна энергии взаимодействия i-го атома с центральным атомом.
Для вычисления энергии взаимодействия всех атомов в кристалле с использованием (7)
необходимо определить значения 5 параметров ( , , , , ). Поэтому следует составить систему
из 5 уравнений. Энергия взаимодействия всех атомов равна энергии связи или энергии
сублимации кристаллa Es. Следовательно, первое уравнение для определения параметров будет иметь
следующий вид:
)−
)−
=
ζ ∑ exp (−2
ζ ∑ exp(−
(8)
∑ exp(−
).
− (1 − )
В равновесном состоянии кристалла, когда = , энергия взаимодействия атомов между
собой будет минимальна. На основании этого можно составить следующее уравнение:
)+
)+
=0=−
ζ ∑
exp (−2
ζ∑
exp (−
(9)
+
1
2
(1
N
i
) ND
M i exp (
a0 M i ).
Третье уравнение находим, связав вторую производную энергии от расстояния между
=
атомами со сжатием кристалла. Известно, что
=−
, где
− модуль объемной
упругости, − объемная сжимаемость, − объем кристалла (в случае кубического кристалла
=
, − число элементарных ячеек в кристалле). Если на одну элементарную ячейку
приходится атомов (в ОЦК решетке 2 атома, а в ГЦК 4 атома), то общее число атомов в
кристалле будет
=
. Поэтому
=
,
=
. Соответственно,
=
.
Тогда
−
9
=2
ζ
−
M exp(−2
(1 − )
∑
)−
exp(−
M exp(−
ζ
).
)−
(10)
Четвертое и пятое уравнения составляются на основе модулей упругости кубического
кристалла
и
. Здесь необходимо сделать небольшое отступление, чтобы пояснить, как
модули упругости связываются с энергией взаимодействия атомов в кристалле.
Пусть в недеформированном кубическом кристалле расстояние между атомом, взятом в
качестве центрального, и другим атомом будет R=xi+yj+zk, где x,y,z – проекции радиуса-вектора
на координатные оси (здесь и далее полужирным шрифтом записаны векторные величины).
При деформации кристалла изменится не только расстояние между рассматриваемыми
атомами, но величина ортов и углы между ними. Связь измененных ортов с первоначальными
устанавливается следующими соотношениями:
= (1 + e ) + e + e ,
= e + (1 + e ) + e ,
(11)
= e + e + (1 + e ) ,
где ехх, еху, еух, еуу, ехz, еzx, еzу, еzz, компоненты тензора деформации, которые с точностью до
первого порядка равняются относительным изменениям ортов при деформации кристалла.
При малых деформациях кристалла смещение атома r будет
= ( − ) + ( − ) + ( − ).
Или с учетом (11)
=
·
+ ·
+ ·
+ ·
+ ·
+ ·
+
+ ·
+ ·
+ ·
.
(12)
Плотность упругой энергии деформированного кристалла можно записать в виде [5]:
=
+
+
+
+
+
+
+
+
,
(13)
где
,
,
– модули упругости. Из (13) можно получит следующее соотношение:
=
.
(14)
Формула (14) применима в рамках представления кристалла однородной и непрерывной
средой, т.е. в рамках континуального приближения. В случае кристалла, состоящего из атомов,
расположенных на определенных расстояниях друг от друга, соотношение (14) должно быть
записано в следующем виде:
( )
=∑
( )
=∑
( )
∑
.
( )
=∑
.
=
(15)
Модуль упругости в виде (15) позволяет суммировать вклад всех атомов в плотность
энергии деформации. Далее следует учесть, что смещение i-го атома при деформации
можно
выразить через изменение параметра решетки
. При этом вполне приемлемо принять
≅ 1.
Тогда соотношение (15) можно представить в виде
( )
=∑
( )
=∑
.
(16)
Подобным образом получаем формулу, связывающую другой модуль упругости со второй
производной плотности упругой энергии,
( )
=∑
.
(17)
Плотность энергии – это общая энергия кристалла E, отнесенная к его объему V. Поэтому
четвертое и пятое уравнения для определения параметров будут иметь следующий вид:
( )
∑
=
.
=
( )
∑
.
(18)
Подставляя в (18) ( ), получаем выражения, которые позволяют определить параметры
полной энергии взаимодействия атомов кристалла, а если известны параметры, то вычислить
модули упругости кристалла
и
ζ
=
2ζ
m exp(−2αaM ) −
m exp (−αaM ) −
−
=
ζ
2ζ
(
m
)
∑ m exp(−βaM ).
exp (−2αaM ) −
m n exp (−αaM ) −
(19)
(1 − )
m n exp (−βaM ).
