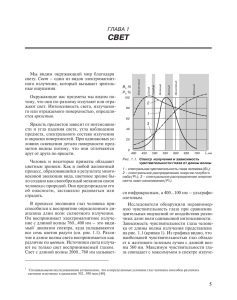
Физико-химические методы анализа и экспертизы товаров народного потребления К.т.н. Гончаров Алексей Иванович 8-903-737-0-738 [email protected] Лекция1 Очень важная • 1. Лирическое отступление – маятник • 2. Учебники • 3. Что это такое – физико-химические методы анализа и зачем они нам нужны • 4. Краткие сведения о строении вещества • 6. Электронная спектрофотометрия • 7. Флуоресцентная спектрофотометрия маятник Маятник — очень интересное устройство. Маятник мог бы качаться вечно, если бы не потери от трения. В верхней точке у маятника скорость равна нулю и потому равна нулю кинетическая энергия, но зато потенциальная энергия максимальна. В нижней точке максимальна скорость маятника и максимальна кинетическая энергия. Так он и качается, перекачивая потенциальную энергию в кинетическую, а кинетическую в потенциальную. Но "вечно" он может двигаться только потому, что не отдаёт своей энергии. А если маятник слегка подтолкнуть, то есть передать ему "кусочек" энергии , то амплитуда его колебаний возрастет — энергия маятника увеличится Вот только толкать маятник нужно с умом. Если подталкивать его "в такт", то амплитуда колебаний возрастет, а если толкать не "в такт" — то амплитуда колебаний уменьшится. А что значит " в такт" и "не в такт"? А вот что: Отклоним покоящийся маятник от положения равновесия и отпустим в свободный полет. Он качнется туда и обратно. Время, нужное ему, чтобы возвратится в исходную точку, называется периодом колебания маятника. Очень часто его измеряют в ГЕРЦах. Герц — это одно колебание в секунду. Обратная величина — это частота колебания. Чем чаще маятник возвращается в исходную точку, тем больше частота его колебаний. Период колебаний маятника зависит от его характеристик. В простейшем случае — к нитке подвешен груз, масса которого сосредоточена в очень-очень небольшом объеме (в точке) — период колебания зависит лишь от одной характеристики маятника — от его длины. Это «математический маятник». Чем длинней маятник, тем больше период его колебаний. И тем меньше частота колебаний. И если мы будем подталкивать маятник каждый раз, когда он вернулся в исходную точку — в то самое место, куда мы его "отклонили", — то энергия его будет увеличиваться всё больше и больше. В этом случае частота нашего подталкивания совпадает с собственной частотой маятника. Это РЕЗОНАНС. Если частота нашего подталкивания вдвое выше собственной частоты маятника или вдвое ниже его собственной частоты, то маятник всё равно будет раскачиваться сильнее — его энергия будет расти за счет энергии наших подталкиваний. Но если частоты подталкивания и собственные частоты маятника будут несоизмеримы, то передачи энергии не будет. Или же подталкивание "не во время" приведет к остановке маятника. В реальности математического маятника нет. Однако все вещи, которые нас окружают, кроме своего прямого предназначения, являются ещё и колебательной системой. И стул, и стол, и воздух в аудитории — это колебательная система. И у каждой — свои собственные колебания Силы гравитации, упругие силы, электромагнитные силы — маятнику всё равно, что заставляет его колебаться. А так как всё вокруг нас состоит из молекул, а молекулы состоят из атомов, связанных пружинками, которые называются химическими связями, то и молекулы и атомы и сами химические связи — это тоже колебательные системы. Свойства этих колебательных систем и используют при физико-химическом анализе Наш основной учебник ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА основаны на зависимости физ. св-в вва от его природы,причем аналитический сигнал представляет собой величину физ. св-ва, функционально связанную с содержанием определяемого компонента. Ф.-х. м. а. могут включать хим. превращения определяемого соед., растворение образца, концентрирование анализируемого компонента, маскирование мешающих в-в и др. В отличие от "классич."химических методов анализа, где аналит. сигналом служит масса в-ва или его объем, в Ф.-х. м. а. в качестве аналит. сигнала используют интенсивность излучения, адсорбционные свойства, взаимодействие вещества с электрическим и магнитным полем, силу тока, электропроводность, разность потенциалов и многое другое. Важное практич. значение имеют методы, основанные на исследовании испускания и поглощения электромагн.излучения в разл. областях спектра. К ним относится спектроскопия (спектроскопия пропускания, рассеяния или излучения) и др. Более половины аналитических работ сейчас выполняют хроматографическими методами. Обычными методами анализа стали хромато-масс-спектрометрические методы. К важным Ф.-х. м. а. принадлежат электрохим. методы, использующие измерение электрич.св-в в-ва (вольтамперометрия, кондуктометрия, кулонометрия, потенциометрия и т. д.). При выполнении Ф.-х. м.а. используют специальную, иногда довольно сложную, измерит. аппаратуру, в связи с чем эти методы часто наз. инструментальными. Многие совр. приборы оснащены встроенными ЭВМ, к-рые позволяют находить оптим. условия анализа (напр., спектральную область получения наиб. точных результатов при анализе смеси окрашенных в-в),выполняют расчеты и т. д. Наука, как и многое другое, родилась в Древней Греции. Благодаря Аристотелю, ученику Платона, написавшему «Аналитики», самым мощным инструментом научного познания мира стала логика. Но современная наука о строении вещества родилась, вероятно, после работ чешского ученого Марци Й. М., оставшихся почти неизвестными, и Ньютона. В 1666 году, направив луч солнечного света с помощью линзы на стеклянную призму, Ньютон получил «спектр» солнечного света, состоящий из красного, оранжевого, желтого, зеленого, голубого, синего, фиолетового цветов, — изображение той самой радужной картинки, которая знакома каждому со средней школы. Опыт И.Ньютона по разложению солнечного света в спектр (1666 г.) Справа - схема стеклянно-водяного объектива И.Ньютона Практически этот опыт происходил, вроде бы, так . Почему это важно? Потому что с простыми вещами легче работать, чем со сложными. Белый свет сложен – он состоит из множества «светов» разного цвета. А свет одного цвета – монохроматический – более прост и лучше подходит для изучения. А призма в этом опыте – это «монохроматор» -- прибор, позволяющий выделять из сложного белого света простые одноцветные (монохромные) света В середине XIX века было установлено (после Максвелла), что распространение света можно описывать почти так же, как описывают распространение звуковых волн или волн, движущихся по колеблющейся струне. И что свет — это электромагнитные колебания, которые можно характеризовать длиной волны. Два представления оптического спектра: сверху «естественное» (видимое в спектроскопе), снизу — как зависимость интенсивности от длины волны. Показан комбинированный спектр излучения солнца. Спектр солнечного света Что такое «спектр»? По латыни spectrum – «представление», образ. В физике спектром называют совокупность всех значений какойлибо физической величины, характеризующей систему или процесс. В 1800 г. английский астроном У.Гершель помещал термометры в разные области спектра и обнаружил, что и за пределами видимого света — за красным светом — тоже есть излучение, которое нагревает термометр. То есть существует ещё и инфракрасное излучение. В 1801 году немецкий ученый И. Риттер помещал в разные области спектра хлорид серебра и открыл ультрафиолетовое излучение — потемнение хлористого серебра было максимальным за фиолетовой областью спектра. А после открытия рентгеновского излучения и и гамма-лучей спектр пришлось расширить и со стороны ультрафиолетового излучения. Нанометр —русское обозначение: нм; международное: nm— единица измерения длины в Международной системе единиц (СИ), равная одной миллиардной части метра (то есть 10−9 метра). Устаревшее название — миллимикрон (10−3 микрона; обозначения: ммк, mμ). В спектроскопии, особенно в инфракрасной области, по оси абсцисс вместо длины волны откладывают «волновое число» (см-1)— число длин волн (в микронах) в 1 см. Так как в 1 см 10000 микрон, то 1000 нм или 1 микрон — это 10000 см-1 , 2000 нм — это 5000 см-1 , 5000 нм —2000 см-1 . Иногда волновое число называют «частотой», хотя частота — это количество колебаний в секунду, то есть скорость света, деленая на длину волны. современный «спектр» простирается от длин волн менее 0.01 нм для гамма-лучей до очень длинных радиоволн что такое длина волны света и откуда она взялась? Вероятно, радужные цвета тонких пленок (слюды, маслянных или мыльных пленок на воде или в мыльных пузырях и т.п.) замечали давным-давно, но лишь в V веке на это обратил внимание ученый — Роберт Гук, которому и приписывают открытие интерференции [лат. inter – между, ferens (ferentis) – несущий, переносящий]. Объяснить интерференцию — радужные цвета на пленке или чередование на пленке (или в других подобных условиях) светлых и темных полос — можно, если предположить, что свет подобен волне. На поверхности воды, например, интерференцию (или наложение) волн, расходящихся от двух источников, наблюдать просто и механизм усиления и уменьшения интенсивности водяных волн при их наложении тоже очень простой — если гребень волны от одного источника накладывается на гребень волны от другого источника, то высота волны возрастает, если накладываются впадины, то возрастает глубина впадины, а если гребень накладывается на волну, то получается гладкое место Что такое интерференция? Рассмотрим волновую картинку, образуемую на воде двумя совершенно одинаково двигающимися поршнями Интерференция волн от двух точечных источников. Рассмотрим два маленьких шарика, колеблющихся на поверхности жидкости. Каждый из шариков возбуждает волну. Налагаясь, эти волны дают интерференционную картину, показанную на анимации. Рассмотрим уравнение, описывающее интерференционную картину. Если пренебречь затуханием, то волна от каждого шарика может быть записана следующим образом: s1=A1cos(wt - kr1); s2=A2cos(wt - kr2); где A1 и A2 - амплитуды волн, r1 и r2 - расстояния соответственно от первого и второго шарика, k = w / v, v - скорость распространения волн. Так как разность D = r2 - r1 много меньше, чем каждое из расстояний r1 и r2, мы можем положить A = A1 = A2. В этом приближении наложение волн s1 и s2 описывается следующим выражением: s = s1 + s2 = 2Acos[ k(r2 - r1)/2 ] cos[ wt - k(r1 + r2)/2 ] Из этого выражения видно, что в точках, для которых r2 - r1 = l (1/2+n) , поверхность жидкости не колеблется. Эти узловые точки (линии) отчётливо видны на анимации. Как мы видим, математический аппарат этого явления очень прост. И позволяет описать не только интерференцию волн на воде, но и объяснить возникновение радужных цветов от пролитого бензина на поверхности воды и образование темных и светлых полос света при многих явлениях Опыт Юнга – 1801 год (объяснение на следующем слайде) Соорудим простейший интерферометр. Возьмем два листа картона. В одном из них прорежем бритвой тонкую щель. В другом прорежем той же бритвой две параллельных щели на небольшом расстоянии друг от друга. Поместим за картоном с одной щелью картон с двумя щелями. А перед картоном с одной щелью поместим источник света. Он будет освещать щель в картоне и эта щель будет сама служить источником света для картона с двумя щелями. И если мы поместим за картоном с двумя щелями белый экран, то в темноте можно будет увидеть на нем чередующиеся темные и светлые полосы. То есть свет будет давать тот же эффект, что и волна на поверхности воды. Типов интерферометров сейчас очень много. Один из них, пожалуй, самый известный и распространенный, изображен на следующем слайде. Картинки, полученные на интерферометре Майкельсона Для монохроматического света интерференционные картины состоят из чередующихся темных и светлых полос, причем для каждой окраски света расстояние между светлыми (или темными) полосами и ширина этих полос разные (при прочих равных). Если принять теорию о волновой природе света, то при расчете этих расстояний, то есть при математической обработке интерференционных картин, получается, что каждому цвету на спектре можно приписать свою длину волны. Не следует только забывать, что последовательность рассуждений, строго говоря, не такая: «Свет имеет волновую природу и потому мы наблюдаем явления интерференции», а такая: «При определенных условиях мы наблюдаем — в радужных пленках или на экране — последовательность темных и светлых полос. Это явление поддается более или менее простому математическому описанию, если предположить, что свет имеет волновую природу, то есть распространяется в пространстве подобно тому, как распространяется волна по поверхности воды». Так как единственная «работающая» теория познания в естественных науках основана на словах Герца: «Теория хороша тогда, когда следствия из модели приводят к моделям следствий», — то есть теория хороша, если следствия из неё можно подвергнуть экспериментальной проверке —то теорию о волновых свойствах света следует считать «хорошей». На рисунке изображен Спектроскоп Кирхгоффа-Бунзена, Annalen der Physik und der Chemie (Poggendorff), Vol. 110 (1860). На котором были выполнены первые исследования по атомной спектроскопии. См. Следующий слайд. Сделаем прибор, почти не отличающийся от того, который сделал Ньютон. Только вместо солнца в качестве источника света будем использовать газовую горелку. Газ (метан, пропан) при горении светит слабым синим светом. Но если поместить в пламя соль натрия или калия или некоторых других металлов, то пламя окрашивается в яркие цвета. Цвет пламени зависит от металла. Помещаем ли мы в пламя сернокислый натрий или хлористый или азотнокислый, пламя от натрия окрашивается в желтый цвет. От бария или стронция -- в красный. Но спектр пламени не похож на солнечный, спектр не сплошной, а полосатый. То есть натрий (и другие металлы) испускают при нагревании свет только в определенной и очень узкой области спектра. Такие спектры называют линейчатыми — спектр испускания металлов состоит из тонких светлых линий , разделенных темными областями. Причем эти линии находятся