")
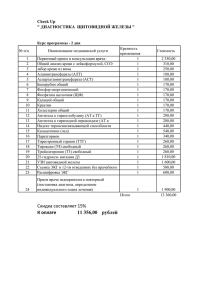
Патент на способ получения трийодпроизводного. Изобретение относится к способу синтеза алифатических трийодпроизводных трийод- N,N-диметилгидразения. Йодосодержащие лекарственные препараты активно используются в лечении зоба и рака щитовидной железы [1]. В их числе препараты тиреоидных гормонов: монокомпонентные: лиотиронин, левотироксин натрия; комбинированные: тиреотом; антитиреоидные средства (снижают уровень гормона щитовидной железы): тиоамиды: пропилтиоурацил, тиамазол; анионные ингибиторы: калия перхлорат; рентгенконтрастные средства (применяются для улучшения визуализации щитовидной органических полийодпроизводных являются:\ железы при рентгеновской диагностике): йопаноевая кислота; радиоактивный йод (131) выпускается в форме Йодотопа; препараты иодидов: иодиды калия (таблетки по 100200 мкг) и натрия (порошок), раствор Люголя (йод в водном растворе йодида калия). Основными методами получения ароматических и жирноароматических полийодпроизводных являются: 1. 2. 3. Подход к алифатическим аналогам более сложен. …………… Наиболее близким к заявляемому является……………………………………………………………………………… ……………………………………………………………………………………….. Однако он имеет определенные недостатки: (п е р е ч и с л и т ь) Мы предлагаем совершенно необычнный способ получения низкомолекулярного трийодпроизводного взаимодействием ………………………………………………………………………………………… … Достоинствами данного подхода являются: 1. 2. 3. Примеры (( разные мольные соотношения, условия) ФОРМУЛА ИЗОБРЕТЕНИЯ Среди них Левротироксин и Лиотиритин - синтетические гормоны щитовидной железы способствуют естественной выработке териоидных гормонов (заместительная терапия) и регуляции синтеза ТТГ (тиреотропного гормона, синтезируемого передней долей гипофиза) (супрессивная терапия). Биологическое действие Левротироксина основано на двух этапах: химическом превращении в Трийодотиронин; активации териоидных рецепторов. На первом этапе одна часть Левротироксина образует комплекс с тироксинсвязывающим белком, другая под действием дейодазы превращается в трийодотиронин. На втором этапе дейодированная форма (трийодотиронин) активирует териоидные рецепторы, открывая «сдерживающий» белок теплового шока. Несмотря на распространённость применения Левротироксина в медицине при гипотериозе (дефиците гормонов щитовидной железы), его эффективность сводится к минимуму, поскольку основная часть исходного соединения связывается с белком (тироксинсвязывающим белком, транстеритином или обладающим наименьшим сродством альбумином) и не оказывает дальнейшего воздействия. Проблема увеличения биодоступности была решена созданием синтетического трийодтиронина - Лиотиронина. Данное соединение не образовывало комплекс с тирозинсвязывающем белком и поэтому в большем количестве способствовало лечению. Однако Лиотиронин оказывает непродолжительный эффект в сравнении с предыдущем аналогом (латентный период Левротироксина 24-48 ч, Лиотиронина - 4-8 ч) и рассчитан на определённую возрастную категорию и группу здоровья (лица до 50 лет, не страдающие сердечно-сосудистыми заболеваниями). Интересным представителем антитериоидных препаратов является Йопановая кислота, также применяющаяся в качестве рентгеноконтрастного средства в холеграфии (рентген желочного пузыря). Данная кислота легко выводится из организма желчегонными средствами. Радиоактивный йод применяется в лечении диффузно-токсического зоба, различных видов онкологии щитовидной железы. При этом возникновение рецидивов и развитие эуториоза после приема йодного изотопа составляет до 15%, что значительно меньше в сравнении с хирургическим лечением, где те же показатели достигают 50%. Основными недостатками данного лекарственного средства является его высокая токсичность и повреждение эпителия щитовидной железы. Йодид калия оказывает на щитовидную железу тереостатическое действие, выражающееся в увеличении концентрации йода внутри железы, что способствует снижению выработки териоидных гормонов и, соответственно, снижению размеров щитовидной железы. Однако данный препарат не