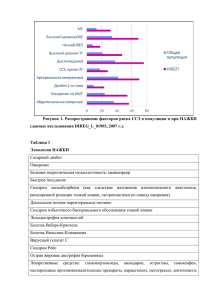
АУТОСОМНОДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ БОЛЕЗНЬ ГЕНТИНГТОНА БОЛЕЗНЬ ГЕНТИНГТОНА • В настоящее время от хореи Хантингтона в США страдает около 7000 человек. • Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100 000, • Частота встречаемости заболевания среди остальных рас 1:1 000 000. БОЛЕЗНЬ ГЕНТИНГТОНА • Начальный этап заболевания диагностируется обычно у лиц 30-50 лет • Данному недугу больше подвержены мужчины, чем женщины. • Длительность заболевания — около 15 лет. • Результатом болезни обычно является атрофия некоторых отделов головного мозга, включая полосатое тело мозга (стриатум — отдел, отвечающий за движения) и кору больших полушарий. БОЛЕЗНЬ ГЕНТИНГТОНА СИМПТОМЫ • Расстройство интеллекта — способность к логическому и абстрактному мышлению снижается, внимание ослабевает, происходят изменения в структуре личности человека. • Хореическая гиперкинезия — возникновение непроизвольных быстрых и беспорядочных движений в разных группах мышц, обусловленное снижением тормозного влияния коры на двигательные нервы БОЛЕЗНЬ ГЕНТИНГТОНА СИМПТОМЫ • Повышенная возбудимость, эмоциональные нарушения. Больной подвержен внезапным немотивированным приступам ярости, паники, тревоги, депрессии. У больного может развиться гиперсексуальность или появиться суицидальные тенденции. • Речевые расстройства, также сопровождающиеся приступами непроизвольных движений в мышцах. Больной при разговоре причмокивает, гримасничает, шмыгает носом, всхлипывает, у него могут беспорядочно двигаться глазные яблоки. На поздних стадиях речь становится совершенно невнятной, глотание затрудняется. • Нарушения сна. • Эндокринные расстройства. • Слабоумие (деменция) — это состояние развивается на последних стадиях заболевания. Могут появиться бред и навязчивые состояния. БОЛЕЗНЬ ГЕНТИНГТОНА ЛЕЧЕНИЕ (СИМПТОМАТИЧЕСКОЕ) • трифтазин, • тетрабеназин для купирования гиперкинезов; • различные нейролептики. Кроме купирования хореи, они помогают справиться с психическими нарушениями; • назначают антидепрессанты, на ранних стадиях болезни, для борьбы с депрессией и суицидальным поведением; • в случае прогрессирования болезни с повышением мышечного тонуса, применяется леводопа. БОЛЕЗНЬ ГЕНТИНГТОНА ПРОФИЛАКТИКА АУТОСОМНОРЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ СИНДРОМ МАРДЕНА-УОКЕРА – редкое заболевание соединительной ткани, которое наследуется по аутосомно-рецессивному признаку. Пациенты с этим расстройством, как правило, имеют отчетливое выражение лица, заячью губу или высокое арочное небо, маленькую челюсть, суставы в фиксированном положении, задержку роста и ограниченный контроль в движениях мышц. СИНДРОМ МАРДЕНА-УОКЕРА Очень редкое заболевание, которое развивается у мужчин чаще, чем у женщин с соотношением 11 к 3. В медицинской литературе описано около двадцати случаев. СИНДРОМ МАРДЕНА-УОКЕРА СИМПТОМЫ И ПРОЯВЛЕНИЯ • аномалии в челюсти, висящие веки, плоскую переносицу, низкие уши и выражение лица в фиксированном положении. • искривление позвоночника в результате чего развивается горб, контрактуры суставов, расселины или высокое арочное небо, задержку роста и медленное движение мышц. СИНДРОМ МАРДЕНА-УОКЕРА СИМПТОМЫ И ПРОЯВЛЕНИЯ • небольшая окружность головы, аномалии сердца, аномалии в мочевыделительной системе, снижение костной массы, аномально маленькие глаза, короткую шею, маленький рот и/или низкую линию роста волос. Девятимесячный мальчик с синдромом МарденаУокера. Тяжелые соматическое и психомоторное развитие, вес 2,5 кг, высота 55 см, окружность головы 43 см, гипотония, высунутый язык, полуоткрытые глаза, контрактуры суставов, арахнодактилия и двусторонние паховые грыжи СИНДРОМ МАРДЕНА-УОКЕРА ЛЕЧЕНИЕ • Генетическое консультирование может быть полезным для пациентов и их семей. • Лечение только симптоматическое и поддерживающее. • Консервативное ортопедическое