там, где в непрерывном солнечном спектре находятся темные полосы. Эти темные полосы были открыты 1802 году Фраунгофером. Спектр соли натрия Линии Фраунгофера Фраунго́феровы ли́нии — линии поглощения, видимые на фоне непрерывного спектра звёзд. Были открыты в 1802 году английским физиком и химиком Уильямом Волластоном и исследованы и подробно описаны немецким физиком Йозефом Фраунгофером в 1814 году[1] при спектроскопическихнаблюдениях Солнца. Фраунгофер выделил и обозначил свыше 570 линий, причём сильные линии получили буквенные обозначения от A до K, а более слабые были обозначены оставшимися буквами. В настоящее время астрономы выделяют в спектре Солнца тысячи фраунгоферовых линий. В 1859 году физик Густав Кирхгоф использовал совпадение спектров излучения и поглощения для калибровки оптического инструмента. Он пропускал через призму сначала свет от раскаленного натрия, а затем солнечный свет, добиваясь совпадения спектральных линий натрия с темными линиями в спектре Солнца. И тут он провел опыт, в результате которого выяснилось, что, если солнечные лучи пропустить через окрашенное натрием пламя горелки, темные линии натрия в спектре Солнца становятся еще более темными и выраженными. Иными словами, выяснилось, что раскаленный натрий не только испускает свет определенных спектральных частот, но и поглощает свет тех же длин волн, причем более интенсивно, если источник излучения разогрет до более высоких температур, чем натрий. И тут Кирхгоф понял, что атом химического элемента способен излучать и поглощать свет лишь одних и тех же частот. Иными словами, если атом излучает свет какой-либо частоты, он обязательно способен и поглощать свет этой частоты. (И такая схема единственная была способна объяснить дальнейшее затемнение линий Фраунгофера в спектре Солнца: продолжая излучать на своих спектральных частотах, атомы раскаленного натрия поглощали еще больше энергии излучения на них же.) С этого и началась современная наука о строении атома Полосатый спектр водорода В 1885 году Бальмер вывел формулу, описывающую положение линий в самом простом спектре — спектре излучения водорода . =b(n2/(n2-22)) где – длина волны n = 3, 4, 5, 6; b = 3645,6 Å, Серия Бальмера — длины волн, соответствующие линиям в спектре излучения водо Обозначе ние Hα Hβ Hγ Hδ Hε Hζ Hη Гран ица сери и n 3 4 5 6 7 8 9 ∞ Длина волны, нм 656, 3 486, 1 434, 1 410, 2 397, 0 388, 9 383, 5 364, 6 в преобразованном Ритцем и Ридбергом виде — при замене длины волны на обратную величину («волновое число» или частота) — формула позволяет с высокой точностью вычислить положение спектральных полос и в ультрафиолетовой (серия Лаймана) и в инфракрасной областях (серия Пашена, Брэккета и др.): 1/RZ2(1/n21 – 1/n22) Где — длина волны света в вакууме; R— постоянная Ридберга для рассматриваемого химического элемента; Z— атомный номер, или число протонов в ядре атома данного элемента; n1 и n2 — целые числа, такие что n1 n2 В 1858 году немецкий стеклодув Генрих Гейсслер создал интересный прибор — «трубку Гейсслера». (http://chemistrychemists.com/N2_2012/U1-1/ChemistryAndChemists_2_2012-U112.html) Он впаял в стеклянную трубку два электрода и откачал из трубки воздух. При подаче на электроды электрического напряжения газ в трубке начинал светиться, причем цвет свечения зависил от природы газа в трубке. Исследования продолжили физики Плюккер и Крукс. Было установлено, что от катода трубки исходят невидимые лучи, которые, несмотря на их невидимость, давали тень на противоположной катоду стороне, если на пути этих лучей устанавливали металлическую преграду. Если преграда была подобна многолепестковому пропеллеру, то она начинала вращаться, что доказывало, что лучи несут определенную энергию. Лучи отклонялись магнитом, что указывало на их заряд. В 1895 году французский физик Перрен доказал, что катодные лучи несут отрицательный электрический заряд, что подтверждало их неволновую природу. В том же году английский физик Томсон начал свои исследования катодных лучей. Он создал прообраз электроннолучевой трубки (http://novmysl.finam.ru/Electrodynamics/Thomson_Electron.html), с помощью которой установил, что катодные лучи являются материальными заряженными частицами с отношением заряда к массе в 1840 раз большим, чем у самого легкого иона — водорода, заряд которого был определен из других опытов. Если допустить, что по модулю эти заряды равны, то масса частиц, из которых состояли катодные лучи, была в 1840 раз меньше, чем масса атома водорода. Через некоторое время было доказано, что эта величина (соотношение заряда к массе) является универсальной, так как не зависит от природы заполняющего трубку газа, то есть. То есть частицы, из которых состоят катодные лучи, являются составной частью атомов всех веществ. Так был открыт электрон В 1910 году Милликен с помощью остроумного эксперимента (измерение скорости падения микроскопических капель масла, заряженных ионизирующим излучением, в электрическом поле между пластинами конденсатора) установил заряд электрона. А так как соотношение заряда к массе Томсоном было уже установлено, то с этого времени можно было определять «настоящую» массу атомов и молекул (до этого времени химики могли пользоваться только «эквивалентами». Историю этого понятия можно посмотреть здесь: http://chemistrychemists.com/N7_2010/6-31.pdf). Открытие радиоктивности и выполненные в 1909-1911 годах Резерфордом, Гейгером и Марсденом опыты по рассеянию альфачастиц на атомах золота позволили сделать вывод, что почти вся масса атома сосредоточена в компактном и очень массивном центре — ядре. Так как атом в целом нейтрален и кроме компактного ядра, состоит ещё и из легких электронов, предстояло объяснить, как всё это устроено. Предположений было несколько, но ни одно из них, в том числе планетарная модель атома, серьезной критики не выдерживало. Планетарная модель атома. Резерфорд, после экспериментов по рассеянию альфа-частиц на золотой фольге, доказывающих, что основная масса атома сосредоточена в компактном массивном ядре, радиус которого значительно меньше радиуса атома, предположил, что атом устроен подобно Солнечной планетной системе. В центре атома — положительно заряженное массивное ядро, вокруг которого с огромной скорости движуться отрицательно заряженные электроны. Если планеты вокруг Солнца удерживают силы гравитации, то электроны вокруг ядра — кулоновские силы притяжения. До этого Джозеф Джон Томсон предлагал свою модель устройства атома — «модель сливового пудинга», согласно которой отрицательно заряженные электроны помещены внутрь положительно заряженного атома. Объяснить опыты по рассеянию альфа частиц эта модель не могла. Предположение, что атом устроен подобно нашей Солнечной системе, вначале не было принято всерьез Планетарная модель атома Предположение, что атом устроен подобно нашей Солнечной системе, вначале не было принято всерьез потому, что электричество в это время уже прочно вошло в быт (во многом благодаря работавшему с восьмидесятых годов XIX века в Германии нашему соотечественнику, М.О. Доливо-Добровольскому, создавшему систему электроснабжения, которой мы до сих пор успешно пользуемся —трехфазный ток и многие устройства для его получения и преобразования). И спорить с подтверждавшимися каждый день законами электродинамики никто не собирался — заряженная частица при ускоренном движении в электрическом поле теряет энергию и обязательно должна упасть на источник поля, то есть электрон в планетарной модели атома должен упасть на ядро и атом должен прекратить существование, что в корне противоречило существующей стабильной Вселенной, во многом построенной на электромагнитных взаимодействиях. Общепринятой «планетарная» модель стала благодаря «упрямству» Резерфорда и Нильсу Бору, применившем идеи Макса Планка и А. Эйнштейна к планетарной модели. Общепринятой «планетарная» модель стала благодаря «упрямству» Резерфорда и Нильсу Бору, применившем идеи Макса Планка и А. Эйнштейна к планетарной модели. Макс Планк и «Ультрафиолетовая катастрофа». В конце XIX века, когда казалось, что теоретическая физика близка к своему завершению, при исследовании излучения «абсолютно черного тела» произошла так называемая «ультрафиолетовая катастрофа». В 1900 году Рэлей, а потом и Джинс вывели формулу излучения, названную законом Рэлея — Джинса. Формула хорошо описывала излучение в области длинных волн, но в области средних приводила к большому расхождению с экспериментальными данными, а в области коротких волн (ультрафиолетовых) приводила к парадоксальным результатам (плотность излучения, в соответствии с этой формулой, должна быть бесконечно большой). Планк предположил, что излучение не может быть непрерывным — абсолютно черное тело, как и любое другое, должно выдавать излучение дискретными порциями, «квантами». С учетом этой идеи удалось преодолеть «ультрафиолетовую катастрофу» и дать адекватное описание интенсивности излучения при всех длинах волн. Красный порог фотоэффекта Альберт Эйнштейн и красный порог фотоэффекта. Фотоэффект — это испускание электронов веществом при поглощении им электромагнитного излучения в видимой и ультрафиолетовой областях спектра. Были определены три закона фотоэффекта: 1.Сила фототока прямо пропорциональна плотности светового потока. 2. максимальная кинетическая энергия вырываемых светом электронов линейно возрастает с частотой света и не зависит от его интенсивности. 3 для каждого вещества существует красная граница фотоэффекта, то есть минимальная частота света 0 (или максимальная длина волны λ0), при которой ещё возможен фотоэффект, и если 0, то фотоэффект уже не происходит.. Фотоэффект был открыт ещё до открытия электронов, и после их открытия сам факт фотоэффекта поддавался простому объяснению — излучение выбивало из атомов электроны. Схема экспериментальной установки для изучения фотоэффекта В демонстрационной модели рассматривается явление фотоэффекта. В ней представлен вакуумный фотоэлемент, катод (пластину из цезия) которого освещают светом определенной длины волны. В результате фотоэффекта вылетевшие из катода фотоэлектроны могут достигать второй пластины (анода), создавая электрический ток (фототок). В это время общепринятым было приписывание электромагнитному излучению, и свету в том числе, волновой природы, а энергия волны, что каждый может заметить при наблюдении волн на воде, «размазана» в пространстве — брошенный в воду камень вызывает расходящуюся кругообразно волну, которая и уносит энергию камня. Но объяснить законы фотоэффекта — безъинерционность и «красный порог», например — волновой природой излучения в то время было невозможно. Эйнштейн предположил, что энергия излучения не размазана в пространстве, а сосредоточена в очень небольшом объеме, то есть излучение состоит из потока частиц, «квантов», «фотонов», которые и являются переносчиками энергии и которые при «столкновении» выбивают электрон из облученного вещества. Энергия фотона связана с частотой (или длиной волны) — чем больше частота (чем меньше длина волны), тем больше энергия кванта (у волн на воде или звуковых волн энергия зависит от аплитуды) . После определенной длины волны (в красной области спектра солнечного света) энергия кванта слишком мала, чтобы выбить электрон из атома. Что и объясняет «красный порог» фотоэффекта Мысленный опыт с океанскими волнами, показывающий связь между длиной (l ), частотой (n ) и энергией (Е) волны.Чем меньше длина волны (l) тем больше частота подъемов на гребень (n ) и энергия волны (E). Таким образом, энергия волны E = kn , где k – коэффициент пропорциональности. Когда гребень волны прокатывается под кораблем, тяжелое судно поднимается вверх. Значит, волна способна совершать работу. Допустим, морская волна в верхней части рисунка достаточно пологая, а морская волна в нижней части рисунка - более частая, похожая на зыбь. При этом пусть высота волн в обоих случаях будет одинаковой, чтобы одинаковой была и высота подъема судна на волне. Тогда получится, что нижняя волна совершает больше работы: на ее гребнях корабль поднимается в единицу времени чаще. Чем больше расстояние между гребнями волны, тем меньшее число раз поднимается корабль в единицу времени. Расстояние между гребнями волны называется длиной волны и обозначается греческой буквой l (лямбда). Частота волны - это число подъемов судна на гребень волны в единицу времени, обозначается буквой n (ню). Итак, поднимая корабль вверх, волна совершает работу. Значит, чем меньше длина волны (или чем больше частота), тем большую энергию несет волна. Энергия волны Е пропорциональна ее частоте: Е = kn , где k - некая константа, которую можно определить экспериментально. Следует отметить, что для волн на воде или звуковых волн это не так – энергия волны зависит от её амплитуды То есть идея «квантов» была вполне плодотворна и формула Бальмера свидетельствовала о применимости этой идеи к устройству атома. Бор постулировал, что электроны в атоме могут находится на определенных орбитах, не излучая энергию. При облучении вещества потоком фотонов электрон может поглотить энергию фотона и перескочить на более высокую «стационарную» орбиту, где он тоже не будет излучать энергию. Однако на такой орбите электрон при обычных условиях не может удерживаться долго и вновь перескакивает на свою «собственную» орбиту. При этом переходе происходит излучения порции энергии, кванта света. Формула, описывающая серию Бальмера (стр. 6), получала простое объяснение — электрон, находящийся на устойчивой безъизлучательной орбите, связанной с числом n1, которое определяло расстояние электрона до ядра и, следовательно, энергию электрона, при поглощении кванта энергии переходил на более высокую орбиту n2, в «возбужденное» состояние. В этом «возбуждённом» состоянии электрон не мог находиться долго и, излучая квант, возвращался на свою орбиту Боровское объяснение излучения Энергия излученного света, то есть его длина волны, была связана с разницей энергетических уровней орбит. В Боровском атоме положение электрона в атоме (радиус его орбиты, расстояние от ядра атома), то есть его энергия, описывалось главным радиальным числом n. Но по мере того, как выяснялось, что линейчатые спектры — это мощный инструмент для установления структуры атома, совершенствовались способы изучения этих спектров. Спектры регистрировали со все большим разрешением, а более высокое разрешение показывало более тонкую «структуру» спектров — некоторые толстые спектральные линии распадались на группу более тонких. Ещё до появления квантовой механики Зоммерфельд внес дополнения в Боровскую модель атома, указав, что круговые орбиты — всего лишь частный случай и постулаты Бора пригодны и для элиптических орбит электрона. Почти так же, как в формуле Ридберга, характеризовать эти элептические орбиты можно было некоторым числом (l), связанным с главным квантовым числом ограничением l = 0, 1, 2, 3,..., n-1. В этом случае «квантоваться» должен был орбитальный момент импульса. И «тонкая» структура спектральных полос это подтверждала. Положение электрона в атоме можно было охарактеризовать четырьмя квантовыми числами: 1. Главное квaнтовое число n, которое определяет общую энергию электрона и степень его удаления от ядра (номер энергетического уровня). Принимает любые целочисленные значения, начиная с 1 (n = 1, 2, 3, . . .) 