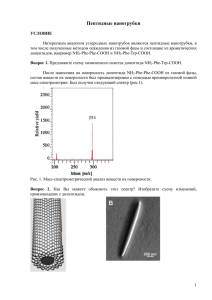
Лекция 3. Химия и теоретические основы оксиалкилирования по атомам кислорода, серы и азота. Химия и теоретические основы синтезов из α-окисей. Алкилирование по атомам O, S и N представляет собой основной метод синтеза соединений с простой эфирной связью, меркаптанов и аминов. O-алкилирование: Наибольшее значение имеют две группы процессов: 1) Межмолекулярная дегидратация спиртов: 2) В ROH + ROH ↔ ROR+ H2O промышленности имеет значение только О-алкилирование хлорпроизводными. Известно, что взаимодействие хлорпроизводных RCl со спиртами ROH является обратимым и весьма медленным процессом, поэтому его проводят в присутствии щелочей, которые переводят спирты и фенолы в алкоголяты и феноляты: ROH +NaOH ↔ RONa + H2O Равновесие реакции тем больше сдвинуто вправо, чем выше кислотность гидроксилпроизводных (фенолы > гликоли > одноатомные спирты) и чем меньше концентрация воды в смеси. Для фенолов, обладающих более высокой кислотностью, равновесие почти полностью сдвинуто в сторону фенолята. В промышленности О- алкилирование в наиболее крупных масштабах используют для производства этил- и бензилцеллюлозы. В начале целлюлозу обрабатывают NaOH, затем нагревают в автоклаве под давлением с соответствующим RCl (этил- или бензилхлоридом). Интересным продуктом такого же типа является карбоксилметилцеллюлоза (КМЦ), которую получают взаимодействием щелочной целлюлозы с монохлорацетатом натрия. Соли КМЦ способны сильно увеличивать вязкость среды(1%-ный водный р-р имеет вязкость в 2000 раз большую, чем у воды). КМЦ применяют как загуститель латексов и смазочных масел, стабилизатор эмульсий и суспензий. Ее добавки к СМС позволяют избежать обратного оседания загрязнений на ткань. В случае нерастворимых в Н2 О RCl реакционная масса является гетерофазной, поэтому большое значение имеет ее эмульсирование. В зависимости от реакционной способности RCl синтез осуществляют в интервале 60÷200 ℃ иногда с повыш. давлением. При периодическом методе процесс проводят в обычном автоклаве с мешалкой и рубашкой для обогрева и охлаждения. S-алкилирование: Хлорпроизводные при взаимодействие с гидросильфид Na (NaSH) образуют меркаптаны: 1 RCl + NaSH → RSH + NaCl В частности, синтез этилмеркаптана: C2H5Cl + NaSH→ C2H5SH + NaCl C2H5SH используют для синтеза инсектицида меркаптофоса. Высшие первичные меркаптаны (С10 − С15 ) используют при синтезе неионогенных моющих веществ на основе окиси этилена: RSH + H2C RS(CH2CH2O)n n CH2 O Они же применяются в качестве регуляторов полимеризации в производстве синтетических каучуков. Другое важное направление: синтез меркаптанов из олефинов и сероводорода. Эта реакция сходна с прямой гидратацией олефинов водой и так же является обратимой: RCH = CH2 + H2S ↔ RCH – CH3 SH Однако ее равновесие в большой степени смещено вправо, а 𝐻2 𝑆 обладает значительно более высокой реакционной способностью по сравнению с Н2 О. Возможно образование сульфидов, но оно нежелательно, поэтому реакцию проводят в избытке 𝐻2 𝑆 по отношению к олефину (~1,5:1). Наиболее практическое применение имеет изододецилмеркаптан С12Н25SН, применяемый как регулятор низкотемпературной полимеризации при синтезе каучука. N-алкилирование: Для алкилирования аммиака или аминов по атому азота в качестве алкилирующих агентов чаще всего используют: - RCl - ROH Следует иметь ввиду, что при N-алкилировании NH3 и аминов хлорпроизводными образуется вначале амин, который в свою очередь способен реагировать с RCl. В результате получаются последовательно первичный, вторичный и третичный амин, а последний дает соль четырех замещенного аммония: NH3 →RNH2→R2NH→R3N→R4NCl Взаимодействие спиртов с аммиаком и аминами: AlkOH+NH3→AlkNH2+H2O Является экзотермическими и практически необратимым процессом. В подавляющем большинстве спирты реагируют с аммиаком только в