может приниматься монотерапевтически и используется, как правило, совместно с другими лекарственными препаратами, например, с радиоактивным йодом для снижения его токсичности. Однако тереостатическое действие данной соли непродолжительное, ко второй-восьмой неделе щитовидная железа перестаёт отвечать на терапию выработкой йода, и гипертериоидное состояние возникает вновь, часто в более тяжёлой форме. Синтез йодированных производных имеет огромное значение в химии и терапии лекарств (зоб, рак щитовидной железы). На сегодняшний день существует множество способов йодирования ароматических и алифатических соединений. Например, введение йода в ароматические кольца мезитилена, пара-ксилола, трет-бутилбензола осуществляются в присутствии йодноватистой и уксусной кислот в водном растворе при нагревании реакционной смеси в течение 15 минут. В случае бензола реакция проводится при 200оС в запаянной трубке. В качестве катализатора прямого йодирования бензола также выступает надсернокислый натрий в среде уксусной кислоты, реакция проводится при непродолжительном нагревании в течение 15 ч. 5-йодо-2-хлор-параксилол получают экстракцией четырёххлористым углеродом из уксусной кислоты. Сначала при температуре 135оС в течение 2 минут прибавляется смесь серной и азотной кислоты (1:1), затем реакционная смесь нагревается до 250оС в течение 45 минут, что приводит к целевому продукту [3]. Алифатическое йодированное производное – метил-йод получают в среде метанола в присутствии йода и смеси красного и жёлтого фосфора при температуре 70-75оС или в присутствии диметилсульфата и йодида калия при температуре 60-65оС [4]. Способ йодирования ароматических соединений через соли диазония [5] осуществляется в два этапа: диазотирование по Гриссу и реакцию Зондмейера. Диазотирование по Гриссу проводится в водном растворе соляной кислоты и ариламина. При 0-5оС по каплям прибавляется раствор нитрита натрия. Необходимо следить, чтобы температура реакционной смеси не превышала 5оС и переодически добавлять кусочки льда в раствор во избежание взрыва азотистой кислоты и диазосоединения. Избыток азотистой кислоты нейтрализуют мочевиной до предотвращения выделения пузырьков газа (азота и углекислого газа). Затем для осуществления реакции Зондмейера соль диазония в воде приливают к предварительно охлажденному водному раствору йодида калия, после чего реакционную смесь оставляют на 2 часа. Затем раствор нагревают, предварительно присоединив к колбе обратный холодильник, для равномерного испарения азота из раствора. Раствор охлаждают и приливают к нему концентрированный щелочной раствор. Йодбензол перегоняют с водяным паром, отделяют от воды на делительной воронке, сушат над хлоридом кальция. Выход – 90%. Несмотря на относительную лёгкость в диазотировании и последующем йодировании ароматического соединения, к недостаткам данного способа можно отнести строгое соблюдение низкой температуры ввиду взрывоопасности азотистой кислоты и образующейся соли диазония, необходимость контроля бурного выделения азота. Также стоит отметить, что синтез предполагает выполнение большого количества операций для устранения побочных продуктов и выделения целевых: добавление мочевины для разрушения азотистой кислоты, нагревание водного раствора с обратным холодильником для удаления азота, добавление щелочи к раствору целевого продукта, отгонка йодобензола с водяным паром с последующей экстракцией. Сложность в осуществлении синтеза также состоит в сооружении сложных химических установок – с обратным холодильником (для удаления азота при нагревании реакционной смеси), установки для перегонки с водяным паром (для выделения йодбензола). Наиболее близким к нашему способу является синтез йодистого метила [4]. Способ основан на взаимодействии диметилсульфата, йодида калия в водном растворе в присутствии карбоната кальция. Реакционная установка состоит из трёхгорлой колбы, снабжённой ректификационной колонкой для отделения целевого йодистого метила, кипящего при 42оС и побочных метанола (t(кип.)=64оС) и триоксида серы (t(кип.)