2. Орбитальное (побочное или азимутальное) квантовое число l определяет форму орбиты электрона. Оно может принимать целочисленные значения от 0 до n-1 (l = 0, 1, 2, 3,..., n-1). Каждому значению l соответствует орбита особой формы. Орбитали с l = 0 называются s-орбиталями, l = 1 – называются р-орбиталями (3 типа, отличающихся магнитным квантовым числом m), l = 2 – называются d-орбиталями (5 типов), l = 3 – называются f-орбиталями (7 типов). 3. Магнитное квантовое число m определяет ориентацию орбиты в пространстве относительно внешнего магнитного или электрического поля. Его значения изменяются от +l до –l, включая 0. Например, при l = 1 число m принимает 3 значения: +1, 0, -1, поэтому существуют 3 типа р-АО: рx, рy, рz. 4. Спиновое квантовое число s может принимать лишь два возможных значения +1/2 и -1/2. Они соответствуют двум возможным и противоположным друг другу направлениям собственного магнитного момента электрона, называемого спином (от англ. веретено). Для обозначения электронов с различными спинами используются символы: Что значит «стационарные орбиты»? Эйнштейн, объясняя фотоэффект, предположил, что свет, кроме волновых свойств, имеет и свойства частиц. Световое излучение – это и волна и «квант», то есть материальная частица, в которой сосредоточена порция энергии электромагнитного излучения. В 1924 г. французский физик Луи де Бройль предположил, что не только излучение, но и материальные частицы обладают двойственной природой, т. е. свойствами волны и частицы. Размышляя над природой квантования, де Бройль предположил, что для электрона характерны свойства электромагнитных волн, и для него можно рассчитать длину волны. Кроме того, де Бройль предположил, что длина волны электрона укладывается целое число раз на орбите, т.е. сопоставил её со стоячей волной. Стоячая волна. Смоделировать стоячие волны даже легче, чем смоделировать интерференцию. Для этого нужно налить в таз или в ванну воды и начать с помощью какого-нибудь «механизма» — хотя бы собственной руки — возбуждать на поверхности воды волны. Волны будут отражаться от бортов ванны и возвращаться обратно. Можно подобрать частоту колебания так, что бегущие от руки волны будут накладываться на волны, отраженные от бортов, таким образом, что они перестанут перемещаться по водной поверхности. Волна будет стоять на месте, но это будет по прежнему волна — гребень будет сменяться впадиной, а впадина гребнем. При благоприятных условиях стоячие волны могут образоваться в результате наложения волн от противоположных бортов ванны, то есть после первичного возбуждения колебаний дальнейшего поддерживания колебаний будет не нужно. И если бы не потери энергии в результате неидеальности системы, то такие колебания самоподдерживались бы вечно, потому что энергии стоячие волны не переносят. С некоторым приближением маятник тоже можно назвать стоячей волной — маятник тоже качался бы вечно, если бы не потери на трение в точке подвеса и сопротивление воздуха Атом де Бройля В атоме электрон не один. Количество электронов в атоме равно номеру элемента в таблице Менделеева. Электроны, окружающие ядро, образуют как бы электронное облако. И кванты света, падающие на вещество, реагируют с «внешними» электронами этого облака. Электрон может поглотить квант. В этом случае его энергия увеличится и он «перепрыгнет» на более высокую «орбиту», на более высокий энергетический уровень. Но только на такой, где его новая длина волны будет снова укладываться целое число раз. Это состояние неустойчиво. И электрон очень быстро прыгнет обратно, испустив квант света. Заставить электрон прыгнуть на более высокий энергетический уровень может не только квант света, но и просто достаточно сильный удар — например, тепловой удар соседнего атома или удар другого электрона, разогнанного каким-то способом. Именно потому, например, светит электрическая лампочка — при прохождении тока вольфрамовый волосок нагревается, тепловые удары соседних атомов заставляют электрон прыгнуть на более высокий энергетический уровень, а при прыжке обратно электрон испускает квант света. А какого света — ультрафиолетового, видимого или инфракрасного — это зависит от температуры вольфрамовой нити При таких «странных» волновых свойствах электрон может двигаться вокруг ядра атома, не падая на него. Но это значит еще и то, что раз «орбиты» электрона могут быть только такими, на которых волна укладывается целое число раз, то существуют «запретные» области орбит. Те, на которых волна не укладывается целое число раз. Для нашего «механического» мира все очень просто. Стакан с водой мы можем поднять на любую высоту. Энергетический уровень стакана, стоящего на стуле один, а стоящего на столе другой. Можем взять стул и стол повыше и пониже. Но для электрона это невозможно. Существую только определенные «высоты», на которые можно «поднять» электрон. А так как каждой «орбите» соответствует свой энергетический уровень, своя энергия подъема, то существуют только определенные количества энергии, которые электрон может поглотить. Между этими орбитами электрон никакими силами не загонишь. Образно выражаясь, электрон может взять 10 рублей, может 20, может 30, но 15 или 25 он брать не может. Этим объясняется то, почему существуют прозрачные вещи — стекло, алмаз, соль, сахар и т.д. — потому что во всей области спектра квантам излучения не хватило энергии для переброски электронов на более высокий энергетический уровень. А ту энергию, которую несло излучение, электроны не могли поглощать — не существует энергетических уровней с такой энергией, потому что электронная волна не укладывается на них целое число раз. А если вещество окрашено, то оно (или примеси в нем) поглощает лишь в определенной области спектра. Другие лучи проходят и мы видим окраску. Как может и как не может прыгать электрон в атоме Термин «орбита» применим лишь к «точечному» объекту, движущемуся вокруг своего «хозяина» -- центрального тела • А раз электрон «волна», то и термин «орбита» применять к его движению не стоит – вызовет неправильные ассоциации. Поэтому был предложен другой термин – «орбиталь». Атомная орбиталь (АО) - область наиболее вероятного пребывания электрона (электронное облако) в электрическом поле ядра атома. Положение элемента в Периодической системе определяет тип орбиталей его атомов (s-, p-, d-, f-АО и т.д.), различающихся энергией, формой, размерами и пространственной направленностью. Для элементов 1-го периода (Н, He) характерна одна АО - 1s. В элементах 2-го периода электроны занимают пять АО на двух энергетических уровнях: первый уровень 1s; второй уровень - 2s, 2px, 2py, 2pz. (цифры обозначают номер энергетического уровня, буквы - форму орбитали). Какие бывают орбитали (электронные облака) А в молекуле всё немного подругому Если атом можно представить себе в виде изолированного шарика, то молекула подобна системе таких шариков, соединенных пружинками — «химическими связями» — плотностью электронного заряда между шариками. Вся эта «система» может вращаться в различных направлениях, «пружинки» могут растягиваться и сжиматься, углы между ними могут деформироваться или оставаться постоянными И потому у молекулы возможностей увеличения своей энергии гораздо больше. Так же, как и у атома, переданная молекуле энергия (при столкновении с квантом излучения или любой другой частицей) может перевести электроны молекулы в возбужденное состояние или покинуть молекулу. Но эта энергия может привести и к усилению вращения молекулы и к усилению колебания атомов молекулы относительно равновесного состояния, а большая энергия (выше энергии химической связи) может разорвать молекулу на части. Всё, что нас окружает (молекула, воздух, заполняющий объем аудитории, стол и всё остальное) кроме своего собственного «предназначения», является ещё и колебательной системой. И у каждой этой колебательной системы есть «свои» собственные колебания. Возбуждая систему с частотой этих колебаний, её можно привести в состояние резананса. А если частота «не собственная», то возбуждения не получится. То есть энергия колебательных уровней «квантуется». Но колебательных уровней много. Энергетические уровни в молекуле Энергетические уровни E r вращательные уровни (j) колебательные уровни ( v) электронные уровни (n) Так же как в газе скорости молекул не одинаковы, а подчиняются распределению Максвелла, то есть при одной и той же температуре в надутом водородом воздушном шарике существуют молекулы водорода и с низкими и с высокими скоростями, а температура воздушного шарика определяет наиболее вероятную скорость молекулы, так же и в любой совокупности молекул существует целый набор различающихся по своим энергиям (и вращательныи, и колебательным, и поступательным) молекул .В молекуле (как и в атоме) имеются дискретные энергетические состояния отдельных электронов (молекулярные орбитали) с их самосогласованным движением в поле друг друга и всех ядер молекулы. Предполагается, что все электроны данной молекулы (как и в атоме) распределяются по соответствующим орбиталям. Каждая орбиталь характеризуется своим набором квантовых чисел, отражающих свойства электронов в данном энергетическом состоянии. В отличие от одноцентровых орбиталей атомов, орбитали молекул многоцентровые, то есть молекулы имеют общие орбитали для двух или более атомных ядер. По аналогии с атомными s-, p-, d-, f- орбиталями молекулярные орбитали обозначают греческими буквами σ-, π-, δ-, γ-. МО образуются при комбинировании атомных орбиталей при достаточном сближении. Из исходных атомных орбиталей возникают МО. В зависимости от способа перекрывания и симметрии образующегося облака различают σ-, π- и δ-связи. Химическая связь, наибольшая электронная плотность которой сосредоточена на оси, соединяющей атомы, называется σ-связью. Её могут образовывать «перекрывания» любых атомных орбиталей — s-s, p-p, d-d, f-f, s-p, p-d, d-f, s-d, p-f, s-f. π-Связь (Пи-связь) возникает при перекрывании электронных облаков по обе стороны от линии соединения атомов (рис. 9, 10). δ-Связь (Дельта-связь) обязана перекрыванию всех четырех лопастей d-электронных облаков, расположенных в параллельных плоскостях (рис. 11). Исходя из условий симметрии, можно сказать, что электроны s-орбиталей могут участвовать лишь в σ-связывании, p-электроны – уже в σ- и π-связывании, а d-электроны – как в σ- и π, так и в δ-связывании. Максимальное перекрывание облаков осуществляется при σ-связи. . σ-Связь между s-орбиталями . σ-Связь между d-орбиталями σ-Связь между p-орбиталями π-Связь между p-орбиталями π-Связь между d-орбиталями δ-Связь между d-орбиталями Между двумя атомами в химической частице возможна только одна σ-связь. Все σ-связи обладают осевой симметрией относительно межъядерной оси. Фрагменты химических частиц могут вращаться вокруг межъядерной оси без нарушения степени перекрывания атомных орбиталей, образующих σ-связи. Совокупность направленных, строго ориентированных в пространстве σ-связей создает структуру химической частицы. При дополнительном перекрывании атомных орбиталей, перпендикулярных линии связи, образуются π-связи. В результате этого между атомами возникают кратные связи: Одинарная (σ) Двойная (σ +π) Тройная (σ + π + π) F−F O=O N≡N С появлением π-связи, не имеющей осевой симметрии, свободное вращение фрагментов химической частицы вокруг σ-связи становится невозможным, так как оно должно привести к разрыву π-связи. Помимо σ- и π-связей, возможно образование еще одного вида связи — δ-связи. Совокупность из положительных (ядра атомов) и отрицательных (электроны) частиц может быть устойчивой лишь в том случае, если плотность отрицательного заряда согласованно меняется так, чтобы при любых смещениях ядер от некоторого начального равновесного взаимного расположения возникали возвращающие силы, стремящиеся это равновесие восстановить. То есть равновесие должно быть динамическим — молекула должна находиться в состоянии незатухающих колебаний. Если плотность электронного заряда («где электрон бывает чаще») в молекуле распределена таким образом, что в одном месте образуется излишек отрицательного заряда, а в другом месте — избыток положительного, то молекулу можно охарактеризовать «дипольным моментом», который равен произведению величины заряда на расстояние от центра отрицательного заряда до центра положительного. Дипольный момент молекулы работает как антена, которая способна поглощать излучение за красным порогом фотоэффекта (инфракрасное), неспособное возбуждать электронные оболочки молекулы. В результате поглощения инфракрасного излучения повышается колебательная энергия молекулы. Полярная молекула воды Зачем всё это нужно? Даже поверхностные знания о строении вещества помогут нам, хотя бы на самом поверхностном уровне, иметь представление о том, что лежит в основе тех физикохимических методах анализа и экспертизы, которые мы будем изучать. А что мы будем изучать? Спектроскопия в видимой, ультрафиолетовой и инфракрасной областях. Флюоресцентная спектроскопия. Спектроскопия в ближней инфракрасной области. (Эмиссионная спектроскопия: пламенная фотометрия и индукционно-связанная плазма.) Атомно-абсорбционная спектроскопия. Хроматография газовая, газо-жидкостная, жидкостная. Хромато-масс-спектрометрия Капиллярный электрофорез Начнем с самых простых и реальных вещей. И постараемся понять, как нам могут помочь методы физикохимического анализа Отвертка после попытки ввернуть шуруп в твердую древесину Произведено отечественной промышленностью около 10 лет назад А как делают сталь? 