присутствии катализаторов H2SO4, 2 или чаще гетерогенных катализаторов кислотного типа: Al2O3, алюмосиликаты, фосфаты алюминия, фосфат аммония. Реакция NH3 cо спиртами также является последовательнопараллельным процессом, сопровождающимся замещением одного за другим всех атомов водорода при азоте и образования смеси, первичных, вторичных и третичных аминов: NH3 → RNH2 → R2NH → R3N Метил- и этиламины в промышленности производятся из спиртов и аммиака. Они применяются в качестве топлива для жидкостных ракетных двигателей и как промежуточные продукты нефтехимического синтеза. Химия и теоретические основы синтезов из α-окисей. Оксиалкилирование – введение оксиалкильных групп в молекулу. Большое значение α-окиси приобрели для производства следующих веществ: - гликолей; - глицерина; - этаноламинов; - неионогенных ПАВ; - ВМС и т.д. Важнейшая группа реакции α-окисей заключается во взаимодействии их с веществами, содержащими достаточно подвижный атом водорода (H2O, ROH, ArOH, H2S, HCN, RCOOH). При этом окисный цикл раскрывается таким образом, что атом водорода присоединяется к кислороду α-окиси, а остаток – к углеводородному остатку α-окиси. I Реакции с раскрытием цикла 1) В результате взаимодействия α-окисей с водой, спиртами и фенолами образуются гликоли или простые эфиры: H2C CH2 + ROH О-алк. O HOCH2 CH2OR оксиэтильная группа 2) С сероводородом и меркантанами – монотиогликоли или тиоэфиры: H2C CH2 + RSH S-алк. O 3 HOCH2 CH2SR 3) С карбоновыми и неорганическими кислотами – сложные эфиры гликолей: H2C C-алк. CH2 + RCO2H RCO2CH2 CH2OH O 4) С аммиаком, с аминами и амидами – этаноламины или этаноламиды: H2C CH2 + N-алк. N H N CH2CH2OH O Во всех перечисленных реакциях в молекулу неорганического или органического вещества вводится β-оксиэтильная (β-оксиалкильная) группа. Эти реакции, которые можно квалифицировать, как О-, S- и N-алкилирования необратимы и сильно экзотермичны. Катализ и механизм реакции I группы Эти реакции α-окисей, протекающие с раскрытием цикла, часто идут в отсутствии катализатора, но сильно ускоряются кислотами или щелочами. Так, если не каталитическая реакция с H2O или ROH происходит при 170-200 оС, то в присутствии небольших количеств щелочи достаточна температура 100-120 оС. Кислотный катализ применяется реже, т.к. α-окиси реагируют с кислотами: HO CH2CH2OSO3H CH2 + H2SO4 H2C O Чаще всего используется щелочной катализ (NaOH, Na2CO3 и т.д.). Фактически катализатором является не сама щелочь, а ее производные – фенолят, алкоголят Na. Так катализатором является основание, сопряженное кислоте-реагенту А (HО¯ , RO¯ , CN¯ , ArO¯ , HS¯ , RСOO¯и т.д.) Добавляемые в реакционную массу основания (NaOH, Na2CO3) быстро образуют производные исходного реагента (т.е. алкоголят, соль кислоты): HA + NaOH H2O NaA H2O + Na+ C2H5ONa + + Например: C2H5OH + NaOH C2H5ONa C2H5O- + Na+ кат-р (RO-) 4 H2O + A- Таким образом, катализатором становится анион реагента: А (HО-, RO-, CN-, ROO-, и т.д.). Механизм реакции включает предварительное активирование α-окиси по ее кислородному атому, что облегчает последующую атаку атома углерода нуклеофилом, идущего с раскрытием цикла: H2C CH2 +HA δ+ δ+ H2C O CH2 Oδ +A- H2C - CH2 δ- - A -A- O водородная связь δ- H HA A HOCH2CH2A Пример: H2C CH2 +ROH δ+ δ+ H2C O CH2 Oδ + RO- H2C - + - OR O водородная связь δ- H HOR HOCH2CH2OR CH2 δ- OR OR- Итак, роль исходного реагента в недиссоциированной форме (HA) состоит в активировании α-окисей за счет возникновения водородных связей. Образующийся при этом комплекс подвергается затем атаке анионом, причем новые связи восстанавливаются синхронно с разрывом старых. По такому же механизму протекает и некаталитическая реакция – с тем отличием, что нуклеофилом является сама молекула реагента. При взаимодействии с веществами, которые сами являются достаточно сильными нуклеофилами (NH3 и