2
Таким образом, из соотношений (8)-(10) и (19) можно найти формулы, позволяющие
вычислить искомые параметры
, , , и . Однако этими соотношениями можно
воспользоваться, если
, и M изменяются монотонно при увеличении N. Ранее было показано
[4], что M с увеличением числа атомов, окружающих центральный атом, меняется не монотонно.
Так, для ГЦК решетки, первые 12 атомов, окружающих центральный атом, имеют одинаковый
−
=
√
. У следующих 6 атомов
= 1, затем у 24 атомов
=
и т.д. Поэтому суммы,
входящие в уравнения (8)-(10) и (19), должны иметь следующий вид:
exp(−2
=
exp(−
=
),
M exp(−2
),
=
M exp(−
),
=
=
m
exp(−
=
),
M exp(−
=
=∑ ∑
),
),
exp(−2
=
=
=∑ ∑
),
M exp(−
),
M exp(−
),
m exp(−2αa M ) ,
=
m exp(−αa M ) ,
=
m exp(−βa M ) ,
exp(−2αa M ) ,
=
m
=
m
(20)
exp(−αa M ) ,
exp(−βa M ).
Здесь L- номер конфигурационного слоя атомов, находящихся на одинаковом расстоянии от
центрального атома. С помощью j задается суммирование. Для ГЦК решетки при j=1
суммирование осуществляется с 1-го по 12-ый атом, окружающих центральный атом, при j=2 – c
13-го по 18-ый атом, при j=3 – с 19-го по 42-ой атом и т.д. С учетом сумм (20) уравнения (8)-(10) и
(19) трансформируются в следующую систему:
−18
=4
−2
−
(1 − )
,
(21)
Система уравнений (21) аналитически не решается. Для вычисления параметров
используется такой алгоритм:
1) задаются , и
для выбранного металла;
2) вычисляются суммы (20);
3) первые три уравнения системы (21) преобразуются в систему линейных алгебраических
уравнений, решение которых проводится методом Гаусса;
4) из найденных решений находится ;
5) определяется
6) определяются
=
и .
;
Энергия
взаимодействия, Е, эВ
После определения всех параметров они вставляются в формулу (7) и проводится расчет
энергии взаимодействия = 1061 атома в металле.
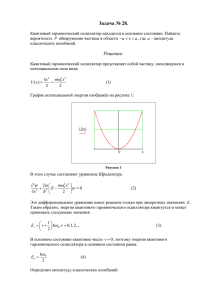
На рис. 1 показана энергия модельного кристалла Al при отсутствии вакансий в металле и
при ее наличии в различных областях металла, определенных в виде конфигурационных сфер,
окружающих центральный атом. После вычисления энергии взаимодействия атомов при наличии
и отсутствии вакансий, с помощью метода, описанного в [4], была вычислена энергия миграции
вакансий
в Al. Было получено значение
= 0,54 эВ, которое лучше согласуется с
экспериментальными значениями, находящимися в интервале 0,58-0,62 эВ [6], чем
значение
, вычисленное в [4]. Это объясняется тем, что в данной работе при определении
энергии взаимодействия атомов в металле помимо потенциала парного взаимодействия атомов
был учтен вклад электронов проводимости через энергию погруженного атома, а в [4]
использовался только потенциал парного взаимодействия Морзе.
-21,2
-21,4 0
5
10
15
-21,6
-21,8
Al
-22
-22,2
N конфигурационной сферы
Рис. 1. Зависимость энергии взаимодействия атомов в кристалле от номера конфигурационной сферы,
содержащей вакансию.
ЛИТЕРАТУРА
1. Dow M.S., Baskes M.I. Embedded atom method: Derivation and application to impurities, surfaces and
other defects in metals. //Phys. Rev. 1984, v. B26, No 12, p. 6443-6453.
2. Dow M.S. Model of metallic cohesion: The embedded atom method. //Phys. Rev. 1989, v. B39, No 11, p.
7441-7452.
3. Foiles S.M., Baskes M.I, Dow M.S. Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni,
Pd, Pt, and their alloys. //Phys. Rev. 1986, v. B33, No 12, p.7983-7991.
4. Искаков Б.М. Метод определения энергии вакансий в ГЦК металлах. //Перспективные материалы.
2011. №6. С. 91-97.
5. Киттель Ч. Введение в физику твердого тела. М., Наука. 1978, 792 с.
6. Орлов А.Н., Трушин Ю.В. Энергии точечных дефектов в металлах. М., Энергоатомиздат, 1983. 80
с.
Резюме
Жаңа әдістеменің қолдануымен кубты кристалдағы өзара әсерлесу энергиясын есептеу алгоритмі
көрсетілген.
Summary
The algorithm for computing the interaction energy of a cubic crystal have been shown, using a new
technique.
КазНТУ им. К.И. Сатпаева
Поступила 10.02.12 г.