1. В мартеновской печи Мартеновский процесс зависит от состава шихты, используемой при плавке. Различают такие разновидности мартеновского процесса: скрап-процесс, при котором шихта состоит из стального лома (скрапа) и 25-45% чушкового предельного чугуна; процесс применяют на заводах, где нет доменных печей, но расположенных в промышленных центрах, где много металлолома скрап-рудный процесс, при котором шихта состоит из жидкого чугуна (55-75%), скрапа и железной руды; процесс применяют на металлургических заводах, имеющих доменные печи. 2 Получение стали в кислородном конвертере Схема получения стали в кислородном конвертере: а — загрузка металлолома; б — заливка чугуна; в — продувка; г — выпуск стали; д — слив шлака. • И в том и в другом случае в сталь добавляют металлолом. В большинстве случаев единственным способом «сортировки» лома в отечественной промышленности очень длительное время был электромагнитный подъемник. И если электромагнит подхватил лом, содержащий, кроме железа, и другие металлы, то все это пойдет в печь или в конвертор. • Через 20-40 лет, после нескольких кругооборотов лома, вся сталь, идущая в «ширпотреб», будет очень плохой. • И девать ее будет некуда. • И потому в цивилизованных странах металлический лом, идущий в переработку, подвергали спектральному анализу. • Именно потому шведская сталь гораздо лучше российской Когда начали широко применять химические средства борьбы защиты растений (а это было более 50 лет назад — посмотрите, что такое ДДТ в Википедии), современных методов анализа, позволяющих определить остаточные содержания ДДТ и других пестицидов в пищевых продуктах не было. Позже было обнаружено, что они накапливаются в организмах животных и людей, потребляющих продукты, произведенные с использованием пестицидов. Во многих случаях это приводило к крайне неблагоприятным последствиям. Аналитический контроль позволяет уменьшить негативные последствия. Так, например, около десяти лет назад были проведены исследования миграции пестицидов при производстве виноградного вина. Было обнаружено, что пестициды скапливаются на поверхности виноградных ягод. И при производстве красного вина, которое настаивают на мезге, пестициды поступают в вино. А при производстве белого вина этого не происходит Как правило, отсутствие аналитического контроля — особенность слаборазвитых стран. Но это не всегда так. Так, например, американские учёные протестировали средства для стирки белья и освежители воздуха нескольких ведущих мировых брендов. Все образцы показали присутствие как минимум одного токсичного компонента. Полученные результаты могут вылиться в расследование со стороны властей США. (Ссылку могу дать) Биолог Кэрол Иверсен (Carol Iversen) из британского университета Nottingham Trent и её коллеги обнаружили опасные бактерии во многих образцах детского питания, произведённого самыми разными компаниями в семи европейских странах, Южной Корее, США и ЮАР. Всего было исследовано 200 взятых наугад упаковок сухой молочной смеси, других видов комбинированного сухого детского питания. 20 порошков содержали опасно высокие уровни болезнетворных бактерий десятков разновидностей, в том числе — вызывающие менингит Необходимость в объективных методах Потребительские свойства отвертки – её способность ввинчивать шурупы в твердую древесину – определяются, следовательно, качеством стали. А качество стали определяется её химическим составом и физической структурой. То есть, имея «паспорт» «отверточной» стали, в которой указаны её марка, а, стало быть, состав и структура, мы, проанализировав сталь той партии отверток, которую нам предлагают для продажи, можем сразу сказать, будут нас ругать покупатели или нет. Нужно знать, какие химические и физические свойства товара связаны с его потребительскими свойствами, и какими методами анализа можно объективно установить эти свойства. Что значит «объективно»? «На глазок» марку стали можно определить по искре, которую она образует при обточке на точильном круге. Особенно распространен этот метод анализа – «на глазок» - в пищевой промышленности, где «органо-лептический метод» используют очень широко. При достаточном опыте и таланте этот метод дает хорошие результаты. При отсутствии опыта и таланта или в том случае, когда опытные дегустаторы не заинтересованы в истинном положении дел, основывать какие-либо выводы на органо-лептическом анализе нельзя. Необходимы объективные, не зависящие от эксперта воспроизводимые и точные методы. Методы инструментального анализа 22. МЕТОДИКА АНАЛИЗА ОБЪЕКТА АНАЛИТИЧЕСКОГО КОНТРОЛЯ (КОНТРОЛЬ ОБЪЕКТА АНАЛИТИЧЕСКИЙ ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ. ГОСТ Р 52361-2005 Дата введения - 1 января 2006 года) документированная совокупность операций и правил, выполнение которых обеспечивает получение результата анализа объекта аналитического контроля с установленными характеристиками Погрешности или неопределенностью, а для методик качественного анализа - с установленной достоверностью (Предусматривает градуировку с использованием Стандартных Образцов) Аттестованный прибор, выдающий воспроизводимые результаты с заданной воспроизводимой погрешностью при анализе стандартного образца Персонал, квалификация которого соответствует обусловленным требованиям ПОЛУЧЕНИЕ ОБЪЕКТИВНОГО РЕЗУЛЬТАТА Однако любой анализ начинается с пробоотбора. Мы не будем обсуждать пробоотбор, но вы должны знать, что для продуктов питания существуют международные документы, устанавливающие процедуры пробоотбора для самых разнообразных случаев и товаров. Это «GENERAL GUIDELINES ON SAMPLING CAC/GL 50-2004» (Кодекс Алиментариус) Смысл документа в том, что пробы, отобранные для контроля, должны быть «представительными» и операции их отбора должны быть воспроизводимы. В большинстве случаев отобранные пробы невозможно непосредственно подвергать анализу. Например, яблоко, которое необходимо проверить на содержание пестицидов, нельзя засунуть в хромато-масс-спетрометр. Яблоко должно пройти «пробоподготовку». Его нужно размельчить и из размельченной массы выделить пестициды. Чаще всего это делают с помощью экстракции (гексаном, ацетонитрилом и т.д.). Экстракт очищают от посторонних примесей, упаривают и лишь потом полученный концентрат вводят в хроматограф масс-спектрометра. А если нужно определить в яблоке токсичные металлы, то измельченное яблоко нужно подвергнуть процедуре «мокрого сожжения» Лишь в некоторых случаях можно обойтись без пробоподготовки. Например, тогда, когда вам нужно идентифицировать полимерную пленку — лавсан это или полиэтилен или еще что-то. КОНТРОЛЬ ОБЪЕКТА АНАЛИТИЧЕСКИЙ ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ ГОСТ Р 52361-2005 Дата введения 1 января 2006 года 4. МЕТОДИКА АНАЛИТИЧЕСКОГО КОНТРОЛЯ (ОБЪЕКТА): документированная совокупность операций и правил проведения аналитического контроля конкретных объектов Примечание - Методика аналитического контроля объекта может быть представлена в виде совокупности нескольких документов: методики отбора проб, методики подготовки проб, методики химического анализа, методики испытаний, методики выполнения измерений, правил приемки и т.п. 9. ПРЕДСТАВИТЕЛЬНАЯ ПРОБА ВЕЩЕСТВА representative [МАТЕРИАЛА] (ОБЪЕКТА АНАЛИТИЧЕСКОГО sample КОНТРОЛЯ): проба вещества [материала], которая по химическому составу, и/или свойствам, и/или структуре принимается идентичной объекту аналитического контроля, от которого она отобрана Электронная спектрофотометрия Что может произойти с квантами света, упавшими на вещество? I0 — падающий на вещество свет; I — прошедший через вещество свет; А — рассеянный веществом свет с той же длиной волны, что и падающий (релеевское рассеяние); B — рассеянный свет с большей длиной волны (комбинационное рассеяние); С — отражённый свет (4%); D — свет, излучённый веществом. И рассеяние и отражение происходит по всем направлениям Энергия кванта электромагнитного излучения фиксирована и равна E = hν, где h = 4·10–15 эВ·с = 6·10–34 Дж·с — постоянная Планка, еще одна фундаментальная физическая величина, определяющая свойства нашего мира. С отдельным электроном при фотоэффекте взаимодействует отдельный квант, и если его энергии недостаточно, он не может выбить электрон из металла. Давний спор о природе света — волны это или поток частиц — разрешился в пользу своеобразного синтеза. Одни явления описываются волновыми уравнениями, а другие — представлениями о фотонах, квантах электромагнитного излучения, которые были введены в оборот двумя немецкими физиками — Максом Планком и Альбертом Эйнштейном. Энергию квантов в физике принято выражать в электрон-вольтах. Это внесистемная единица измерения энергии. Один электрон-вольт (1 эВ) равен энергии, которую приобретает электрон, когда разгоняется электрическим полем напряжением 1 вольт. Это очень небольшая величина, в единицах системы Си 1 эВ = 1,6·10–19 Дж. Но в масштабах атомов и молекул электрон-вольт — вполне солидная величина. Квант электромагнитного излучения может «удариться» о молекулу вещества и отскочить от неё. В этом случае, как и при соударении биллиардных шаров, возможны два варианта. Первый вариант похож на соударение биллиардного шара со стенкой биллиардного стола — шар после такого соударения отлетает с той же скоростью (если не учитывать потери, связанные с неидеальностью системы), с какой он ударил стенку, а стенка, естественно, остается неподвижной. То есть она не отбирает у ударившего шара энергию. Это упругое рассеяние. Упругое рассеяние возможно не только в результате столкновения со стенкой. Если два одинаковых биллиардных шара движутся навстречу друг другу с одинаковыми скоростями, то есть с одинаковыми энергиями, то и после столкновения они разлетятся с теми же скоростями, то есть с одинаковыми энергиями. Для кванта света — это релеевское рассеяние, рассеяние с такой же длиной волны, что и у падающего на вещество света. Однако в биллиарде шары редко имеют одинаковые скорости. И если шар ударяет неподвижный шар или ударяет шар, догоняя его, то чаще всего он отдаёт часть своей энергии другому шару. И потому отлетает после столкновения с меньшей энергией. А если он ударяет шар, движущийся ему навстречу, то энергия ударяющего шара после столкновения может возрасти. Для кванта света — это так называемое «комбинационное» (или рамановское*) рассеяние. В этом случае, в случае обмена энергиями, длина волны рассеянного света будет или больше, чем длина волны падающего света или меньше её. Использованием этого явления в физико-химических методах анализа занимается спектрофотометрия комбинационного рассеяния. Но на долю рассеяния (релеевского или комбинационного) приходится лишь малая часть (10-7) падающего на вещество света Судьба основной части квантов, падающих на вещество, зависит от возможности их взаимодействия с веществом. Если энергия падающего кванта света такова, что он может «перебросить» электрон внешней электронной оболочки на более высокий энергетический уровень, то квант света будет поглощен веществом. Использованием этого явления в физико-химических методах анализа занимается электронноколебательная (электронная) спектрофотометрия. Это спектрофотометрия в области ультрафиолетового и видимого излучения — в области от 200 нм до 900 нм. Многие современные спектрофотометры регистрируют спектры от 190 нм до 1100 нм. Молекула вещества при поглощении фотона переходит в неустойчивое возбужденное состояние. Выйти из этого состояния молекула может двумя путями. Для большинства веществ переход в основное, невозбужденное состояние связано с отдачей «лишней» энергии соседним молекулам. То есть на увеличение теплового движения молекул, на повышение температуры вещества. Для некоторых, относительно немногих веществ, переход в основное состояние связан не только с отдачей излишка энергии соседним молекулам, но и с излучением. Это явление — свечение вещества под действием возбуждающего излучения, называется люминисценцией. Если свечение происходит только во время облучения возбуждающим излучением, то это флуоресценция, если свечение происходит и после прекращения возбуждения, то это фосфоресценция. Использованием этого явления в физико-химических методах анализа занимается флуоресцентная спектрофотометрия. И в этом случае нас интересует область излучения ультрафиолетового (УФ) и видимого света — чаще всего от 220 нм до 900 нм Электронно-колебательная (или просто электронная) спектрофотометрия — это метод исследования веществ, основанный на поглощении электромагнитного излучения с длиной волны от 185 нм до 1100 нм. Поглощением более коротковолнового (менее 185 нм) ультрафиолетового излучения занимается вакуумная спектрофотометрия, так как это излучение поглощают газы, входящие в состав воздуха, и потому прибор для изучения поглощения в этой области должен быть вакуумирован. Поглощением длинноволнового излучения (от 1100 нм до 2500 нм) занимается ближняя инфракрасная спектрофотометрия. Электронная спектроскопия используется как для качественного, так и для количественного анализа. Качественный анализ основан на связи структуры вещества со спектрами поглощения. Количественный анализ — на зависимости величины поглощения электромагнитного излучения от концентрации анализируемого вещества В атомах поглощение излучения происходит в результате возбуждения электронов на внешних атомных орбиталях и переходе этих электронов на более «высокую» орбиталь, откуда они снова переходят в основное состояние с излучением кванта. В молекуле химическая связь обусловлена распределением электронной плотности, которую квантовая механика описывает, как образование молекулярных орбиталей, и поглощение излучения в ультрафиолетовой и видимой областях связано с возбуждением электронов этих орбиталей и усилением колебаний всей молекулы. Для ионов металлов электромагнитное излучение возбуждает d- и fорбитали. Но для товароведения более интересны органические вещества. В молекуле органического вещества можно выделить четыре типа электронов. Во-первых, это электроны заполненных внутренних оболочек. Они не участвуют в химической связи, энергия их возбуждения очень высока, и в ультрафиолетовой и видимой областях они неактивны. Во-вторых, это электроны одинарных -связей — таких, например, как в предельных углеводородах Метан Этан -связи предельных углеводородов В третьих, это электроны электронных пар валентной оболочки атомов, которые не участвуют в химической связи, например в атомах азота, кислорода, серы, галогенов. Их называют n-электроны. Энергия их возбуждения меньше, чем у электронов -связей, и они могут поглощать в ультрафиолетовом и видимом диапазонах. И, в четвертых, это электроны -орбиталей (-электроны) — электроны двойных и тройных связей. Энергия возбуждения этих связей самая меньшая из перечисленных и поэтому вещества, содержащие эти связи, поглощают в ультрафиолетовой и видимой областях Следовательно, могут существовать следующие электронные переходы при поглощении электромагнитного излучения: * n * n * * Энергетическая диаграмма электронных переходов, ответственных (в большинстве случаев) за поглощение электромагнитного излучения в области 185 нм-1100 нм Энергетические уровни электронных переходов с учетом колебательно-вращательных составляющих Шкала электромагнитных волн Длина Название Частота более 100 км Нзкочастотные электрические колебания 0-3 кГц 100 км - 1 мм Радиоволны 3 кГц - 3 ТГц 100-10 км мириаметровые (очень низкие частоты) 3 - 3-кГц 10 - 1 км километровые (низкие частоты) 30 - 300 кГц 1 км - 100 м гектометровые (средние частоты) 300 кГц - 3 МГц 100 - 10 м декаметровые (высокие частоты) 3 - 30 МГц 10 - 1 м метровые (очень высокие частоты) 30 - 300МГц 1 м - 10 см дециметровые (ультравысокие) 300 МГц - 3 ГГц 10 - 1 см сантиметровые (сверхвысокие) 3 - 30 ГГц 1 см - 1 мм миллиметровые (крайне высокие) 30 - 300 ГГц 1 - 0.1 мм децимиллиметровые (гипервысокие) 300 ГГц - 3 ТГц 2 мм - 760 нм Инфракрасное излучение 150 ГГц - 400 ТГц 760 - 380 нм Видимое излучение (оптический спектр) 400 - 800 ТГц 380 - 3 нм Ультрафиолетовое излучение 800 ТГц - 100 ПГц 10 нм - 1пм Рентгеновское излучение 30 ПГц - 300 ЭГц <=10 пм Гамма-излучение >=30 ЭГц Для информации Внесистемные единицы измерения: микрон, равный 1 мкм, и ангстрем (Å), (0,1нм) Кратные Дольные величина название обозначение величина название 101 м декаметр дам 102 м гектометр 103 м обозначение dam 10−1 м дециметр дм dm гм hm 10−2 м сантиметр см cm километр км km 10−3 м миллиметр мм mm 106 м мегаметр Мм Mm 10−6 м микрометр мкм µm 109 м гигаметр Гм Gm 10−9 м нанометр нм nm 1012 м тераметр Тм Tm 10−12 м пикометр пм pm 1015 м петаметр Пм Pm 10−15 м фемтометр фм fm 1018 м эксаметр Эм Em 10−18 м аттометр ам am 1021 м зеттаметр Зм Zm 10−21 м зептометр зм zm 1024 м йоттаметр Им Ym 10−24 м йоктометр им ym О том, что свет выбивает из щелочных металлов электроны, мы знаем в последнюю очередь. А в первую очередь мы знаем о том, что через стекло, например, свет проходит, а через дерево – нет. И что за тонированным стеклом в машине сидеть менее жарко, чем за безцветным. Почему? Как свет (электромагнитные колебания) взаимодействует с веществом? Остановимся на области 200-1000 нм. Это области ультрафиолетовых (200350 нм), видимых лучей (350-800нм) и часть инфракрасной области. Здесь нам нужно вспомнить строение атомов и молекул Это очень грубая «боровская» модель. Как планеты вокруг Солнца, движутся отрицательно заряженные электроны вокруг положительного ядра. Но реально так быть не может, потому что заряженные частицы в электрическом поле должны терять энергию, то есть электроны должны упасть на ядро. Эйнштейн, объясняя фотоэффект, предположил, что свет, кроме волновых свойств, имеет и свойства частиц. Световое излучение – это и волна и «квант», частица, в которой сосредоточена порция энергии электромагнитного излучения. В 1924 г. французский физик Луи де Бройль предположил, что не только излучение, но и материальные частицы обладают двойственной природой, т. е. свойствами волны и частицы. Размышляя над природой квантования, де Бройль предположил, что для электрона характерны свойства электромагнитных волн, и для него можно рассчитать длину волны. Кроме того, де Бройль предположил, что длина волны электрона укладывается целое число раз на орбите, т.е. сопоставил её со стоячей волной. Примером стоячей волны могут служить колебания скрипичной струны, закрепленной на обоих концах. Струна может колебаться только с определенными частотами. Когда волна колеблется как одно целое, то издает основной тон, при колебаниях с более короткими длинами волн издаются обертоны. В атоме электрон не один. Количество электронов в атоме равно номеру элемента в таблице Менделеева. Электроны, окружающие ядро, образуют как-бы электронное облако. И кванты света, падающие на вещество, реагируют с «внешними» электронами этого облака. Электрон может поглотить квант. В этом случае его энергия увеличится и он «перепрыгнет» на более высокую «орбиту», на более высокий энергетический уровень. Но только на такой, где его новая длина волны будет снова укладываться целое число раз. Это состояние неустойчиво. И электрон очень быстро прыгнет обратно, испустив квант света. Заставить электрон прыгнуть на более высокий энергетический уровень может не только квант света, но и просто достаточно сильный удар — например, тепловой удар соседнего атома или удар другого электрона, разогнанного каким-то способом. Именно потому, например, светит электрическая лампочка — при прохождении тока вольфрамовый волосок нагревается, тепловые удары соседних атомов заставляют электрон прыгнуть на более высокий энергетический уровень, а при прыжке обратно электрон испускает квант света. А какого света — ультрафиолетового, видимого или инфракрасного — это зависит от температуры вольфрамовой нити При таких «странных» волновых свойствах электрон может двигаться вокруг ядра атома, не падая на него. Но это значит еще и то, что раз «орбиты» электрона могут быть только такими, на которых волна укладывается целое число раз, то существуют «запретные» области орбит. Те, на которых волна не укладывается целое число раз. Для нашего «механического» мира все очень просто. Стакан с водой мы можем поднять на любую высоту. Энергетический уровень стакана, стоящего на стуле один, а стоящего на столе другой. Можем взять стул и стол повыше и пониже. Но для электрона это невозможно. Существую только определенные «высоты», на которые можно «поднять» электрон. А так как каждой «орбите» соответствует свой энергетический уровень, своя энергия подъема, то существуют только определенные количества энергии, которые электрон может поглотить. Между этими орбитами электрон никакими силами не загонишь. Образно выражаясь, электрон может взять 10 рублей, может 20, может 30, но 15 или 25 он брать не может. Этим объясняется то, почему существуют прозрачные вещи — стекло, алмаз, соль, сахар и т.д. — потому что во всей области спектра квантам излучения не хватило энергии для переброски электронов на более высокий энергетический уровень. А ту энергию, которую несло излучение, электроны не могли поглощать — не существует энергетических уровней с такой энергией, потому что электронная волна не укладывается на них целое число раз. А если вещество окрашено, то оно (или примеси в нем) поглощает лишь в определенной области спектра. Другие лучи проходят и мы видим окраску. Как может и как не может прыгать электрон Всё это очень понятным языком написано здесь: http://fiz.1september.ru/2005/02/14.htm Беллур Сиварамия Чандрасекар Почему всё вокруг такое, какое оно есть? Термин «орбита» применим лишь к «точечному» объекту, движущемуся вокруг своего «хозяина» -- центрального тела • А раз электрон «волна», то и термин «орбита» применять к его движению не стоит – вызовет неправильные ассоциации. Поэтому был предложен другой термин – «орбиталь». Атомная орбиталь (АО) - область наиболее вероятного пребывания электрона (электронное облако) в электрическом поле ядра атома. Положение элемента в Периодической системе определяет тип орбиталей его атомов (s-, p-, d-, f-АО и т.д.), различающихся энергией, формой, размерами и пространственной направленностью. Для элементов 1-го периода (Н, He) характерна одна АО - 1s. В элементах 2-го периода электроны занимают пять АО на двух энергетических уровнях: первый уровень 1s; второй уровень - 2s, 2px, 2py, 2pz. (цифры обозначают номер энергетического уровня, буквы - форму орбитали). Какие бывают орбитали (электронные облака) .В молекуле (как и в атоме) имеются дискретные энергетические состояния отдельных электронов (молекулярные орбитали) с их самосогласованным движением в поле друг друга и всех ядер молекулы. Предполагается, что все электроны данной молекулы (как и в атоме) распределяются по соответствующим орбиталям. Каждая орбиталь характеризуется своим набором квантовых чисел, отражающих свойства электронов в данном энергетическом состоянии. В отличие от одноцентровых орбиталей атомов, орбитали молекул многоцентровые, то есть молекулы имеют общие орбитали для двух или более атомных ядер. По аналогии с атомными s-, p-, d-, f- орбиталями молекулярные орбитали обозначают греческими буквами σ-, π-, δ-, γ-. МО образуются при комбинировании атомных орбиталей при достаточном сближении. Из исходных атомных орбиталей возникают МО. Несмотря на то, что энергетические уровни колебания и вращения молекул дискретны (они «квантуются»), их очень много. И поэтому спектр поглощения молекул, в отличие от спектров атомов, состоит из суммы множества узких полос, дающих в результате широкую полосу. Чаще всего исследуют растворы веществ, а в растворе молекула вещества сольватирована, то есть окружена молекулами растворителя, связанными с ней межмолекулярными связями, и это тоже влияет на энергетическое состояние молекулы. В результате этого возбуждение электронных переходов происходит не в узкой области (практически при одной длине волны), как это было с атомными спектрами (линии Фраунгофера или спектры испускания в опытах Кирхгофа – Бунзена), а в некотором спектральном интервале. Наглядно это можно представить следующим образом: колеблющийся в молекуле атом, «привязанный» к молекуле химической связью, а, значит, окруженный электронным облаком, может двигаться навстречу летящему фотону и тогда возбуждение перехода может произойти при меньшей энергии фотона (энергия возбуждения складывается из энергии фотона и энергии движущегося ему навстречу атома), а если атом при колебании двигается от летящего на него фотона, то энергия фотона для возбуждения перехода должна быть большей. Но это один и тот же электронный переход, хотя в спектре поглощения вместо тонкой линии возникает широкая полоса поглощения излучения — от минимальной до максимальной длины волны, при которой происходит возбуждение электронного перехода. Именно потому, что возбуждение переходов осуществляется при взаимодействии фотонов излучения с колеблющейся и вращающейся молекулой, спектроскопия в УФ- и видимой областях, называется «электронной вращательно-колебательной спектроскопией». Энергия Е электромагнитного излучения определяется уравнением Планка: E= h ν где ν—частота света (ν=С /) h — постоянная Планка, С— скорость света, — длина волны Для видимой части спектра величина энергии составляет ~ 158-300 кДж/моль (такой энергией обладает число фотонов, равное числу Авогадро), для ультрафиолетовой области —более 300 кДж/моль. Для того чтобы молекула поглощала в видимом диапазоне, то есть вещество было бы окрашенным, энергия возбуждения его молекулы – (Е0и E' - энергия молекулы соответственно в основном и возбужденном состояниях) должна лежать в этих пределах (158-300 кДж/моль) При > 300 кДж/моль поглощение происходит в УФ, при < 158 кДж/моль — в инфракрасной области спектра. Форма молекулярных орбиталей. s- и p-МО Форма молекулярных орбиталей определяется геометрией перекрывания атомных орбиталей. Возможны два типа перекрывания атомных орбиталей: •осевое или s (сигма)-перекрывание; •боковое или p (пи)-перекрывание. • По типу перекрывания исходных АО образующиеся молекулярные орбитали относят к sМО или p-МО. Для того, чтобы перебросить электрон с σ-орбитали на более высокий энергетический уровень, нужна очень большая энергия. То есть очень маленькая длина волны — около 130нм.