амины), раскрытие цикла может происходить и без помощи электрофила: δ+ CH2 + R2NH H2C O H2C CH2 Oδ - 5 NHR2 HOCH2CH2NR2 При наличии в смеси H2O или другими веществами с подвижным атомом Н они активируют прежним способом окисный цикл, и реакция существенно ускоряется, протекая с достаточной скоростью при 40-100 оС. К несимметричным α-окисям присоединение может протекать в двух направлениях: H3C CH3CHCH2A H C CH2 + HA OH O CH3CHCH2OH A Реакции с раскрытием цикла: Раскрытие эпоксидного цикла с его расширением до более устойчивых циклических систем. Этим путем α-окиси взаимодействуют с СО2, СS2, карбонилными соединениями и т.д., например: H2C H3C CHO + H2C CH2 O O HC CH2 O CH3 2-метил-1,3-диоксолан II Процессы β-оксиалкилирования как последовательно-параллельные реакции. При реакциях α-окисей с H2O, ROH, NH3 и др. всегда получаются продукты, которые содержат гидроксильные группы, способные к дальнейшему присоединению αокисей. В результате этого происходят последовательно-параллельные реакции с образованием продуктов все более высокой степени оксиэтилировнаия: 6 H2C + H2 C H2 C CH2 + H2O O CH2 O CH2 H 2C H2C H2 C CH2 O C H2 HO H2 C O O OH диэтиленгликоль (ДЭГ) этиленгликоль (МЭГ) + CH2 OH OH OH CH2 H2C C H2 H2 C OH и т.д. C H2 O триэтиленгликоль (ТЭГ) Получение целлозольвов и карбитолов: H2C + O ROH H2C CH2 CH2 + H2C O CH2 H2C CH2 O OH OR OH OR CH2 H2C карбитол целлозольв H2C + H2 C CH2 O C H2 RO H2 C O C H2 H2 C O OH C H2 и т.д. Эти процессы иногда нежелательны (например, при производстве МЭГ или целлозольва), но в других случаях наоборот используют для получения ДЭГ, карбитолов и неионогенных ПАВ с длинной цепочкой оксиэтильных групп: RC6H4OH + n H2C CH2 R C6H4 O (CH2CH2O)n H O Взаимодействие α-окисей с NH3, аминами, амидами, H2S, RSH также сопровождается последовательными присоединениями молекул α-окиси, но в первую очередь за счет замещения атомов водорода, связанных с N или S: 7 O H2C CH2 + NH3 O OH CH2 + H2C CH2 (HOCH2CH2)2NH диэтаноламин (ДЭА) NH2 моноэтаноламин (МЭА) O + H2C H2C CH2 (HOCH2CH2)3N триэтаноламин (ТЭА) O H2 C CH2 + H2S O H 2C OH + H2C CH2 CH2 H2 C OH SH CH2 H2C CH2 S OH тиодигликоль тиогликоль Закономерности Зависимости состава продуктов оксиэтилирования от соотношения реагентов б) реакции с фенолом или карбоновой кислотой (более сильные кислотные свойства) а) реакции с H2O или спиртами C, % C, % n n n - мольное отношение окись этилена : реагент 1 – реагент, 2 – монозамещенные (МЭГ), 3 – дизамещенные (ДЭГ), 4 – тризамещенные (ТЭГ), 5 – тетра-замещенные 8 В случае а первая стадия присоединения всегда протекает значительно медленнее второй, но последующие имеют примерно такую же скорость, как вторая. При таких последовательно-параллельных процессах образование первого промежуточного продукта 2 невелико и его максимум смещен в сторону низких мольных соотношений окиси этилена и реагента. В случае б первая стадия присоединения четко отделена от последующих и первый промежуточный продукт удается получить с выходом ~100% и только после исчерпания исходного вещества образуются последующие продукты присоединения α-окисей. Для достижения высокого выхода целевого продукта в каждом случае нужно выбирать оптимальное соотношение исходных реагентов с учетом экономических затрат на отгонку и рециркуляцию избыточного реагента. Так, при производстве этилен- и пропиленгликолей или целлозольвов (т. е. при введении одной оксиэтильной группы) реакция всегда проводится при недостатке окиси этилена (мольное отношение от 7:1 до1:15 и более; величина ß1 на рис. а). Для получения монооксиэтилированных производных карбоновых кислот, фенолов, меркаптанов и других веществ с кислотными свойствами мольное отношение исходных реагентов может быть близким к единице (величина (ß 2 на рис. б). Небольшой избыток непревращенного кислотного реагента обеспечивает