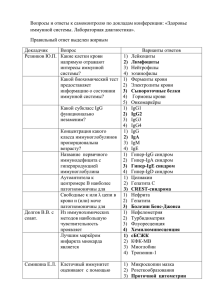
СПИСОК ПРИНЯТЫХ СОКРАЩЕНИЙ
реклама

Е. Л. НАСОНОВ ПРОТИВОВОСПАЛИТЕЛЬНАЯ ТЕРАПИЯ РЕВМАТИЧЕСКИХ БОЛЕЗНЕЙ МОСКВА Издательство "М-СИТИ" 1996 В книге суммированы современные данные о фармакологических свойствах, механизмах действия, клиническом применении и побочных эффектах основных групп противоревматических препаратов, включая нестероидные противовоспалительные средства, глюкокортикоиды, цитотоксики, препараты "базисного" действия, циклоспорин А и др. Особое внимание уделяется новым подходам к воздействию на иммунопатологические процессы, лежащие в основе ревматических заболеваний, в том числе методам иммунотерапии и экстракорпорального очищения крови. Книга рассчитана на ревматологов, терапевтов, специалистов в области клинической фармакологии и иммунологии. ООО "М-СИТИ" 121609, г. Москва, Осенний бульвар, д. 5. к. 1 Лицензия ¹ 064486 серия л.р от 11 марта 1996 г. Отпечатано в АО "Красногорская типография" 143400, г. Красногорск, Коммунальный кв., д. 2 Заказ ¹ 883. Тираж 5.000 экз. ОГЛАВЛЕНИЕ Список принятых сокращений Предисловие Введение. Современные классификации противоревматических препаратов Е. Л. Насонов Глава 1. Иммунитет и воспаление при ревматических заболеваниях Е. Л. Насонов Глава 2. Нестероидные противовоспалительные препараты. Е. Л. Насонов, Н. В. Чичасова, О. И. Лебедева Глава 3. Глюкокортикоиды Е. Л. Насонов, С. К. Соловьев. Глава 4. Соли золота Í. В. Чичасова, Е. Л. Насонов Глава 5. D-ïåíèöèëëàìèí Е. Л. Насонов, Н. В. Чичасова Глава 6. Метотрексат Е. Л. Насонов Глава 7. Антималярийные препараты Е. Л. Насонов Глава 8. Цитотоксические препараты Е. Л. Насонов Глава 9. Сульфасалазин Е. Л. Насонов, Н. В. Чичасова Глава 10. Циклоспорин А и другие препараты с близкими механизмами действия. Е. Л. Насонов, В. 3. Штутман Глава 11. Тенидап Е. Л. Насонов Глава 12. Внутривенный иммуноглобулин Е. Л. Насонов, С. К. Соловьев, А. В. Шайков, М. В. Богданова Глава 13. Экстракорпоральное очищение крови, дренаж грудного лимфатического протока, общее облучение лимфатических узлов Е. Л. Насонов Глава 14. Методы иммунотерапии Е. Л. Насонов Глава 15. Другие препараты и методы лечения Е. Л. Насонов Заключение. Песпективы фармакотерапии ревматических заболеваний Приложение Предметный указатель СПИСОК ПРИНЯТЫХ СОКРАЩЕНИЙ Азатиоприн АЗ Артериальная гипертензия АГ Адренокортикотропный гормон АКТГ Антигенпрезентирующие клетки АПК Ангиотензин-превращающий фермент АПФ Антифосфолипидный синдрома АФС Антифосфолипидные антитела ФЛ Глюкокортикоиды ГК Главный комплекс гистосовместимости ГКГ Диффузные болезни соединительной ткани ДБСТ Дерматомиозит ДМ Естественные киллерные клетки ЕК-клетки Желудочно-кишечный тракт ЖКТ Идиопатические воспалительные миопатии ИВМ Интерлейкины ИЛ Интерферон ИФ Клеточные молекулы адгезии КМА Лейкотриены ЛТ Матриксные металлопротеиназы ММП Метотрексат МТ Макрофаг МФ Моноциты МЦ Нестероидные противовоспалительные препараты НПВС Острофазовые белки ОФБ Простагландины ПГ Полимиозит ПМ Рецептор Р Ревматоидный артрит РА Ревматоидный фактор РФ Системная красная волчанка СКВ С-реактивный белок СРБ Системная склеродермия ССД Т-клеточные рецепторы ТКР Трансформирующий фактор роста ТФР Фактор активации тромбоцитов ФАТ Фактор некроза опухоли ФНО Хлорамбуцил ХБ Циклооксигеназы ЦОГ Циклоспорин ЦсА Циклофосфамид АЦФ Эндотелиальная клетка ЭК ПРЕДИСЛОВИЕ Воспалительные ревматические заболевания, основными формами которых являются ревматоидный артрит (РА), диффузные болезни соединительной ткани (ДБСТ), системные васкулиты, серонегативные и микрокристаллические артропатии, относятся к числу наиболее тяжелых форм хронической патологии человека. Фармакотерапия этих заболеваний продолжает оставаться одной из наиболее сложных проблем современной клинической медицины. Этиология многих болезней неизвестна, что делает невозможным проведение эффективной этиотропной терапии. Однако в расшифровке их патогенеза в последние годы наметился очевидный прогресс, что в первую очередь обусловлено расширением знаний о структурных и функциональных особенностях системы иммунитета, механизмах развития иммунного ответа и воспаления. В настоящее время для лечения ревматических заболеваний используется большое число лекарственных средств с различной химической структурой и фармакологическими механизмами действия, общим свойством которых является способность подавлять развитие воспаления. К ним относятся нестероидные противовоспалительные препараты, глюкокортикоиды, обладающие противовоспалительной активностью и так называемые базисные противоревматические препараты (соли золота, D-ïåíèöèëëàìèí, антималярийные препараты, цитотоксики и др.), которые, как полагают, оказывают более глубокое воздействие на иммунную систему и воспалительные процессы, лежащие в основе ревматических заболеваний. Интенсивно разрабатываются новые подходы к лечения, основанные на использовании иммунотерапевтических методов. В нашей стране было опубликовано несколько монографий, посвященных фармакотерапии ревматических заболеваний (В. А. Насонова, Я. А. Сигидин. Патогенетическая терапия ревматических заболеваний, 1985; В. А. Насонова, М. Г. Астапенко. Клиническая ревматология, 1989; Я. А. Сигидин, Н. Г. Гусева, М. М. Иванова. Диффузные болезни соединительной ткани, 1994). Однако в последние годы появилось очень большое количество новых клинических и экспериментальных данных, касающихся механизмов действия, тактики применения и эффективности как ранее известных противоревматических препаратов, так и новых лекарственных средств и методов лечения. В книге систематически изложены современные сведения о наиболее важных противовоспалительных препаратах, однако основной задачей явилось ознакомление с новыми тенденциями в развитии фармакотерапии воспалительных ревматических заболеваний. Мы надеемся, что книга окажется полезной практическим врачам в лечении больных ревматическими заболеваниями и стимулирует интерес к фармакологическим аспектам ревматологии у специалистов, занимающихся разработкой теоретических проблем медицины, иммунологов, биохимиков, фармакологов. Выражаем огромную признательность Е. Н. Литвак и Н. Ю. Милосердовой, а также Компании "АГАБ"" и рекламно-издательской группе "М-СИТИ" за помощь в подготовке книги. Е. Л. Насонов ВВЕДЕНИЕ СОВРЕМЕННЫЕ КЛАССИФИКАЦИИ ПРОТИВОРЕВМАТИЧЕСКИХ ПРЕПАРАТОВ Одним из наиболее часто встречающихся и тяжелых ревматических заболеваний является РА, для лечения которого используется весь арсенал противоревматических препаратов и методов терапии (В. А. Насонова и М. Г. Астапенко, 1989). Именно поэтому классификации противоревматических препаратов разрабатываются с точки зрения их места в лечении РА. На основании различий в фармакологических свойствах противоревматические препараты подразделяются на противовоспалительные анальгетики ( НПВС); противовоспалительные глюкокортикоиды (ГК), иммуномодулирующие/иммуносупрессивные агенты (соли золота, D-ïåíèöèëëàìèí, антималярийные препараты, цитотоксики и др.). Согласно другой классификации НПВС рассматриваются как симптоматические, не влияющие на механизмы развития заболевания, в противоположность модифицирующим болезнь или медленно действующим антиревматическим препаратам, которые, как полагали, влияют на этиопатогенез болезни. Для классификации противоревматических препаратов был также использован подход, учитывающий в первую очередь их токсичность, по которой они подразделяются на препараты первой, второй и третьей линии. Предлагалось классифицировать противоревматические препараты на основании быстроты наступления терапевтического эффекта и его длительности после прекращения лечения. НПВС и ГК в отличие от модифицирующих болезнь/медленно действующих противоревматических препаратов проявляют свой эффект очень быстро (в течение несколько часов или дней). Кроме того, предполагалось, что если после отмены НПВС и ГК обострение развивается достаточно быстро, то эффект медленно действующих противоревматических препаратов сохраняется в течение более длительного времени. Однако в настоящее время стало очевидным, что традиционные классификации не отвечают современным требованиям в отношении как терминологии, так и подразделения на фармакологические категории. Фактически только НПВС и ГК представляют собой относительно гомогенные в плане фармакологической и терапевтической активности группы лекарственных средств. Начиная с 1991 года под эгидой ВОЗ и Международной лиги по борьбе с ревматическими заболеваниями создается новая классификация противоревматических препаратов (H. E. Paulus и соавт., 1992; J. P. Edmonds и соавт., 1993), согласно которой эти препараты подразделяются на две основные категории: I. Модифицирующие симптомы антиревматические препараты, оказывающие положительное влияние на симптомы и клинические проявления воспалительного синовита: 1). нестероидные противовоспалительные препараты 2). глюкокортикоиды 3). медленно-действующие препараты: антималярийные, соли золота, D-ïåíèöèëëàìèí, антиметаболиты, цитотоксические агенты II. Контролирующие болезнь антиревматические препараты, влияющие на течение РА, которые должны соответствовать следующим требованиям: а. улучшать и поддерживать функциональную способность суставов в сочетании со снижением интенсивности воспалительного синовита; б. предотвращать или существенно снижать скорость прогрессирования структурных изменений в суставах. При этом перечисленные эффекты должны проявляться в течение не менее 1 года от момента начала терапии; в процессе классификации препарата должен указываться срок, (не менее 2 лет) в течение которого его терапевтический эффект соответствует перечисленным критериям. Эта классификация отличается от предыдущих более реалистичным подходом к оценке терапевтической эффективности препаратов при РА. В настоящее время стало очевидным, что общим доказанным свойством всех существующих противоревматических препаратов является способность вызывать клиническое улучшение, в то время как их возможность влиять на прогрессирование и исходы ревматоидного процесса нельзя считать строго доказанной. Поэтому ни один противоревматический препарат в настоящее время не может быть классифицирован как "контролирующий заболевание". Это, впрочем, не исключает возможности переноса в процессе дальнейших исследований тех или иных лекарственных средств из первой группы во вторую. Это положение представляется принципиальным, так как должно способствовать расширению фармакологических и клинических исследований в ревматологии в плане разработки критериев эффективности лечения, а также создания новых, более эффективных антиревматических средств или их рациональных комбинаций. Список литературы к введению "Современные классификации противоревматических препаратов" 1. Насонова В. А., Астапенко М. Г.: Клиническая ревматология. М., 1965, 589 стр. 2. Paulus Н. Е., Scott D. L, Edmonds J. P.: Classification of antirheumatic drugs: A new proposal. Arthritis Rheum. 1992; 35: 364-365. 3. Edmonds J. P.. Scott D. L., Furst D. E., Brooks p., Paulus Í. Å.: Antirheumatic drugs: a proposal for new classification (editorial). Arthritis Rheum. 1993; 36: 336-339. ГЛАВА 1 ИММУНИТЕТ И ВОСПАЛЕНИЕ ПРИ РЕВМАТИЧЕСКИХ ЗАБОЛЕВАНИЯХ Иммунная система играет важнейшую роль в защите организма от чужеродных агентов и поддержании постоянства внутренней среды. Характерной особенностью иммунной системы является специфичность иммунного ответа. В то же время эффективное предотвращение потенциально негативных последствий воздействия факторов внешней среды на организм человека во многом зависит и от неспецифических механизмов иммунной защиты. Основными компонентами иммунной системы, обеспечивающими клеточные и гуморальные иммунные реакции и развитие воспалительных процессов, являются лимфоциты и мононуклеарные фагоциты, а также клетки сосудистого эндотелия. 1.1. Иммунный ответ Иммунный ответ включает в себя 3 основные фазы: 1) фазу распознавания, во время которой макрофаги и лимфоциты реагируют с чужеродным антигеном (или аутоантигеном); 2) фазу активации, заключающуюся в клональной пролиферации и дифференцировке лимфоцитов, распознающих соответствующие антигены; 3) эффекторную фазу, которая приводит к элиминации чужеродного агента из организма. Нормальный иммунный ответ и формирование генетически детерминированной предрасположенности к развитию воспалительных ревматических болезней определяется генами главного комплекса гистосовместимости (ГКГ). ГКГ включает несколько сотен индивидуальных генов, которые кодируют экспрессию антигенов систем HLA человека (Human Leukocyte Antigen). Гены ГКГ занимают около 1% короткого плеча шестой хромосомы человека, включая 3000 kb ДНК. Система HLA и её продукты в соответствии со структурными и функциональными свойствами разделяется на классы. К классу 1 относятся HLA-A, -В и -С локусы и их серологически определяемые антигены. В локусе А установлено 24 специфичности, в локусе В — 52 специфичности, в локусе С — 11 специфичностей. Класс II включает специфичности, кодируемые HLA-DR, -DQ, -DP, -DN и -DO генами. К классу III относятся гены, кодирующие белки системы комплемента Bf, С2, С4, гены цитокинов (ФНО, ИЛ-1 α/β, антагонист ИЛ-1 рецептора) , стрессорных белков (Hsp70). В центромерной части В-локуса обнаружено 5 добавочных генов, которые получили название "В-ассоциированные транскрипты". Предполагается, что эти гены составляют семейство класса IV ГКГ (Bodmer W. A.). Антигены классов I и II ГКГ являются гликопротеинами и имеют доменную структуру. Антигены класса 1 присутствуют на мембранах всех ядерных клеток и тромбоцитов. Они состоят из субъединиц "тяжелых" цепей, соединяющихся с клеткой посредством трансмембранного гидрофобного "хвоста", и внеклеточных гидрофильных участков, составляющих 3 домена (α1,-2,-3). Тяжелые цепи нековалентно ассоциируются с β2-микроглобулином — неполиморфным белком, гены которого находятся вне пределов ГКГ. (α-1 и α-2 домены весьма полиморфны и имеют детерминанты, специфичные для каждого индивидуума. Антигены класса II состоят из двух нековалентно связанных цепей, образующих α/β гетеродимер. Антигены класса II в норме экспрессируются только на так называемых антигенпрезентирующих клетках (АПК), функцию которых выполняют макрофаги, моноциты, дендритные клетки, В-лимфоциты, клетки Лангерганса, купферовские клетки, активированные Т-лимфоциты и, вероятно, эндотелиальные клетки (ЭК). Аминотерминальные домены α/β цепей обладают выраженной гипервариабельностью, которая определяет полиморфизм молекул класса II ГКГ. Важной функцией молекул ГКГ класса I и II является связывание и представление (презентирование) процессированных пептидных фрагментов чужеродных молекул для распознавания Т-лимфоцитами (более подробно механизмы "процессинга" антигенов будут рассмотрены ниже). При этом только клетки, имеющие одинаковый тип молекул ГКГ, могут реагировать друг с другом; этот феномен получил название "ограничение" (рестрикция) по ГКГ. Установлено, что процессированные пептидные фрагменты находятся как бы в "полости", образованной полиморфными аминокислотными остатками, составляющими гипервариабельные участки молекул ГКГ, и лишь в такой форме способны связываться с клонотипическими антигенными рецепторами Тлимфоцитов (ТКР). Таким образом, в отличие от иммуноглобулиновых молекул В-лимфоцитов, которые способны непосредственно реагировать с антигенами, особенностью Т-клеточного иммунного ответа является необходимость образования тримолекулярного комплекса, состоящего из молекулы ГКГ, процессированного антигенного пептидного фрагмента и ТКР. Кроме того, имеются различия в стабильности связывания пептидов с тяжелой цепью молекулы класса I или гетеродимером класса II ГКГ (K. Dormair и соавт., 1991). Молекулы класса I образуют комплекс с цитозольными и ядерными пептидами, состоящими примерно из 9 аминокислотных остатков, который распознается специфическими рецепторами CD8+ цитотоксических Тлимфоцитов, Молекулы класса II ГКГ связывают пептиды, происходящие из мембранных белков (в том числе других молекул ГКГ), которые состоят из 10-34 аминокислотных остатков (T. Elliot и соавт., 1990), и распознаются CD4+ хелперными Т-лимфоцитами. Существование аллельных форм HLA оказывает выраженное влияние на характер иммунного ответа. Вопервых, различия аминокислотной последовательности молекул HLA может приводить к селективному связыванию процессированных антигенных фрагментов только с определенными аллелями ГКГ. Этот процесс получил название "селекции детерминанты". Во-вторых, молекулы ГКГ оказывают влияние на "репертуар" ТКР в период созревания иммунной системы. Полагают, что молекулы ГКГ принимают участие в селекции Тклеточных клонов, экспрессирующих определенный тип ТКР. Этот процесс позитивной и негативной селекции, с одной стороны, определяет направленность иммунного ответа к собственным молекулам ГКГ, а с другой — формирование толерантности к собственным антигенам (аутоантигенам). Его развитие связывают с презентацией аутологичных антигенных фрагментов собственными молекулами ГКГ в период раннего онтогенеза. Итак, в процессе Т-зависимого иммунного ответа антигенные пептиды, ассоциированные с молекулами класса II ГКГ, экспрессирующимися на мембране АПК, реагируют с ТКР CD4+ Т-лимфоцитов, обладающими свойствами Т-хелперов. ТКР состоит из двух пептидных α и β цепей (с молекулярной массой примерно 45 kD), которые имеют вариабельные и константные участки и образуют комплекс с другим мембранным белком, определяемым как CD3. Небольшая популяция Т-лимфоцитов экспрессирует другой тип ТКР, состоящих из полиморфных цепей γ/σ типа. Полагают, что Т-лимфоциты, имеющие этот тип ТКР, играют важную роль в защите слизистой оболочки кишечника и мочеполовой системы, а также патогенезе некоторых ревматических заболеваний. Например, γ/σ Т-клетки распознают стрессорные белки бактерий в отсутствие молекул ГКГ, некоторые из них могут реагировать с аутоантигенами поврежденных или подвергнутых стрессу клеток. Активация Т-клеток, опосредуемая взаимодействием ТКР с процессированными АПК антигенными фрагментами, усиливается добавочными молекулами, такими, как CD4, CD8 и LFA-1. Важное значение имеют и так называемые костимуляторные молекулы, к которым относится CD2, взаимодействующий с LFA-3, и CD28, взаимодействующий с В7 белком, специфичным для В-лимфоцитов. Начальная активация Т-клеток происходит за счет перекрестного связывания ТКР, что в присутствии костимулирующих сигналов обеспечивает активацию фосфатидилинозитольного пути. Ключевую роль в этом процессе играет тирозинкиназа, активация которой опосредуется цитоплазматическим компонентом молекулы CD3. Активированные Т-клетки экспрессируют рецепторы для ИЛ-2 и ИЛ-4, связывание которых с соответствующими цитокинами обеспечивает аутокринную стимуляцию пролиферации Т- и В-лимфоцитов. Кроме того, АПК начинают синтезировать растворимые медиаторы, в частности ИЛ-1, который, связываясь с ИЛ-1 рецепторами на АПК, вызывает дополнительную активацию последних, стимулирует Т-лимфоциты и индуцирует разнообразные системные реакции (острофазовый ответ), выходящие за рамки иммунной системы. Большинство антигенов инициирует развитие как клеточного, так и гуморального типов иммунного ответа, требующего кооперации СD4+Т-лимфоцитов и В-лимфоцитов. Цитокины, синтезируемые Т-клетками обеспечивают антигеннеспецифическую стимуляцию В-лимфоцитов. Т-лимфоциты, экспрессирующие на мембране молекулы CD8, связывают антигенные пептиды, ассоциированные с молекулами класса I ГКГ, и обладают цитотоксической активностью. Эти клетки играют важную роль в защите организма от вирусных инфекций и других внутриклеточных микроорганизмов. Кроме того, антиген CD8 экспрессируется на Т-супрессорных лимфоцитах. Предполагается, что в реализации активности этих клеток принимает участие ТФР-β. В-лимфоциты происходят из костномозговых предшественников и в отличие от Т-клеток не подвергаются дифференцировке в тимусе. В -лимфоциты обнаруживаются во всех лимфоидных органах, особенно в большом количестве в миндалинах, зародышевых центрах лимфатических узлов и селезенки. В-лимфоциты экспрессируют Fc-ðåöåïòîð (CD32), молекулы класса II ГКГ и другие детерминанты (CD19, CD20 и др.). Основной функцией В-клеток является синтез антител. При этом В-клетки принимают участие в "презентировании" антигенов Т-лимфоцитам. Для реализации этих функций В-лимфоциты должны активироваться. При соответствующей стимуляции В-клетки подвергаются быстрой дифференцировке и пролиферации, что приводит к секреции специфических антител. Некоторые В-лимфоциты, синтезирующие иммуноглобулины, имеют морфологию плазматических клеток, другие — остаются похожими на лимфоциты. В-клеточный ответ на так называемые Т-зависимые антигены предполагает их взаимодействие с CD4+Tлимфоцитами. Это взаимодействие опосредуется CD40 и другими мембранными молекулами В-лимфоцитов, обладающими костимуляторной активностью. Добавочный сигнал, необходимый для В-клеточной активации, предоставляется самим антигеном, особенно если он способен вызывать перекрестное связывание В-клеточных рецепторов. Это позволяет объяснить способность молекул с множественными антигенными детерминантами (бактериальный липополисахарид, вирус Эпштейна-Барр и др.) вызывать антигеннеспецифическую активацию В-клеток. Последняя получила название "поликлональная В-клеточная активация", и для ее развития необходимость в Т-хелперах (CD4+T-лимфоциты) отсутствует. 