Клинические испытания медицинских изделий
реклама

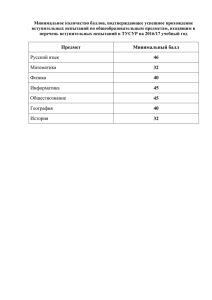
Клинические испытания медицинских изделий Нормативные документы Постановление Правительства российской Федерации от 27.12.2012 №1416 «Об утверждении правил государственной регистрации медицинских изделий» Приказ Минздрава России от 9 января 2014 г. N 2н «Об утверждении Порядка проведения оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий» Приказ Минздрава России от 08.02.2013 №58н «Об утверждении Положения о совете по этике в сфере обращения медицинских изделий» Приказ Минздрава России от 01.03.2013 №108 «Об утверждении состава совета по этике в сфере обращения медицинских изделий» Национальный стандарт «Руководство по проведению клинических испытаний медицинских изделий» ГОСТ Р ИСО 14155-1-2008 и ГОСТ Р ИСО 14155-2-2008 Национальный стандарт «Оценка функциональных характеристик медицинских изделий для диагностики in vitro» ГОСТ Р ЕН 13612-2010 Правила государственной регистрации медицинских изделий (ПП РФ №1416 от 27.12.2012) п. 4 Клинические испытания - разработанное и запланированное систематическое исследование, предпринятое, в том числе с участием человека в качестве субъекта для оценки безопасности и эффективности медицинского изделия п. 26 Клинические испытания медицинского изделия проводятся на основании: - разрешения на проведение клинических испытаний, - заключения об этической обоснованности проведения клинических испытаний, выданного советом по этике п. 27 Перечень медицинских организаций, имеющих право проводить клинические испытания медицинских изделий, и реестр выданных разрешений на проведение клинических испытаний медицинских изделий опубликовываются и размещаются на официальном сайте п. 29 О клинических испытаниях медицинского изделия заявитель уведомляет регистрирующий орган в течение 5 рабочих дней с начала их проведения Приказ Минздрава России от 09.01.2014 №2н п. 36 Клинические испытания медицинских изделий для оценки их безопасности и эффективности проводятся: • в форме исследований (анализ и оценка клинических данных); • в форме испытаний с участием человека п. 37 Испытания МИ с участием человека проводятся в следующих случаях: • новый вид медицинского изделия; • применение новых сложных и (или) уникальных и (или) специальных методов профилактики, диагностики и лечения заболеваний и состояний, а также применение новых сложных медицинских технологий; • если при проведении анализа и оценки клинических данных не подтверждены эффективность и безопасность медицинского изделия. Программа клинических испытаний п.39 При рассмотрении представленной документации на медицинское изделие согласовываются программа и продолжительность клинических испытаний медицинского изделия. Продолжительность клинических испытаний определяется назначением и сложностью медицинского изделия. Программа клинических испытаний составляется заявителем совместно с медицинской организацией, осуществляющей проведение клинических испытаний медицинского изделия, в соответствии с требованиями, указанными в технической и эксплуатационной документации производителя, а также требованиями нормативной документации. Оценка соответствия медицинских изделий для диагностики in vitro п.47. Клинические испытания медицинских изделий для диагностики in vitro проводятся в лабораторных условиях с применением образцов биоматериала пациентов, взятых в ходе лечебно-диагностического процесса (далее - клинико-лабораторные испытания) для проверки функциональных свойств и (или) эффективности медицинского изделия при использовании его в соответствии с назначением, предусмотренным документацией производителя. Клинико-лабораторные испытания медицинских изделий для диагностики in vitro в виде аналитических систем, проводимые в отношении медицинского изделия вместе с принадлежностями, наборами реагентов и калибраторов, необходимыми для применения медицинского изделия по назначению могут проводиться в рамках одного испытания. Приложения №4, 5 к порядку проведения оценки соответствия медицинских изделий - акты оценки результатов КИ МИ • утвержденная программа КИ МИ; • протоколы КИ или результаты оценки и анализа данных, включая графики, снимки, выписки из историй болезни, табулированный, статистически обработанный материал; • подробные данные по использованию МИ в медицинской практике, данные отдаленных результатов наблюдения (при наличии). Основные замечания по протоколам клинических замечаний для МИ ИВД • программа испытаний не охватывает все аспекты назначения или не отвечает назначению МИ ; • не обоснован объем выборки; • не обоснован выбор референтного метода; • не подтверждена правильность полученных результатов; • не проведен или не полностью проведен анализ полученных результатов и имеющихся данных: - не определены диагностические характеристики (или, наоборот, определены только диагностические характеристики); - не проведена статистическая обработка данных; - в анализ не включены имеющиеся данные производителя Статистическая обработка материалов клинических испытаний МИ ИВД • (1) Когда вообще применим статистический подход? • (2) Какую статистическую обработку результатов необходимо выполнить? • (3) Какое количество лабораторных исследований (тестов) должно быть выполнено в ходе испытаний? Оценка диагностической чувствительности при клинических испытаниях наборов реагентов для диагностики in vitro, предназначенных для выявления целевых маркеров в пробе • Диагностическая чувствительность – число лиц, точно классифицированных по результатам исследования, как находящихся в определенном состоянии, деленное на число лиц в этом состоянии[1]. Допускается применение термина «диагностическая чувствительность» как характеристики способности диагностического исследования in vitro идентифицировать присутствие в пробах пациента целевого маркера, связанного с целевым состоянием [2, 3] При этом в информации, предоставляемой потребителю, должно быть в явной форме указано, к определению какого маркера (аналита) относится характеристика диагностической чувствительности, а также должны быть приведены данные о степени связи (корреляции) присутствия целевого маркера с целевым состоянием пациента. 1. ГОСТ Р 53022.3-2008 «Технологии лабораторные клинические. Требования к качеству клинических лабораторных исследований. Часть 3. Правила оценки клинической информативности лабораторных тестов.» 2. ISO 18113-1:2009 (ГОСТ Р ИСО 18113-1 проект, одобренный техническим комитетом по стандартизации ТК 380 "Клинические лабораторные исследования и диагностические тест-системы ин витро") 3. Decision 2009/108/EC – Решение Еврокомиссии по общим техническим спецификациям медицинских изделий для диагностики in-vitro Статистические оценки применимы для случайных процессов! В диагностики in vitro случайными являются, в частности: - матричные эффекты (интерференты, кросс-реактивность и т.п.); - вариабельность аналита (различная активность|концентрация – разные стадии заболевания, разные генотипы, мутации и т.п.); - случайные составляющие погрешности аналитической аппаратуры/аналитического процесса. Как следствие, результаты изменений (исследований) содержат статистическую погрешность, которая должна быть оценена для принятия обоснованного решения о клинической эффективности медицинского изделия для диагностики in vitro. Типичный случай №1: исследуется эффективность выявления инфекционного агента в пробах пациентов; правильный результат в 100% случаев. Какова статистическая достоверность результатов испытаний? • Формула, описывающая нижнюю границу интервала, в котором находится «истинное» значение диагностической чувствительности с доверительной вероятностью C = 95%: D% = (1-C/100)(1/n) * 100% , где n – число измеренных проб. Примеры значений: n D% 5 10 20 50 100 400 1000 4000 54.9 74.1 86.1 94.2 97.0 99.3 99.7 99.9 Форма записи (для испытаний с n=100): диагностическая чувствительность с доверительной вероятностью 95% (97.0% - 100%) Типичный случай №2: исследуется эффективность выявления инфекционного агента в пробах пациентов; правильный результат получен в D0% случаев (D0% не равно 100%). Какова статистическая достоверность результатов испытаний? • Приближенная формула, описывающая интервал, в котором находится «истинное» значение диагностической чувствительности с доверительной вероятностью C = 95%: D% = D0% ± 1.96 * [D0% *(100- D0%)/n](1/2) проб. Примеры значений: n D0 % 100 400 1000 , где n – число измеренных D-% D+% 92 86.7 97.3 92 89.3 94.7 92 90.3 93.7 Форма записи (для испытаний с n=400): диагностическая чувствительность с доверительной вероятностью 95% (89.3% - 94.7%) Точное решение для нижней границы интервала для разных объемов выборки Требования к количеству проб при клинических испытаниях в Европейском союзе для МИ класса риска 3 • COMMISSION DECISION of 3 February 2009 amending Decision 2002/364/EC on common technical specifications for in vitro-diagnostic medical devices Anti-HIV-1/2 Пример: Определение диагностической чувствительности Определение диагностической специфичности 500 проб, включая различные субтипы + не менее 20 сероконверсионных панелей 5000 проб доноров + 200 проб больных + 100 проб с потенциальными интерферентами Пример: результаты клинических испытаний ВОЗ. МИ: INSTI HIV-1/HIV-2 Antibody Test ( bioLytical™ Laboratories), отчет № PQDx 0002-002-00 Формулировка результатов с учетом статистической погрешности: «В испытаниях на 1079 образцах проб пациентов, получена диагностическая чувствительность (с доверительной вероятностью 95% ) 100% (99.1% - 100%) и диагностическая специфичность (с доверительной вероятностью 95%) 99.7% (98.9% - 100%) в сравнении с референтным методом»