=45оС), (колонка необходима, поскольку синтез проводится в диапазоне температур 60-75оС), холодильника Либиха и приёмника, помещённого в ледяную баню. В водный раствор в трёхгорлой колбе добавляют йодид калия и карбонат кальция в качестве катализатора. Реакционную смесь нагревают до 60-65оС, с помощью капельной воронки добавляют диметилсульфат в течение двух часов, что необходимо ввиду высокой экзотермичности и скорости реакции (после смешения образовавшийся метил-йод быстро отделяется от реакционной смеси, так как его температура кипения 42оС). По окончании добавления метилирующего агента температуру реакционной смеси повышают до 65-70оС на 40 минут для ускорения дистилляции йодистого метила. Вещество отделяют от воды, сушат над хлоридом кальция, перегоняют над кристаллами йодида калия. Выход целевого продукта составляет 90-94%. Основным недостатком данного способа является токсичность диметилсульфата, что создаёт необходимость работы в нитриловых перчатках в вытяжном шкафу и максимального ограничения контакта с данным веществом. Синтез предполагает сбор сложной реакционной установки, контроль и изменение высоких температур (с 60 до 70оС), длительность добавления диметилсульфата, что обусловлено быстрым выделением из реакционной смеси йодистого метила и сложностью его фиксации. Мы предполагаем принципиально новый синтетический подход к йодированию соединений, содержащий утилизацию подлежащего к уничтожению некондиционного ракетного топлива «Гептил» (несимметричного диметилгидразина (НДМГ)). В отличие от описанных выше синтезов в нашем способе все стадии синтеза проводились при комнатной температуре, без необходимости сбора сложных химических установок. Кроме того, разработанное нами трийодпроизводное будет обладать сравнительно низкой себестоимостью - 0,01 г трийодида N,N-диметилгидразения составляет 50 рублей, за вычетом стоимости расворителей. В то время как стоимость 0,0125 г (125 мкг по 100 таблеток) Левротироксина (L-Тироксина) составляет 180 р., 0,01г (100 мкг по 100 таблеток ) Лиотиронина (Тибона) – 8900 р (на сайте Авис Фарм), 35 г (350 мг в 100 мл) Йопановой кислоты (Омнипак) - 17689 р (на сайте apteka.ru), 0,0224 г (200 мкг по 112 такблеток), йодида калия - 136 р. (на сайте ЕАптека). Цель изобретения – разработка простой эффективной методики синтеза трийодида N,N-диметилгидразения. Техническим результатом является относительно низкая себестоимость реактивов, упрощение способа получения трийодида N,N-диметилгидразения, выгодная цена потенциального лекарственного препарата с одновременной переработкой некондиционного ракетного топлива «Гептил» (несимметричного диметилгидразина (НДМГ)). Технический результат достигается модифицированной реакцией Видеквиста [2], предполагающей смешение диметилгидразона глиоксаля, броммалоннитрила, йодида калия в водно-диоксановом растворе, температура варьируется от 20 до 40оС в зависимости от выбранных условий реакции. Сущность изобретения состоит во взаимодействии двух эквивалентов броммалоннитрила, одного эквивалента N,N-диметилгидразона глиоксаля и трёх эквивалентов йодида калия в водно-диоксановой среде. Реакция проводится при стандартных условиях (20оС, 1 атм.) 72 часа, под действием ультразвука при частоте 90-100 Гц, температуре 35-40оС 5 минут, при микроволновом излучении 150W 8 минут. Целевой продукт, выпавший в осадок, отфильтровывают на фильтре Шота, промывают холодным этилацетатом. Схема модифицированной реакции Видеквиста с N,N-диметилгидразоном глиоксаля: Для получения используют следующие соединения: некондиционное, подлежащее утилизации ракетное топливо на основе НДМГ («Газпром Нефтехим» г. Салават), глиоксаль 40% (ГОСТ 4517-87), дистиллированная вода (ГОСТ 58144-2018), дихлорметан (ГОСТ 9968-86), сульфат натрия (ГОСТ 4166-76), малоннитрил (СКТБ Технолог, CAS № 109-77-3), бром (ГОСТ 4109-79), йодид калия (ГОСТ 4232-74), диоксан (ГОСТ 10455-80), этилацетат (ГОСТ 8981-78). Глиоксаль 40%, дихлорметан, малоннитрил, бром были приобретены у коммерческих поставщиков и использовались без дальнейшей очистки. НДМГ был перегнан при