1.2. Иммуногенетическая предрасположенность С помощью современных генетических методов изучены аминокислотные последовательности гипервариабельных участков молекул. ГКГ и получены доказательства их участия в предрасположенности к развитию ревматических болезней и продукции аутоантител с определенной специфичностью. За исключением анкилозирующего спондилита и близких серонегативных спондилоартропатий, связанных с носительством HLA-B27 ("болезни круга HLA-B27"), все остальные аутоиммунные ревматические заболевания ассоциируются с определенными гаплотипами HLA класса II или молекулами ГКГ класса III (T. A. Dalton и J. C. Bennet, 1992). Установлено, что гипервариабельная область β1 домена, который, как предполагается, непосредственно контактирует с процессированным антигеном, обладает выраженным полиморфизмом. 80-90% больных РА являются носителями HLA-DR1 или одного из трех вариантов HLA-DR4 (Dw4, Dwl4, Dwl5). Эти гаплотипы имеют очень сходные аминокислотные последовательности (эпитопы) в гипервариабельном участке DRβ1-äîìåíà в положении 65-74. Имеется определенная связь между носительством HLA-DR4 и тяжестью РА, гиперпродукцией РФ, нарушением функции легких и быстрым развитием эрозивных изменений в суставах. Носительство HLA-DR3-y больных РА ассоциируется с развитием побочных реакций при применении солей золота (глава 4) и D-ïåíèöèëëàìèíà (глава 5). Предполагается, что при СКВ носительство тех или иных антигенов ГКГ в большей степени коррелирует не с самим заболеванием, а с продукцией определенных типов аутоантител (F. C. Arnett и J. D. Reveille, 1992). С4 нулевой аллель (C4*QO) обнаруживается у 50-80% больных СКВ и дает относительный риск, равный 3. Гомозиготный дефицит С4А (C4A*QO, *QO) выявляется у 10-15% больных СКВ и у 2-3% -в контроле (относительный риск 17). При этом практически все больные с полным дефицитом Ñ4 страдают СКВ или близкими аутоиммунными заболеваниями. Кроме того, частичный дефицит С4А и/или С4В ассоциируется с другими аутоиммунными заболеваниями, такими, как хронический активный гепатит, диффузный токсический зоб, синдром Шегрена, синдром Шенлейна-Геноха, IgA-íåôðîïàòèÿ, инсулинзависимый сахарный диабет. Необычный вариант Ñ4 2.9 (С4ВЗ) с высокой частотой встречается при РА и находится в неравновесном сцеплении с HLA-DR4. Приблизительно 75% лиц с гомозиготным С2 дефицитом имеют волчаночноподобный синдром или другие аутоиммунные заболевания, включая ДМ/ПМ или васкулит. Предполагается, что повышенный риск аутоиммунной патологии у больных с дефицитом компонентов комплемента связан с нарушением клиренса иммунных комплексов. Обсуждается значение генетического полиморфизма цитокинов в развитии ревматических болезней. Имеются данные о том, что особенности полиморфизма длины рестрикционных фрагментов ДНК гена ФÍÎ-α у NZBxNZW F1 мышей коррелирует с синтезом ФНО-α и развитием нефрита (C. D. Jacob и H. O. McDevitt, 1988). Полиморфные замены в промоторном участке гена ФНО-α ассоциируются с "аутоиммунным гаплотипом" HLA-A1, HLA-B8, HLA-DR3 (L. J. Abracham и соавт., 1993), носительство которого отмечено при нескольких аутоиммунных заболеваниях. Некоторые аллели ФНО-α и полиморфизм промоторных участков гена ФНО-α ассоциируются с развитием СКВ (M. P. Bettinotti и соавт., 1993) и синтезом антител к Ro- и La-àíòèãåíàì. Другим важным генетическим элементом является группа генов, кодирующих синтез ИЛ-1 α/β и антагониста ИЛ-1 рецептора, которые локализованы на длинном плече второй хромосомы. Отмечена связь между полиморфизмом ИЛ-1 генов и развитием СКВ (A. I. F. Blakemora и соавт., 1993), ювенильного хронического артрита (T. L. McDowell и соавт., 1993), ССД. Наряду с ГКГ важную роль в предрасположенности к аутоиммунным ревматическим болезням играют гены ТКР, кодирующие синтез иммуноглобулинов и гены, связанные с половыми гормонами (таблица 1.1.) Таблица 1.1. Роль генетических факторов в развитии аутоиммунных болезней (по D. A. Carson, 1992) Фактор Механизм предрасположенности к заболеванию Гены ГКГ Селективное связывание аутоантигенных пептидов; экспансия аутореактивных Т-клеток; делеция Т-клеток, контролирующих инфекцию Гены ТКР Увеличение количества аутореактивных Т клеток; снижение способности контролировать инфекцию, индуцирующую аутоиммунную патологию Гены иммуноглобулинов Нарушение презентации аутоантигенов; дефекты анергии; увеличение синтеза аутоантител Гены, кодирующие процессинг антигенов и транспорт пептидов Недостаточная экспрессия молекул ГКГ, предотвращающих периферическую анергию; нарушение процессинга аутоантигенов Гены комплемента Нарушение клиренса иммунных комплексов Гены, связанные с полом Иммунные эффекты половых гормонов Гены цитокинов Иммунные эффекты цитокинов Гены стрессорных белков ? Гены HLA-B ассоциированных транскриптов ? 1.3. Воспаление Воспаление является физиологической защитной реакцией организма в ответ на тканевое повреждение. Основу развития воспалительного процесса составляет каскад биохимических и иммунологических процессов, направленных на элиминацию повреждающего фактора, заживление пораженных тканей и восстановление нарушенной функции. Таким образом, воспаление, как и иммунный ответ, играет важнейшую роль в защите организма от патогенных возбудителей, способствует репарации тканей после физического или химического повреждения. Воспаление может быть острым или хроническим, однако в процессе развития воспалительной реакции, как правило, наблюдаются признаки воспаления обеих форм. Начальная фаза воспаления чаще всего протекает остро и характеризуется сосудистым ответом, участием нейтрофилов и тучных клеток. Хроническое воспаление обычно ассоциируется с мононуклеарными клетками (макрофаги, лимфоциты, плазматические клетки) и фибробластами. Присутствие последних отражает пролиферацию соединительной ткани. Хроническое воспаление может следовать за острым, но иногда воспалительный процесс с самого начала имеет черты хронического. При аутоиммунных ревматических заболеваниях нормальный воспалительный процесс не приводит к элиминации этиологических повреждающих факторов, которые в большинстве случаев неизвестны. Более того, персистенция повреждающего фактора, часто сочетающаяся с нарушением нормальных механизмов, ограничивающих выраженность воспалительных реакций, приводит к интенсификации и хронизации воспалительного процесса, а следовательно, к прогрессирующему повреждению тканей и нарушению функции различных органов и систем организма. В целом, в развитии воспаления принимают участие различные клетки — лейкоциты (в первую очередь нейтрофилы), моноциты/макрофаги, тромбоциты, лимфоциты, а также ЭК, а регуляция сосудистых и клеточных воспалительных реакций осуществляется очень большим числом биологически активных веществ. Важным начальным этапом воспаления является фагоцитоз, который заключается в поглощении и деструкции инородных частиц фагоцитирующими клетками. Среди лейкоцитов периферической крови способностью к фагоцитозу обладают гранулоциты (главным образом нейтрофилы, в меньшей степени — другие клетки) и моноциты. Процесс фагоцитоза подразделяется на несколько этапов — прилипание, хемотаксис, собственно фагоцитоз и внутриклеточное разрушение патогенных субстанций — и регулируется различными хемотаксическими стимулами, такими, как анафилотоксины (С5а), иммунные комплексы, ИЛ-8 и др. 1.4. Нейтрофилы На мембране нейтрофилов экспрессируются рецепторы, способные связывать компоненты комплемента С3b (CR1), iC3b (CR3; Mac-1 или CD11b/CD18) и С5а компоненты комплемента, а также рецепторы для Fcôðàãìåíòà иммуноглобулинов. Иммунные комплексы и активированные компоненты комплемента, связываясь с этими рецепторами, индуцируют секрецию воспалительных медиаторов, вызывающих тканевое повреждение. CR1 и CR3 принимают участие в процессе опсонизации бактерий. Кроме того, CR3 выступает в качестве молекулы адгезии, определяющей прилипание нейтрофилов к эндотелию и другим нейтрофилам. Адгезия активированных нейтрофилов к сосудистому эндотелию является одним из начальных этапов воспалительного процесса. Существует 3 основных класса Fc-ðåöåïòîðîâ. FcRI обладает высокой аффинностью к мономерному lgG, обнаруживается преимущественно на нейтрофилах и других мононуклеарных клетках. FcII и FcIII рецепторы присутствуют на нейтрофилах и мононуклеарных клетках и связывают иммунные комплексы с более высокой аффинностью, чем мономерный lgG. FcII обнаруживаются также на В-лимфоцитах и тромбоцитах. FcIII присутствуют на нейтрофилах, макрофагах, тромбоцитах и Т-лимфоцитах. Нейтрофилам придают важное значение в развитии тканевой деструкции при остром и хроническом воспалении. (H. L.Malech и J. I. Gallin, 1987). Развитие воспаления, независимо от причины и локализации, определяется способностью нейтрофилов образовывать широкий спектр биологически активных молекул, способных разрушать нормальные клетки и соединительную ткань. Нейтрофилы образуют не менее 50 субстанций, которые могут принимать участие в развитии воспаления и выступать в качестве медиаторов тканевого повреждения (R. I. Lehner и соавт. 1988). Роль этих веществ заключается в первую очередь в защите организма от патогенных воздействий. Однако, поскольку сами нейтрофилы не обладают внутренней способностью дифференцировать чужеродные и собственные клетки-мишени, а их специфическая активность зависит от действия других компонентов иммунной системы (антитела, комплемент, цитокины и др.), в случае ошибочного распознавания нормальных тканей как чужеродных активация нейтрофилов вызовет повреждение собственных клеток. Основные секреторные продукты нейтрофилов: Протеиназы Супероксидные анионы Окись азота Цитокины Антагонисты цитокинов Растворимые цитокиновые рецепторы Н+ лактат Эйкозаноиды Катионные белки Гликозидаза, липаза, сульфаты ФАТ 1.5. Макрофаги Макрофагами являются тканевые фагоциты, происходящие из циркулирующих моноцитов. Эти клетки играют важную роль в развитии хронического воспаления, характерного для большинства аутоиммунных ревматических болезней, в первую очередь РА. В процессе активации макрофаги секретируют очень большое количество субстанций, начиная от свободных радикалов (супероксидные анионы) и кончая веществами с высокой молекулярной массой (фибронектин). Некоторые из них секретируются макрофагами только под влиянием воспалительных стимулов, в то время как образование других не зависит от наличия воспаления. Секреторные продукты мононуклеарных фагоцитов (по C. F. Nathan, 1987) Цитокины и полипептиды: ИЛ-1 α/β ФНО-α ИЛ-8 ИФ-α ИФ-γ(?) тромбоцитарный фактор роста (?) ТФР-β β-эндорфин Компоненты комплемента: классический путь: С2, С4, СЗ, С5 альтернативный путь: фактор В, фактор D, пропердин ингибиторы: С3b инактиватор, β1Н Коагуляционные факторы: внутренний путь: факторы IX, X, V и протромбин внешний путь: фактор VII поверхностная активность: тканевый фактор, протромбиназа протромболитическая активность: ингибиторы активатора плазминогена, ингибитор плазмина Биоактивные липиды: продукты циклооксигеназы: ПГЕ2, ПГF2α, ПГI2, тромбоксан продукты липоксигеназы: лейкотриены В4, С, D, Е. ФАТ Протеиназы и их ингибиторы Супероксидные анионы Окись азота Большинство макрофагальных медиаторов увеличивает интенсивность воспаления и вызывает разрушение патогенных микроорганизмов, но они, как и нейтрофилы, могут индуцировать повреждение нормальных тканей. Наряду с участием в воспалительных реакциях макрофаги функционируют как АПК. Как уже отмечалось, ключевым событием иммунного ответа является фагоцитоз и расщепление антигенов макрофагами и представление ("презентация") подвергнутых процессингу антигенных пептидов CD4+ Т-лимфоцитам. "Презентация" антигена — сложный процесс, включающий интернализацию и частичное переваривание "чужеродных" белков в лизосомах АПК с образованием комплексов, состоящих из α/β цепей молекул класса II ГКГ и образующихся в результате переваривания интернализованных белков, пептидных фрагментов, α и βцепи ГКГ синтезируются независимо друг от друга в эндоплазматическом ретикулуме. При нейтральном рН (α/β цепи не могут образовать стабильный α/β димер и быстро связываются с инвариантной Ii -цепью. Этот процесс имеет важное физиологическое значение, так как предотвращает связывание α/β цепей с другими белками эндоплазматического ретикулума. Кроме того, инвариантная цепь выполняет роль "чаперона", направляющего перемещение α-Ii и β-Ii комплексов к более кислым везикулам (эндосомам) и сигнальной молекулы, указывающей направление движения α/β цепям, но не Ii-α и Ii-β комплексам, к клеточной мембране. Из эндоплазматического ретикулума α/Ii и β/Ii комплексы мигрируют к первичным эндосомам. Путем слияния интрацитоплазматических пузырьков содержимое эндосом смешивается с содержимым лизосом, образуя вторичные эндосомы, содержащие α-Ii и α/β-Ii расщепленные пептиды и кислые гидролазы. В кислом микроокружении вторичных эндосом пептиды конкурируют с Ii цепью за связывание с α/β цепями ГКГ. Если аффинность пептида к α/β цепям выше, чем Ii цепей, он способен вытеснять Ii и формировать комплекс с α/β цепями ГКГ. Образование такого комплекса стабилизируется при повышении рН, после чего он начинает перемещаться по направлению к клеточной поверхности. Таким образом, кислое содержимое очень важно для образования α/β пептидного комплекса, особенно, если афинность процессированного пептиды низкая. В дальнейшем комплексы, состоящие из пептидов и α/β цепей, экспрессируются на мембране макрофагов, где распознаются СD4+Т-лимфоцитами. 1.6. Тромбоциты Тромбоциты происходят из костномозговых мегакариоцитов и принимают участие в гемостазе, заживлении ран и других реакциях организма на повреждение. В процессе активации и агрегации на фоне воспаления тромбоциты секретируют большое количество разнообразных веществ, неполный перечень которых и установленные биологические эффекты представлены в таблице 1.2. Таблица 1.2. Тромбоцитарные продукты воспалительных реакций (по F. H. Valone. 1993) Класс Медиатор Биологическая активность Тромбоксан А2 Вазоконстрикция; агрегация; усиление прилипания нейтрофилов Тромбоксан В2 Более стабильный аналог ПГD2, Е2, F2 Модуляция сосудистой проницаемости, гемостаза и функции нейтрофилов ННТ Хемотаксис 12-НРЕТЕ Вазокоистрикция; ингибиция ЦОГ; стимуляция синтеза лейкотриена В4 лейкоцитами 12-НЕТЕ Хемотаксис ФАТ Агрегация. увеличение сосудистой проницаемости; активация нейтрофилов и моноцитов Плотные тельца Серотонин Вазоконстрикция; увеличение сосудистой проницаемости; фиброз α-гранулы Тромбоцитарный фактор роста Хемотаксис; митогенез клеток соединительной ткани; фактор, трансформирующий клетки Тромбоглобулин-β Агрегация; хемотаксис; индукция синтеза гистамина базофилами Липиды Циклооксигеназные Липоксигеназные Фосфолипиды Áåëêè/ïåïòèäû "Гранулярное" содержимое ИЛ-1-β Пироген; тканевое воспаление; модуляция пролиферации фибробластов Катионный фактор проницаемости Стимуляция высвобождения гистамина тучными клетками; хемотаксис Эластаза Нейтральная протеиназа Коллагеназа Нейтральная протеиназа α1-антитрипсин Ингибитор протеаз α2-макроглобулин Ингибитор протеаз α2-антиплазмин Ингибитор плазмина Ингибитор активатораплазминогена-I Ингибитор активаторов плазминогена Пептид, активирующий соединительную ткань III Пролиферация фибробластов Фактор роста эндотелия Пролиферация эндотелия Эпидермальный фактор роста Эпидермальная и эпителиальная пролиферация Наряду с перечисленными веществами тромбоциты вырабатывают еще ряд субстанций, тесно связанных с развитием воспалительных реакций. К ним относятся фибронектин, фибриноген, тромбоспондин, тромбоцитарный фактор 4, β-тромбоглобулин, фактор Виллебранда, факторы D и Н, присутствующие в αгранулах, а также серотонин, аденозиндифосфат (АДФ) и ионы кальция, являющиеся содержимым плотных гранул. Например, тромбоцитарный фактор 4 и β-тромбоглобулин обладают свойствами хемоаттрактантов и активируют моноциты и нейтрофилы. Тромбоспондин (гликопротеин с молекулярной массой 450 kD) секретируется как тромбоцитами, так и ЭК в зоне воспаления и наряду с CD11/CD18 молекулами играет важную независимую роль в прилипании нейтрофилов к сосудистому эндотелию. АДФ обладает вазоконстрикторной способностью и активирует связывание интегрина gpIIb/IIIa и серотонина. Активация тромбоцитов в процессе сосудистого повреждения определяется двумя основными механизмами: прилипанием к поврежденной поверхности сосудов и непосредственной активацией растворимыми факторами. Эти процессы регулируются разнообразными медиаторами как иммунной, так и неиммунной природы. Например, адгезия тромбоцитов к сосудистой стенке индуцируется коллагеном, фибронектином, ламинином, витронектином, кристаллами уратов и др. Активация тромбоцитов в кровяном русле может быть связана с воздействием гемостатических факторов (тромбин, коллаген, метаболиты арахидоновой кислоты), вазопрессина, серотонина, а в процессе иммунных реакций с ФАТ, иммунными комплексами, субстанцией Р, компонентами комплемента. Тромбоциты экспрессируют два типа молекул адгезии. gpIIb/IIIa является представителем семейства интегринов β-З, которые связываются с фибронектином, витронектином, фактором Виллебранда. Р-селектин (GMP-140) — мембранный гликопротеин, локализующийся в α-гранулах тромбоцитов и тельцах Weibel-Palade ЭК, который в процессе клеточной активации (под влиянием тромбина) быстро транслоцируется на мембрану тромбоцитов и служит рецептором для нейтрофилов и моноцитов. Последнее имеет очень важное значение для привлечения нейтрофилов в зону тромбоза и воспаления (B. Cronstein и G. Weissman, 1993). Имеются многочисленные доказательства активации тромбоцитов при системных ревматических болезнях, таких, как РА, СКВ, ССД, системные васкулиты и др. (M. H. Ginsberg, 1986). 1.7. Эндотелиальные клетки Сосудистый эндотелий является высокоспециализированным метаболически активным монослоем клеток, которые выстилают все сосуды организма человека. Ранее эндотелий рассматривался только как пассивный, хотя и селективный барьер между кровью и тканями. Однако в настоящее время стало очевидным, что ЭК, специфически реагируя на различные молекулярные сигналы, генерирующиеся как локально, так и дистантно, выполняют многообразные функции, в том числе селективные транспортные и барьерные, участвуют в метаболизме внеклеточного матрикса, биосинтезе разнообразных цитокинов, ангиогенeзе, регулируют процессы свертывания и агрегации тромбоцитов, сосудистый тонус и иммуновоспалительные реакции. (К. В. Саложин и соавт. 1992; B. A. Cronstein и G. Weissman, 1993). Основные типы биологически активных продуктов, которые обнаружены на мембране ЭК или синтезируются эндотелием, суммированы ниже (таблица 1.3.). Таблица 1.3. Молекулы, синтезирующиеся и экспрессирующиеся сосудистым эндотелием Факторы роста: тромбоцитарный трансформирующий β ФАТ Антикоагулянтные белки: тканевый активатор плазминогена антитромбин III тромбомодулин белок S Цитокины: ИЛ-1 ИЛ-6 ИЛ-8 колониестимулирующие фактор(ы) Вазоактивные пептиды: эндотелиальный фактор релаксации/окись азота эндотелины ПГI2 ПГЕ2 Мембранные рецепторы и белки: класс I/II ГКГ АВО антигены VCAM семейство IСАМ семейство CD32 (IgG Fc) CD35 (С3b) Clq CD31/13 (гранулоцитарно/ макрофагальный антиген) CDw29 (антиген 120-130kDa) CD34 (миелоидный антиген) CD41 (IIb/IIIa гликопротеин) CDw42-ïoäoáíûé гликопротеиновый рецептор CD73 (экто-5-нуклеотидаза) ИЛ-1, ИФ-γ/β ФНО-α/β плазминоген тромбомодулин С-белок IX/Ixa гепаринсвязывающий фактор роста серотонин инсулин и инсулиноподобный фактор роста P2-g пуринорецептор Внеклеточный матрикс: коллаген тип III, IV, V протеогликаны фибронектин Другие: фактор Виллебранда тромбоспондин антиген Кавасаки Антитромбоцитарная и фибринолитическая активность составляет основу тромборезистентности сосудистого эндотелия. В норме антикоагулянтный и прокоагулянтный эффекты уравновешены, однако на фоне развития иммуновоспалительных процессов этот баланс может быть нарушен. Например, ИЛ-1 и ФНО обладают выраженной прокоагулянтной активностью, которая определяется способностью этих цитокинов усиливать экспрессию тканевых факторов, снижать экспрессию тромбомодулина, стимулировать синтез ингибитора 1 тканевого активатора плазминогена. ЭК участвуют во всех фазах острого и хронического воспаления, таких как начальная вазодилатация, увеличение сосудистой проницаемости, прилипание, трансмиграция и активация лейкоцитов, ангиогенез и фиброплазия. 