( Для того, чтобы наблюдать это поглощение, нужны специальные приборы — их нужно вакуумировать). Для рутинных анализов такие приборы не используют. А для того, чтобы перебросить на более высокий энергетический уровень электрон, принадлежащий к -орбитали, нужна более низкая энергия. А если двойная связь «сопряженная» (как в бензоле, например), то для ее возбуждения нужна еще более низкая энергия Существуют структурные группы, которые называют хромофорами, присутствие которых в молекуле вызывает поглощение излучения. Некоторые из этих групп приведены в таблице Структурная группа >C=N>C=O -N=N>C=S -N=O Максимум полосы поглощения 240 280 350 500 660 (Хромофорные группы, введение которых в молекулу вызывает поглощение света в результате n* перехода ) Хромофорные группы—группы, введение которых в вещество вызывает поглощение света Некоторые структурные группы, оказывающие влияние на положение полосы поглощения Ауксохромы (электродоноры) Антиауксохромы (электроакцепторы) -CH3 -COO -OCH3 -NHCH3 -N(CH3)2 -NHC6H5 -NO2 При увеличении энергии связи полоса поглощения сдвигается в коротковолновую область (гипсохромный сдвиг). На положение полос поглощения оказывают влияние и растворители. Взаимное влияние различных факторов на положение полос поглощения накладывает существенные ограничения на использование электронно-колебательных спектров для установление структуры соединения, хотя во многих случаях предположения о наличии сопряженных групп и других хромофоров в исследуемом веществе по его электронному спектру могут быть относительно легко проверены другими методами, то есть ультрафиолетовая и видимая спектрофотометрия может рассматриваться, как начальный и самый простой этап качественного анализа. • То есть способность вещества поглощать излучение зависит от его структуры, от наличия тех или иных «функциональных» групп. Благодаря которым в молекуле существуют молекулярные орбитали с энергетическими уровнями, которые соответствуют (могут быть возбуждены) электромагнитному излучению, которое мы используем. Только надо учитывать, что важна не столько структура молекулы, сколько тип связи, вид электронного облака, которое взаимодействует с электромагнитным излучением. • Конечно, для каждой молекулы найдется такое излучение, которое она будет поглощать. И очень коротковолновое излучение (гамма-лучи) ионизирует вещество – выбивает электроны так, что они улетают в пространство • Но нас сейчас интересует только область от 190нм до 1100нм – та область, которую используют при исследованиях и анализе на самых «обычных» спектрофотометрах. Которые не надо вакуумировать и при работе на которых можно использовать для кювет такие доступные материалы, как кварц (для УФ и видимой области) и стекло (для видимой области) Очень коротко и более или менее понятно о том, какие виды химической связи существуют, можно прочесть здесь. http://www.chemport.ru/chemicalbond.shtml http://www.hemi.nsu.ru/ucheb132.htm http://www.krugosvet.ru/articles/113/1011313/1011313a1.htm Для нас важно, что существует «красный порог» для взаимодействия света (электромагнитного излучения) с электронами вещества. И потому вся область взаимодействия излучения, если не прибегать к каким-то особым приемам и не рассматривать излучение, разрушающее вещество, ограничена относительно узкой спектральной областью – от «полужесткого» ультрафиолета (жесткий, коротковолновый ультрафиолет вещество уже разрушает) до 800нм. То есть вся совокупность химических связей, если говорить о возможности электронов этих связей «прыгать» на более высокие энергетические уровни, укладывается в эту область. Их не так уж много, типов этих связей. И потому электронные спектры мало пригодны для идентификации вещества. Они «маловыразительные». И приходится прибегать к специальным приемам, чтобы использовать их в аналитической химии. Чтобы сделать какие-то выводы о структуре вещества, нам нужно знать, какое электромагнитное излучение оно поглощает, а какое нет. То есть надо получить зависимость поглощения веществом излучения от длины волны. То есть нам нужно знать, свет какой волны поглощает исследуемое вещество. Эта зависимость называется спектром поглощения Попробуем это сделать. Так как это пригодится нам и для количественного анализа, вспомним, что это такое Количественный анализ Для количественного анализа электронно-колебательная спектрофотометрия используется очень широко. До шестидесятых годов прошлого века основной объем аналитических работ был связан со спектрофотометрией и лишь с развитием хроматографических методов спектрофотометрия отошла на второй план. На связь интенсивности окрашивания раствора с количеством растворённого вещества обратили внимание очень давно. А приборы, позволяющие установить более или менее точную количественную связь на основе визуальных наблюдений, были созданы в 1870 году. Появление электронных устройств, позволяющих усиливать и измерять слабые токи, возникающие в результате фотоэффекта, позволили перейти от измерений, основанных на визуальных наблюдений, к более точным инструментальным определениям. Приборы первого поколения, как и визуальные приборы, были основаны на светофильтрах. Цвет прозрачного раствора обусловлен тем, какой свет он поглощает. Если раствор поглощает оранжевый (600 нм) свет, то «на просвет» мы видим синий цвет раствора (490 нм), то есть раствор окрашивается в дополнительный к поглощенному цвет. (Дополнительные цвета — цвета, которые при суммировании дают белый цвет) То есть по окраске раствора можно судить о длине волны поглощенного излучения. И для того, чтобы лучше всего заметить поглощение излучения, нужно облучать раствор тем излучением, которое раствор лучше всего поглощает, то есть через светофильтр дополнительного цвета относительно цвета раствора. То есть синий «на просвет» раствор нужно облучать через оранжевый светофильтр. Для определения вещества спектрофотометрией в видимой области стараются получить, в результате какой-либо химической реакции, его окрашенное производное, по поглощению которого и судят о наличии и количестве аналита. Разработка методики определения заключается в поиске реактива, дающего с аналитом цветную реакцию. Реакция должна быть селективна и образовавшееся цветное производное должно иметь очень сильное поглощение. Для видимой области без получения цветных производных можно определять лишь немногие окрашенные вещества — такие, как иод, бром, хромат- и перманганат- ионы (K2CrO4, KMnO4) и др.. Но в УФ области часто вещества определяют непосредственно, по собственному поглощению. Потому что в УФ, особенно менее 200нм, поглощают все вещества. А теперь можно попытаться сконструировать спектрофотометр – прибор, с помощью которого мы будем исследовать, как вещества поглощают электромагнитное излучение. То есть определять, какое (с какой длиной волны, с какой энергией) излучение вещество поглощает, а какое нет. И в какой степени. Нам нужен источник излучения, приемник излучения – фотоэлемент и устройство, с помощью которого на фотоэлемент мы будем направлять излучение с определенной длиной волны. Потому что фотоэлементу все равно, какая длина волны у излучения. На любое излучение (до «красного порога») фотоэлемент будет реагировать появлением электрического тока. Сконструируем прибор для измерения поглощения света Блок-схема устройства для измерения поглощения электромагнитного излучения 1 — источник излучения; 2 — луч света от источника излучения; 3 —устройство, позволяющее выбирать свет с определенной длиной волны – светофильтр или призма; 4 — кювета, в которую помещают исследуемое вещество; 5 — устройство, измеряющее интенсивность прошедшего через раствор излучения (фотоэлемент или фотоэлектронный умножитель). Однако при реализации такой простой схемы возникают две проблемы. Это неравномерность энергии излучения источника при различных длинах волн и неравномерность чувствительности приемника излучения в различных волновых диапазонах. Что мы хотим? Мы хотим получить объективные характеристики нашего товара при взаимодействии его (или того, что мы из него выделим, того, что мы из него получим) со световым излучением. А с каким «световым излучением»? У солнца это излучение одно, у водорода, когда мы заставили его светиться — другое. Если мы выбрали такой «полосатый» источник излучения, то очень может быть, что фотоэлемент будет говорить нам не о поглощении света анализируемым веществом, а о том, что света просто нет. Это первая проблема при построении спектрофотометра Различные источники излучения: ксеноновая лампа, дейтериевая лампа и галогеновая лампа накаливания А вот вторая проблема. Фотоэлементы на разные участки спектра реагируют по разному Чувствительность приемников излучения А это значит, что при решении вопроса, как реагирует наше вещество на световое излучение, мы должны учитывать, какой спектр того источника, которым мы пользуемся, и какие свойства у нашего приемника излучения На каждом участке спектра мы должны быть уверены, что измеренные нами характеристики — это характеристики нашего образца, а не источника света и не приемника излучения. Для того, чтобы учесть это, добавляют еще один луч — эталонный, который не проходит через образец. И в каждом участке спектра измеряют отношение энергий эталонного луча и луча, прошедшего через образец. По этому принципу устроены все спектрофотометры Принципиальная схема спектрофотометра 1- источник излучения; 2,3,4,5-зеркала, служащие для формирования двух одинаковых лучей; 6-кювета с образцом, 8кювета сравнения, 7-оптический клин; 9- прерыватель луча; 10-монохроматор; 11- приемник излучения На схеме показан только один источник излучения. На самом деле их два – один для УФ области (дейтериевая лампа), другой для видимой (лампа накаливания, галогенная) Современный УФ-Вид спектрофотометр Количественное определение Применим общеупотребительный метод • Разрежем нашу кювету на очень-очень тонкие слои (в идеале – на бесконечнотонкие). И предположим, что каждый такой слой поглощает определенную долю излучения. Например 10% Разрежем кювету на тонкие слои Номер слоя Интенсивность излучения 1 1000 2 900 3 810 1200 4 729 1000 5 656 800 6 590 7 531 600 400 8 478 9 430 200 10 387 0 11 348 12 313 13 282 14 254 1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39 Теперь перейдем к бесконечно тонким слоям Уменьшение интенсивности излучения в этом слое (-dI ) зависит от числа фотонов, то есть от интенсивности падающего излучения, и от числа молекул, которые может встретить фотон, то есть от концентрации n0 и от толщины слоя. Кроме того, вероятность поглощения фотона при столкновении с молекулой зависит от энергии фотона, то есть от длины волны, и от структуры вещества. Учтем эту зависимость введением коэффициента, зависящего от длины волны — k(). Полученное уравнение имеет следующий вид (знак минус показывает, что интенсивность уменьшается): -dI()=I()k()n0dL После разделения переменных получим легко интегрируемые (табличные) функции: -dI()/I() = k()n0dL После интегрирования: -dI()/I()=-lnI + C1 k()n0dL = Lk()n0 +C2 -lnI + C1= Lk()n0 +C2 -lnI = Lk()n0 + (-C1+C2) где С1 и С2 — постоянные интегрирования, которые можно объединить и найти, приравняв толщину поглощающего слоя к нулю. В этом случае, естественно (на пути луча ничего нет), интенсивность излучения не будет ничем ослаблена и будет равна I0 -lnI0= (-C1+C2) После подстановки: -lnI = Lk()n0 - lnI0 После преобразования: -lnI + lnI0= Lk()n0 ln(I0/I)= Lk()n0 То есть логарифм отношения интенсивности падающего излучения к интенсивности прошедшего излучения линейно зависит от концентрации и толщины слоя, через которого проходит излучение. Эта величина — ln(I0/I) — называется оптической плотностью и обозначается буквой A (adsorbtion). Так как натуральные и десятичные логарифмы связаны простым соотношением (ln x 2.30259 lg x и lg x 0.43429 ln x), и проще пользоваться десятичными логарифмами, их и используют. При фиксированной толщине слоя (часто используют толщину 1см) связь концентрации и оптической плотности выражается простым соотношением: lg(I0/I)=A A = k()n0 Что измеряет спектрофотометр? Как и каждый прибор, которым мы пользуемся (компьютер, телевизор, холодильник, стиральная машина) спектрофотометр – электротехнический прибор. И измеряет он электрический ток. Этот ток функционально связан с отношением излучения, прошедшего через исследуемое вещество и излучения, прошедшего через эталонную кювету. За это отвечает фотоэлемент (фотоумножитель или фотодиод) и электронная схема и программное обеспечение. Чем меньше света прошло через вещество, тем меньше измеренный нами сигнал. Если эталон поглотил очень много света, то эталонный сигнал равен нулю. А на ноль делить нельзя. То есть при очень большом поглощении эталона измерить мы ничего не можем. Это раз И два. Измеренный нами сигнал, который мы называем «пропусканием» (а это отношение энергии контрольного луча к эталонному), как следует из нашего вывода, связан с концентрацией вещества не линейно. Линейно связан с концентрацией вещества логарифм отношения энергии эталонного луча к энергии луча, прошедшего через образец. Эта же величина линейно связана и с толщиной кюветы. Называется она оптической плотностью (илиAbsorbence) Все старые спектрофотометры выдавали результат в пропускании Пусть пропускание в минимуме 10% Вычислим оптическую плотность: 100% делим на 10% Получаем 10. Это безразмерная величина. Логарифм 10 равен 1. Тоже величина безразмерная Современный спектрофотометр работает с компьютером и выдает результат в той форме, которая нам удобна Если пропускание равно 50%, то оптическая плотность равна 0.30 Если пропускание 1%, то оптическая плотность равна 2 Если пропускание 0.1%, то оптическая плотность равна 3 Оптическая плотность Оптическая плотность связана с концентрацией линейно (закон Бугера-ЛамбертаБера). То есть в идеале график функции, определяющей зависимость оптической плотности от концентрации – прямая линия, проходящая через начало координат. В том случае, когда в системе происходит химическая реакция, наблюдаются отклонения от закона Бугера-Ламберта-Бера. Отклонения могут быть и при очень больших концентрациях (в результате межмолекулярного взаимодействия), и при образовании «лаков», но в большинстве случаев закон выполняется очень хорошо. И при установлении параметров градуировочного графика, то есть при определении коэффициента в уравнении: A =k C L Где С – концентрация, А – оптическая плотность при длине кюветы L, а k – коэффициент пропорциональности, то есть тангенс угла наклона прямой к оси абсцисс, приходится решать лишь одну проблему. Проблема эта состоит в том, что ни один из измеряемых параметров ( в любой области) мы не можем определить точно. Все экспериментальные значения любой величины могут быть определены лишь с погрешностью. Экспериментальные точки Но сначала определим физический смысл коэффициента пропорциональности в уравнении A =k CL Концентрация — это г/объем, А — величина безразмерная, L – длина То есть размерность коэффициента — площадь, деленая на количество вещества. Если количество вещества выразить в молях и вспомнить, что в моле 610 в 23 степени, то коэффициент можно определить, как «эффективную» площадь молекулы, поглощающей свет. Принцип градуировки по экспериментальным точкам http://chemstat.com.ru/node/14 Соболев В.