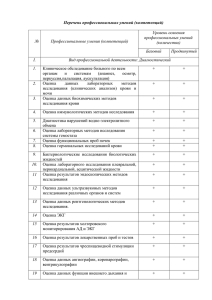
дальнейшее повышение эффективности и прозрачности
advertisement

МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ УКРАИНЫ ГОСУДАРСТВЕННЫЙ ЭКСПЕРТНЫЙ ЦЕНТР ДАЛЬНЕЙШЕЕ ПОВЫШЕНИЕ ЭФФЕКТИВНОСТИ И ПРОЗРАЧНОСТИ СИСТЕМЫ РЕГИСТРАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ В УКРАИНЕ развитие Процессы гармонизации законодательства Украины в системе регистрации лекарственных средств Политика в сфере регулирования национального фармацевтического сектора осуществляется с учетом евроинтеграционных процессов Регуляторные требования, принятые в Украине, по регистрации лекарственных средств разработаны в соответствии с директивами и регламентами ЕС руководствами ЕМA, ICH рекомендациями WHO рекомендациями ICDRA Упрощенная процедура государственной регистрации лекарственных средств - проект новой редакции гармонизация Порядка проведения экспертизы регистрационных материалов Препараты, лицензированные ЕМА по централизованной процедуре, Препараты для лечения ВИЧ/СПИД, туберкулеза, вирусных гепатитов, препаратов-сирот, зарегистрированных в странах со строгими регуляторными Препараты, лечения ВИЧ/СПИД, туберкулеза, преквалифицированные ВОЗ. Регистрационное досье подается в электронном виде, дополненное экспертным отчетом регуляторного органа или ВОЗ. Предусмотрен отдельный подход к оценке досье на ЛС, зарегистрированных ЕМА по централизованной процедуре: подтверждается соответствие инструкции для медицинского применения, короткой характеристики, текста маркировки упаковок и МКЯ, предоставленных заявителем, материалам регистрационного досье без проведения лабораторных испытаний (ранее – предрегистрационного контроля качества) Тем самым упрощается процедура регистрации и сокращается время экспертизы. Упрощена процедура регистрации вакцин и анатоксинов, которые прошли преквалификацию ВОЗ достижения Упрощенная процедура государственной регистрации лекарственных средств - исключения из общей процедуры регистрации ИЗМЕНЕНИЯ К ЗАКОНУ «О ЛЕКАРСТВЕННЫХ СРЕДСТВАХ» При регистрации лекарственного средства, предназначенного исключительно для лечения туберкулеза, ВИЧ/СПИД, вирусных гепатитов, онкологических и орфанных заболеваний, зарегистрированного регуляторным органом США, Швейцарии, Японии, Австралии, Канады или ЕС к заявлению прилагаются только МКК, инструкция для медицинского применения и макет упаковки. РЕШЕНИЕ О РЕГИСТРАЦИИ ПРИНИМАЕТСЯ МИНИСТЕРСТВОМ ЗДРАВООХРАНЕНИЯ В ТЕЧЕНИЕ 7 ДНЕЙ. достижения Процедура государственной перерегистрации ЛС – ПРОЦЕССЫ ГАРМОНИЗАЦИИ Директива 2001/83/ЕС Закон Украины Европейского Парламента и Совета ЕС «О лекарственных средствах» Продление торговой лицензии Статья 24 Торговая лицензия • действует в течении 5 лет; • через 5 лет лицензия может быть продлена на неограниченный период на основании обновленного досье; • может быть применен дополнительный 5-летний период обновления Перерегистрация лекарственного средства Статья 9 (изменения от 20.10.2014) «Після закінчення строку, протягом якого було дозволено застосування лікарського засобув Україні, його подальше застосування можливе за умови перереєстрації. Після перереєстрації строк застосування в Україні лікарського засобу не обмежується» достижения Процедура государственной перерегистрации ЛС – ПРОЦЕССЫ ГАРМОНИЗАЦИИ На основании экспертной оценки обновленных данных о соотношении польза/риск при применении лекарственного средства. Предусмотрен переходный период 2 года, в течение которого заявитель может подавать PSUR. Комплект сопроводительных документов не включает модуль 3. Процессы гармонизации - государственной регистрации лекарственных средств - проект новой редакции гармонизация Порядка проведения экспертизы регистрационных материалов Во время первой регистрации ЛС в Украине необходимо подавать План управления рисками; мастер-файл системы фармаконадзора Порядок внесения изменений в регистрационные материалы (досье) разработан в полном соответствие с руководством ЕС (Communication from the Commission — Regulation (EC) No 1234/2008 of 24 November 2008 «Guideline on the details of the various categories