1.8. Клеточные молекулы адгезии Рециркуляция и рекрутирование (накопление) лейкоцитов в зоне воспаления опосредуются специфическим взаимодействием лиганд — рецептор между молекулами адгезии ЭК и лейкоцитов, поверхностная экспрессия которых регулируется воспалительными медиаторами и цитокинами. Клеточные молекулы адгезии (КМА) являются белками, гликопротеинами или пектинами, обеспечивающими межклеточные контакты. На ЭК и клетках иммунной системы присутствуют 3 семейства КМА: семейство селектинов, семейство интегринов и суперсемейство генов иммуноглобулинов (таблица 1.4.). Таблица 1.4. Основные типы молекул адгезии и их лиганды Семейства молекул адгезии Основные представители Селектины Е-селектин Р-селектин L-ñåëåêòèí Интегрины β1 СЕмейство VLA-4β1, α4, β2 семейство LFA-1β2, α1 Mac-1β2, αm р 150 95, β2, αx Суперсемейство IСАМ-1 IСАМ-2 иммуноглобулина IСАМ-3 VCAM-1 MedCAM-l Лиганды Сиалированные, фукозилированные тетрасахариды (например, Lewis х) VCAM-1, фибронектин IÑÀÌ-1, IÑÀÌ-2, IÑÀÌ-3 IÑÀÌ-1, фибриноген, С3bi C3bi LFA-I. Mac-1 LFA-I LFA-I LFA-4 β7, α4 L-ñåëåêòèí PECAM-I PECAM-I Селектины — гликопротеины, экспрессирующиеся на лейкоцитах, тромбоцитах и ЭК, обладают Nòåðìèíàëüíûì лектиноподобным доменом, за которым следует последовательность, напоминающая эпидермальный фактор роста, и короткий повтор, напоминающий последовательность в белках, регулирующих активацию комплемента. Описано 3 типа селектинов, принимающих участие в связывании лейкоцитов с ЭК в зоне воспаления: 1). L-ñåëåêòèí (лейкоцитарная молекула адгезии) экспрессируется на лейкоцитах, рассматривается как основной "homing" рецептор для миграции лейкоцитов, которая очень быстро освобождается с клеточной поверхности после активации Т-лимфоцитов и нейтрофилов; 2). Р-селектин (гранулярный мембранный белок 140) хранится в α-гранулах тромбоцитов и секреторных гранулах (тельца Weibel-Palade) ЭК, индуцируется гистамином и тромбином и имеет важное значение в связывании нейтрофилов и моноцитов с ЭК; 3). Е-селектин (эндотелиальная лейкоцитарная молекула адгезии 1; ELAM-1) экспрессируется на клеточной поверхности ЭК под влиянием ИЛ-1 (α/β, ФНО-α и ИФ-γ и играет важную роль в инициации связывания лейкоцитов и ЭК в зоне воспаления. Гиперэкспрессия Е-селектина обнаружена на ЭК синовиальной ткани при РА (A. E. Koch и соавт., 1991), а также в кровяных сосудах кожи при ССД и СКВ (H. M. Belmont и соавт., 1994). Интегрины — семейство родственных молекул адгезии, опосредующих связывание клеток с матриксом и межклеточные взаимодействия. Они обнаруживаются на лимфоцитах, моноцитах, тромбоцитах, эпителиальных и ЭК. По структуре интегрины являются гетеродимерами, состоящими из α- и β-субъединиц. В зависимости от структуры β-цепи интегрины подразделяются на 3 субкласса, каждый из которых имеет общую β-цепь, нековалентно связанную с α-цепью. Описано 3 подсемейства интегринов (β1, β2, β3). β1-èíòåãðèíû (CD29 или VLA белки), имеют общую β1-öåïü (мол. масса 130 KD), ассоциирующуюся с одной из 6 возможных α-цепей (мол. масса 150-200 kD). β1-èíòåãðèíû и β3-интегрины выполняют функцию рецепторов для фибронектина, ламинина, витронектина, GPII/IIIa антигена тромбоцитов, участвуют в адгезии фактора Виллебранда. β2интегрины (CDIIa, b, с / CD18) обнаруживаются на мембране лейкоцитов, участвуют в адгезии лейкоцитов к эндотелию, их миграции, связываются с компонентами комплемента или факторами свертывания. Они состоят из 3 различных альфа-цепей (CD11a, b, c) и общей β-2 субъединицы (CD18). Большинство интегринов связываются с общей аминокислотной последовательностью аргинин-глицин-аспарагин, присутствующей в составе различных белковых молекул. Суперсемейство молекул адгезии, относящихся к генам иммуноглобулинов, экспрессируется на ЭК и, являясь лигандами для лейкоцитарных интегринов, играет важную роль в процессах взаимодействия ЭК и лейкоцитов. К ним относятся межклеточная молекула адгезии 1 (ICAM-1; CD54), межклеточная молекула адгезии 2, тромбоцитарно-эндотелиальная молекула адгезии 1 (РЕСАМ-1; CD31), сосудистая молекула адгезии 1 (VCAM-1), а также молекулы ГКГ классов I и II. Экспрессия VCAM-1 и ICAM-1 индуцируется ИЛ-1 α/β, ФНО-α и ИФ-γ. При СКВ и РА наблюдается гиперэкспрессия CD11b/CD18 на мембране лейкоцитов, что, как полагают, отражает внутрисосудистую активацию нейтрофилов провоспалительными стимулами (например, С5а). Увеличение экспрессии CD11b/CD18 коррелирует с активностью СКВ. Имеются данные о гиперэкспрессии ICAM-1 и VCAM-1 на микрососудах синовии у больных РА, но не остеоартритом. 1.9. Взаимодействие ЭК и лейкоцитов Взаимодействие лейкоцитов и эндотелия лежит в основе физиологической рециркуляции и направленной трансмиграции лейкоцитов в лимфоидные ткани и участки воспаления; рассматривается как комплексный динамический процесс, регулирующийся каскадом молекулярных реакций, напоминающих реакции, составляющие основу активации системы комплемента или системы свертывания крови. Для перехода из кровяного русла в ткани лейкоциты должны в начале прилипнуть к сосудистому эндотелию. Прилипание и трансэндотелиальная миграция имеет место преимущественно в специализированных участках сосудистого русла, а именно в посткапиллярных венулах нелимфоидных тканей и высокоэндотелиальных венулах лимфатических узлов. ЭК очень быстро реагируют на некоторые вещества (гистамин, тромбин), однако только воздействие цитокинов вызывает выраженное изменение функциональных свойств ЭК, проявляющееся гиперэкспрессией генов и синтезом белка. Этот процесс нарастает постепенно (в течение нескольких часов), но сохраняется длительное время. Индуцированное провоспалительными цитокинами функциональное перепрограммирование сосудистого эндотелия приводит к изменению его функции в направлении, способствующем формированию протромботического и провоспалительного состояния, которое получило название "активация ЭК". В отсутствие воспаления спонтанная адгезия лейкоцитов к эндотелию и их миграция через эндотелиальный барьер ограничены. Однако при активации ЭК развивается каскадный процесс взаимодействия ЭК и лейкоцитов, который условно подразделяется на несколько этапов (таблица 1.5.). Таблица 1.5. Молекулы, принимающие участие в процессе взаимодействия между лейкоцитами и эндотелием (по D. H. Adams и S. Shaw, 1994) Ограничение Запуск Функция Транзиторная адгезия Активация интегрина Остановка движения клеток Молекулы Селектины Цитокины Интегрины Лиганд Углеводы G-рецептор IСАМ-1, IСАМ-2, VCAM-1, LFA-1, Мас-1 Прототип для нейтрофилов Е-селектин (на ЭК) ИЛ-8 LFA-1, Мас-1 Прототип для Т-клеток L-ñåëåêòèè (на Т-клетках) МIР-1 β Сильная адгезия VLA-4, LFA-1 Фаза ограничения (tethering) проявляется замедлением скорости движения лейкоцитов в кровяном русле. Клетки начинают как бы перекатываться (rolling) вдоль стенки сосуда, что связывают со сравнительно слабой адгезией лейкоцитов к эндотелию, которая обеспечивается только L-ñåëåêòèíàìè. В участках активации ЭК наблюдается более тесная адгезия лейкоцитов к эндотелию, опосредуемая взаимодействием L-ñåëåêòèíà и Рселектина или Е-селектина и углеводного компонента мембраны лейкоцитов. Фаза запускания (trigering) характеризуется активацией молекул адгезии лейкоцитов (интегринов) цитокинами, синтезирующимися непосредственно в сосудистой стенке. В фазе сильной адгезии происходит интегринзависимое связывание лейкоцитов с молекулами адгезии ЭК, в результате чего движение лейкоцитов замедляется, а затем и приостанавливается. Фаза миграции лейкоцитов в ткани связана с влиянием цитокинов, обладающих свойствами хемотаксических факторов, большинство из которых индуцирует сильную адгезию. Более подробно процессы, лежащие в основе взаимодействия ЭК и лейкоцитов, представлены в обзоре J. M. Harlan и D. Y. Liu (1992) 1.10. Цитокины и факторы роста Цитокины являются низкомолекулярными белковыми клеточными регуляторами, участвующими в процессах межклеточной коммуникации, рассматриваются как медиаторы нормальных биологических процессов, таких, как рост и дифференцировка гемопоэтических, лимфоидных и мезенхимальных клеток, иммунные реакции и воспаление. К цитокинам относятся колониестимулирующие факторы, факторы роста, интерлейкины и интерфероны (K. Arai и соавт., 1990). Цитокины составляют сеть взаимодействий, в которой каждый цитокин обладает перекрещивающейся и синергической активностью с другими цитокинами и системой саморегуляции, нарушение которой приводит к избыточному или недостаточному синтезу определенных цитокинов. Все цитокины имеют общие свойства, к которым относятся следующие: 1) низкая молекулярная масса (менее 80kD); 2) аутокринный и паракринный способы клеточной регуляции; 3) участие в регуляции иммунного ответа; 4) связывание с высокоаффинными рецепторами, специфичными для каждого цитокина или групп цитокинов; 5) влияние на синтез ДНК, РНК и белка в клетках; 6) плеотропная регуляторная активность; 7) сложное взаимодействие друг с другом и факторами роста в рамках цитокиновой сети; 8) участие в развитии воспалительных заболеваний человека, в том числе ревматических. В настоящее время идентифицировано большое число молекул, которые классифицируются как цитокины (таблица 1.6.). Таблица 1.6. Общая характеристика цитокинов КСФ ГМ-КСФ (мономер 127 ак*.) Синтез: МФ, Т-клетки, ЭК, нейтрофилы и др. Эффекты: способствует росту и дифференцировке мультипотентных клеток-предшественников Г-КСФ (мономер 174 ак.) КСФ Эритропоэтин (иономер 165 ак.) Синтез: почки, печень, МФ? Эффекты: стимулирует рост и дифференцировку клетокпредшественников эритроцитов Èíòåðëåéêèíû Синтез: МЦ/МФ, ЭК, ФБ, и др. Эффекты: регулирует функциональную активность клеток, участвующих в воспалении и иммунном ответе: регуляция нервной и эндокринной ИЛ-1 α (мономер 159 ак.) систем. ИЛ-1β (мономер 153 ак.) Те же ИЛ-2 (мономер 133 ак.) Синтез: Т-клетки Эффекты: стимулирует пролиферацию Т-, В-, и ЕК-клеток, активацию ЕК клеток ИЛ-3 (мономер 133 ак.) Синтез: Т-клетки, и др. Эффекты: способствует пролиферации и дифференцировке гемопоэтических клеток ИЛ-4 (мономер 129 ак.) Синтез: Т- и В-клетки, МФ, тучные клетки, базофилы, стромальные костномозговые клетки. Эффекты: индуцирует дифференцировку CD4+ Т-лимфоцитов в Th2 клетки, пролиферацию и дифференцировку В-клеток; различные эффекты на Т-клетки, моноциты, фибробласты. и ЭК ИЛ-5 (гомодимер 115 ак.) Синтез: Т-клетки, тучные клетки Эффекты: стимулирует рост и дифференцировку эозинофилов; активирует функциональную активность эозинофилов и хемотаксис ИЛ-6 (мономер 184 ак.) Синтез: Т клетки, МФ/МЦ, фибробласты, гепатоциты, ЭК, нейрональные клетки Эффекты: индуцирует дифференцировку В-клеток, стимулирует активацию Т-клеток, является кофактором роста и созревания клетокпредшественников, регулирует синтез ОФБ ИЛ-7 (мономер 152 ак.) Синтез: стромальные костномозговые клетки Эффекты: индуцирует пролиферацию В-клеток-предшественников; поддерживает пролиферацию Т-клеток; стимулирует пролиферацию и цитотоксическую активность Т-клеток ИЛ-8 (димер 69-79 ак.) Синтез: моноциты, Т-клетки, фибробласты, ЭК, и др. Эффекты: хемотаксическая активность; активация нейтрофилов, вызывающая высвобождение лизосомальных ферментов; индуцирует адгезию нейтрофилов к ЭК ИЛ-9 (мономер 126 ак.) Синтез: Т-клетки Эффекты: индуцирует пролиферацию Т-клеток ИЛ-10 (гомодимер 160 ак.) Синтез: Т-клетки, МФ, В-клетки, и др. Эффекты: ингибирует функцию макрофагов; подавляет синтез провоспалительных цитокинов активироваными МФ/МЦ; усиливает пролиферацию В-клеток и секрецию Ig ИЛ-11 (мономер 178 ак.) Синтез: стромальные фибробласты, трофобласты. Эффекты: кофактор ИЛ-3 в отношении стимуляции мегакариоцитов; стимулирует синтез ОФБ гепатоцитами ИЛ-12 (гетеродимер р35-197 ак. и Синтез: В-клетки, МФ Эффекты: стимулирует образование Thl-êëåòîê; стимулирует рост и р40-306 ак.) функциональную активность ЕК-клеток и Т-клеток ИЛ-13 (мономер 132 ак.) Синтез: Т-клетки Эффекты: подавляет активность макрофагов, экспрессию интегринов, CD23 и молекул класса II ГКГ; индуцирует рост и дифференцировку Вклеток ИЛ-14 (мономер 486 ак.) Синтез: Т-клетки Ýôôåêòû: èíäóöèðóåò пролиферацию активированных В-клеток, но не покоящихся В-клеток Фактор некроза Синтез: нейтрофилы, активированные лимфоциты, ЕК-клетки, ЭК и др. Эффекты: разнообразные эффекты, связанные со способностью регулировать экспрессию генов факторов роста и цитокинов, факторов транскрипции, воспалительных медиаторов, ОФБ; иммуностимулятор и медиатор воспаления α (тример 157 ак.) β (тример 171 ак.) Интерфероны ИФ-α/β (мономер) Синтез: Т- и В-клетки, МФ/МН, фибробласты Эффекты: антивирусная и антипролиферативная активности; стимуляция ЕК-клеток; модуляция экспрессии I и II классов ГКГ; стимуляция активности макрофагов; регуляция специфического иммунного ответа ИФ-γ (гомодимер 143 ак.) Синтез: Т-клетки, ЕК-клетки Эффекты: антивирусная и антипролиферативная активности; усиление экспрессии Fc-ðåöåïòîðîâ и класса II ГКГ: регуляция специфического иммунного ответа IP-10 (мономер? 77 ак.) Синтез: МЦ ЭК, фибробласты. Эффекты: хемоаттрактант для моноцитов и Т-клеток; способствует Тклеточной адгезии к ЭК Лейкемический ингибиторный фактор (мономер 179 ак.) Синтез: костномозговые стромальные клетки, фибробласты, Т-клетки, МФ/МЦ Эффекты: потенцирует ИЛ-3-зависимую пролиферацию клеток предшественников гемопоэза Моноцитарный хемотаксический Синтез: МФ/МЦ, фибробласты, В-клетки, ЗК и др. Эффекты: моноцитарный хемоаттрактант, регулирует экспрессию белок-1 (мономер? 76 ак.) молекул адгезии, синтез цитокинов моноцитами Макрофагальный воспалительный белок-1 α (мономер? 66 ак.) Ñèíòåç: Ò- и В-клетки, МН, тучные клетки, фибробласты. Эффекты: хемоаттрактант для моноцитов Т-клеток, эозинофилов; ингибирует раннюю пролиферацию гемопоэтических стволовых клеток Тромбоцитарный эндотелиальный фактор роста мономер 471 ак.) Синтез: тромбоциты и другие клетки. Эффекты: стимулирует рост и хемотаксис ЭК и ангиогенез Тромбоцитарный фактор роста (гомо- и гетеродимеры двух цепей: А-цепь -125 или 119 ак., В-цепь -160 или 109 ак.) Синтез: тромбоциты, ЭК, МФ/МЦ, и др. Эффекты: митоген для ЭК и других клеток; хемодимеры, таксис фибробластов, ЭК и МН; ингибирует ЕК-клетки; стимулирует нейрофилы и МН и синтез коллагена Трансформирующий фактор роста-α (мономер 50 ак.) Синтез: МФ, и другие клетки Эффекты: митоген для фибробластов Трансформирующий фактор Синтез: хондроциты, остеобласты, остеокласты, тромбоциты, роста β (гомо и гетеродимер 3112 фибробласты, МЦ Эффекты: ингибитор роста многих клеток; стимулирует остеобласты, ак. изоформ) ингибирует остеокласты; ингибирует ЕК-клетки, пролиферацию Т- и Вклеток; вместе с ИЛ-4 стимулирует секрецию IgA *àê. — аминокислота Многие цитокины присутствуют в биологических жидкостях в достаточно высоких концентрациях; изменение их уровня ассоциируется с активностью и прогрессированием различных патологических процессов при ревматических заболеваниях (воспаления, фиброз, аутоиммунитет и др.). Некоторые цитокины, полученные генноинженерным путем, такие, как эритропоэтин, КСФ, ИФ, ИЛ-2, используются в клинической практике для стимуляции гемопоэза (глава 15) или противоопухолевого иммунитета. Для лечения воспалительных заболеваний недавно начали применять ингибиторы провоспалительных цитокинов (J. J. Oppenheim и соавт., 1993) и моноклональные антитела, блокирующие активность соответствующих цитокинов. (глава 14). 1.10.1. КСФ КСФ обладают активностью факторов роста гемопоэза и принимают участие в созревании лимфоцитов, нейтрофилов, моноцитов, макрофагов (D. Metcalf, 1991). ГМ КСФ и ИЛ-3 потенцируют рост нескольких типов костномозговых клеток-предшественников. Их активность усиливается в присутствии ИЛ-1 и ИЛ-6. Кроме того, ГМ-КСФ оказывает влияние на функциональную активность зрелых моноцитов и гранулоцитов, увеличивает чувствительность нейтрофилов, эозинофилов и базофилов к воздействию триггерных факторов, стимулирующих хемотаксис, продукцию кислородных радикалов и фагоцитоз, усиливает цитотоксичность эозинофилов и стимулирует высвобождение базофилами гистамина. Все эти свойства ГМ-КСФ определяют его важную роль в развитии острого воспаления при ревматических болезнях. Кроме того, ГМ-КСФ усиливает способность моноцитов и макрофагов презентировать антиген (увеличение экспрессии мембранной формы ИЛ-1 и класса II ГКС), а следовательно, принимает участие в развитии иммунного ответа. С другой стороны, ГМ-КСФ индуцирует синтез антагониста ИЛ-1 рецептора. Особенно важную роль придают ГМ-КСФ в развитии PA (J. A. Hamilton, 1993), при котором в зоне суставного повреждения обнаруживается иРНК ГМ-КСФ, а сам ГМ-КСФ в избыточном количестве присутствует в синовиальной жидкости. Полагают, что одним из патогенных эффектов ГМ-КСФ при РА является увеличение экспрессии антигенов класса II ГКГ на мембране синовиальных макрофагов. Установлено, что ИЛ-1 и ФНО-α увеличивают синтез ГМ-КСФ моноцитами, фибробластами и ЭК. Имеются данные о том, что введение рекомбинантного ГМ-КСФ больным РА вызывает обострение суставного процесса (глава 15). 1.10.2. Иммунорегуляторные цитокины в процессе иммунного ответа Т-лимфоциты синтезируют ИЛ-2, ИЛ-4 ИЛ-5, ИЛ-7, ИЛ-9, ИЛ-10, которые в свою очередь регулируют интенсивность этого ответа, а некоторые из них (ИЛ-4, ИЛ-10 и ИФ-γ) оказывают воздействие на функциональную активность моноцитов и макрофагов. Как уже отмечалось, взаимодействие АПК с СD4+Т-лимфоцитами стимулирует синтез последними ИЛ-2. Связывание ИЛ-2 со специфическими ИЛ-2 рецепторами (ИЛ-2Р), экспрессирующимися на различных клетках иммунной системы, вызывает клональную экспансию Т-лимфоцитов, усиливает рост В-клеток и функциональную активность ЕК-клеток, приводит к активации макрофагов. Имеются данные об увеличении синтеза ИЛ-2 при аутоиммунных ревматических болезнях in vivo (G. Kroemer и G. Wick, 1989). Например, отмечено увеличение уровня ИЛ-2 в сыворотках больных с диффузной и лимитированной формами ССД. Примечательно, что моноклональные антитела к ИЛ-2 рецепторам подавляют развитие экспериментального коллагенового артрита и спонтанного аутоиммунного волчаночноподобного заболевания у мышей. Эти данные свидетельствуют о важной роли ИЛ-2-зависимого Т-клеточного ответа в развитии воспалительных ревматических заболеваний. На мембране клеток обнаружено два типа гликопротеиновых молекул, обладающих свойствами ИЛ-2 рецепторов (ÈË-2Ð): Тас (ÈË-2Ð α, р55) и р75 (ÈË-2Ð β), которые связываются с ИЛ-2 с низкой или промежуточной аффинностью. При нековалентном связывании эти гликопротеины образуют высокоаффинный рецепторный комплекс, который опосредует передачу внутриклеточного сигнала от молекулы ИЛ-2. В сыворотках обнаруживаются растворимые (р) формы ÈË-2Ð, которые являются компонентами (55kD) ÈË-2Ð. Основными источниками рИЛ-2Р в кровяном русле являются активированные в процессе иммунного ответа Тлимфоцитами В-лимфоциты и макрофаги. Для определения рИЛ-2Р в сыворотке разработан иммуноферментный метод, который широко используется в клинической практике. Увеличение концентрации ðÈË-2Ð в сыворотке обнаружено при многих ревматических и неревматических заболеваниях, таких, как РА, СКВ, ССД, ПМ/ДМ, системные васкулиты, лимфопролиферативные опухоли, паразитарные заболевания, СПИД (L. A. Rubin, 1990), дилатационная кардиомиопатия, миокардит (M. Y. Samsonov и соавт., 1995). При РА и гранулематозе Вегенера гиперпродукция рИЛ-2Р коррелирует с активностью заболевания, а при СКВ — с активностью процесса и уровнем антител к ДНК. Сывороточный уровень ðÈË-2Ð возрастает по мере прогрессирования IgA-íåôðîïàòèè. ИЛ-4 влияет на функциональную активность В-клеток (усиливает синтез IgG1 и IgE) и тучных клеток, стимулирует экспрессию молекул класса II ГКГ, а также является фактором роста Т-лимфоцитов и индуцирует цитотоксическую активность Т-лимфоцитов. Важной особенностью ИЛ-4 является то, что он оказывает как стимулирующее, так и ингибирующее действие на систему мононуклеарных фагоцитов. С одной стороны, он индуцирует экспрессию молекул класса II ГКГ на мембране макрофагов и, таким образом, усиливает антигенпрезентирующую способность этих клеток, а с другой — на уровне транскрипции соответствующих генов подавляет синтез провоспалительных цитокинов (ИЛ-1, ИЛ-6 и ФНО-α), а также усиливает синтез антагониста ИЛ-1 рецепторов. Кроме того, ИЛ-4 ингибирует экспрессию молекул адгезии на мембране ЭК, что приводит к подавлению прилипания нейтрофилов к эндотелию, но усиливает миграцию лимфоцитов в ткани. Предполагается, что дефицит синтеза ИЛ-4 является одним из важных патогенетических факторов, обусловливающих гиперпродукцию провоспалительных цитокинов при PA (P. Moisec, 1993). В экспериментальных исследованиях было показано, что ИЛ-4 подавляет индуцированную провоспалительными цитокинами деструкцию костной ткани и снижает активность и прогрессирования поражения суставов при коллагеновом артрите и артрите, индуцированном мембраной стрептококка (P. Miossec, 1993; K. Watanabe и соавт., 1990). С другой стороны, способность ИЛ-4 усиливать синтез коллагена фибробластами позволяет предположить его участие в патогенезе ССД. Имеются данные о том, что ГК усиливают синтез IgE лимфоцитами в присутствии ИЛ-4, что объясняет увеличение продукции IgE у больных аллергическими заболеваниями, леченных ГК, а также гиперпродукцию IgE при стрессе (инфаркт миокарда). ИЛ-5, ИЛ-7, ИЛ-9 и ИЛ-11 функционируют преимущественно как факторы роста и дифференцировки. Кроме того, ИЛ-5 является самым мощным эозинофильным цитокином, индуцирует хемотаксис и синтез супероксидных радикалов, принимает участие в дифференцировке и пролиферации эозинофилов и Влимфоцитов и изотипическом переключении синтеза IgA. Обнаружено увеличение концентрации ИЛ-5 в сыворотках больных с синдромом эозинофилии-миалгии. ИЛ-10 гомодимерный цитокин с мол. массой 35kD, синтезируется Т- и В-лимфоцитами и моноцитами. Его основной эффект связан с ингибицией синтеза нескольких Т-клеточных цитокинов (ИФ-γ, ГМ-КСФ, ИЛ-4 и ИЛ-5) и подавлением антигенспецифической Т-клеточной пролиферации. Кроме того, ИЛ-10 обладает противовоспалительной активностью, так как ингибирует синтез провоспалительных цитокинов (ФНО-α, ИЛ-1, ГМ-КСФ) (J. F. Fiorentino и соавт., 1991) и экспрессию антигенов класса II ГКГ на мембранах моноцитов. Другой стороной биологической активности ИЛ-10 является активация В-лимфоцитов и стимуляция синтеза иммуноглобулинов. Кроме того, ИЛ-10 увеличивает выживаемость В-клеток, индуцируя синтез bcl-2, предохраняющего клетки от апоптоза (Y. Levi и J-C. Broet, 1994). Предполагается, что ИЛ-10 играет важную роль в механизмах, определяющих доминирование гуморальных (синтез антител) или клеточных (гиперчувствительность замедленного типа) иммунных реакций в процессе развития иммунного ответа. Введение нейтрализующих антител к ИЛ-10 NZB/WF1 мышам замедляет развитие аутоиммунных нарушений, в то время как введение самого ИЛ-10 ускоряет развитие болезни (H.Ishida и соавт., 1994). Установлено также, что у мышей В-клетки, синтезирующие ИЛ-10, принадлежат к субпопуляции CD5+ В-лимфоцитов, обладающей способностью синтезировать некоторые типы аутоантител (A. J'Garra и соавт. 