А. // СОЖ 2000, №4, с. 117. Основным требованием при градуировке по экспериментальным точкам является минимизация суммы квадратов отклонения этих точек от той градуировочной функции, которую мы хотим построить. Метод построения этой функции так и называется — метод наименьших квадратов. В приведенных ссылках можно прочитать о нем, но самый простой вывод в книге Р.С. Гутер и Б. В. Овчинский Элементы численного анализа и математической обработки результатов опыта Москва, 1962 Программа «Анализ данных»-Регрессия в Excel все сделает за вас. Но нужно помнить, что при задании данных в эту программу за Х принимают те экспериментальные данные, ошибка которых меньше (это заложено в вывод МНК). Раньше ошибка определения веса была меньше, чем ошибка измерения оптической плотности. И за Х принимали концентрацию. Теперь ошибки определения веса и оптической плотности одного порядка. И не будет большой ошибкой менять их местами (вместо прямой регрессии использовать обратную. Но в общем случае этого делать нельзя). В «обычном» спектрофотометре приемник регистрирует излучение в очень маленьком интервале спектра. И чем этот интервал меньше, тем лучше «разрешение» (способность прибора регистрировать различие спектра в соседних точках) прибора. И для того, чтобы зарегистрировать спектр по всей области, монохроматор необходимо поворачивать так, чтобы он последовательно направлял на приемник весь спектр от начала и до конца. Кроме того, чтобы учитывать и сравнивать энергию источника излучения и чуствительность приемника, необходимо направлять на приемник попеременно луч, проходящий через измеряемый образец и проходящий через эталон. Следовательно, спектрофотометр, построенный по такой схеме, должен содержать подвижные части. Которые от времени изнашиваются, разбалтываются и за счет этого вносят в измерение ошибки. От этого недостатка свободны спектрометры на диодных матрицах. Спектрометр на линейной диодной матрице Что такое линейная диодная матрица? Это маленькая тонкая пластинка, состоящая из нескольких сотен фотодиодов. Аналогичные стоят в современных фотоаппаратах и видеокамерах и телефонах. Этих фотодиодов (элементов матрицы) может быть 256. Или 512. Или 1024. Или 2048. Или еще больше. И монохроматор (в этом случае он называется полихроматор) направляет весь спектр целиком на диодную матрицу. Так как источника излучения два – дейтериевая лампа для УФ и галогенная лампа для видимой области — то спектрометр, в сущности, состоит из двух спектрометров. Один на УФ, другой на видимую область. Но никаких движущихся частей в оптической схеме нет, и регистрация спектра происходит сразу по всей области. Разве что кюветы приходится двигать. Потому что все равно сначала необходимо зарегистрировать эталонный спектр, а потом уже спектр исследуемого образца. Компьютер эти спектры запоминает и потом выдает нам их отношение. Теперь мы знаем все, что необходимо для работы на спектрофотометре. Очень просто на спектрофотометре определить свободный хлор в водопроводной воде. Как вы знаете, водопроводную воду для уничтожения болезнетворных микроорганизмов хлорируют. Воду плавательных бассейнов тоже хлорируют. Хлор вообще вреден, и необходимо следить, чтобы больших излишков хлора в воде не было. Методика основана на вытеснении свободным хлором иода из его солей (школьный курс химии) 2 KJ + Cl2 = J2+2KCl Иод плохо растворим в воде и хорошо в бензине. Если в водный раствор иода налить бензин и встряхнуть несколько раз, то весь иод перейдет в бензин. Если водного раствора было 200 мл, а бензина 20 мл, то произойдет десятикратное концентрирование. Для градуировки необходимо приготовить серию градуировочных растворов. Взять навеску иода и растворить в определенном объеме бензина. Очень большую навеску брать бессмысленно. Потому что измерить оптическую плотность очень окрашенных растворов нельзя (почему?). А очень маленькую навеску взять точно не получится. Взвесить мы можем не менее ??? (весы взвешивают до четвертого знака после запятой). Это будет рабочий раствор. Из рабочего раствора надо приготовить не менее пяти градуировочных. То есть взять пять мерных колб и мерной пипеткой внести в них различное количество градуировочного раствора. Прилить бензин до метки. Определить в этих растворах оптическую плотность, построить градуировочную функцию и с ее помощью определить иод в экстракте. И пересчитать на хлор. Измерять оптическую плотность следует в области от 0.3 до 0.8 единиц. В этом случае ошибка измерения будет минимальной. Можно снимать спектры непрозрачных образцов. Таких, как окрашенные ткани. Так можно опредялять стойкость красителя. Или качество моющего средства, используемого для стирки ткани — отстиралась ткань или нет. Спектры регистрируют при помощи специальных приспособлений. Это основное, что нужно знать о спектрофотометрии поглощения • Без знания этого делать практические задачи очень сложно. • Но существует ещё и спектрофотометрия флуоресценции Флуоресцентная спектроскопия Применение флуоресценции для аналитических целей включает широкую область использования ее для идентификации веществ, для обнаружения малых концентраций веществ, для контроля изменений, претерпеваемых веществом, для определения степени чистоты веществ. Широко применяются измерения флуоресценции при изучении кинетики обычных химических реакций. Высокая чувствительность метода позволяет фиксировать малую степень превращения веществ, а иногда по люминесценции промежуточных соединений становится возможным установить механизм химической реакции. Флуоресцентные методы используются в биологии, в частности для исследования структуры белков методом флуоресцентных зондов и меток. Механизм флуоресценции очень сложен. При поглощении молекулой кванта света его энергия затрачивается не только на забрасывание электрона на более высокий энергетический уровень, но ещё и на возбуждение колебаний и вращение молекулы. Эти явления создают очень сложную картину при переходе электрона на основной невозбужденный уровень. В некоторых случаях переход безизлучательный – энергия уходит на разогрев системы. В других случаях безилучательно расходуется часть энергии. А другая часть расходуется на излучение. Спектр этого излучения всегда сдвинут в более длинноволновую область по отношению к спектру возбуждения (то есть поглощения) Флуоресцируют в основном вещества биологического происхождения. Но если уж вещество флуоресцирует, то чуствительность его определения очень высока. d – толщина кюветы, – молярный коэффициент поглощения, с – концентрация В современном флуоресцентном спектрометре два монохроматора. Это позволяет регистрировать одновременно спектр возбуждения и спектр флуоресценции Флуоресцентная спектрометрия используется при определении микотоксинов, бензпирена и других полиароматических углеводородов. Несколько лет назад был принят ГОСТ по определению идентификации водок этим методом. Но флуоресцентные спектрометры дороже УФ-ВИД спектрофотометров • То есть способность вещества поглощать излучение зависит от его структуры, от наличия тех или иных «функциональных» групп. Благодаря которым в молекуле существуют молекулярные орбитали с энергетическими уровнями, которые соответствуют (могут быть возбуждены) электромагнитному излучению, которое мы используем. Только надо учитывать, что важна не столько структура молекулы, сколько тип связи, вид электронного облака, которое взаимодействует с электромагнитным излучением. • Конечно, для каждой молекулы найдется такое излучение, которое она будет поглощать. И очень коротковолновое излучение (гамма-лучи) ионизирует вещество – выбивает электроны так, что они улетают в пространство • Но нас сейчас интересует только область от 200нм до 1000нм – та область, которую используют при исследованиях и анализе на самых «обычных» спектрофотометрах. Которые не надо вакуумировать и при работе на которых можно использовать для кювет такие доступные материалы, как кварц (для УФ и видимой области) и стекло (для видимой области) Очень коротко и более или менее понятно о том, какие виды химической связи существуют, можно прочесть здесь. http://www.chemport.ru/chemicalbond.shtml http://www.hemi.nsu.ru/ucheb132.htm http://www.krugosvet.ru/articles/113/1011313/1011313a1.htm Для нас важно, что существует «красный порог» для взаимодействия света (электромагнитного излучения) с электронами вещества. И потому вся область взаимодействия излучения, если не прибегать к каким-то особым приемам и не рассматривать излучение, разрушающее вещество, ограничена относительно узкой спектральной областью – от «полужесткого» ультрафиолета (жесткий, коротковолновый ультрафиолет вещество уже разрушает) до 800нм. То есть вся совокупность энергий химических связей, если говорить о возможности электронов этих связей «прыгать» на более высокие энергетические уровни, укладывается в эту область. Их не так уж много, типов этих связей. И потому электронные спектры мало пригодны для идентификации вещества. Они «маловыразительные». И приходится прибегать к специальным приемам, чтобы использовать их в аналитической химии. Для определения вещества спектрофотометрией в видимой области стараются получить, в результате какой-либо химической реакции, его окрашенное производное, по поглощению которого и судят о наличии и количестве аналита. Разработка методики определения заключается в поиске реактива, дающего с аналитом цветную реакцию. Реакция должна быть селективна и образовавшееся цветное производное должно иметь очень сильное поглощение. Для видимой области без получения цветных производных можно определять лишь немногие окрашенные вещества — такие, как иод, бром, хромат- и перманганат- ионы (K2CrO4, KMnO4) и др.. Но в УФ области часто вещества определяют непосредственно, по собственному поглощению. Потому что в УФ, особенно менее 200нм, поглощают все вещества. ЭНЕРГИИ ВОЗБУЖДЕНИЯ И ДЛИННОВОЛНОВЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ АЛИФАТИЧЕСКИХ И АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ Соединение кДж/ моль нм Цвет Бутадиен СН2 = СНСН= СН2 553 217 Бесцв. Гексатриен СН2 = СНСН= СНСН= СН2 481 260 То же Октатетраен СН2 = СН(СН= СН)2СН= СН2 397 302 То же Ликопин {(СН3)2С = СН(СН2)2С(СН3) = СН[СН = СНС(СН3) = СН]2СН = }2 237 506 Яркокрасный бензол 471 255 Бесцв. нафталин 383 275 То же коронен 292 411 Желтый ДЛИННОВОЛНОВЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ РАЗЛИЧНЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ Соединение Бензол C6Н6 Нитрозобензол C6H5NO Анилин С6Н5NН2 Диметиланилин C6H5N(CCH3)2 4-Нитрозодиметиланилин (CH3)2NC6H4NO нм 255 280 282 297 420,5 Хромофорные группы—группы, введение которых в вещество вызывает поглощение света А это группы, оказывающие влияние на положение полос поглощения Таблица взята из этой книги А теперь можно попытаться сконструировать спектрофотометр – прибор, с помощью которого мы будем исследовать, как вещества поглощают электромагнитное излучение. То есть определять, какое (с какой длиной волны, с какой энергией) излучение вещество поглощает, а какое нет. И в какой степени. Нам нужен источник излучения, приемник излучения – фотоэлемент и устройство, с помощью которого на фотоэлемент мы будем направлять излучение с определенной длиной волны. Потому что фотоэлементу все равно, какая длина волны у излучения. На любое излучение (до «красного порога») фотоэлемент будет реагировать появлением электрического тока. Только сначала обратим внимание на две вещи. Вот первая Спектр солнечного излучения, полученный с помощью простейшего спектрометра Полученный спектр «сплошной», «непрерывный» Спектр испускания раскаленного атомарного водорода. Водород в так называемой водородной лампе разогревается сильным электрическим разрядом. Свет лампы, пройдя через призму, дает спектр, состоящий из отдельных линий Что мы хотим? Мы хотим получить объективные характеристики нашего товара при взаимодействии его (или того, что мы из него выделим, того, что мы из него получим) со световым излучением. А с каким «световым излучением»? У солнца это излучение одно, у водорода, когда мы заставили его светиться — другое. Если мы выбрали такой «полосатый» источник излучения, то очень может быть, что фотоэлемент будет говорить нам не о поглощении света анализируемым веществом, а о том, что света просто нет. Это первая проблема при построении спектрофотометра А вот вторая проблема. Фотоэлементы на разные участки спектра реагируют по разному А это значит, что при решении вопроса, как реагирует наше вещество на световое излучение, мы должны учитывать, какой спектр того источника, которым мы пользуемся, и какие свойства у нашего приемника излучения На каждом участке спектра мы должны быть уверены, что измеренные нами характеристики — это характеристики нашего образца, а не источника света и не приемника излучения. Для того, чтобы учесть это, добавляют еще один луч — эталонный, который не проходит через образец. И в каждом участке спектра измеряют отношение энергий эталонного луча и луча, прошедшего через образец. По этому принципу устроены все спектрофотометры Принципиальная схема спектрофотометра 1- источник излучения; 2,3,4,5-зеркала, служащие для формирования двух одинаковых лучей; 6-кювета с образцом, 8кювета сравнения, 7-оптический клин; 9- прерыватель луча; 10-монохроматор; 11- приемник излучения На схеме показан только один источник излучения. На самом деле их два – один для УФ области (дейтериевая лампа), другой для видимой (лампа накаливания, галогенная) Современный УФ-Вид спектрофотометр Количественное определение Что измеряет спектрофотометр? Как и каждый прибор, которым мы пользуемся (компьютер, телевизор, холодильник, стиральная машина) спектрофотометр – электротехнический прибор. И измеряет он электрический ток. Этот ток функционально связан с отношением излучения, прошедшего через исследуемое вещество и излучения, прошедшего через эталонную кювету. За это отвечает фотоэлемент (фотоумножитель или фотодиод) и электронная схема и программное