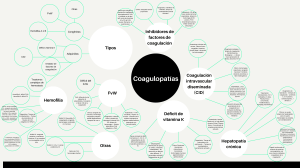
Otras Anticuerpos dirigidos a factores de coagulación (principalmente al VIII), el inhibidor es de tipo IgG contra los dominios A2 y C2 de FVIII; causas: fármacos, E. autoinmunes FvW Hemofilia A o B Clínica: Sangrado severo y equimosis Tratamiento: 1/3 de los casos remite de forma espontánea, detener la hemorragia (FVIII porcino, FVIII recombinante activo), inhibir síntesis de anticuerpos Inhibidores de factores de coagulación Congénitas Tipos Déficit vitamina K Diagnóstico: Identificar el inhibidor, tiempo de tromboplastina parcial activado + Adquiridas Tratamiento: Eliminar las causas, Plasma fresco congelado, Heparina (puede llegar a aumentar las hemorragias) Diagnóstico biológico: + tiempo de coagulación, trombocitopenia, fibrinógeno +, + dímero D por generación de fibrina, descenso AT-III CID Inhibidor de factores de coagulación Trastornos complejos de hemostasia FvW Hemofilia A: déficit FVIII; Hemofilia B: déficit FIX Hemofilia Clínica: Severa (actividad <1%, sangrado espontáneo antes de 6 mes o intracraneal en parto); Moderada (actividad 1-5%, sangrado antes de 2 años con leve traumatismo); Leve (niveles >5% y <40%, sangrado en traumatismos importantes Diagnóstico: Tiempo de tromboplastina +; Tiempo de protrombina N; Niveles de FvW antigénico N; RIPA N Tratamiento: Hemofílicos leve y moderados (DDAVP y antifibrinolíticos); Hemofílicos graves (tratamiento con FVIII o IX); analgesia (paracetamol, sin AINES) Coagulopatías Déficit del FvW Clínica: Hemorragias (equimosis, sangrado mucoso); Trombosis (lesiones necróticas o fallo multiorgánico) Coagulación intravascular diseminada (CID) Etiopatogenia: + en la expresión de factor tisular en sepsis, inducidos por IL-6 y TNFalfa; exposición FT por traumatismo o quemaduras; Neoplasias; Hemangiomas gigantes FvW: Adhesión de las plaquetas al subendotelio y transporta el factor VIII de la coagulación Déficit FXI (hemofilia C): coagulopatía congénita. Hemorragias moderadas en mucosas. Déficit FII: sangrado posterior a maniobras invasivas. Si es completa es incompatible con la vida. Clínica: sangrado de mucosas, menorragia, sangrados postparto, puede diagnosticarse después de una cx Diagnóstico: Historial clínico, pruebas de laboratorio (método PFA100: tiempo de hemorragia +; Niveles plasmáticos FvW antigénico y FVIII- Otras Déficit FV, VII y X: similar al déficit de FII Déficit de FXIII: hemorragias tardías. Tratamiento: + FvW circulante y mejorar su función: 1-deamino-8-Darginina-vasopresina; agentes antifibrinolíticos; tratamiento con hemoderivados Causa principal de déficit de factores: II, VII, IX, X (dependientes de K); déficit anticoagulantes naturales (proteínas C y S) Déficit de vitamina K Etiopatogenia: Disminución de aporte, E. celíaca, colestasis intrahepática (- de absorción por falta de sales biliares), E. hemorrágica neonatal, Antagonistas de vit. K Clínica: Equimosis, Hematomas (subcutáneos, musculares), Hemorragias mucosas (gastrointestinal, genitourinario) Activación intravascular generalizada de la coagulación por formación de fibrina y oclusión trombótica en los vasos pequeños y medianos Tratamiento: Aportar vitamina K (10 mg diarios durante 3 días), Plasma fresco, Concentrado de complejo protrombínico activado Diagnóstico: Tiempo de protrombina (vía extrínseca) +; Tiempo de tromboplastina parcialmente activado + Hepatopatía crónica Tratamiento: Desmopresina, Vit. K (10mg/día x 3 días), Plasma fresco concentrado, Antifibrinolíticos, Complejo protrombínico Diagnóstico de laboratorio: - tiempos de coagulación, - proteínas C, S en plasma, trombocitopenia, + dímero D Clínica: no suelen sangrar las hepatopatías, pero el cuadro hemorrágico se precipita por complicaciones. Alteraciones hemostásicas producidas por E. hepática: Hipo coagulabilidad (fibrinógeno, protrombina); Hiperfibrinolisis (síntesis de antifibrinolíticos); Trombocitopenia (relacionada con hiperesplenismo) Hígado involucrado en síntesis de proteínas procoagulantes, así como función inhibidora o reguladora de la coagulación