1992). Все эти данные свидетельствуют о важной роли ИЛ-10 в развитии некоторых аутоиммунных нарушений при ревматических болезнях. (P. D. Katsikis и соавт., 1994; L.Llorente и соавт., 1994). ИЛ-11, наряду с ИЛ-1 и ИЛ-6 индуцирует синтез острофазовых белков в печени и усиливает антигенспецифический ответ В-лимфоцитов. ИФ-γ систезируется вместе с ИЛ-2 антигенстимулированными Т лимфоцитами. Основные эффекты ИФ-γ связаны со стимуляцией экспрессии молекул класса II ГКГ на макрофагах, ЭК, фибробластах и других клетках, что приводит к усилению презентации антигенов Т-лимфоцитам. Кроме того, ИФ-γ является мощным активатором функциональной активности макрофагов, цитотоксических Т-лимфоцитов и ЕК-клеток, стимулирует синтез антител В-лимфоцитами. Предполагается, что ÈÔ-γ играет важную роль в развитии аутоиммунных ревматических заболеваний. ИФ-γ подавляет миграцию макрофагов, способствует дифференцировке супрессорных Т-лимфоцитов и естественных супрессорных клеток (S. Wahl и соавт., 1991). При РА наблюдается дефект синтеза ИФ-γ клетками синовиальной оболочки, что, как предполагают, способствует гиперпродукции ИЛ-1. Этот механизм лежит в основе использования ИФ-γ для лечения РА (глава 14), С другой стороны, имеются данные о том, что гиперпродукция ИФ-γ вызывает аномальную гиперэкспрессию молекул ГКГ на различных клетках и тем самым индуцирует развитие аутоиммунных реакций (G. F. R. Bottazzo и соавт., 1993). Установлено существование 2 типов CD4+ Т-клеток, обладающих хелперной активностью, — Th1 и Th2 (T. R. Mosmann и R. L. Coffman, 1989; G. Del Prete и соавт., 1994), которые синтезируют разные типы цитокинов (таблица 1.7.). Таблица 1.7. Профиль секреции цитокинов CD4+ Т-клетками человека (по G. Del Prete и соавт., 1994) Цитокины Тh1 Th2 Th0 ИЛ-2 ++ - ++ ИФ-γ ++ - ++ ФНО-β ++ - ++ ИЛ-4 - ++ + ИЛ-5 - ++ + ИЛ-3 +/- ++ + ИЛ-6 +/- ++ + ГМ-КСФ +/- ++ + ФНО-α ++ - ++ ИЛ-10 +/- ++ + Th1-клетки продуцируют преимущественно ИЛ-2, ИФ-γ, которые вовлечены в реакции клеточного иммунитета, в то время как Th2 — синтезируют ИЛ-4, ИЛ-5 и ИЛ-6, которые регулируют синтез антител. 1.10.3. Пpовоспалительные цитокины К группе провоспалительных цитокинов, которым придают особенно важное значение в патогенезе воспалительных ревматических заболеваний принадлежат ФНО-α, ИЛ-1, ИЛ-6 и ИЛ-8. ФНО-α и ИЛ-1 синтезируются параллельно, обладают способностью индуцировать продукцию друг друга и проявляют многочисленные общие эффекты. ФНО-α по структуре напоминает трансмембранные молекулы и синтезируется моноцитами, макрофагами и лимфоцитами под влиянием эндотоксинов, вирусов и других цитокинов. Нa клетках-мишенях присутствуют два типа ФНО-рецепторов. Обнаружена растворимая форма рецептора, которая также принимает участие в реализации биологических эффектов ФНО-α. ФНО-α является очень важным провоспалительным цитокином, участвующим также в развитии кахексии при злокачественных новообразованиях. Выраженное увеличение концентрации ФНО-α обнаруживается у больных с сепсисом и коррелирует с неблагоприятным прогнозом. ФНО-альфа наряду с ИЛ-1 играет важную роль в деструкции хряща при РА (глава 14). Однако при СКВ снижение продукции ФНО-α ассоциируется с носительством HLA-DR4 и низкой частотой развития нефрита. Введение рекомбинантного ФНО-α мышам со спонтанно развивающимся волчаночноподобным заболеванием (NZBxNZW F1) подавляет активность болезни. Таким образом, ФНО-α может принимать участие как в развитии, так и в предотвращении аутоиммунной патологии. ИЛ-1 семейство состоит из трех молекул: ИЛ-1 α, ИЛ-1 β и антагониста ИЛ-1 рецепторов. ИЛ-1 α и ИЛ-1 β синтезируются макрофагами и моноцитами, а также ЭК, эпителиальными клетками, фибробластами, активированными Т-лимфоцитами и др. При этом ИЛ-1 β может находиться в экстрацеллюлярном пространстве, а ИЛ-1 α существует преимущественно в мембраносвязанной форме. Описано 2 типа ИЛ-1 рецепторов: тип 1 ИЛ-1Р присутствует на Т-клетках, ЭК, фибробластах, в то время как тип II экспрессируется на В-клетках, моноцитах и нейтрофилах (S. K. Dower и J. E. Smith, 1990). Экспрессия ÈË-1Ð подавляется ТФРβ, что и определяет иммуносупрессивную активность этого цитокина. ИЛ-1 проявляет не только локальный, но и системный эффект, к которым относятся лихорадка, мышечная слабость, синтез острофазовых белков (наряду с ИЛ-6 и ИЛ-11) и многие другие (C. A. Dinarello, 1989; Е. Л. Насонов, 1987) ИЛ-6 синтезируется многими клетками, включая клетки синовиальной оболочки сустава, и стимулирует образование ИЛ-1 и ФНО-альфа. ИЛ-6 участвует в дифференцировке стимулированных В-лимфоцитов в иммуноглобулинсекретирующие плазматические клетки и регуляции острофазового ответа (T. Hirano и соавт., 1990). Увеличение концентрации ИЛ-6 в сыворотке выявлено при многих воспалительных заболеваниях, оно коррелирует с лабораторными маркерами активности воспаления: СОЭ и особенно концентрацией СРБ, Высокий уровень ИЛ-6 в сыворотке обнаружен при системном варианте (болезнь Стилла) ювенильного хронического артрита, при РА, в спинномозговой жидкости при волчаночном цереброваскулите, в синовиальной жидкости при РА. Сывороточный уровень ИЛ-6 коррелирует с тяжестью процесса при миеломе. Гиперпродукция ИЛ-6 играет важную роль в развитии гипергаммаглобулинемии и продукции аутоантител при предсердной миксоме, локальном синтезе РФ при РА и синтезе аутоантител при СКВ. Предполагают, что РА и миелома относятся к так называемым ИЛ-6-зависимым заболеваниям человека. ИЛ-6 убыстряет прогрессирование процесса у мышей линии NZB/NZW F1 с волчаночноподобным синдромом (B. K. Finch и соавт., 1994). Введение моноклональных антител к ИЛ-6 подавляет активность процесса при PA (D. Wendling и соавт. 1993) и прогрессирование болезни у NZB/NZW F1 мышей (B. K. Finch и соавт. 1994). ИЛ-8 (4q12-q21 моноцитарный фактор) является членом семейства пептидов с мол. массой 8kD, участвующих в специфическом хемотаксисе, регуляции воспаления и клеточного роста (M. Baggiolini и соавт., 1989). ИЛ-8 вызывает активацию Т-лимфоцитов и нейтрофилов, хемотаксис и образование отека, подавляет прилипание нейтрофилов к цитокинактивированным ЭК и тем самым ослабляет опосредуемое нейтрофилами повреждение ЭК в зоне воспаления. ФНО-α и ИЛ-1 стимулируют синтез ИЛ-8 моноцитами, макрофагами, ЭК, фибробластами и другими клетками. Полагают, что ИЛ-8 играет важную роль в развитии артритов, направляя движение нейтрофилов в полость сустава. Кроме того, ИЛ-8 усиливает функциональную активность нейтрофилов, в том числе экспрессию молекул адгезии, образование кислородных радикалов и высвобождение лизосомальных ферментов. 1.10.4. Факторы роста и дифференцировки Факторы роста и дифференцировки, свойствами которых наряду с тромбоцитарным и эпидермальными факторами роста, ТФР-β и фактором роста фибробластов и др. обладают некоторые цитокины, играют важную роль в пролиферации фибробластов и ангиогенезе при хронических заболеваниях человека, в том числе ревматических. Полагают также, что ТФР-β принимает участие в развитии острого воспаления. Тромбоцитарный фактор роста синтезируется главным образом тромбоцитами и в меньшей степени макрофагами, эндотелиальными и другими клетками. Эпидермальный фактор роста образуется многими клетками и наряду с фактором роста фибробластов играет важную роль в ангиогенезе. Кроме того, оба эти фактора индуцируют пролиферацию и рост различных эпителиальных и мезенхимальных клеток. Установлено, что эти факторы роста присутствуют в синовиальной жидкости при РА и синтезируются синовиальными макрофагами. Предполагается, что пролиферация синовиальных фибробластов ревматоидного синовиума связана с действием всех трех перечисленных факторов роста, а резкое усиление роста новых капилляров в ревматоидном синовиуме связано с воздействием двух последних. Тканевый фиброз, являющийся характерной особенностью ССД, вероятно, является результатом неконтролируемой продукции тромбоцитарного, эпидермального факторов роста и фактора роста фибробластов. Очень большое значение в развитии ревматических болезней придают ТФР-β, который обладает как провоспалительной, так и антивоспалительной активностью (W. A. Border и N. Noble, 1994). ТФР-бета стимулирует аккумуляцию моноцитов в тканях, регулирует функциональную активность лимфоцитов и макрофагов и стимулирует тканевой фиброз. Примечательно, что в зависимости от присутствия других цитокинов, ТФР β способен как подавлять, так и стимулировать рост и дифференцировку фибробластов. ТФРбета стимулирует синтез коллагена и фибронектина фибробластами, а ИФ-γ и ФНО-α оказывают противоположное действие. В присутствии тромбоцитарного фактора роста, эпидермального фактора роста и фактора роста фибробластов ТФР-β подавляет синтез коллагеназы и других нейтральных протеаз и увеличивает продукцию ингибиторов этих ферментов. Предполагается участие ТФР-β в развитии фиброза при ССД. Показано, что моноциты, инфильтрирующие кожу и ткани при ССД, содержат иРНК ТФР-бета. Кроме того, ТФР-β присутствует в зоне кожного фиброза недалеко от фибробластов. Важным свойством ТФР-β является способность модулировать некоторые активности моноцитов и лимфоцитов. Показано, что ТФР-β является самым мощным из известных в настоящее время хемотаксических агентов для моноцитов, вызывает усиление экспрессии FcIII-ðåöåïòîðîâ, но ингибирует синтез цитокинов, подавляет ИЛ-1-индуцируемую пролиферацию Тлимфоцитов, рост и синтез иммуноглобулинов В-лимфоцитами, ингибирует активность ЕК-клеток. С одной стороны, ТФР-β, вызывая аккумуляцию моноцитов, отек, покраснение и гиперплазию синовиальных фибробластов, индуцирует развитие воспаления, а с другой — обладает способностью снижать экспрессию HLA-Dr и синтез кислородных радикалов моноцитами. 1.10.5. Регуляция цитокиновой сети Цитокиновая сеть рассматривается как саморегулирующаяся система, основу функционирования которой составляют продукция специфических антагонистов цитокиновых рецепторов, растворимых цитокиновых рецепторов, антител к цитокинам, связывание цитокинов с некоторыми ингибиторными белками и, наконец, противоположные эффекты различных цитокинов на регуляторные компоненты, обеспечивающие развитие иммунного ответа и воспаления (J-M. Dayer и H. Fenner, 1992). Биологическая активность провоспалительных цитокинов зависит от соотношения между уровнем синтеза цитокинов и ингибирующих молекул. Описано несколько ингибиторов ИЛ-1. К ним относится специфический антагонист ИЛ-1 рецепторов (ИЛ-1ра), который синтезируется моноцитами, макрофагами и нейтрофилами и имеет структурное сходство с ИЛ-1; он обладает способностью связываться ИЛ-1 рецепторами, но не индуцирует развитие биологических эффектов, характерных для ИЛ-1 (С. А. Dinarello, 1991; W. P. Arend, 1991). В различных экспериментальных системах было показано, что ÈË-1ðà блокирует провоспалительные эффекты ИЛ-1 (C. A. Dinarello и R. C. Thompson, 1991). ÈË-1ðà эффективно подавляет развитие экспериментального коллагенового артрита, но не влияет на течение антигениндуцированного артрита (P. Wooley и соавт., 1993). Имеются данные о положительном влиянии ÈË-1ðà при септическом шоке, РА и других заболеваниях. Недавно было показано, что при РА наблюдается дефицит синтеза ÈË-1ðà синовиоцитами по сравнению с общей продукцией ИЛ-1, что может иметь значение в прогрессировании суставной деструкции, индуцированной ИЛ-1 (G. S. Firestein и соавт., 1994). Предполагается, что антивоспалительная активность ИЛ-1ра связана с подавлением ИЛ-1 индуцированного синтеза ИЛ-8. Примечательно, что синтез ÈË-1ðà усиливается ИЛ-4, который, как уже отмечалось, обладает способностью подавлять синтез самого ИЛ-1 (E. Vannier и соавт., 1992). Другой механизм саморегуляции цитокиновой сети обусловлен продукцией растворимых цитокиновых рецепторов, которые, связываясь с цитокинами в кровяном русле, блокируют их активность (R. FernandezBotran, 1991). Описаны растворимые формы ИЛ-1, ИЛ-2, ИЛ-4, ИЛ-6 и ФНО рецепторов. Например, ФНО-α и ФНО-β связываются с двумя высокоаффинными клеточными поверхностными рецепторами с молекулярной массой соответственно 55 kD и 75 kD, экспрессирующимися на мембранах практически всех клеток, включая Т-лимфоциты, макрофаги и нейтрофилы. Растворимые формы ФНО-рецепторов (рФНО-Р) представляют собой белки (30 kD). Увеличение уровня рФНО-Р обнаружено в сыворотке и синовиальной жидкости больных РА, оно коррелирует с воспалительной активностью заболевания. При остеоартрозе также отмечено увеличение уровня рФНО-Р в сыворотке, но не в синовиальной жидкости. Предполагается, что определение концентрации рФНО-Р может иметь значение для оценки активности процесса и диагностики РА. Кроме того, существует несколько ярких примеров противоположного действия различных цитокинов на одни и те же иммуновоспалительные процессы. Например, ФНО-β в некоторых случаях проявляет антагонистическую активность с ИЛ-1-α и ФНО-α, a ÒÔÐ-β действует с противоположной направленностью с ИФ-γ и ФНО-α. ИЛ-4 и ИЛ-10 блокируют синтез ИЛ-1, ФНО-α, ИЛ-6 и других цитокинов моноцитами. Наконец, в сыворотках нормальных индивидуумов обнаружены антитела к цитокинам, включая ИЛ-1-α, ФНО-α и ИЛ-6, которые блокируют биологические эффекты цитокинов (K. Bendtzen и соавт., 1990). Эффекты многих цитокинов блокируются при связывании с α-2 макроглобулином. 1.11. Система комплемента Система комплемента, состоящая из более чем 20 биохимически различающихся белков, играет важную роль в развитии воспаления и иммунном ответе (S. K. Law, 1988). Компоненты комплемента присутствуют в кровяном русле в виде неактивных предшественников (зимоген). При воздействии различных иммунологических и неиммунологических стимулов отдельные компоненты комплемента вступают в серию взаимодействий с активирующими субстанциями и друг с другом, что приводит к образованию биологически активных форм, обладающих мощной провоспалительной и литической активностью. Существуют два основных пути активации комплемента: классический и альтернативный, которые функционируют независимо друг от друга. К белкам классического пути активации комплемента относятся Clq, C1s, C1r, C4, C2, СЗ; белки альтернативного пути включают пропердин, факторы В, D и С3. С3-компонент, являясь одним из основных гликопротеинов плазмы (присутствует в сыворотке в концентрации 1.3 мг/мл), занимает центральное место в обоих путях активации комплемента. Белки С5-С9 обозначаются как терминальные компоненты (мембраноатакующий комплекс) и также являются общими для обоих путей, осуществляют лизис и повреждение клеток-мишеней. Контроль за активацией комплемента осуществляется семью контролирующими белками, основными из которых являются C1INH, I (C3bINA), Н (В1Н), С4ВР, а также мембранными белками и клеточными рецепторами. Активация классического пути происходит при взаимодействии Clq с иммунными комплексами, содержащими антитела IgG1, IgG2, IgG3, IgM или в отсутствие антител под воздействием различных полианионов, некоторых полисахаридов, вирусных мембран и других субстанций. Активация альтернативного пути может происходить в отсутствие антител за счет воздействия липополисахаридов бактерий, вирусов и вирусинфицированных клеток. Анафилотоксины (С3а, С4а, С5а) являются биологически активными фрагментами компонентов комплемента (С3, C4, С5), принимающими участие в развитии воспаления и вазоспазме. Связываясь со специфическими рецепторами, экспрессирующимися на мембране различных клеток, они индуцируют клеточную активацию (хемотаксис и активацию фагоцитов, синтез цитокинов и др.). У человека описано 2 полиморфных тесно связанных структурных локуса C4, которые называются Ñ4À и С4В и располагаются на коротком плече 6-й хромосомы. Каждый из этих локусов имеет несколько экспрессирующихся аллелей (в настоящее время выявлено 13 аллельных продуктов C4 локуса и 21 аллельный продукт Ñ4 локуса), а также неэкспрессирующихся (нулевых). Наличие неэкспрессирующихся аллелей Ñ4À и Ñ4 определяет широкие колебания концентрации C4 в сыворотке, предрасполагает к развитию аутоиммунных заболеваний, включая СКВ, лекарственную волчанку, ССД, РА, синдром Шегрена, склерозирующий панэнцефалит, IgA-èììóíîäåôèöèò. Полагают, что нулевой аллель Ñ4À создает предпосылки для нарушения клиренса иммунных комплексов и, таким образом, играет важную роль в развитии аутоиммунных и иммунокомплексных заболеваний человека. Подавление C4 гидралазином и изониазидом может иметь значение в развитии лекарственной волчанки. 1.12. ПГ, Л Т и другие медиаторы воспаления ПГ, открытые независимо друг от друга M. W. Goldblatt и U. S. von Euler в 1934 году, являются естественными медиаторами воспаления (таблица 1.8.). Все основные типы клеток, принимающие участие в развитии воспалительных и иммунных реакций, способны образовывать ПГ, однако наиболее важными источниками ПГ, вероятно, являются моноциты и макрофаги. Таблица 1.8. Роль ПГ в развитии основных признаков воспаления (J. J. F. Belch, 1989) Признаки Роль ПГ Покраснение (Rubor) ПГ обладают вазодилататорной активностью Отек (Tumor) Большинство ПГ проявляют синергизм с другими медиаторами, вызывающими отек Боль (Dolor) ПГ проявляют синергизм с брадикинином и гистамином в отношении гипералгезии и боли Покраснение (Calor) ПГ индуцируют повышение температуры Нарушение функции (Functio ПГ изменяют функциональную активность клеток, принимающих участие в laesia) развитии воспаления Арахидоновая кислота является С20 полиненасыщенной органической жирной кислотой, принадлежащей к группе органических кислот с длинными водородными цепочками. Арахидоновая кислота присутствует в мембранных фосфолипидах нескольких классов, но особенно широко она представлена в составе фосфатидилхолина (лейцитин). К продуктам метаболизма арахидоновой кислоты, образующимся в процессе взаимодействия кислорода с этой полиненасыщенной жирной кислотой, относятся ПГ, тромбоксаны, простациклин, эндопероксиды ПГ, гидропероксиэйкозатетрановые кислоты (НРЕТЕ), гидроксиэйкозатетрановые кислоты (НЕТЕ), ЛТ и др. Все эти вещества, имеющие общее название "эйкозаноиды", т.е. производные эйкозатетраноиковой (арахидоновой) кислоты, принимают участие в развитии воспалительных реакций. ПГ представляют собой полиненасыщенные жирные кислоты, имеющие в молекуле 20 атомов углерода. В соответствии со структурой они разделены на семейства, члены которых обладают разными биологическими свойствами. Семейства обозначаются буквами (А, В, Е, F и т.д.) и дифференцируются в зависимости от числа двойных связей в боковых цепочках. Продукция ПГ связана с высвобождением арахидоновой кислоты под воздействием различных ферментов (фосфолипазы А2, фосфолипазы С, диацилглицерола, циклооксигеназы), Существуют несколько путей метаболизма арахидоновой кислоты, в каждом из которых функции катализаторов выполняют определенные ферменты. Один из этих путей — циклооксигеназный — обеспечивает синтез эндопероксидов ПГ, самих ПГ, простациклина и тромбоксана. Другой путь — липоксигеназный — связан с продукцией НРЕТЕ, НЕТЕ и ЛТ. Эйкозаноиды не хранятся в клетках, но быстро синтезируются в ответ на различные стимулы (гормоны, химические медиаторы, повреждение клеток и др.), претерпевают очень быстрый метаболизм и превращение в неактивные продукты и выводятся из кровообращения. Все клетки в той или иной степени обладают способностью синтезировать эйкозаноиды, однако их специфические формы преимущественно образуются в клетках различных типов. Например, тромбоциты являются основным источником тромбоксана À2, а ЭК продуцируют простациклин I2. Для стимуляции синтеза ПГ требуется активация фосфолипазы, которая обеспечивает образование достаточного количества свободной арахидоновой кислоты для действия ферментов арахидонового каскада: циклооксигеназы и липоксигеназы. Циклооксигеназа (ЦОГ), известная также как PGH эндопероксид синтетаза, бифункционально связанный с мембраной гемопротеин, располагающийся вблизи места высвобождения арахидоновой кислоты из мембранных фосфолипидов и имеющий двойную каталитическую активность. ЦОГ катализирует оксигенацию арахидоновой кислоты, т.