of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products (2010/C 17/01) Отмена обязательного предрегистрационного контроля качества, а только в некоторых случаях воспроизведение в лаборатории методов контроля. Особенные подходы к регистрации подобных биологических препаратов, которые отличаются от подходов к регистрации обыкновенных генериков. достижения Регистрация биоподобных препаратов (биосимиляров) Государственным экспертным центром МЗУ, согласно с Концепцией адаптации законодательства Украины к законодательству ЕС (постановление КМУ от 16.08.1999 № 1496), разработаны и утверждены нормативные документы, которые регламентируют подходы к разработке, планированию и проведению исследований подобных биологических препаратов (биосимиляров) и экспертизы результатов таких исследований с целью установления подобности биосимиляра к референтному препарату. На основании Руководства EMEA/CHMP/BMWP/42832/05 (пересмотренная версия) Методические рекомендации Центра «Общие принципы доклинических и клинических исследований биологически подобных лекарственных средств, содержащих в качестве активной субстанции белки, полученные методами биотехнологий» http://www.dec.gov.ua/site/file_uploads/ua/biosimilars/3.pdf достижения Нормативные документы и руководства по разработке и производству биологических/биотехнологических продуктов и биосимиляров, утвержденные в Украине ICH Q6B Настанова СТ-Н МЗУ 42 – 8.3:2013 «Спецификации: методы испытаний и критерии приемлемости для биотехнологических/биологических продуктов» EMEA/CНMP/BWP/328/99 «Development Pharmaceutics for Biotechnological and Biological Products (Annex to Note for Guidance on Development Pharmaceutics)» Настанова СТ-Н МЗУ 42 – 8.1:2013 «Фармацевтическая разработка биотехнологических и биологических продуктов» ICH Q5С Настанова СТ-Н МЗУ 42 – 8.2:2013 «Испытания стабильности битехнологических/биологических продуктов» CH Q5E Настанова СТ-Н МЗУ 42 – 8.4:2013 «Сравнимость биотехнологических/биологических продуктов при изменениях в процессе их производства» EMEA/CHMP/BWP/49348/2005 Настанова СТ-Н МЗУ 42-8.0:2013 «Подобные биологические лекарственные препараты, которые содержат как активные вещества протеины, полученные биотехнологическим путем» перспективы План управления рисками для биосимиляров Guideline on good pharmacovigilance practices (GVP) Настанова СТ-Н МОУ «Лекарственные средства. Надлежащая практика фармаконадзора» (планируется к утверждению – декабрь 2014 года) Module V – Risk management systems (Rev 1) Требования к Плану управления рисками в Модуле V – система управления рисками Выявление всех клинически значимых признаков иммуногенности и других нежелательных эффектов после регистрации препарата Для адекватного учета информации по нежелательным эффектам необходимо использование не МНН, а именно торгового названия препарата, номер серии, названия производителя ПРОЦЕССЫ ГАРМОНИЗАЦИИ обеспечение прозрачности проведения экспертизы С 17.03.2014 с целью обеспечения прозрачности деятельности Государственного экспертного Центра МЗ относительно процедуры государственной регистрации лекарственных средств приказом Государственного экспертного Центра МЗ утверждена форма «Экспертного отчета по оценке лекарственного средства для общественности» (разработан в соответствии с формой Summary for the Public of the European Public Assessment Report (EMA). Recommendations on transparency related to agendas/minutes on product related issues (implementation of Article 126b of Directive 2001/83/EC as amended and Article 80 of Regulation (EC) No 726/2004 November 2010 EMA/484118/2010 HMA/EMA recommendations on transparency После утверждения МЗУ решения о государственной регистрации лекарственного средства Отчет для общественности подается для публикации на официальном сайте Государственного экспертного Центра МЗ http://www.dec.gov.ua/index.php/zviti-dlya-gromadskosti МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ УКРАИНЫ ГОСУДАРСТВЕННЫЙ ЭКСПЕРТНЫЙ ЦЕНТР СПАСИБО ЗА ВНИМАНИЕ! ГП «Государственный экспертный центр МЗ Украины» ДОРОШУК Л.В. doroshuk@dec.gov.ua Киев 2014