обеспечение. Чем меньше света прошло через вещество, тем меньше измеренный нами сигнал. Если эталон поглотил очень много света, то эталонный сигнал равен нулю. А на ноль делить нельзя. То есть при очень большом поглощении эталона измерить мы ничего не можем. Это раз И два. Измеренный нами сигнал, который мы называем «пропусканием» (а это отношение энергии контрольного луча к эталонному), как следует из нашего вывода, связан с концентрацией вещества не линейно. Линейно связан с концентрацией вещества логарифм отношения энергии эталонного луча к энергии луча, прошедшего через образец. Эта же величина линейно связана и с толщиной кюветы. Называется она оптической плотностью (илиAbsorbence) Все старые спектрофотометры выдавали результат в пропускании Пусть пропускание в минимуме 10% Вычислим оптическую плотность: 100% делим на 10% Получаем 10. Это безразмерная величина. Логарифм 10 равен 1. Тоже величина безразмерная Современный спектрофотометр работает с компьютером и выдает результат в той форме, которая нам удобна Если пропускание равно 50%, то оптическая плотность равна 0.30 Если пропускание 1%, то оптическая плотность равна 2 Если пропускание 0.1%, то оптическая плотность равна 3 Оптическая плотность Оптическая плотность связана с концентрацией линейно (закон Бугера-ЛамбертаБера). То есть в идеале график функции, определяющей зависимость оптической плотности от концентрации – прямая линия, проходящая через начало координат. В том случае, когда в системе происходит химическая реакция, наблюдаются отклонения от закона Бугера-Ламберта-Бера. Отклонения могут быть и при очень больших концентрациях (в результате межмолекулярного взаимодействия), и при образовании «лаков», но в большинстве случаев закон выполняется очень хорошо. И при установлении параметров градуировочного графика, то есть при определении коэффициента в уравнении: A =k C L Где С – концентрация, А – оптическая плотность при длине кюветы L, а k – коэффициент пропорциональности, то есть тангенс угла наклона прямой к оси абсцисс, приходится решать лишь одну проблему. Проблема эта состоит в том, что ни один из измеряемых параметров ( в любой области) мы не можем определить точно. Все экспериментальные значения любой величины могут быть определены лишь с погрешностью. Экспериментальные точки Но сначала определим физический смысл коэффициента пропорциональности в уравнении A =k CL Концентрация — это г/объем, А — величина безразмерная, L – длина То есть размерность коэффициента — площадь, деленая на количество вещества. Если количество вещества выразить в молях и вспомнить, что в моле 610 в 23 степени, то коэффициент можно определить, как «эффективную» площадь молекулы, поглощающей свет. Принцип градуировки по экспериментальным точкам http://chemstat.com.ru/node/14 Соболев В.А. // СОЖ 2000, №4, с. 117. Основным требованием при градуировке по экспериментальным точкам является минимизация суммы квадратов отклонения этих точек от той градуировочной функции, которую мы хотим построить. Метод построения этой функции так и называется — метод наименьших квадратов. В приведенных ссылках можно прочитать о нем, но самый простой вывод в книге Р.С. Гутер и Б. В. Овчинский Элементы численного анализа и математической обработки результатов опыта Москва, 1962 Программа «Анализ данных»-Регрессия в Excel все сделает за вас. Но нужно помнить, что при задании данных в эту программу за Х принимают те экспериментальные данные, ошибка которых меньше (это заложено в вывод МНК). Раньше ошибка определения веса была меньше, чем ошибка измерения оптической плотности. И за Х принимали концентрацию. Теперь ошибки определения веса и оптической плотности одного порядка. И не будет большой ошибкой менять их местами (вместо прямой регрессии использовать обратную. Но в общем случае этого делать нельзя). В «обычном» спектрофотометре приемник регистрирует излучение в очень маленьком интервале спектра. И чем этот интервал меньше, тем лучше «разрешение» (способность прибора регистрировать различие спектра в соседних точках) прибора. И для того, чтобы зарегистрировать спектр по всей области, монохроматор необходимо поворачивать так, чтобы он последовательно направлял на приемник весь спектр от начала и до конца. Кроме того, чтобы учитывать и сравнивать энергию источника излучения и чуствительность приемника, необходимо направлять на приемник попеременно луч, проходящий через измеряемый образец и проходящий через эталон. Следовательно, спектрофотометр, построенный по такой схеме, должен содержать подвижные части. Которые от времени изнашиваются, разбалтываются и за счет этого вносят в измерение ошибки. От этого недостатка свободны спектрометры на диодных матрицах. Спектрометр на линейной диодной матрице Что такое линейная диодная матрица? Это маленькая тонкая пластинка, состоящая из нескольких сотен фотодиодов. Этих фотодиодов (элементов матрицы) может быть 256. Или 512. Или 1024. Или 2048. Или еще больше. И монохроматор (в этом случае он называется полихроматор) направляет весь спектр целиком на диодную матрицу. Так как источника излучения два – дейтериевая лампа для УФ и галогенная лампа для видимой области — то спектрометр, в сущности, состоит из двух спектрометров. Один на УФ, другой на видимую область. Но никаких движущихся частей в оптической схеме нет, и регистрация спектра происходит сразу по всей области. Разве что кюветы приходится двигать. Потому что все равно сначала необходимо зарегистрировать эталонный спектр, а потом уже спектр исследуемого образца. Компьютер эти спектры запоминает и потом выдает нам их отношение. Теперь мы знаем все, что необходимо для работы на спектрофотометре. Очень просто на спектрофотометре определить свободный хлор в водопроводной воде. Как вы знаете, водопроводную воду для уничтожения болезнетворных микроорганизмов хлорируют. Воду плавательных бассейнов тоже хлорируют. Хлор вообще вреден, и необходимо следить, чтобы больших излишков хлора в воде не было. Методика основана на вытеснении свободным хлором иода из его солей (школьный курс химии) 2 KJ + Cl2 = J2+2KCl Иод плохо растворим в воде и хорошо в бензине. Если в водный раствор иода налить бензин и встряхнуть несколько раз, то весь иод перейдет в бензин. Если водного раствора было 200 мл, а бензина 20 мл, то произойдет десятикратное концентрирование. Для градуировки необходимо приготовить серию градуировочных растворов. Взять навеску иода и растворить в определенном объеме бензина. Очень большую навеску брать бессмысленно. Потому что измерить оптическую плотность очень окрашенных растворов нельзя (почему?). А очень маленькую навеску взять точно не получится. Взвесить мы можем не менее ??? (весы взвешивают до четвертого знака после запятой). Это будет рабочий раствор. Из рабочего раствора надо приготовить не менее пяти градуировочных. То есть взять пять мерных колб и мерной пипеткой внести в них различное количество градуировочного раствора. Прилить бензин до метки. Определить в этих растворах оптическую плотность, построить градуировочную функцию и с ее помощью определить иод в экстракте. И пересчитать на хлор. Измерять оптическую плотность следует в области от 0.3 до 0.8 единиц. В этом случае ошибка измерения будет минимальной. Можно снимать спектры непрозрачных образцов. Таких, как окрашенные ткани. Так можно опредялять стойкость красителя. Или качество моющего средства, используемого для стирки ткани — отстиралась ткань или нет. Спектры регистрируют при помощи специальных приспособлений. Флуоресцентная спектроскопия Применение флуоресценции для аналитических целей включает широкую область использования ее для идентификации веществ, для обнаружения малых концентраций веществ, для контроля изменений, претерпеваемых веществом, для определения степени чистоты веществ. Широко применяются измерения флуоресценции при изучении кинетики обычных химических реакций. Высокая чувствительность метода позволяет фиксировать малую степень превращения веществ, а иногда по люминесценции промежуточных соединений становится возможным установить механизм химической реакции. Флуоресцентные методы используются в биологии, в частности для исследования структуры белков методом флуоресцентных зондов и меток. Механизм флуоресценции очень сложен. При поглощении молекулой кванта света его энергия затрачивается не только на забрасывание электрона на более высокий энергетический уровень, но ещё и на возбуждение колебаний и вращение молекулы. Эти явления создают очень сложную картину при переходе электрона на основной невозбужденный уровень. В некоторых случаях переход безизлучательный – энергия уходит на разогрев системы. В других случаях безилучательно расходуется часть энергии. А другая часть расходуется на излучение. Спектр этого излучения всегда сдвинут в более длинноволновую область по отношению к спектру возбуждения (то есть поглощения) Флуоресцируют в основном вещества биологического происхождения. Но если уж вещество флуоресцирует, то чуствительность его определения очень высока. d – толщина кюветы, – молярный коэффициент поглощения, с – концентрация В современном флуоресцентном спектрометре два монохроматора. Это позволяет регистрировать одновременно спектр возбуждения и спектр флуоресценции Флуоресцентная спектрометрия используется при определении микотоксинов, бензпирена и других полиароматических углеводородов. Несколько лет назад был принят ГОСТ по определению идентификации водок этим методом. Но флуоресцентные спектрометры дороже УФ-ВИД спектрофотометров • Как пример прозрачного вещества рассмотрим стекло. Оно является диэлектриком, и мы качественно представляем себе, как выглядит электронная структура стекла, если использовать понятие энергетических зон. На рис. 9-7 показаны две энергетические зоны, достаточные для объяснения явления. Нижняя зона заполнена: каждое квантовое состояние занято электроном. Над этой зоной находится отделённая энергетической щелью (зоной запрещённых энергий) пустая зона, в которой нет электронов. Расстояния по вертикали на рисунке соответствуют значениям энергий. Начальный фотон с энергией, соответствующей длине отрезка АВ, которая меньше, чем величина энергетической щели, не может быть поглощён ни одним из электронов в заполненной зоне, т.к. конечное состояние электрона должно находиться либо в заполненной зоне, где все места заняты, либо попадает в щель, где вообще не может быть никаких квантовых состояний электронов. Поэтому такой фотон будет проходить сквозь вещество. Пусть теперь фотоны имеют энергию большую, чем ширина энергетической щели, например, CD. Тогда электрон в нижней зоне может поглотить фотон и перейти в незанятое состояние в верхней зоне. Энергетические зоны электронов в прозрачном веществе типа стекла. Фотоны с энергиями АВ (величине энергии соответствует длина отрезка АВ), меньшими ширины щели между занятой и незаполненной зонами, не могут поглотиться электронами, т.к. нет таких конечных состояний, в которое те могли бы перейти. Такие фотоны проходят сквозь вещество. Если же энергии фотонов превышают ширину щели (CD на рисунке), то такие фотоны поглощаются веществом •Теперь можно понять, почему стекло прозрачно для видимого света. Зонная структура стекла такова, что у фотонов видимого света не хватает энергии, чтобы перебросить электроны из нижней зоны в верхнюю, преодолев щель. Поэтому фотоны видимого света проходят сквозь стекло без поглощения. Но фотоны ультрафиолетового света имеют более высокую частоту, следовательно, большую энергию, которой оказывается достаточно, чтобы вырвать электроны из заполненной зоны и перебросить их в незанятую зону. Поэтому такой свет будет поглощаться стеклом. На рис. 9-7 отрезку АВ соответствует фотон видимого света, а отрезку CD – ультрафиолетового. В кварце энергетическая щель шире, чем у обычного стекла, поэтому фотоны не только видимого, но и ультрафиолетового света не обладают достаточной энергией, чтобы электроны в заполненной зоне могли их поглотить и перейти в верхнюю пустую зону. В этом причина того, что ультрафиолетовые лампы делаются не из обычного стекла, а из кварца. Рассмотрим теперь случай света, падающего на поверхность металла. Мы знаем, что металл хорошо проводит электрический ток, т.к. внешние электроны каждого атома свободно путешествуют по всему металлу. На рис. 9-8 показан пучок света, отражающийся от поверхности металла. Ближайшие к поверхности электроны испытывают действие силы со стороны колеблющегося электрического поля световой волны и начинают сами колебаться с той же частотой, что и свет. Один из этих электронов, помеченный цифрой 1, показан на рисунке. Такой колеблющийся слой электронов отбирает энергию у световой волны, поэтому она ослабляется и заставляет колебаться следующий слой электронов (помеченный цифрой 2) с меньшей частотой и т.д., до тех пор, пока на определённой глубине от поверхности колебания поля в световой волне и соответствующие колебания электронов не затухнут. В большинстве металлов эта глубина очень мала и составляет величину порядка 10–6см. Теперь понятно, почему даже очень тонкий слой металла не пропускает свет . Поглощение и отражение света от металла. Осциллирующее электрическое поле световой волны заставляет колебаться электроны проводимости (показаны четыре электрона, причём два первых пронумерованы). Амплитуды колебаний электронов показаны стрелками. Эти стрелки уменьшаются при продвижении в глубь металла, что соответствует затуханию колебаний. Таким образом, электрон частично поглощает энергию падающего фотона, а частично переизлучает её в окружающее пространство Вы можете задать вопрос: откуда же берётся свет, отражённый от поверхности металла, т.к. создаётся впечатление, что вся энергия падающего фотона переходит в энергию колебаний электронов? Однако вспомним, что электромагнитные волны порождаются колеблющимися зарядами. Электроны на поверхности металла колеблются с той же частотой, что и падающий свет. Именно эти осцилляции и порождают электромагнитные волны той же частоты, что и падающая волна, вот они-то и образуют отражённый свет