е. присоединение молекулы кислорода к арахидоновой кислоте в положении 9, 11 è 15, что приводит к конверсии арахидоновой кислоты в ПГG2. За счет своей пероксидазной активности ЦОГ конвертирует ПГG2 в ПГН2, которые являются предшественниками всех типов ПГ и тромбоксана. Установлено существование по крайней мере двух тесно связанных изоферментов ЦОГ ( PGH-ñèíòåòàçû): ЦОГ-1 и ЦОГ-2. (I. Appleton и соавт., 1994). Эти изоферменты идентичны на 60% и имеют приблизительно одинаковую способность конвертировать арахидоновую кислоту в ПГ, однако существенно различаются по механизмам регуляции и экспрессии активности (таблица 1.9.) Таблица 1.9. Сравнительная характеристика ЦОГ-1 и ЦОГ-2 (по D. L. DeWitt и соaвт., 1993) Свойства ЦОГ-1 ЦОГ-2 Регуляция Общая Локальная Ген 2.8kb иРНК 4 kb иРНК Выраженность экспрессии Увеличение в 2-4 раза Увеличение в 10-80 раз Тканевая экспрессия Тромбоциты, ЭК, желудок, почки, другие ткани Предстательная железа, мозг, активированные моноциты и фибробласты, синовиоциты; возможно, экспрессируются в других тканях при их стимуляции гормонами, цитокинами, факторами роста Эффект ГК Отсутствует Выраженное подавление экспрессии Предполагаемая роль фермента Синтез ПГ, регулирующих физиологические функции желудка, почек и сосудов Синтез ПГ, участвующих в развитии воспаления, контроле клеточного митогенеза ЦОГ-1 рассматривается как "конституциональный" фермент, экспрессирующийся во всех клетках, хотя степень экспрессии в различных тканях может быть неодинаковой. Предполагается, что синтез ЦОГ-1 кодируется генами (housekeeping), регулирующими продукцию ПГ в ответ на стимуляцию гормонами, участвующими в обеспечении нормального клеточного цикла. ЦОГ-2 в физиологических условиях присутствует в тканях в крайне низкой концентрации, но на фоне воспаления ее уровень резко возрастает. Таким образом, ЦОГ-2 принимает участие в продукции ПГ, вовлеченных в процессы воспаления, митогенеза и клеточной пролиферации. Установлено, что одним из мощным индукторов гена ЦОГ является ИЛ-1 (J. A. M. Maier и соавт., 1990). Имеются данные об индукции ЦОГ-2 в ЭК микрососудов синовиальной оболочки под действием ИЛ-1 (A. Szczepanski и соавт., 1994). При этом важным элементом регуляции экспрессии ЦОГ-2 является высокая чувствительность к ГК, которые полностью подавляют активность ЦОГ-2. Это позволяет рассматривать ген, кодирующий ЦОГ-2, в качестве представителя семейства ГК-чувствительных воспалительных генов. В отличие от ПГ, которые вырабатываются многими клетками, ЛТ синтезируются преимущественно клетками, принимающими участие в развитии воспаления, такими, как нейтрофилы, моноциты/макрофаги, тучные клетки, базофилы и эозинофилы. Продукция ЛТ определяется активностью ферментов, отличающихся от ЦОГ, которые получили название "липоксигеназы". Последние также катализируют включение молекулы кислорода в специфические связи полиненасыщенных жирных кислот, главным образом арахидоновой (B. Samuelsson и соавт., 1987). Основной формой липоксигеназы является 5-липоксигеназа, которая добавляет кислород в 5-е положение арахидоновой кислоты. Это приводит к образованию 5гидропероксиэйкозатетраеновой кислоты (5-НРЕТЕ). Этот же фермент катализирует образование циклической формы 5,6 эпоксида-ЛТА5. ЛТА5 является предшественником лейкотриенов двух важных классов: дегидроксильного производного ЛТВ4, ЛТС4, ЛТD4 и ЛТЕ4. Последние три субстанции сейчас называются сульфопептидами ЛТ, а ранее определялись как медленно реагирующая субстанция анафилаксии. Установлено, что продукты 5-липоксигеназного пути играют очень важную роль в развитии воспаления (таблица 1.10.). Таблица 1.10. Основные биологические эффекты ЛТ ËÒÂ4 ËÒÑ4, D4, Е4 Активация лейкоцитов: Сокращение гладкой мускулатуры хемокинез Венозный выпот хемотаксис прилипание агрегация дегрануляция Усиление экспрессии СЗb рецепторов Супрессия функции лимфоцитов ËÒÂ4 является очень мощным хемоаттрактантом лейкоцитов, способствует прилипанию лейкоцитов к эндотелию, активирует секрецию кислородных радикалов и протеолитических ферментов нейтрофилами. Сульфидопептиды ЛТ вызывают сокращение гладкой мускулатуры сосудов, дыхательной и кишечной тканей. При этом индуцированная этими ЛТ вазоконстрикция ассоциируется с увеличением сосудистой проницаемости. Обладая способностью вызывать сокращение бронхов и бронхиальную секрецию, ЛТ играют важную роль в патогегезе бронхиальной астмы. Кроме того, сульфидопептиды ЛТ дают отрицательный инотропный и аритмогенный эффекты на миокард, стимулируют сокращение мезангиальных клеток и др. По современным представлением, эйкозаноиды рассматриваются не как медиаторы, а как регуляторы воспаления, а также иммунных реакций (R. P. Phipps и соавт., 1991; R. B. Zurier, 1990). Различные формы ПГ (ПГЕ1, ПГЕ2 и ПГI2) связываются с различными типами клеточных рецепторов и нередко проявляют разнонаправленную биологическую активность. При определенных условиях и в зависимости от концентрации они могут как подавлять, так и стимулировать Т- и В-клеточный иммунные ответы, что опосредуется их влиянием на синтез цитокинов и цитокиновых рецепторов. Известно, что ПГ являются регуляторами концентрации внутриклеточного цАМФ (J. S. Goodwin и соавт., 1981). При этом ÏÃÅ2, связываясь с ПГЕспецифическими рецепторами, ассоциированными с G-áåëêîì, увеличивают концентрацию внутриклеточного цАМФ, что в свою очередь оказывает регулирующее воздействие на экспрессию иРНК цитокинов (T. J. Novae и E. V. Rothenberg, 1990). Имеются данные о том, что ÏÃÅ2 подавляет продукцию Т1 цитокинов, синтезирующихся Th1 лимфоцитами, и не влияет на образование цитокинов, продуцируемых Тh2лимфоцитами (M. Betz и В. S. Fox, 1991; K. Gold и соавт., 1994). Высказано предположение о том, что ÏÃÅ2 может проявлять антивоспалительные эффекты при заболеваниях с Thl-öèòîêèíîâûì профилем, в то время как при заболеваниях, при которых доминирует Тh2-тип иммунного ответа (например, при РА), ÏÃÅ2 обладает провоспалительной активностью. 1.13. ФАТ ФАТ считается единственным фосфолипидом, обладающим очень мощной биологической активностью (G. Camussi и соавт., 1990). Он принадлежит к классу фосфолипидных эфиров, имеющих О-алкиловую эфирную последовательность в положении 1 молекулы глицерина и рассматривается как важный медиатор немедленной гиперчувствительности. ФАТ синтезируется нейтрофилами, макрофагами, тромбоцитами. Он вызывает агрегацию тромбоцитов, усиливает хемотаксис, стимулирует высвобождение протеолитических ферментов из нейтрофилов и макрофагов, вызывает сокращение гладкой мускулатуры, усиливает сосудистую проницаемость. 1.14. Активированные формы кислорода При фагоцитозе любых чужеродных частиц в фагоцитирующих клетках резко возрастает поглощение кислорода, расход глюкозы, выделение углекислого газа и молочной кислоты. Такая активация энергетического метаболизма называется "респираторный" (метаболический, окислительный) взрыв". Для индукции "респираторного взрыва" необходимо связывание различных лигандов с соответствующими рецепторными белками фагоцитов. Передача сигнала активации метаболизма с клеточной поверхности опосредуется системой циклических нуклеотидов и происходит с участием ионов Са2+. В момент "респираторного взрыва", нейтрофилы генерируют высокоактивные нестабильные продукты восстановления кислорода: супероксиданион O2-, перекись водорода H2O2, радикал ОН- и синглетный кислород (1O2). Кислородные свободные радикалы, обладая способностью реагировать с различными компонентами тканей (липиды, белки и др.), являются важными медиаторами воспаления, вызывают тканевое повреждение и необратимую модификацию многих макромолекул (C. F. Nathan, 1987). К основным эффектам кислородных радикалов относятся: 1. Разрушение микроорганизмов (вирусы, бактерии, грибы, простейшие) 2. Стимуляция секреции тромбоцитов, тучных клеток, эндотелиальных клеток, клеток почечных канальцев 3. Образование хемотаксических липидов из арахидоната 4. Активация лейкоцитарной коллагеназы и желатиназы 5. Инактивация хемотаксических ЛТ, других хемотаксических пептидов, (α-1-антипротеаз, метэнкефалина, лейкоцитарных гидролаз, бактериальных токсинов. 1.15. Протеиназы и их ингибиторы Как уже отмечалось, деструкция нормальных тканей, их замещение воспалительной и фибротической тканями ведет к нарушению функции органов и считается характерной чертой воспалительных ревматических заболеваний. Важными медиаторами повреждения внеклеточного матрикса в процессе развития воспаления являются протеазы, которые подразделяются на 4 основных класса. Представители двух из них (аспарагиновая и цистеиновая протеиназы) проявляют активность при кислых значениях рН, а представители двух других классов (сериновые и металлопротеиназы) активны при нейтральном рН. Протеиназы вырабатываются различными типами клеток, включая нейтрофилы, моноциты, макрофаги и фибробласты. В настоящее время описано около 20 протеиназ. Аспарагиновые протеиназы. Большая часть внутриклеточных белков переваривается в лизосомах при кислых значениях рН. Наиболее важной лизосомальной протеиназой, действующей при кислом рН, является катепсин D, принадлежащий к семейству генов ренина и пепсина. Высокая активность катепсина D присуща фагоцитирующим клеткам. В процессе воспаления наблюдается внеклеточная продукция катепсина D макрофагами и клетками соединительной ткани в форме профермента. Цистеиновые протеиназы. Цистеиновые протеиназы (катепсин В и катепсин L) ассоциируются с воспалением и активностью остеокластов в костной ткани, Сериновые протеазы. Сериновые протеазы наиболее активны при нейтральных значениях рН. К ним относятся многие белки коагуляционного каскада, систем фибринолиза и комплемента, а также ферменты поджелудочной железы. Существует 2 типа активаторов плазминогена (АП): тканевый тип АП продуцируется главным образом ЭК, а урокиназный тип АП участвует в активации плазминогена (зимоген плазмина). АП секретируются макрофагами, фибробластами, синовиальными клетками, ЭК и сегментоядерными лейкоцитами. Эластаза сегментоядерных лейкоцитов присутствует в азурофильных гранулах лейкоцитов и моноцитов в качестве предшественника, содержащего 2 добавочных аминокислоты (GluGly). Она способна разрушать протеогликан хряща, а также эластин, являющийся структурным белком артериальной стенки, легких, сустава, капсул и кожи. Кроме того, эластаза лейкоцитов разрушает фибронектин, ламинин, коллаген IV типа базальных мембран. Катепсин G является хемотрипсиновым ферментом лейкоцитов, который структурно связан с химазой тучных клеток. Действие катепсина G на протеогликан хряща более ограничено, чем у эластазы. Он не действует на эластин и коллаген типа I, но эффективно солюбилизирует коллаген хряща и принимает участие в образовании активных продуктов компонентов комплемента. Кроме того, катепсин G является активатором металлопротеиназ. Металлопротеиназы. Матриксные металлопротеиназы (ММП), являющиеся цинкзависимыми ферментами, играют наиболее важную роль в деградации макромолекул внеклеточного матрикса (ВКМ) соединительной ткани (M. P. Vincenti и соавт., 1994). ММП подразделяются на три основных класса. К классу I относятся интерстициальная коллагеназа (ММР-1) и нейтрофильная коллагеназа (ММР-8), основным субстратом которых является коллаген типов I, II и III. Класс II составляют желатиназа с мол. массой 72 kD (ММР-2) и желатиназа В с мол. массой 92 kD (ММР-9), которые разрушают желатин и коллаген типа IV базальной мембраны. К классу III относится стромелизин-1 (ММР-3), стромелизин-2 (ММР-2) и стромелизин-3 и матрилизин (ММР-7), которые проявляют активность против широкого спектра субстратов, включая протеогликаны, ламинин, фибронектин и некоторые типы коллагена. Предполагается, что в развитии суставной патологии (РА и остеоартрит) наиболее существенную роль играет коллагеназа (ММР-1), стромелизин 1 (ММР3) (E. J. Harris, 1990). Индукторами синтеза этих ферментов являются цитокины, в том числе ИЛ-1 α/β, эпидермальный фактор роста, тромбоцитарный фактор роста и ФНО-α, а также кристаллы уратов. Показано, что уровень ферментной активности в хряще коррелирует с тяжестью поражения суставов. Дополнительным доказательством патогенетического значения этих ферментов является обнаружение гиперэкспрессии их иРНК в воспаленной ткани с помощью техники in situ гибридизации. Существует несколько естественных ингибиторов протеолитических ферментов, к которым относится (α2-макроглобулин, тканевые ингибиторы металлопротеиназ (ТИМП) 1, 2,3 и др. Предполагается, что при РА и остеоартрите имеет место локальное нарушение баланса между продукцией активированных форм ММП и ТИМП. Введение ТИМП мышам с экспериментальным коллагеновым артритом приводит к уменьшению тяжести заболевания. Существует большое количество субстанций, которые увеличивают локальную продукцию ТИМП. К ним относятся транс-ретиноидные кислоты, синтетические аналоги ретиноидов (витамин А) и некоторые цитокины, в том числе ТФР-β, ИЛ-6, ИЛ-11, лейкемический ингибиторный фактор и онкостатин М. В таблице 1.11. представлены некоторые вещества, регулирующие экспрессию металлопротеиназ. Таблица 1.11. Факторы, регулирующие экспрессию металлопротеиназ Стимулирующие факторы Ингибирующие факторы Взаимодействие клеток с матриксом посредством интегринов, фибронектина, растворимого коллагена и др. ТФР-β Протеиназы ГК Факторы роста (эпидермальный фактор роста, тромбоцитарный фактор роста, ТФР-β и др.) Ретиноиды Паратиреоидный гормон Эстрогены, прогестереон 1,25-дигидрокси витамин Д ÈÔ-γ Сывороточный амилоидный белок А НПВС β2-микроглобулин Индукторы ТИМП Фагоцитоз ПГЕ В последние годы большое внимание уделяется изучению связи между деструктивной активностью некоторых нейтрофильных протеиназ (протеиназа-3, коллагеназа и эластаза) и образованием реактивных метаболитов кислорода. Предполагается, что перечисленные выше ферменты проявляют более выраженный деструктивный эффект в присутствии активных форм кислорода. Это связывают со способностью последних вызывать инактивацию естественных ингибиторов протеиназ (S. J. Weiss, 1994) 1.16. Вазоактивные амины, окись азота и эндотелины Важными медиаторами воспаления являются гистамин, серотонин и аденозин. Гистамин — продукт декарбоксилирования гистидина, хранится в гранулах тучных клеток и образуется в процессе активации последних медиаторами немедленной гиперчувствительности, в первую очередь IgE. Гистамин, взаимодействуя со специфическими клеточными рецепторами, вызывает вазодилатацию, увеличение проницаемости посткапиллярных венул, бронхоспазм, увеличение секреции бронхиальной слизи. Серотонин (5-гидрокситриптамин) хранится в плотных гранулах тромбоцитов, обладает сосудосуживающей активностью, усиливает сосудистую проницаемость. Важным свойством серотонина является способность стимулировать синтез коллагена фибробластами, что способствует фиброзообразованию. Аденозин — пуриновый нуклеозид, образующийся после внутриклеточного расщепления АТФ (B. N. Cronstein и соавт., 1990), обладает способностью подавлять агрегацию тромбоцитов и модулировать иммунные и воспалительные реакции. На мембранах различных клеток экспрессируются 2 типа аденозиновых рецепторов. В низких концентрациях аденозин реагируют с А1 типом рецепторов нейтрофилов. Это вызывает усиление Fcçàâèñèìîãî фагоцитоза иммунных комплексов, образования супероксидных анионов и хемотаксиса нейтрофилов (F. R. Rose и соавт., 1988; J. E. Salmon и соавт., 1993), то есть дает провоспалительный эффект. Напротив, в высоких концентрациях аденозин связывается преимущественно с А2 рецепторами активированных нейтрофилов, что приводит к антивоспалительным эффектам, в том числе ингибиции Fc-çàâèñèìîãî фагоцитоза, образованию супероксидных анионов и прилипанию нейтрофилов к сосудистому эндотелию (C. H. Jurgensen и соавт., 1990). Связывание аденозина с À2 рецепторами ингибирует экспрессию CD11b/CD18 на активированных нейтрофилах (A. Wollner и соавт., 1993). Недавно было показано, что аденозин и À2 рецепторспецифические аденозиновые аналоги подавляют синтез ФНО-α, ИЛ-6 и ИЛ-8 активированными моноцитами (M. J. Parmely и соавт., 1993; M. G. Bouma и соавт., 1994). Полагают, что стимуляция продукции аденозина является одним из механизмов противовоспалительного действия МТ (глава 6). Большой интерес вызывает изучение роли окиси азота, впервые идентифицированной как эндотелиальный фактор релаксации. Это вещество представляет собой неорганический газообразный свободный радикал (*N=O), который образуется в различных клетках из аргинина под действием синтетазы окиси азота. Существует несколько изомеров окиси азота, основными из которых являются "констутивная" и "индуцируемая" формы. Эндотелий, макрофаги, нейтрофилы, гепатоциты, хондроциты, синовиоциты и нейрональные клетки синтезируют "индуцируемую" форму окиси азота. Установлено, что формы биологической активности окиси азота многообразны: она участвует в регуляции функции тромбоцитов, нейтротрансмиссии, проявляет токсическую активность в отношении внутриклеточных патогенов и др. Это свидетельствует об участии окиси азота в развитии иммунных и воспалительных процессах (S. Moncada и A. Higgs, 1993). Некоторые эффекты окиси азота, которые могут иметь значение при ревматических заболеваниях, обобщены M. Stefanovic-Bacic и соавт. (1993): 1. Вазодилатация, снижение АД. 2. Повреждение тканей. 3. Подавление активности остеокластов и резорбции костной ткани. 4. Синтез ПГЕ2 хондроцитами: в низкой концентрации стимулирует; в высокой концентрации ингибирует. 5. Ингибиция синтеза тромбоксана и ЛТВ4. 6. Ингибиция синтеза ИЛ-6. 7. Ингибиция адгезии лейкоцитов к эндотелию. Описаны как провоспалительные, так и антивоспалительные эффекты окиси азота. Например, будучи вазодилататором и усиливая кровоток, окись азота может участвовать в развитии таких признаков воспаления, как покраснение и локальная гипертермия, отек. С другой стороны, окись азота подавляет адгезию нейтрофилов к эндотелию, конкурируя с CD11/CD18 или, выступая в роли скавенджера супероксидных радикалов, ингибирует синтез ÏÃÅ2, тромбоксана, ИЛ-6 и продукцию супероксидных радикалов нейтрофилами, то есть проявляет антивоспалительную активность. Кроме того, окись азота, вероятно, обладает анальгетическим свойством. Однако на модели индуцированного стрептозоцином диабета было показано, что ингибиторы окиси азота подавляют развитие деструкции клеток поджелудочной железы. Предполагается, что окись азота дает протективный эффект при остром воспалении и стимулирует тканевое повреждение при хроническом воспалении (M. Stefanovic-Racic и соавт., 1993). О роли окиси азота в развитии артритов свидетельствуют данные о способности ингибиторов окиси азота подавлять развитие экспериментальных артритов: индуцированного клеточной мембраной стрептококка (N. McCartney-Francis и не хватает стр. 52-55 ным на защиту организма от потенциально патогенных воздействий. Однако при определенных условиях ИК могут играть важную роль в развитии ревматических болезней (Е. Л. Насонов, 1984; K. A. Davies, 1990). Классическими проявлениями иммунокомплексного процесса, связанными с нарушением клиренса и отложением ИК в тканях, являются васкулит, нефрит и артрит, которые относятся к числу ведущих форм органной патологии при многих ревматических заболеваниях. При ревматических заболеваниях развитие иммунокомплексной патологии связывают со следующими факторами: 1) нарушение механизмов нормального клиренса иммунных комплексов из кровяного русла: а) генетически детерминированная или приобретенная патология системы комплемента, ведущая к нарушению процесса ингибиции иммунной преципитации и солюбилизации комплексов антиген—антитело, что способствует циркуляции комплексов с более выраженным воспалительным потенциалом и возможности их отложения в органах-мишенях; б) врожденное или приобретенное нарушение эритроцитарного клиренса ИК в связи с патологией CR1 рецепторов эритроцитов; в) блокада функциональной активности Fc-ðåöåïòîðîâ мононуклеарных фагоцитирующих клеток, локализованных в печени и селезенке; 2) Гиперпродукция циркулирующих ИК с определенной структурой и зарядом, обладающих способностью связываться с заряженными биомолекулами органов-мишеней. В целом при системных ревматических заболеваниях аутоиммунные и иммунокомплексные патологические процессы находятся в тесной взаимосвязи, которая определяется общей генетической предрасположенностью к нарушениям иммунорегуляции и ослаблению клиренса ИК и сходными механизмами развития воспаления и тканевой деструкции, опосредуемой аутоантителами и ИК. Список литературы к главе "Иммунитет и воспаление при ревматических заболеваниях" 1. Жарова Е. А., Горбачева О. Н., Насонов Е. Л., Карпов Ю. А: Эндотелин, физиологическая активность, роль в сердечно-сосудистой патологии. Терапевт, архив, 1990, No. 8, стр. 140-144. 2. Насонов Е. Л.: Иммунные комплексы при ревматических болезнях. В книге: Иммунология заболеваний соединительной ткани. Москва. 1984, стр. 104-158. Насонов Е. Л.: Интерлейкин 1 и его роль в патологии человека. Терапевт, архив 1987;12: 112-117. 