лечение и физиотерапия будут обязательно указаны. Проблемы с развитием будут постоянными и тяжелыми, но контрактуры не прогрессируют и будут уменьшаться с возрастом и физиотерапией. Х-СЦЕПЛЕННЫЙ ДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ НЕДЕРЖАНИЕ ПИГМЕНТА (СИНДРОМ БЛОХА — СУЛЬЦБЕРГЕРА) Системное заболевание, наследуемое доминантно, Хсцеплено, проявляющееся своеобразными стадийными изменениями кожи в сочетании с патологией глаз, зубов, волос и ногтей, ЦНС и костномышечного аппарата. НЕДЕРЖАНИЕ ПИГМЕНТА (СИНДРОМ БЛОХА — СУЛЬЦБЕРГЕРА) • Распространенность составляет приблизительно 1 / 143,000. • Соотношение женщин и мужчин составляет 20: 1. • Заболевание существует с рождения или проявляется в первые недели жизни. • Имеет летальный эффект для эмбрионов мужского пола. НЕДЕРЖАНИЕ ПИГМЕНТА (СИНДРОМ БЛОХА — СУЛЬЦБЕРГЕРА) Стадия I (пузырьковая, или воспалительная). Она возникает в первые часы жизни в виде отечной эритемы с везикулобуллезными, реже уртикарными элементами, которые склонны к линейному расположению. Стадия II (веррукозная). Она развивается приблизительно через 2-3 мес и характеризуется лентикулярными ороговевающими папулами, расположенными преимущественно линейно в зоне бывших везикул или беспорядочно, напоминая бородавчатый невус. НЕДЕРЖАНИЕ ПИГМЕНТА (СИНДРОМ БЛОХА — СУЛЬЦБЕРГЕРА) Стадия III (гиперпигментация). Она наступает через 3-6 мес от начала заболевания. На месте регрессировавших очагов развивается пигментация коричневатого или темно-серого цвета, напоминающая брызги грязи на светлом фоне, в виде полосок и завихрений. Стадия IV (гипопигментация). Она проявляется на 2-3-м десятилетии жизни. Пигментация у части больных сменяется депигментацией, умеренно выраженной атрофией, очаговым склерозом. НЕДЕРЖАНИЕ ПИГМЕНТА (СИНДРОМ БЛОХА — СУЛЬЦБЕРГЕРА) • Лечение симптоматическое. • На ранних стадиях назначают : o 10% мазь цинка оксида, o мази с глюкокортикоидами, o анилиновые красители, при инфицировании - антибиотики. • При развитии массивных веррукозных изменений используют изотретиноин. Х-СЦЕПЛЕННЫЙ РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ СИНДРОМ ЛОУ генетическая патология человека, передающаяся по Хсцепленному рецессивному тип у наследования, относящаяся к группе цилиопатий. Характеризуется значительными аномалиями глаз, мозга, почек. СИНДРОМ ЛОУ • Синдром встречается редко. Встречается с частотой 1:500000 новорожденных. • Начальные симптомы заболевания выявляются в грудном и дошкольном возрасте, но не всегда могут быть выражены полностью, поэтому предельный возраст описания синдрома колеблется от 1 мес до 19-22 лет. • Болеют только мальчики, однако описаны случаи заболевания у женщин. СИНДРОМ ЛОУ СИМПТОМЫ ПОРАЖЕНИЯ ПОЧЕК • Патология проксимальных почечных канальцев: o гипераминоацидурия, протеинурия, o метаболический канальцевый ацидоз, o фосфатурия, o кальцийурия, глюкозурия. • В дальнейшем возможно поражение клубочков и развитие нефротического синдрома. • Прогрессирование хронической почечной недостаточности СИНДРОМ ЛОУ СИМПТОМЫ ПОРАЖЕНИЯ ГЛАЗ • двусторонняя катаракта, микрофакия, • сужение зрачков и отсутствие их реакции на свет, • повышение внутриглазного давления, врожденная глаукома, • голубые склеры, • вторичное помутнение роговицы, • хориоретинальные очаги и снижение светочувствительности сетчатки, • горизонтальный нистагм, • расходящееся косоглазие, • пальцеглазной синдром Фанческетта СИНДРОМ ЛОУ СИМПТОМЫ ПОРАЖЕНИЯ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ • отставание в психическом развитии, • выраженная мышечная гипотония, сопровождающаяся гиперподвижностью суставов, • снижение или отсутствие глубоких сухожильных рефлексов СИНДРОМ ЛОУ ЛЕЧЕНИЕ • Своевременно назначенная диета с ограничением поваренной соли и галактозы, • терапия синдрома Фанкони и рахита улучшают состояние больных. • Кроме того, необходима коррекция метаболического ацидоза.