образование монооксиэтильного производного с выходом, близким к 100%. В отличие от этого, при синтезе полиоксиэтилированных соединений (полигликоли, неионогенные ПАВ) необходим избыток α-оксида, соответствующий желаемой длине цепи. К графику на рисунке б: 9 ArOH + H2C H2C CH2 O CH2 OAr OH ArOH + NaOH ArONa + H2O катализ ArO- + Na+ ArONa H2C CH2OH + OAr смещение влево OAr - ArO + H2C CH2 CH2O- ArOH + H2C смещение вправо H2C O CH2O- OAr Продукты получаемые из окисей этилена и пропилена Из процессов β-оксиалкилирования, гидратации и других превращений α-окисей промышленное значение приобрели реакции со следующими соединениями: O O O CH3 Cl окись этилена окись пропилена окись эпихлоргидрина Окись этилена (tкип=10,6 оС) и пропилена (tкип=33,9 оС) являются летучими жидкостями, обладают заметной токсичностью дают с воздухом взрывоопасные смеси Продукты, получаемые из α-оксидов: Гликоли и их простые эфиры – наибольшее количество α-оксидов расходуется на получение гликолей и их простых эфиров. Этиленгликоль (МЭГ) СН2ОН—СН2ОН (вязкая бесцветная жидкость, tкип=197°С, полностью смешивается с водой). Используется в: - производстве антифризов (смеси с водой, не замерзающие при низких температypax и используемые для охлаждения двигателей в зимних условиях); - производстве полимеров (ПЭТФ полиуретанов, алкидных полимеров и т. д.). 10 (лавсан), ненасыщенных полиэфиров, Промышленный синтез этиленгликоля: гидратация этиленоксида без катализаторов при 170-200°С и 15-кратном избытке воды. Диэтиленгликоль (ДЭГ) НОСН2СН2-О-СН2СН2ОН (бесцветная, смешивающаяся с водой жидкость, , tкип=245°С). Используется для синтеза: - полиэфиров; - сложные эфиры ДЭГ с монокарбоновыми кислотами С7-С10 служат пластификаторами и смазочными маслами; - взрывчатого вещества — диэтиленгликольдинитрата; - в нефтеперерабатывающей промышленности для осушки газов и экстракции ароматических углеводородов. Диэтиленгликоль — второй продукт оксиэтилирования воды, и его получают при меньшем мольном избытке воды (от 4:1 до 5: 1), возвращая промежуточный этиленгликоль на реакцию. Триэтиленгликоль (ТЭГ) и полигликоли (ПГ) – побочные продукты в производстве МЭГ и ДЭГ, густые, смешивающиеся с водой жидкости. В виде сложных эфиров с карбоновыми кислотами С6-С10 используются как пластификаторы и смазочные масла. ПГ получают оксиэтилированием этиленгликоля в присутствии щелочи при 100130°С: ПГ с М менее 600 – вязкие жидкости; с М=4000-6000 – твердые воскоподобные вещества («карбовакс») с низкой температурой размягчения (40—60 °С). Полигликоли имеют значение в качестве смазок, высокотемпературных теплоносителей, пеногасителей, мягчителей. Пропиленгликоль СНзСН(ОН)СН2ОН может заменять этиленгликоль. Получают гидратацией окиси пропилена: Образующиеся побочно ди- и полипропиленгликоли можно использовать для приготовления полиэфиров, пластификаторов и смазочных масел. Целлозольвы — простые моноэфиры ЭГ ROCH2—CH2OH обладают хорошим растворяющим свойствам по отношению к эфирам целлюлозы. В качестве растворителей чаще всего используют этилцеллозольв (ЭЦ), реже – метилцеллозольв (МЦ) и бутилцеллозольв (БЦ): СН3-О-СН2СН2ОН, C2H5-О-СН2СН2ОН, 11 н-С4Н9-О-СН2СН2ОН метилцеллозольв этилцеллозольв бутилцеллозольв БЦ и другие высшие целлозольвы в виде их сложных эфиров с дикарбоновыми кислотами применяют в качестве пластификаторов. Целлозольвы получают взаимодействием окиси этилена с соответствующими спиртами при температуре ~200°С и мольном отношении спирта к а-оксиду от (7-8): 1. Побочными продуктами являются карбитолы используются как растворители, а также для синтеза пластификаторов. Этаноламины. диэтаноламин (ДЭА) Моноэтаноламин NH(CH2CH2OH)2 (МЭА) NH2CH2CH2OH (tкип=268°С) и (tкип=172,2°С), триэтаноламин (ТЭА) N(СН2СН2ОН)3 (tкип=360 °С) – вязкие жидкости, смешивающиеся с водой. Применение – очистка газов от