3. Саложин К. В., Насонов Е. Ë., Беленков Ю. Н.: Роль эндотелиальной клетки в иммунопатологии. Терапевт, архив. 1992; 3: 150-157. 4. Adams D., Shaw S.: Leucocyte-endothelial interaction and regulation of leucocyte migration. Lancet 1994; 343: 831-836. 5. Appleton I, Tomlinson A., Willoughby D. A.: Inducible cyclooxygenase (COX-2): a safer therapeutic target. Brit. J. Med., 1994; 33: 410-412. 6. Arai К., Lee F., Miyajima A., et al.: Cytokines: coordinators of immune and inflammatory responses. Ann. Rev. Biochem. 1990; 59: 783. 7. Arend W. P.: Interleukin-I receptor antagonist: a new member of the interleukin-I family. J. Clin. Invest. 1991; 88: 1445-1451. 8. Arend W. P., Dayer J-M.: Inhibition of the production and effects of interleukin-I and tumor necrosis (actor alpha in rheumatoid arthritis. Arthritis Rheum. 1995; 38: 151-160. 9. Arnett FC, Reveille JD: Genetics of systemic lupus erytheatosus. Rheumatic Dis. Clin. North. Amer. 1992; 18: 865-892. 10. Baggiolini M., Walz A., Kunkel S. L.: Neutrophil-activating peptide-l/interleukin-8: a novel cytokine that activates neutrophils. J. Clin. Invest. 1989; 84: 1045-1049. 11. Battistini В, D'Orleans-Juste P, Sirois P: Endothelin: Cir culating plasma levels and presence in other biological fluids. Lab. Invest. 1993; 68: 600-628. 12. Belch J. J. F.: Eicosanoids and rheumatology: Inflammatory and vascular aspects. Prostaglandins, Leukotriens and essential fatty acids. 1989; 36: 219-234. 13. Bendtzen К.. Svenson M., Jonsson V., et al.: Autoantibodies to cytokines: friends or toes. Immunol. Today 1990; II: 167-169. 14. Betz M., Fox B. S.: Prostaglandin E2 inhibits production of Th 1 lymphokine but not of Th2 lymphokine. J. Immunol. 1991; 146: 108-113. 15. Bodmer J. G., Marsh S. G. E., Albert E.: Nomenclature for factors of the HLA system, 1989. Immunol. Today 1990; II: 3-10. 16. Border W. A., Noble N. A.: Transforming growth factor beta in tissue fibrosis. New Engl. J. Med. 1994; 331: 1286-1292. 17. Bottazzo G. F., Borrell-Pujol R., Hanafusa Т., Feidman M.: Role of abberant HLA-DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet 1983; 2: 1115-1117. 18. Camussi G., Terra C., Bagilioni C.: The role of platelet-activating factor in inflammation. Clin. Immunol. Immunopathol. 1990; 57: 331-338. 19. Carson D. A.: Genetic factors in the etiology and pathogenesis of autoimmunity. FASEB J. 1992; 6: 28002805. 20. Cronstein В., Weissman G.: The adhesion molecules of inflammation. Arthritis Rheum. 1993; 36: 147-157. 21. Dalton T. A., Bennet J. C: Autoimmune Disease and the Major Histocompatibility Complex: Therapeutic Implication. Amer. J. Med. 1992; 92: 183188. 22. Davies К. A.: Immune complexes and disease. Eur. J. Intern. Med. 1992; 3: 95-108. 23. Dayer J-M., Fenner H. Cytokines and their inhibitors in arthritis. In: Bailliere's clinical rheumatology. 1992; 6: 485-516. 24. Del Prete G., Maggi E., Romagnani S.: Human Th1 and Th2 cells: functional properties, mechanism of regulation and role in disease. Lab. Invest. 1994; 70: 299-306. 25. DeWitt D. L, Smith W. L: PGH synthase isoenzyme selectivity: the potential for safer nonsteroidal antiinflamatory drugs. Amer. J. Med., 1993: 95 (suppl. 2A): 40S-44S. 26. Dinarello C. A.: Interleukin-I and its biologically related cytokines. Adv. Immunol. 1989; 44: 153-205. 27. Dinarello С. A., Thompson R. C.: Blocking IL-I: Interleukin-I receptor antagonist in vivo and in vitro. Immunol. Today 1991; 12: 404-410. 28. Dormair К., Clark В. R., McConnell H. M.: In vitro peptide binding to the heavy chain of the class I molecule of the major histocompatibility complex molecule HLA-A2. Proc. Natl. Acad. Sci USA. 1991; 88: 1335-1338. 29. Dower S. K., Sims J. M.: Molecular characterisation of cytokine receptors. Ann. Rheum. Dis. 1990; 49: 452459. 30. Duff G. W.: Cytokines and acute phase proteins in rheumatoid arthritis. Scand. J. Rheumatol. 1994; 23 (suppl. 100): 9-19. 31. Elliott Т., Towsend A., Cerundolo V.: Antigen presentation. Naturally processed peptides. Nature 1990; 348: 195-197. 32. Fernanses-Botran R.: Soluble cytokine receptor: their role in immunoregulation. FASEB J. 1991; 5: 25672574. 33. Fioremtino J. F., Ziotnik A., Mosmann T. R., et al.: IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991; 147: 3815-3822. 34. Firestein G., Boyle D. L., Yu C., et al.: Synovial interleukin-I receptor antagonist and interleukin-I balance in rheumatoid arthritis. Arthritis Rheum, 1994: 5: 644-652. 35. Ginsberg M. H.: Role of platelets in inflammatory and rheumatic disease. Adv. Immun. Res. 1986: 2: 53-71. 36. Gold К. N., Weyand C. M., Goronzy J. J. Modulation of helper T cell function by prostaglandins. Arthritis Rheum. 1994; 37: 925-933. 37. Hamilton J. A.: Rheumatoid arthritis: opposing action og haemopoietic growth factors and slow-acting antirheumatic drugs. Lancet 1993; 342: 536-539. 38. Harlan J. M., Liy D. Y.: Adhesion: Its role in Inflammatory disease. W. H. Freeman and Co. New York, 1992. 39. Harris E. J.: Rheumatoid arthritis: pathophysiology and implications for therapy. New Engl. J. Med. 1990; 322: 1277-1289. 40. O'Garra A., Chang R., Go N., et al.: Ly-I В (B-l) cells are the main source of B-cell-derived interleukin-10. Eur. J. Immunol. 1992; 22: 711-717. 41. Hirano Т., Abira S., Taga Т., et al.: Biological and clinical aspects of interleukin-6. Immunol. Today 1990; II: 443-449. 42. lalenti A., Moncada S., DiRosa M.: Modulation of adjuvant arthritis by endogenous nitric oxide. Br. J. Pharmacol. 1993; 110: 701-706. 43. Ishida H., Muchamuel T., Sakaguchi S., et al.: Continious administration of antiinterleukin-10 antibodies delays onset of autoimmunity in NZB/WF1 mice. J. Exp. Med. 1994; 179: 305-310. 44. Katsikis P. D.. Chu C-O., Brennan F. M., et al.: Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J. Exp. Med. 1994; 179: 1517-1527. 45. Kroemer G., Wick G.: The role of interleukin-2 in autoimmunity. Immunol. Today. 1989; 10: 246-251. 46. Law SKA, Reid KBV: Complement. IRL Press. Oxford. Washington DC 1988, 73p. 47. Levy Y., Brouet J-C.: Interleukin-10 prevents spontaneous death of germinal center В cells by induction of the bcl-2 protein. J. Clin. Invest. 1994; 93: 424-428. 48. Llorente L., Richaud-Patin Y., Fior R., et al.: In vitro production of interleukin-10 by non-T cells in rheumatoid arthritis, Sjogren's syndrome, and systemic lupus erythematosus. Arthritis Rheum. 1994; II: 16471655. 49. Maier J. A. M., Hia T., Maciag Т.: Cyclooxigenase is an immediate-early gene induced by Interleukin-I in human endothelial cells. J. Biol. Chem. 1990; 265: 10805-10808. 50. Malech H. L., Gallin J. I.: Neutrophils in human disease. New Engl. J. Med.: 1987; 317: 687-694. 51. McCartney-Francis N., Allen J. B., Mizel D. E., et al.: Suppression of arthritis by an inhibitor of nitric oxide synthase. J. Exp. Med. 1993; 178: 749-754. Metcalf D.: Control of granulocytes and macrophages: Molecular, cellular and clinical aspects. Science 1991; 254: 529-533. 52. Miyasaka N., Hirata Y., Ando K., et al.: Increased production of endothelin-1 in patients with inflammatory arthritides. Arthritis Rheum. 1992; 35: 397-400. 53. Moisec P.: Interleukin 4. A potent anti-inflammatory agent. Rev. Rheum. 1993; 60: 87-91. 54. Moncada S., Higgs A. H.: The L-arginine-nitric oxide path way. New Engl. J. Med. 1993; 329: 2002-2012. 55. Mosmann T. R., Coffman R. L.: TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annual Rev. Med. 1989; 7: 145-173. 56. Nathan C. F.: Secretory products of macrophages. J. Clin. Invest. 1987; 79: 319-326. 57. Novak T. J., Rothenberg E. V.: cAMP inhibits induction of interleukin 2 but not of interleukin 4 in T cells. Proc. Natl. Acad. Sci. USA. 1990; 172: 95-103. 58. Oppenheim J., Rossio J., Gearing A. J. H.: Clinical applications of cytokines: Role in diagnosis, pathogenesis, and therapy. Oxford Unuversity Press. 1993. 59. Parmely M. J., Zhou W-W., Edwards C. K. III, et al.: Adenosine and related carbocyclic nucleoside analogue selectively inhibit tumor necrosis factor-alpha production and protect mice against endotoxon challenge. J. Immunol. 1993; 151: 389-396. 60. Phipps R. P., Stein S. H., Roper R. L.: A new view of prostaglandin E regulation of immune response. Immunol. Today 1991; 12: 349-352 61. 61. Rubin L. A.: The soluble interleukin-2 receptor in rheumatic disease. Arthritis Rheum. 1990: 33: 1145-1148. 62. Samuelsson B., Dahlen S-E., Lingbergen J. A., et al.: Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 1987; 237: 1171-1176. 63. Schwartz R. S., Datta S. K.: Autoimmunity and autoimmune disease. In. Fundamental Immunol. Ed. W. E. Paul. Raven Press, New York, 1989: 819-866. 64. Sinha A. S., Lopez M. T., McDevitt H. 0.: Autoimmune disease; the failure of self tolerance. Science 1990; 248: 1380-1388. 65. Stefanovic-Racik M., Stadler J., Evans C. H.: Nitric oxide and arthritis. Arthritis Rheum. 1993; 36: 1036-1044. 66. Stefanovic-Racik M., Meyers K., Meschter C., et al.: N-monomethyl arginine, an inhibitor of mnitric oxide synthase, suppresses the development of adjuvant arthritis in rats. Arthritis Rheum. 1994; 37: 1062-1069. 67. Szczepanski A., Moatter Т., Carley W. W., Gerritsen M.: Induction of cyclooxygenase II in human synovial microvessel endothelial cells by interleukin-I. Arthritis Rheum. 1994; 37: 495-503. 68. Valone F. H. Platelets. In: Textbook of rheumatology. Ed. W. N. Kelley, E. D. Harris, S. Ruddy, C. B. Sledge. W. B. Saunders Company. Philadelphia, London, Toronto. 1994; 1: 319-326. 69. Vincenti M. P., Clark 1. M., Brinckerhoff C. E.: Using inhibitors of metalloprofeinases to treat arthritis. Arthritis Rheum. 1994; 37: 1115-1126. 70. Wahl S. M., Allen J. B., Ohura K., et al.: IFN-gamma inhibits inflammatory cell recruitment and the evolution of bacterial cell induced arthritis. J. Immunol. 1991; 146: 95. 71. Weiss S. J.: Mechanism of tissue destruction by neutrophils. Rheumatology Review course. Minneapolis, Minnesota, 23 October, 1994. 72. Wooley P., H., Whalen J. D., Charman D. L. et al.: The effect of an interleukin-I receptor antagonist protein on type II collagen-induced arthritis and antigen-induced arthritis in mice. Arthritis Rheum. 1993; 36: 1305-1314. 73. Zurier R. B.: Prostaglandin El: Is it usefull?. J. Rheumatol. 1990; 171439-1441. ГЛАВА 2 НЕСТЕРОИДНЫЕ ПРОТИВОВОСПАЛИТЕЛЬНЫЕ ПРЕПАРАТЫ Нестероидные противовоспалительные препараты ( НПВС) представляют собой класс фармакологических агентов, терапевтическая активность которых связана с предотвращением развития или снижением интенсивности воспаления. В настоящее время существует более 50 различающихся по химической структуре лекарственных форм, классифицирумых как НПВС, которые в свою очередь подразделяются на несколько основных подклассов (таблица 2.1.). Таблица 2.1. Классификация НПВС (по P. I. Clements и H. E. Paulus. 1994) I. ПРОИЗВОДНЫЕ КИСЛОТ 1. Арилкарбоновые кислоты Салициловая кислота: Антраниловая кислота (фенаматы): • аспирин • флуфенамовая кислота • дифлунисал • мефенамовая кислота • трисалицилат • меклофенамовая кислота • бенорилат • нифлумиковая кислота • салицилат натрия 2. Арилалкановые кислоты Арилпропионовая кислота: • ибупрофен • флурбипрофен • кетопрофен • напроксен • оксапрозин Гетероарилуксусная кислота • фенопрофен • толметин • фенбуфен • зомепирак • супрофен • клоперак • индопрофен • кеторолак триметамин • тиапрофеновая кислота • беноксапрофен • пирпрофен Индол/инден уксусные кислоты: • индометацин • сулиндак • этодолак • ацеметацин Арилуксусная кислота: • диклофенак • фенклофенак • алклофенак • фентиазак Пиразолидиндионы: • фенилбутазон • оксифенилбутазон 3. Эноликовая кислота Оксикамы: • пироксикам • изоксикам • азапропазон • фепразон • судоксикам • мелоксикам II. НЕКИСЛОТНЫЕ ПРОИЗВОДНЫЕ • проквазон • тиарамид • буфексамак • эпиразол • набуметон • • • • флюрпроквазон флуфизон тиноридин колхицин III. КОМБИНИРОВАННЫЕ ПРЕПАРАТЫ • артротек (диклофенак+мизопростол) НПВС являются одними из наиболее часто используемых в клинической практике лекарственных средств (P. M. Brooks, 1989). Их назначают приблизительно 20% стационарных больных, страдающих различными заболеваниями внутренних органов. Механизм действия За исключением набуметона (пролекарство в форме основания), НПВС являются органическими кислотами со сравнительно низкой рК. Благодаря этому они активно связываются с белками плазмы и накапливаются в очаге воспаления, в котором в отличие от невоспаленной ткани наблюдается повышение сосудистой проницаемости и сравнительно низкая рН. НПВС сходны по фармакологическим свойствам, биологической активности и механизмам действия (P. J. Clements, H. E. Paulus, 1993). В 1971 г. J. Vane впервые обнаружил, что ацетилсалициловая кислота и индометацин в низких концентрациях проявляют свое противовоспалительное, анальгетическое и жаропонижающее действие за счет подавления активности фермента ЦОГ, принимающего участие в биосинтезе ПГ. С тех пор точка зрения, согласно которой противовоспалительный и другие эффекты НПВС связаны в первую очередь с подавлением синтеза ПГ, является общепринятой. Действительно, практически все синтезированные в настоящее время НПВС in vitro блокируют ЦОГ в составе ПГ-эндопероксидсинтетазного комплекса, в меньшей степени не влияя на активность других ферментов, участвующих в метаболизме арахидоновой кислоты (фосфолипаза А2, липоксигеназа, изомераза). Предполагается также, что подавление синтеза ПГ в свою очередь может приводить к многообразным вторичным фармакологическим эффектам, выявляемым у больных, леченных НПВС, в том числе связанным с изменением функции нейтрофилов, Т- и В-лимфоцитов, синтезом ЛТ и др. Кроме того, антипростагландиновая активность НПВС объясняет их некоторые сосудистые эффекты (снижение интенсивности индуцированного ПГ отека и эритемы), анальгетическое действие и причины развития основных побочных реакций (пептическая язва, нарушение функции тромбоцитов, бронхоспазм, гипертензия, нарушение клубочковой фильтрации). Возможные точки приложения фармакологической активности НПВС (no P. M. Brooks, 1993). • • • • • • • • • • • • • • • Синтез ПГ Синтез ЛТ Образование супероксидных радикалов Высвобождение лизосомальных ферментов Активация клеточных мембран: Ферменты NAPDH окисление Фосфолипазы Трансмембранный транспорт анионов Захват предшественников ПГ Агрегация и адгезия нейтрофилов Функция лимфоцитов Синтез РФ Синтез цитокинов Метаболизм хряща Однако в последние годы представления о точках приложения НПВС в регуляции синтеза ПГ существенно расширились и уточнились. Ранее считали, что ЦОГ является единственным ферментом, ингибиция которого снижает синтез ПГ, участвующих в развитии воспаления, и "нормальных" ПГ, регулирующих функцию желудка, почек и других органов. Но недавно были открыты две изоформы ЦОГ (ЦОГ-1 и ЦОГ-2), играющие различную роль в регуляции синтеза ПГ (глава 2). Как уже отмечалось, именно ЦОГ-2 регулирует синтез ПГ, индуцированный различными провоспалительными стимулами, в то время как активность ЦОГ-1 определяет продукцию ПГ, принимающих участие в нормальных физиологических клеточных реакциях, не связанных с развитием воспаления. Предварительные результаты, полученные пока только в опытах in vitro, показали, что некоторые НПВС в равной степени ингибируют ЦОГ-1 и ЦОГ-2, в то время как другие в 10-30 раз сильнее подавляли ЦОГ-1, чем ЦОГ-2 (E. A. Meade и соавт., 1993). Эти результаты, хотя и являются предварительными, имеют очень важное значение, так как позволяют объяснить особенности фармакологической активности НПВС и причины развития некоторых побочных эффектов, наиболее присущих сильным ингибиторам ЦОГ. Действительно, хорошо известно, что ПГЕ2 и ПГI2 оказывают протективное действие на слизистую желудка, что связывают с их способностью снижать желудочную секрецию соляной кислоты и увеличивать синтез цитопротективных веществ. Предполагается, что желудочно-кишечные осложнения НПВС связаны с подавлением именно ЦОГ-1. Другим циклооксигеназным продуктом является тромбоксан À2, ингибиция синтеза которого НПВС нарушает агрегацию тромбоцитов и способствует кровоточивости. Кроме того, ПГ играют важную роль в регуляции клубочковой фильтрации, секреции ренина и поддержании водно-электролитного баланса. Очевидно, что ингибиция ПГ может приводить к разнообразным нарушениям функции почек, особенно у больных с сопутствующей почечной патологией. Полагают, что именно способность ГК селективно ингибировать ЦОГ-2 обусловливает существенно более низкую частоту язвенного поражения желудка на фоне лечения этими препаратами по сравнению с НПВС, отсутствие влияния на свертываемость крови и функцию почек. Наконец, подавление циклооксигеназной активности может потенциально способствовать переключению метаболизма арахидоновой кислоты на липоксигеназный путь, вызывая гиперпродукцию ЛТ. Именно этим объясняют развитие у некоторых больных, получающих НПВС, бронхоспазма и других реакций немедленной гиперчувствительности. Полагают, что гиперпродукция ЛТВ4 в желудке может являться одной из причин развития сосудистого воспалительного компонента язвенного поражения желудочно-кишечного тракта (J. L. Wallace и D. N. Granger, 1992). Известно, что ËÒÂ4 вызывает активацию и гиперэкспрессию лейкоцитарной молекулы адгезии CD11b/CD18. При этом антитела к CD11b/CD18 способны предотвращать развитие индуцированного НПВС изъязвления желудка. С этих позиций хорошо объясним мощный профилактический эффект синтетических ПГ серии Е1 при НПВСиндуцированных гастропатиях. Известно, что ПГЕ1 обладают способностью подавлять активацию нейтрофилов, предотвращать прилипание нейтрофилов к ЭК, стимулированное НПВС, ингибировать синтез ËÒÂ4 нейтрофилами. В целом все эти результаты создают теоретическую основу для целенаправленной разработки новых химических соединений, способных селективно ингибировать ЦОГ-2, что позволит подойти к созданию препаратов с более высокой противовоспалительной активностью и низкой токсичностью. В последние годы стало очевидным, что в простагландиновую гипотезу удовлетворительно укладываются терапевтические эффекты только низких доз НПВС, но она не может полностью объяснить механизмы действия высоких доз препаратов. Оказалось, что противовоспалительная и анальгетическая активность НПВС часто не коррелирует с их способностью подавлять синтез ПГ (J. Y. Jeremy и D. P. Mikhalidis, 1990). Например, "противовоспалительная" доза аспирина значительно выше, чем та, которая необходима для подавления синтеза ПГ, а салициловый натрий и другие неацетилированные салицилаты, которые очень слабо подавляют активность ЦОГ, не уступают по противовоспалительной активности НПВС, являющихся сильными ингибиторами синтеза ПГ. (Multicenter salsalate/aspirin comparison study group, 1989; S. J. Preston и соавт., 1989; R. D. Altman, 1990). Полагают, что именно эти особенности определяют более низкую токсичность неацетилированных салицилатов в отношении ЖКТ, отсутствие действия на тромбоциты и хорошую переносимость этих препаратов даже у больных с гиперчувствительностью к аспирину (J. M. Scheiman и G. H. Elta, 1990). Некоторые токсические реакции, такие, как гепатит, неврологические расстройства (шум в ушах, депрессия, менингит, дезориентация), интерстициальный нефрит, также, вероятно, не связаны с ПГзависимыми механизмами действия НПВС. К эффектам НПВС, которые, как полагают, непосредственно не связаны с их антипростагландиновой активностью, относятся следующие: 1. подавление синтеза протеогликана клетками суставного хряща. 