температуре 64оС. Протекание реакции и чистоту продуктов контролируют методом ТСХ на пластинах Sorbfil (пятна визуализировались в УФ-свете, при обработке парами йода или при нагревании). Температура плавления и разложения определена на приборе Optimelt MPA100. Спектры ЯМР 1Н и 13 С записаны в ДМСО-d6 с внутренним стандартом ТМС на спектрометре Bruker AVANCE400 WB при рабочей частоте 400,13 МГц для 1H и 100,61 МГц для 13C. Данные рентгеновской дифракции монокристаллов были собраны на дифрактометре Bruker Smart Apex II CCD. Структуры расшифрованы прямым методом с использованием программы SHELXT-2014/5 и уточнены полноматричным методом наименьших квадратов по F2 с использованием программы SHELXL-2017/1. Расчеты проводились с использованием пакета программ WinGX-2014.1. Неводородные атомы уточнены анизотропно. Синтез диметилгидразона глиоксаля: К 4 г 40% раствора глиоксаля (0,07 моль) в 50 мл воды по каплям прибавляли 4,14 г (0,07 моль) НДМГ. Водный раствор перемешивали в течение 1 часа, затем экстрагировали продукт реакции дихлорметаном (25мл*3). Органический слой сушили над сульфатом натрия в течение 1,5 часа. Дихлорметан отогнали на роторе при температуре 35оС, 150 об./мин. Диметилгидразон глиоксаля перегнали под вакуумом. Получили жидкий продукт светло-жёлтого цвета, выход 70% (4,8 г). Синтез броммалоннитрила: К 1,32 г (0,02 моль) малоннитрила в 10 мл воды добавили 3,2 г (0,02 моль). Раствор перемешивали на магнитной мешалке. Через несколько минут после обесцвечивания реакционной смеси в осадок выпала жёлтая эвтетика (продукты диспропорционирования броммалоннитрила – малоннитрил и диброммалоннитрил). Её использовали в дальнейшем синтезе, загрузки брали как на броммалоннитрил. Синтез трийодида N,N-диметилгидразения Пример 1: К 5,97 г (0,04 моль) двум эквивалентам броммалононитрила и 10,25 г (0,06 моль) йодида калия в водном диоксане прикапывают 2,06 г (0,02 моль) диметилгидразона глиоксаля . Реакционную смесь оставляют при комнатной температуре на 72ч. После выпадения игл осадка (трийодпроизводного) реакционную смесь фильтруют, осадок промывают этилацетатом. Выход 78% (8,19 г). Пример 2: Загрузки и последовательность добавления реагентов в водно-диоксановый раствор аналогично примеру 1. Реакционную смесь помещают в ультразвуковой реактор с частотой 90-100 Гц на 5 минут. Выпавший аморфный осадок трийодпроизводного фильтруют, промывают этилацетатом. Выход 81% (8,51 г). Пример 3: Загрузки и последовательность добавления реагентов в водно-диоксановый раствор аналогично примеру 1. Реакцию проводят в микроволновой печи при излучении 150W в течение 8 минут. Мелкокристаллический осадок трийодпроизводного фильтруют, промывают этилацетатом. Выход 77% (8,13 г). Данные трийодида N,N-диметилгидразения: т. пл. 168оС с разложением. ЯМР 1Н , м. д., J, Гц (ДМСО-d6): 3.59 (d, J = 14.5 Hz, 6H, N(CH35,4)2), 3.31 (d, J = 23.8 Hz, 6H, N(CH36,2)2), 2.53 (s, 1H, CH7). ЯМР 13 С м. д., (ДМСО-d6): 69.56 (CH17), 47.72 (N(CH35,4)2, 44.88 (N(CH35,4)2). Найдено, C, 11.58; H, 2.77; I, 74.42; N, 11.01. C5H13N4I3. Вычислено,%: C, 11.78; H, 2.57; I, 74.66; N, 10.99. Таким образом, разработана эффективная методика получения потенциального лекарственного препарата в отношении щитовидной железы с одновременной переработкой некондиционного ракетного топлива. Список литературы [1] Рождественский, Д. А. Препараты гормонов щитовидной железы и антитиреоидные средства / Д. А. Рождественский // Вестник фармации. - 2003. - № 3. - С. 61-79. [2] Бардасов, И. Н. Синтез 2, 2, 3, 3-тетрацианоциклопропилкетонов и взаимодействие их с о-нуклеофилами / И. Н. Бардасов; О. В. Каюкова; Я. С. Каюков; О. В. Ершов; О. Е. Насакин; В. А. Тафеенко// Журнал органической химии. - 2009. - Т. 45, вып: вып. 9. - С. 1340-1351. Библиогр.: с. 1350-1351 (23 назв. ) . - ISSN 0514-7492 [3] Хиккинботтом В. Реакции органических соединений. – Рипол Классик, 1939. [4] Гарольд С. К., Луис Ф. Ф. Органические синтезы, Сборник, том 2, стр.399 (1943); Том 13, стр.60 (1933) [5] О. А. Голубчиков Органический практикум Санкт-Петербург Нии химии сПбГу – 2012