кислотных примесей (H2S, С02). При низкой температуре они образуют с кислотными примесями соли, которые при нагревании разлагаются с регенерацией этаноламинов: NH(CH2CH2OH)2 + H2S 20 oC 80 oC H2S HN(CH2CH2OH)2 Соли этаноламинов обладают поверхностно-активными и пенообразующими свойствами и могут использоваться как компоненты моющих и смачивающих средств. Из этаноламинов синтезируют также морфолин, этиленимин и некоторые взрывчатые вещества. Получают ЭА реакцией этиленоксида с концентрированным водным раствором аммиака при 40-60 °С. По более новой технологии ЭА получают из аммиака и этиленоксида только с небольшой добавкой воды, катализирующей начальную стадию реакции и снимающей индукционный период, при 100-130 °С, давление 7-10 МПа, чтобы сохранить реакционную смесь в жидком состоянии. Способ отличается высокой эффективностью и заметным снижением затрат на отгонку и рециркуляцию воды. Тиогликоли – тиодигликоль: При взаимодействии этиленоксида с меркаптанами получаются тиоэфиры, например, ß-гидроксидиэтилсульфид: Последний служит промежуточным продуктом в производстве пестицида 12 (ядохимиката) меркаптофоса. Сложные эфиры гликолей – карбонаты 1,2-гликоли (алкиленкарбонаты). Из них: CH3 O O C O O O C O этиленкарбонат пропиленкарбонат В последнее время приобрели важное практическое значение в качестве растворителей с высокой диэлектрической проницаемостью, а также как промежуточные продукты органического синтеза. Реакция образования циклических карбонатов 1,2гликолей протекает под давлением при ≈150°С в среде соответствующего алкиленкарбоната и, в отличие от других превращений α-оксидов, является обратимой. β-фениловый спирт – действующее начало розового масла: H2C H2 C AlCl3 CH2 + OH C H2 O Неионогенные поверхностно-активные вещества (ПАВ). ПАВ классифицируют на следующие основные группы: 1) полиоксиэтилированные алкилфенолы (изооктил-, нонил-, додецилфенолы), называемые ОП, с цифрой, отвечающей числу введенных оксиэтильных групп (ОП-7, ОП10): RC6H4OH + n H2C CH2 RC6H4-O-(CH2CH2O)n-H O 2) продукты полиоксиэтилирования высших карбоновых кислот (смеси высших жирных кислот, получаемых окислением парафина): RCOOH + n H2C CH2 O 13 RCOO-(CH2CH2O)n-H продукты полиоксиэтилирования высших спиртов и соответствующих 3) меркаптанов: ROH + n H2C CH2 RO-(CH2CH2O)n-H CH2 RS-(CH2CH2O)n-H O RSH + n H2C O 4) продукты полиоксиэтилирования амидов высших кислот, сульфамидов и аминов: (CH2CH2O)x H RCONH2 + (x+y) H2C CH2 RC O N (CH2CH2O)y H O (CH2CH2O)x H RNH2 + (x+y) H2C CH2 R N (CH2CH2O)y H O 5) полимерные неионогенные моющие вещества, где гидрофобной группировкой служат пропиленгликоли: CH3 + HO H2C H C CH2OH CH O HO CH3 H C CH2O CH2CHOH CH3 CH3 CH3 + nH2C CH O HO H C CH2 --OCH2CH--O-CH2 CHOH CH3 CH3 n CH3 При М ˃˃ 800 в результате наличия СН3–групп обладают достаточной гидрофобностью. При их оксиэтилировании по концевым гидроксильным группам получаются моющие вещества. 14 O HO H + (x+y) C CH2 --OCH2CH--O-CH2 CHOH CH3 CH3 H2C CH2 CH3 n H H-[O-CH2-CH2]x- O C CH2 --OCH2CH--O-CH2 CH -[O-CH2-CH2]y-OH CH3 CH3 n CH3 Все эти продукты являются вязкими жидкостями, пастами или воскообразными веществами, растворимыми в воде. Пенообразующая способность неионогенных моющих веществ, как правило, меньше, чем у ионогенных (например, у алкиларилсульфонатов), и зависит от природы гидрофобной части и длины оксиэтилированной цепочки. Моющая способность неионогенных ПАВ является высокой даже без всяких добавок, сохраняющих моющие свойства в жесткой воде, способны препятствовать обратному оседанию загрязнений на ткань, совместимы с большинством красителей и прочих реагентов. Получают неионогенные ПАВ при 150-180 °С в присутствии оснований в качестве катализаторов (около 0,3% NaOH или метилата натрия) при атмосферном или несколько повышенном давлении (3-5 кгс/см2 или 0,3-0,5 МПа). 15