2. подавление периферического воспаления за счет центральных механизмов; 3. усиление Т-клеточной пролиферации и синтеза ИЛ-2 лимфоцитами; 4. подавление активации нейтрофилов; 5. нарушение адгезивных свойств нейтрофилов, опосредуемых CD11b/CD18? В частности, показано, что ацетилсалициловая кислота и салициловый натрий (но не индометацин) подавляют развитие воспалительного отека конечностей при введении препаратов в латеральный желудочек мозга. Это не связано с системными антипростагландиновыми эффектами, так как аналогичные дозы салицилатов и индометацина в кровяном русле не оказывали противовоспалительного действия. Эти данные свидетельствуют о том, что салицилаты могут подавлять нейрогенные (центральные) механизмы развития периферического воспаления (A. Catania и соавт., 1991). По данным K. K. Wu и соавт. (1991), салицилаты подавляют индуцированную ИЛ-1 экспрессию гена ЦОГ в культуре ЭК. Кроме того, при определенных экспериментальных условиях, некоторые НПВС обладают способностью усиливать пролиферативную активность Т-лимфоцитов и синтез ИЛ-2, что ассоциируется с повышением уровня внутриклеточного кальция (E. Flescher и соавт., 1991), а также ингибируют хемотаксис и агрегацию нейтрофилов, образование гипохлорной кислоты и супероксидных радикалов лейкоцитами (E. Shacter и соавт., 1991; M.Ip и соавт., 1990), подавляют активность фосфолипазы С и синтез ИЛ-1 моноцитами (D. M. Chang и соавт., 1990). При этом стабильный аналог ПГЕ1мизопростол усиливает ингибирующее действие НПВС на активацию нейтрофилов (E. A. Kirtis и соавт., 1991). Молекулярные механизмы, определяющие эти фармакологические эффекты НПВС, до конца не ясны. Предполагается, что, являясь анионными липофильными молекулами, НПВС могут проникать в фосфолипидный бислой и изменять вязкость биомембран (S. B. Abramson и соавт., 1990). Это в свою очередь приводит к нарушению нормальных взаимодействий между мембранными белками и фосфолипидами и предотвращает клеточную активацию лейкоцитов на ранних этапах воспаления. Этот эффект может реализовываться за счет прерывания передачи активационных сигналов на уровне гуанозинтрифосфатсвязывающего белка (G-áåëîê) (S. B. Abramson и соавт., 1990; 1991). Известно, что G-áåëîê играет важную роль в регуляции процесса активации лейкоцитов под влиянием анафилотоксина (С5а) и хемотакстического пептида формил-метионин-лейцин-фенилаланина (ФМЛФ). Связывание этих лигандов со специфическими мембранными рецепторами лейкоцитов приводит к изменению их конформации. Конформационная перестройка передается через мембрану G-áåëêó, в результате чего он приобретает способность связывать внутриклеточный гуанозинтрифосфат. Это ведет к таким изменениям конформации Gáåëêà, которые индуцируют активацию фосфолипазы А2 и С и генерацию вторичных мессенджеров (диацилглицерол, арахидоновая кислота, инозитолтрифосфат), необходимых для реализации функциональной активности лейкоцитов (A. G. Gilman, 1987). В экспериментальных исследованиях было показано, что НПВС способны блокировать связывание гуанозинтрифосфата с G-белком, что приводит к отмене хемотаксических эффектов Ñ5à и ФМЛФ и подавлению клеточной активации. В свою очередь арахидоновая кислота, высвобождающаяся из мембранных фосфолипидов при клеточной активации, усиливает связывание гуанозинтрифосфата с G-áåëêîì, то есть дает эффект, противоположным действию НПВС (S. B. Abramson и соавт., 1990; 1991). Таким образом, с учетом представленных выше данных можно предположить, что противовоспалительный эффект НПВС опосредуется двумя независимыми механизмами: низкие концентрации НПВС, интерферируя с комплексом арахидонат—ЦОГ, предотвращают образование стабильных ПГ, а в высоких (противовоспалительных) концентрациях — блокируют ассоциацию арахидоната с G-áåëêîì, и таким образом, подавляют клеточную активацию. Совсем недавно Е. Корр и S. Ghosh (1994) обнаружили новый молекулярный механизм действия НПВС, возможно имеющий наиболее важное значение в реализации противовоспалительной и иммуномодулирующей активности этих препаратов. Оказалось, что салициловая кислота и аспирин в терапевтических концентрациях подавляют активацию фактора транскрипции (NF-kB) в Тлимфоцитах. Известно, что NF-kB является индуцируемым фактором транскрипции, присутствующим в цитоплазме эукариотических клеток, который активируется под влиянием различных провоспалительных стимулов (бактериальный липополисахарид, ИЛ-1, ФНО и др.). Эти активационные сигналы приводят к транслокации NF-kB из цитоплазмы в ядро, где NF-kB связывается с ДНК и регулирует транскрипцию нескольких генов, большинство из которых кодируют синтез молекул, принимающих участие в развитии воспаления и иммунных реакциях: цитокины (ИЛ-1, ИЛ-6, ИЛ-8, ИФ-b, ФНО-а) и молекулы клеточной адгезии (молекулы межклеточной адгезии 1 (IСАМ-1), эндотелиально-лейкоцитарная молекула адгезии -1, сосудистая молекула адгезии-1 (VCAM-1). Примечательно, что подобные механизмы действия имеют ГК (глава 3) и ЦсА (глава 10), что позволяет по новому оценить терапевтические возможности использования НПВС. Практически все НПВС обладают способностью уменьшать боль в концентрации меньшей, чем необходимо для подавления воспаления. Ранее полагали, что, поскольку ПГ усиливают болевой ответ, индуцированный брадикинином, подавление их синтеза является одним из основных механизмов анальгетических эффектов НПВС (K. Brune, 1983). С другой стороны, имеются данные о влиянии НПВС на центральные механизмы боли, не связанные с ингибицией синтеза ПГ. Например, ацетоменофен обладает очень высокой анальгетической активностью, несмотря на отсутствие способности ингибировать активность ЦОГ. НПВС эффективно подавляют лихорадку у человека и экспериментальных животных. Известно, что очень многие цитокины, включая ÈË-1a/b, ФНО-a/b, ИЛ-6, макрофагальный воспалительный белок 1 и ИФ-a обладают активностью эндогенных пирогенов, а ИЛ-2 и ИФ-у могут индуцировать лихорадку, увеличивая синтез одного или более из перечисленных выше цитокинов (C. A. Dinarello, 1991). Поскольку развитие лихорадки связано с индуцированным провоспалительными цитокинами синтезом ПГ, предполагается, что антипиретический эффект НПВС обусловлен их антицитокиновой и антипростагландиновой активностью. Под влиянием аспирина и в значительно меньшей степени — других НПВС ослабляется агрегационный ответ тромбоцитов на различные тромбогенные стимулы, включая коллаген, норадреналин, АДФ и арахидонат. Это связано с тем, что в тромбоцитах аспирин блокирует синтез тромбоксана А2, который обладает сосудосуживающей активностью и способствует агрегации тромбоцитов. Механизм действия аспирина на синтез тромбоксана À2 определяется необратимым ацетилированием сериновых остатков (Ser529) и подавлением активности ЦОГ и гидропероксида, необходимых для синтеза тромбоксана À2. Полагают, что, кроме антиагрегационного действия, аспирин может иметь и другие точки приложения в механизмах свертывания крови: подавление синтеза витамин-К-зависимых факторов свертывания, стимуляция фибринолиза и подавление липоксигеназного пути арахидонового метаболизма в тромбоцитах и лейкоцитах. Установлено, что тромбоциты особенно чувствительны к аспирину: однократный прием 100 мг аспирина приводит к снижению сывороточной концентрации тромбоксана В2 (продукт гидролиза тромбоксана À2) на 98% в течение 1 ч, и только 30 мг в день эффективно подавляют синтез тромбоксана. В то же время антитромбогенный эффект аспирина ограничен способностью подавлять продукцию простациклина (ПГ12), обладающего влиянием на сосудистый тонус и состояние тромбоцитов, противоположным таковому у тромбоксана À2, Однако в отличие от тромбоцитов синтез простациклина ЭК после приема аспирина очень быстро восстанавливается. Все это вместе взятое создало предпосылки к использованию именно аспирина для профилактики тромботических нарушений при различных заболеваниях. Клиническое применение В ревматологии НПВС наиболее часто используются по следующим показаниям: • РА • Артрит и серозит при СКВ • Артрит при ССД • Артрит при синдроме Шегрена • Артрит и серозит при смешанном заболевании соединительной ткани • Остеоартрит • Анкилозирующий спондилит • Синдром Рейтера • Псориатический артрит • Микрокристаллический артрит • Бурсит, тендинит, теносиновит • Артрит при васкулитах • Ювенильный хронический артрит Кроме того, НПВС часто используют для уменьшения выраженности менструальных спастических реакций; они способствуют более быстрому закрытию ductus arteriosus; НПВС нашли применение при воспалительных офтальмологических заболеваниях, шоке, периодонтите, спортивных травмах и лечении осложнений химиотерапии злокачественных новообразований. Имеются сообщение о антипролиферативном действии аспирина и НПВС на слизистую кишок, что позволило обсуждать потенциальную возможность их применения у больных со злокачественными новообразованиями толстой кишки (M. J. Thun и соавт., 1991; R. F. Logan и соавт., 1993). По данным F. M. Giardello и соавт. (1993), сулиндак подавляет развитие аденоматозного полипоза кишечника. Недавно обнаружена клиническая эффективность индометацина при болезни Альцгеймера (J. Rogers и соавт., 1993). Особенно широко НПВС используются в лечении мигрени (K. M. A. Welch, 1993). Полагают, что они являются средством выбора у больных с умеренными или выраженными мигренозными атаками. Например, в двойном слепом контролируемом исследовании было показано, что напроксен достоверно уменьшает тяжесть и продолжительность головных болей и фотофобии, и что он более эффективен в этом отношении, чем эрготамин. Сходный эффект дает аспирин и другие НПВС. Для достижения более выраженного действия в отношении тошноты и рвоты рекомендуется комбинировать НПВС с метоклопрамидом, который ускоряет абсорбцию препаратов. Для быстрого купирования мигренозных приступов рекомендуется использовать кеторолак, который можно вводить парентерально. Предполагается, что эффективность НПВС при мигрени связана с их способностью, ингибируя синтез ПГ, снижать интенсивность нейрогенного воспаления или, интерферируя с серотонином, уменьшать выраженность сосудистого спазма. Несмотря на сходство химических свойств и основных фармакологических эффектов различных НПВС, у отдельных больных одним заболеванием (например РА) или при разных ревматических заболеваниях наблюдаются существенные колебания в "ответе" на тот или иной препарат (E. C. Huskisson и соавт., 1976). Действительно, на популяционном уровне достоверных различий между аспирином и другими НПВС при РА не выявлено (L. A. Heller и соавт., 1985), однако они становятся очевидными при анализе эффективности различных НПВС у отдельных больных (T. Pincus и соавт., 1989). Это диктует необходимость индивидуального подбора НПВС для каждого больного (Н. В. Бунчук, 1994). Выбор НПВС является, как правило, эмпирическим и во многом основывается на личном опыте врача и прошлом опыте больного. Существует точка зрения о целесообразности в начале лечения использовать наименее токсичные препараты, к которым в первую очередь относятся производные пропионовой кислоты. Необходимо постепенно титровать дозу НПВС до эффективной, но не превышающей максимально допустимую, в течение 1-2 нед. и при отсутствии эффекта попытаться использовать другой или другие препараты. Назначение простых анальгетиков (парацетамола) позволяет уменьшить потребность в НПВС. Рекомендуемые дозы наиболее широко распространенных в клинической практике НПВС представлены в таблице 2.2. Таблица 2.2. Рекомендуемые дозы НПВС при ревматических заболеваниях. Препарат Диапазон доз (мг/день) Кратность приема в течение дня Ацетилсалициловая кислота: 1000-6000 • аспирин • холин салицилат магния 1500-4000 1500-5000 • салсалат 500-1500 • дифлунизал 200-400 • меклофенамат натрия Арилалкановая кислота: 1200-3200 • ибупрофен 1200-3200 • фенопрофен 100-400 • кетопрофен 75-150 • диклофенак 100-300 • флурбипрофен 250-1500 • напроксен Индол/инденуксусная кислота: 50-200 • индометацин 300-400 • сулиндак 600-120 • этодолак Гетероарилуксусная кислота: 800-1600 • толметин 15-150 • кеторолак Эноликовая кислота: 200-800 • фенилбутазон 20-40 • пироксикам Нафтилалканоны: 1000-2000 • набуметон Оксазолпропионовая кислота: 600-1200 • оксапрозин 2-4 2-4 2-4 2 4 3-6 3-4 3-4 2-3 2-3 2 2-4 2 3-4 4-6 4 1-4 1 1-2 1 Особенно ярко различия между НПВС видны при сравнении их клинической эффективности у больных разными ревматическими заболеваниями. Например, при подагре все НПВС более эффективны, чем толметин, а при анкилозирующем спондилоартрите индометацин и другие НПВС более эффективны, чем аспирин. Возможные причины, определяющие различную клиническую эффективность НПВС и спектр токсических реакций у отдельных больных и при разных ревматических заболеваниях, а также практические рекомендации по применению НПВС недавно суммированы в обзорах D. E. Furst (1994) и P. M. Brooks (1993). Важной характеристикой НПВС является продолжительность полужизни в плазме (таблица 2.3.). Таблица 2.3. Средний период полужизни различных НПВС (P. M. Brooks, 1993) Препарат Короткоживущие: • аспирин • диклофенак • этодолак • фенопрофен • флуфенаминовая кислота • флурбипрофен • ибупрофен • индометацин • кетопрофен • пирпрофен • тиапрофеновая кислота • толметин Длительноживущие: • азапропазон • дифлунизал • фенбуфен Время полужизни, ч 0.25 (0.03) 1.1 (0.2) 3.0; 6.5 (0.3)* 2.5 (0.5) 1.4; 9.0 3.8 (1.2) 2.1 (0.3) 4.6 (0.7) 1.8 (0.4) 3.8; 6.8 3.0 (0.2) 1.0 (0.3); 5.8 (1.5)* 15 (4) 13 (2) 11.0 • • • • • • • • набуметон напроксен оксапрозин фенилбутазон пироксикам сулиндак теноксикам салицилаты 26 (5) 14 (2) 58 (10) 68 (25) 57 (22) 14 (8) 60 (11) 2-15** Примечание. В скобках дано стандартное отклонение; одна звездочка — двухфазная элиминация; две звездочки — элиминация имеет зависимый от дозы характер. В зависимости от периода полужизни НПВС разделяются на две основные категории: короткоживущие, имеющие продолжительность полужизни не более 4 ч, и длительноживущие, у которых этот показатель составляет 12 ч и более. Однако необходимо иметь в виду, что кинетические параметры НПВС в синовиальной жидкости и ткани могут существенно отличаться от сывороточных, и в этом случае различия между НПВС по периоду полужизни в синовии становятся менее существенными, чем в кровяном русле. При этом синовиальная концентрация длительноживущих препаратов коррелирует с уровнем в сыворотке, а при приеме короткоживущих препаратов она сначала низкая, но затем существенно нарастает и может превышать сывороточную концентрацию. Это позволяет объяснить длительно сохраняющуюся клиническую эффективность короткоживущих препаратов. Например, имеются данные о том, что при РА прием ибупрофена 2 раза в день столь же эффективен, как и прием 4 раза, несмотря на очень короткий период полужизни ибупрофена в плазме (2.1 ч). Получены данные о различных фармакологических свойствах левовращающих (S) и правовращающих (R) изомеров НПВС. Например, ибупрофен является рацемической смесью лево- и правовращающих изомеров, причем R-èçîìåð в основном и определяет анальгетический потенциал препарата. S-ôîðìà флурбипрофена проявляет сильную анальгетическую активность, но слабо подавляет синтез ПГ, а R-èçîìåð, напротив, обладает более высокой противовоспалительной активностью. Эти данные в будущем могут быть стимулом для создания более мощных и селективных НПВС, однако в настоящее время клиническое значение существования различных энантиомерных форм НПВС неясно. Большее значение имеет белковосвязывающая способность НПВС. Известно, что все НПВС (кроме пироксикама и салицилатов) более чем на 98% связываются с альбумином. Клиническое значение этого свойства НПВС заключается в том, что развитие гипоальбуминемии, печеночной или почечной недостаточности диктует необходимость назначения меньших доз препаратов. В процессе лечения необходимо учитывать суточные колебания выраженности клинических симптомов и воспалительной активности болезни. Например, при РА максимальная интенсивность скованности, болей в суставах и снижение силы сжатия кисти наблюдаются в утренние часы, в то время как при остеоартрите симптомы усиливаются к вечеру. Имеются данные о том, что при РА прием флурбипрофена в ночное время дает более сильный обезболивающий эффект, чем в утреннее, дневное время или днем и вечером (R. Pownal и N. J. Pickvane, 1987). Больным остеоартритом, у которых выраженность болей максимальна вечером и ранним утром, предпочтительнее назначать пролонгированный индометацин перед сном. Примечательно, что такой ритм приема приводил к существенному снижению частоты побочных эффектов. Таким образом, синхронизация назначения НПВС с ритмом клинической активности позволяет повысить эффективность лечения, особенно препаратами с коротким периодом полужизни. Последние следует назначать незадолго до максимального нарастания симптомов. Побочные эффекты НПВС относятся к препаратам, часто вызывающим различные побочные реакции (таблицы 2.4. и 2.5.). Таблица 2.4. Основные побочные эффекты НПВС (по J. G. Hardin и G. L. Longenecker, 1992) Побочные эффекты Частота Комментарий Желудочно-кишечные: Симптомы поражения ++++ Плохо коррелируют с верхних и нижних отделов развитием кровотечения и ЖКТ выявляемым поражением Эрозии и язвы в желудке ++++ Поражение толстой и тонкой +++ Является причиной скрытой кишок потери крови и развития железодефицитной анемии Ототоксичность +++ Салицилаты, дифлунисал; редко при приеме других Гиперчувствительность ++ Кожные +++ Печеночные ++ Неврологические Почечные: Ингибиция ПГ ++ Идиосинкразия + ++ НПВС Астма и крапивница; редко коллапс Зуд, неспецифическая сыпь; могут быть не связаны с НПВС Обычно умеренное бессимптомное повышение уровня печеночных ферментов Обычно индометацин Редко в группах низкого риска; часто у больных с отеками, почечной недостаточностью, гиперкалиемией Интерстициальный нефрит, нефротический синдром; наиболее часто индометацин и фенопрофен Примечание: ++++ >10%; +++ 5-10%; ++ 1-4%; + <1%. Таблица 2.5. Редко встречающиеся побочные эффекты НПВС Побочные эффекты Препараты Острый отек легких Салицилаты (токсические дозы) Гиперчувствительный Напроксен, ибупрофен, сулиндак, пневмонит фенилбутазон и др. Стоматит Любой препарат Сиалоденит Фенилбутазон и др. Лихорадка Ибупрофен Лекарственная волчанка Фенилбутазон, ибупрофен Кардит Фенилбутазон Васкулит Фенилбутазон, индометацин, напроксен Панкреатит Сулиндак и др. Острый проктит Мефенаминовая кислота, аспирин Асептический менингит Ибупрофен, реже другие пропионовые производные (наиболее часто при ДБСТ) Поражение ЖКТ Поскольку ПГ играют важную роль в физиологической регуляции ЖКТ, неудивительно, что желудочнокишечные расстройства являются самым частым побочным эффектом НПВС (D. R. Lichtenstein и соавт., 1995). Прием НПВС ассоциируется с развитием эрозивного поражения желудка, пептических язв, кровотечениями, реже воспалением тонкой и толстой кишок. Частота тяжелого поражения ЖКТ составляет 2 на 10000 больных, принимающих НПВС в течение месяца, а абсолютный риск этих осложнений при РА повышается в 7 раз (M. J. S. Langman, 1989). По данным L. A. G. Rodriguez и H. Jick (1994), относительный риск развития кровотечений из верхних отделов ЖКТ и перфораций на фоне приема современных НПВС составляет в среднем 4.7 (3.8-5.7). Факторы риска поражения ЖКТ у больных, леченных НПВС (J. F. Fries и соавт., 1991; S. E. Gabriel и соавт., 1991; LA. G. Rodriguez и Jick, 1994; M. J. S. Langham и ñîàâò., 1994; D. R. Lichtenstein и ñîàâò., 1996): 1. Определенные: 1. возраст старше 65 лет; 2. наличие патологии ЖКТ в анамнезе; 3. прием высоких доз НПВС или одновременный прием нескольких НПВС (за исключением низких доз аспирина?); 4. сочетанный прием ГК и антикоагулянтов; 5. длительность лечения НПВС (менее трех месяцев) Вероятные: 1. наличие определенного заболевания (РА); 2. женский пол; 3. курение; 4. прием алкоголя; 5. helicobacter pylori. При сравнении отдельных НПВС было установлено, что наибольшей ульцерогенной активностью обладает азапропазон (относительный риск 23.4) и пироксикам (18.0), а наименьшей — ибупрофен, другие производные пропионовой кислоты, включая флурбипрофен, напроксен, а также сулиндак, дифлунисал, этодолак и набуметон (2.9-3.1). Диклофенак, кетопрофен и индометацин занимают промежуточное положение, относительный риск 3.9, 5.4 и 6.3 соответственно (L. A. G. Rodriguez и H. Jick, 1994). По данным M. J. S. Langman и соавт. (1994), относительный риск развития кровотечения у лиц старше 60 лет минимален при приеме ибупрофена и диклофенака (соответственно 2.0 и 4.2). Индометацин (11.3), напроксен (9, 1) и пироксикам (13.7) занимают промежуточное положение, а азапропазон и кетопрофен — наиболее опасные препараты (соответственно 31.5 и 23.7). Для профилактики и лечения язвенного поражения желудка и двенадцатиперстной кишки используется весь арсенал методов противоязвенной терапии. В серии исследований было показано, что блокаторы Н2гистаминовых рецепторов (циметидин, ранитидин) эффективно предотвращают развитие дуоденальных язв, но в меньшей степени влияют на образование язв желудка (M. G. Robinson и соавт., 1989; R. S. Ehsanulian и соавт., 1988), однако прием НПВС ассоциируется с развитием именно язв желудка, С другой стороны, имеются данные о том, что прием ранитидина в дозе 150 мг 2 раза в день в течение 4 нед. ведет к заживлению язвенных дефектов в желудке и двенадцатиперстной кишке, даже на фоне продолжения приема НПВС (G. Tildesley и соавт., 1993). В недавно проведенном двойном слепом контролируемом исследовании было показано, что ранитидин в дозе 300 мг/день более эффективен, чем плацебо, в отношении предотвращения развития пептической язвы желудка (S. Ten Wokde и соавт., 1994). Тем не менее полагают, что наиболее эффективен для профилактики и лечения индуцированного НПВС язвенного поражения желудка и двенадцатиперстной кишки препарат мизопростол — стабильный синтетический аналог ПГЕ1 (У. Дауни, 1992; D. Y. Graham и соавт., 1988; R. P. Walt, 1992; D. R. Miller, 1992). Для лечения язв рекомендуется принимать мизопростол в дозе 200 мг 4 раза в день во время еды и перед сном (Сайтотек, "Searle", США). Прием препарата с пищей уменьшает частоту и тяжесть нарушений стула. Для профилактики гастропатий, вызванных НПВС, следует принимать мизопростол 2-4 раза в день в той же дозе. Предполагается, что мизопростол особенно показан больным, принимающим пролонгированные НПВС в высоких дозах или НПВС в сочетании с пероральными антикоагулянтами или ГК, лицам пожилого возраста и больным, у которых имеется вероятность оперативных вмешательств (G. Pipkin и соавт., 1993). Недавно был разработан комбинированный препарат, содержащий 50 мг диклофенака и 200 мг мизопростола (Артротек, "Searle", США), который оказывает существенно меньшее ульцерогенное действие, чем диклофенак, напроксен и пироксикам (V. Wright, 1993). Предварительные результаты свидетельствуют также о возможности использования ингибитора протонного насоса — омепрозола для заживления язвенных поражений, индуцированных НПВС, даже при продолжении приема этих препаратов (A. Walan и соавт., 1989). НПВС могут индуцировать развитие кишечного воспаления (A. J. Morris и соавт., 1991, 1992). У 60-70% больных, длительно леченных НПВС, развивается бессимптомная энтеропатия, ассоциирующаяся со скрытой кровоточивостью и потерей белка (I. Bjarnason и соавт., 1993). Полагают, что это может приводить к развитию железодефицитной анемии и гипоальбуминении, иногда наблюдаемых у больных, леченных НПВС. Интересно, что сульфасалазин способен подавлять кишечное воспаление и уменьшать потерю крови (глава 9). Редким, но потенциально опасным осложнением является активация предсуществующего латентного воспаления толстой кишки и повреждение тонкой кишки, частота которых нарастает при длительном приеме НПВС (M. C. Allison и соавт., 1992). Ректальное введение НПВС может приводить к развитию проктита (P. Lanthier и соавт., 1987). Описаны больные, у которых НПВС вызвали обострение скрыто протекавших язвенного колита или болезни Крона (T. J. Kauffman и H. L. Taubin, 1987). Очень редким осложнением лечения является коллагенозный колит (R. H. Riddel и соавт., 1992). Поражение почек Существенным нежелательным проявлением антипростагландинового действия НПВС является нарушение функции почек, проявляющееся задержкой жидкости и электролитным дисбалансом. В свою очередь развитие почечной недостаточности и нефротического синдрома связывают с иммунными (идиосинкразия?) эффектами НПВС. Полагают, что ПГ-зависимые почечные эффекты чаще развиваются у больных с предсуществующим поражением почек и гипоальбуминемией. При этом сулиндак и неацетилированные салицилаты реже дают эти побочные эффекты, чем другие НПВС. Что касается интерстициального поражения почек, то наибольшее число описанных в литературе случаев связано с приемом фенопрофена. В недавних эпидемиологических исследованиях было показано, что риск тяжелого поражения почек наиболее высок у больных, длительно получающих ацетоменофен или другие НПВС, в то время как прием аспирина не ассоциируется с нарастанием частоты почечной патологии (T. V. Perneger и соавт., 1994). Практически все НПВС, хотя и крайне редко, могут вызывать развитие гепатита (M. Rabinovitz и D. H. Van Thei, 1993). Наиболее часто это описано на фоне лечения изоксикамом, сулиндаком и пирпрофеном, реже — индометацином и кетопрофеном, и в единичных случаях — при приеме диклофенака и пироксикама (< 1/500000). Наиболее частыми неврологическими побочными эффектами НПВС, развитие которых, как правило, связано с приемом индометацина, являются головные боли. Иногда головные боли сочетаются с другими неврологическими расстройствами, такими, как головокружение, галлюцинация, депрессия, деперсонализация, очень редко — судороги. Перечисленные неврологические симптомы обычно развиваются в течение первых дней приема НПВС и быстро исчезают после отмены препаратов. У лиц пожилого возраста на фоне лечения НПВС иногда наблюдается своеобразный симптомокомплекс, проявляющийся вначале ухудшением памяти, а впоследствии спутанностью сознания, депрессией и сонливостью. Наконец, у некоторых больных СКВ после однократного приема препаратов (наиболее часто ибупрофена, реже толметина и сулиндака) описано быстрое развитие симптомов асептического менингита. Эти проявления исчезают в течение 48 ч после отмены препаратов. Гиперчувствительность Часть больных (1-2%) обладают необычно высокой чувствительностью к индуцированному НПВС подавлению активности ЦОГ. Это приводит к появлению симптомов бронхиальной астмы, крапивницы или более тяжелых проявлений анафилаксии. Наиболее часто развитие гиперчувствительности наблюдается у лиц с клинической триадой, включающей вазомоторный ринит, полипоз носа и астму, и на фоне приема сильных ингибиторов ЦОГ (аспирин, индометацин). Кожные реакции Кожный зуд и небольшая сыпь — хорошо известные побочные эффекты НПВС, однако тяжелые кожные реакции развиваются крайне редко. Фотосенсибилизация чаще наблюдается при приеме пироксикама, а также фенилбутазона, сулиндака и меклофенамата, чем при приеме других НПВС. Имеются отдельные сообщения о развитии узловатой эритемы, гиперчувствительного васкулита, многоформной эритемы (включая синдром Стивенса—Джонсона), эксфолиативного дерматита. Гематологические нарушения Наиболее опасными токсическими реакциями являются гематологические, такие, как агранулоцитоз и апластическая анемия. Фенилбутазон и индометацин фактически являются единственными НПВС, на фоне лечения которыми документировано развитие этих осложнений. Необходимо иметь в виду и антитромбоцитарный эффект НПВС, наиболее выраженный у аспирина. С практической точки зрения эти данные свидетельствуют, во-первых, о необходимости более тщательного гематологического контроля при лечении фенилбутазоном и индометацином, чем при лечении другими НПВС, и во-вторых, о целесообразности использования НПВС короткого действия у больных с желудочным кровотечением в анамнезе. Хотя все НПВС дают сходный спектр побочных эффектов, некоторые из них чаще развиваются при определенных заболеваниях. Например, имеются данные о том, что на фоне лечения напроксеном и диклофенаком изменение показателей печеночных проб чаще наблюдается у больных остеоартритом, чем больных PA, (D. E. Furst и соавт., 1993). Аспирин более гепатотоксичен при ювенильном хроническом артрите и, вероятно, СКВ, чем при других заболеваниях. Асептический менингит описан только при СКВ и близких ДБСТ, он практически никогда не развивается при РА, остеоартрите и серонегативных спондилоартритах. Взаимодействия НПВС с другими препаратами В процессе лечения НПВС необходимо иметь в виду их способность взаимодействовать с другими лекарственными средствами, особенно непрямыми антикоагулянтами, диуретиками, b-блокаторами, ингибиторами ангиотензинпревращающего фермента (АПФ), солями лития и пробенецидом (A. L. Tonkin и L. M. H. Wang, 1988), которые нередко назначают больным ревматическими заболеваниями, особенно в пожилом возрасте (таблица 2.6.). Таблица 2.6. Наиболее важные лекарственные взаимодействия НПВС с другими препаратами (по P. J. Clements, H. E. Paulus, 1993) Лекарственные взаимодействия Эффект Рекомендации Салицилаты/антациды Снижение уровня салицилатов Оценить необходимость антацидов; назначить другой НПВС Антикоагулянты/ фенилбутазон Увеличение протромбинового Назначить ибупрофен, напроксен или времени толметин Антикоагулянты/ НПВС Увеличение риска Использовать низкие дозы ГК? кровотечений (нарушение b-блокаторы/ НПВС Периферические вазодилататоры/ НПВС функции тромбоцитов, повреждение слизистой желудка) Уменьшение гипотензивного эффекта Уменьшение гипотензивного эффекта Уменьшение гипотензивного эффекта Диуретики/ НПВС Уменьшение натрийуретического, диуретического, гипотензивного действия фуросемида; уменьшение гипотензивного, но не мочегонного действия гипотиазида Салицилаты/ГК Снижение уровня салицилатов Метотрексат/салицилаты, Увеличение концентрации МТ фенилбутазон (другие в сыворотке, связанное со НПВС)/пробенецид снижением клиренса Фенитоин/фенилбутазон, Салицилаты Увеличение токсичности фенитоина Пробенецид, Подавление урикозурии сульфинпиразон/салицилаты (низкие дозы) НПВС/пробеницид Подавление экскреции НПВС, увеличение токсичности НПВС/аспирин Снижение концентрации НПВС Дигоксин/аспирин, ибупрофен Увеличение уровня дигоксина Не назначать бутадион, аспирин, индометацин Увеличить дозу гипотензивных препаратов; использовать тиазидовые диуретики Ингибиторы АПФ/ НПВС НПВС/омепразол, Н2-блокаторы, мизопростол Пироксикам/циметидин Снижение секреции желудочного сока Увеличение концентрации пироксикама Использовать другой НПВС При необходимости снизить дозу МТ; использовать другой НПВС Использовать другой НПВС То же Подбирать дозу НПВС Избегать сочетанного приема НПВС и аспирина Мониторировать концентрацию дигоксина, использовать минимальные дозы НПВС Снизить дозу пироксикама Использование НПВС при СКВ Артрит и артральгии относятся к числу частых проявлений СКВ, при умеренной выраженности которых в отсутствие других признаков активности болезни нередко используют НПВС. Однако назначать НПВС при СКВ следует с особой осторожностью из-за возможности развития необычных, тяжелых побочных эффектов, в первую очередь асептического менингита, описанного на фоне лечения ибупрофеном, толметином и сулиндаком. При СКВ НПВС чаще оказывают гепатотоксическое действие (обычно проявляющееся изолированным увеличением уровня трансаминаз), чем при других заболеваниях. Кроме того, эти препараты могут вызывать ослабление клубочковой фильтрации (особенно у больных с предсуществующим поражением почек, застойной сердечной недостаточностью и циррозом печени), снижать эффективность фуросемида и тиазидовых диуретиков, вызывать задержку жидкости, повышение АД, повреждение ЖКТ. Не следует сочетать ГК и салицилаты, поскольку это приводит к снижению уровня ГК и увеличению концентрации салицилатов в сыворотке, а следовательно уменьшает эффективность ГК и увеличивает токсичность салицилатов. Использование аспирина при АФС Агрегация тромбоцитов в ответ на повреждение сосудистой стенки или в связи с воздействием аФЛ на мембрану тромбоцитов является одним из возможных механизмов развития тромбозов при АФС. У больных с акушерской патологией, связанной с АФС, монотерапия аспирином применяется редко, как правило, его назначают в сочетании с преднизолоном или гепарином. Имеются наблюдения об использовании аспирина в дозе от 80 до 1300 мг у больных с поражением ЦНС. На фоне лечения аспирином иногда удается полностью купировать преходящие нарушения зрения, избежать развития повторных инсультов. Описана больная с АФС, проявлявшимся быстропрогрессирующей гемолитической анемией, тромбоцитопенией, тромбозом вен сетчатки, двумя эпизодами церебральных тромбозов и легочной геморрагией, у которой высокие дозы преднизолона, плазмаферез, IVIg и ЦФ не позволяли контролировать течение болезни. Однако назначение аспирина в дозе 100 мг/сутки позволило купировать анемию и тромбоцитопении. Полагают, что эффективность аспирина именно в отношении цитопенических проявлений АФС связана с его способностью подавлять экспонирование отрицательно заряженных фосфолипидных эпитопов на мембранах клеток крови в процессе их агрегации. По мнению Y. Shoenfeld и соавт. (1995), способность аспирина предотвращать акушерскую патологию у больных с АФС связана с тем, что препарат, подавляя синтез ПГ, вызывает гиперпродукцию ЛТ.  свою очередь ЛТВ4 и ЛТС4 могут стимулировать синтез ИЛ-3, необходимого для нормального протекания беременности и развития плода. Использование НПВС в период беременности и лактации НПВС проникают через плаценту и могут вызывать развитие врожденных уродств у экспериментальных животных, однако убедительных данных, указывающих на тератогенность НПВС у человека, не получено. На основании анализа информации, полученной при обследовании более 14 000 матерей и их новорожденных установлено, что прием аспирина в 1 триместре беременности не ассоциируется с увеличением риска врожденных нарушений у новорожденных (A. Schoenfeld и соавт., 1992). У женщин, получавших в период беременности НПВС, отмечено развитие побочных реакций, проявлявшихся в увеличении продолжительности гестации, длительности родов и повышении кровопотери, а у новорожденных иногда наблюдалась задержка роста. Такие нарушения, как церебральные геморрагии, окулоартикулярная дисплазия, преждевременное закрытие дуктус артериолус, неонатальный ацидоз отмечены лишь в единичных клинических наблюдениях, наиболее часто при использовании матерями индометацина. Поскольку НПВС являются слабыми аминокислотами, они не присутствуют в высокой концентрации в молоке; тем не менее, их применение у кормящих матерей нежелательно. При наличии показаний рекомендуется использовать пропионовые производные (ибупрофен, флурбипрофен) или производные фенилуксусной кислоты (диклофенак), которые имеют короткую продолжительность жизни и образуют инертные метаболиты. Список литературы К главе "Нестероидные противовоспалительные препараты" 1. Бунчук Н. В.: Нестероидные противовоспалительные средства в лечении ревматоидного артрита. Клиническая ревматология. 1994; 1; 42-43. 2. Насонова В. А., Сигидин Я. А.: Патогенетическая терапия ревматических заболеваний. М. Москва. 1985. 3. Дауни У: Роль препарата мизопростол в лечении гастропатий, вызываемых нестероидными противовоспалительными препаратами. Ревматология 1992; 24: 40-43. 4. Abramson S. B., Cherksey B., Coode D., et al.: Nonsteroidal antiinflammatory drugs exert different effects on neutrophil function and plasma membrane viscosity. Inflammation. 1990; 14: 11-30. 5. Altman R. D.: Neutrophil activation: an alternative to prostaglandin inhibition as the mechanism of action for NSAIDs. Semin. Arthritis Rheum., 1990; 19 (suppl. 2): 1-5. 6. Bjarnason 1., Hayllar J., Macpherson A. J., Russel A. S.: Side effects of nonsteroidal antiinflammatory drugs on the small and large interstine in humans. Gastroenterol. 1993; 104: 1832-1847. 7. Brooks P. Ì.: Clinical management of rheumatoid arthritis. Lancet. 1993; 341: 286-290. 8. Brooks P. Ì., Day R. 0.: Nonsteroidal antiinflammatory drugs differences and similarities. New Engl. J. Med. 1991; 324: 1716-1725. 9. Brune К.: Prostaglandins and the mode of action of antipiretic analgesic drugs. Amer. J. Med., 1983; 75: 19. 10. Catania A., Arnold J., Macaluso A., et al.: Inhibition of acute Inflammation in the periphery by central action of salicylates. Proc. Natl. Acad. Sci. USA. 1991; 88: 8544-8547. 11. Clements P. J., Paulus Í. E., Nonsteroidal antiinflammatory drugs (NSAIDS). In. Textbook of Rheumatology. Ed. W. N. Kelly, E. D. Harris, S. Ruddy, C. B. Sledge. 1993. B. Saunders Company. Philadelphia, London. Toronto, Montreal, Sydney, Tokyo. V. 1. 700-730. 12. Chang D. M., Baptiste P., Schur P. Í.: The effect of antirheumatic drugs on interleukin I (IL-I) activity and IL-I and IL-I inhibitor production by human monocytes. J. Rheumatol., 1990; 17: 1148-1157. 13. Ehsanulian R. S., Page Ì. C., Tidesley G., Wood J. R.: Prevention of gastroduodenal damage induced by nonsteroidal antiinflammatory drugs: controlled trial of ranitidine. Br. Med. J. 1988; 297: 1017-1021. 14. Flescher E., Fossum D., Gray P. J., et al.: Aspirin-like drugs prime human T cell: modulation of intracellular calcium concentration. J. Immunol., 1991; 146: 2553-2559. 15. Furst D. E. Are there differences among nonsteroidal antiinflammatory drugs. Arthritis. Rheum. 1994; 37: 1-9. 16. Giardello F., M., Hamilton S. R., Krush A. J., et al.: Treatment of colonic and rectal adenomas with sulindac in familial adenoma tous polyposis. N. Engl. J. Med. 1993; 328: 1313-1316. 17. Gilman A. G.: G protein: transduser of receptor generation signals. Ann. Rev. Biochem., 1987; 56: 615-649. 18. Graham D. Y., Agarwal N. A., Roth S. H.: Prevention of NSAID-induced gastric ulcer with misiprostol: multicentre, double-blind, placebo-controlled trial. Lancet 1988; ii: 1277-1280. 19. Hardin J. G., Longenecker G. L. Handbook of drug therapy in rheumatic disease. Pharmacology and clinical aspect. Little, Brown and Company. Boston, Toronto, London. 1992. 20. Huskisson E. C., Woolf D. L., Balme H. W., et al.: Four new antiinflammatory drugs: responses and variations. B. M. J. 1976; i: 1048-1049. 21. Ip M., Lomas A., Shaw J., et al.: Effect of nonsteroidal antiinflammatory drugs on neutrophil chemotaxis: an in vitro and in vivo study. Br. J. Rheumatol., 1990; 29: 363-367. 22. Jeremy J. Y., Mikhalidis D. D.: NSAID efficacy and side-effects: are they wholly prostaglandin-mediated. J. Drug Rev. 1990; 3: 3-4. 23. Kaufman H. J., Taubin H. L.: NSAID activate quescent inflammatory bowel disease. Ann. Int. Med. 1987; 107: 513-516. 24. Kistis E. A., Weissman G., Abramson S. B.: The prostaglandin paradox: additive inhibition of neutrophil function by aspirin-like drugs and the prostaglandin El analog misoprostol. J. Rheumatol. 1991; 18: 14611465. 25. Корр Е., Ghosh S.: Inhibition of NF-kB by sodium salicylate and aspirin. Science. 1994; 265: 956-959. 26. Langham M. J. S., Well J., Wainwright P., et al.: Risk of bleeding peptic ulcer associated with individual nonsteroid antiinflammatory drugs. Lancet. 1994: 343: 1075-1078. 27. Lanthier P., Detry R., debongnie J. C., et al.: Solitary rectal lesions due to suppositories containing acetylsalicylic acid and paracetamol. Gastroenterol. Clin Biol. 1987; II: 250-253. 28. Lichtenstein D. R., Syngal S., Wolfe M.: Nonsteroidal antiinflamatory drugs and the gastrointestinal tract: the double-edged sword. Arthritis Rheum. 1995; 38: 5-18. 29. Logan R. F., Little J., Hawtin P. G., Hardcastle J. D.: Effect of aspirin and nonsteroidal antiinflammatory drugs on colorectal adenomas: case-control study of subjects participating in the Nottingham fecal occult blood screening program. Br. Med. J.: 1993; 307: 285-289. 30. Meade E. A., Smith W. L, DeWitt D. L: Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other nonsteroidal antiinflammatory drugs. J. Biol. Chem. 1993; 268: 6610-6614. 31. Miller R. P.: Misiprostol for the treatment of peptic ulcer and antiinflammatory drug induced gastroduodenal ulceration. New Engl. J. Med.: 1992; 327: 1575-1580. 32. Morris A. J., Madhock R., Sturrock R. D., et al.: Endoscopic diagnosis of small bowel ulceration in patients receiving nonsteroidal antiinflammatory drugs. Lancet 1991; 337: 520. 33. Morris A. J., Wasson L. A., MacKenzie J. F.: Small bowel enteroscopy in undiagnosed gastrointerstinal blood loss. Gut 1992; 33: 887-889. 34. Pipkin G., Mills J. G.: Treatment of nonsteroidal antiinflammatory drug-associated and duodenal damage. Dig. Dis. 1993; II (suppl. ): 1-13. 35. Rabinovitz M., Van Theil D. H.: Hepatotoxicyty of nonsteroidal antiinflammatory drugs. Am. J. Med., 1992; 87: 1696-1704. 36. Riddel R. H., Tanaka M., Mazzoleni G.: Non steroidal antiinflammatory drugs as a possible cause of collagenous colitis: a case-control study. Gut 1992; 33: 683-686. 37. Robinson M. G. Jr, Griffin J. W., Bowers J., et al.: Effect of ranitidine on gastroduodenal damage induced by nonsteroidal antiinflammatory drugs. Dig. Dis. Sci. 1989; 34: 424-428. 38. Rodriguez L. A. G., Jick H.: Risk of upper gastrointestinal bleeding and perforation associated with individual nonsteroid antiinflammatory drugs. Lancet. 1995: 343: 769-772. 39. Rogers J., Kirby L. C., Hempelman S. R., et al.: Clinical trial of indometacin in Aizheimer's disease. Neurology 1993; 43; 1609-1611. 40. Scheiman J. M., Elta G. H.: Gastroduodenal mucosal damage with salsalate versus aspirin: results of experimental models and endoscopic studies in humans. Semin. Arthritis Rheum. 1990; 29: 121-127. 41. Seibert К., Zhang Y., Leahy K., et al.: Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflamation and pain. Proc. Natl. Acad. Sci. USA. 1994; 91: 1203-12017. 42. Shacter E., Lopez R. L., Pati S.: Inhibition of the myeloperoxidase-H202-C1-system of neutrophils by indometacin and other nonsteroidal antiinflammatory drugs. Biochem Pharma col., 1991; 41: 975-984. 43. Shoenfeld Y., Blank M., Fishman P.: Antiphospholipid syndrome: from laboratory bench to the patient's bedside. Lupus 1995; 4 (suppl. 1): S33-S36. 44. The multicenter salsalate/aspirin comparison study group: does the acetil group of aspirin contribute to the antiinflammatory efficacy of salicilic acid in the treatment of rheumatoid arthritis. J. Rheumatol. 1989; 16: 321 327. 45. Ten Wolde S., Dijkmans B. A. C., Janssen M., et al.: A double-blind placebo-controlled study of ranitidine for the prevention of reccurent peptic ulcer disease in rheumatoid arthritis patients on nonsteroidal antiinflammatory drugs. Arthritis Rheum. 1994; 37: S258. 46. Thun M. J., Namsoodiri M. M., Heath C. W.: Aspirin use and reduced risk of fatal colon cancer. N Engl. J. Med. 1991; 325: 1593-1596. 47. Tildesley G., Ehsanullah R. S. B., Wood J. R.: Ranitidine in the treatment of gastric and duodenal ulcers associated with nonsteroidal antiinflammatory drugs. Brit. J. Rheumatol. 1993; 32: 474-478. 48. Vane J.: Inhibition of prostaglandin synthesis as a mechanism of action of aspirin-like drugs. Nature. 1971; 231: 232-234. 49. Walan A., Bader J. P., Classen M., et al.: Effects of omeprasol and ranitidine on ulcer healing and relapse rates in patients with benigh gastric ulcer. N. Engl. J. Med. 1989; 320: 69-75. 50. Wallace J. L., Granger D. N.: Pathogenesis of NSAID gastropathy: are neutrophils the culprits. TIPS 1992; 13: 129-130. 51. Walt D. R.: Treatment of nonsteroidal antiinflammatory drug-induced gastropathy. Clin. Pharm. 1992; II: 690704. 52. Welch К. M. A.: Drug therapy of migrane. New Engl. J. Med. 1993; 329: 1476-1483. 53. Wright V.: Arthrotec: a review of a new concept in NSAID therapy. J. Orthopedic Rheumatol. 1993; 6: 129133. 54. Wu К. К., Sanduja R., Tsai A. L., et al.: Aspirin inhibits interleukin I-induced prostaglandin H synthase expression in cultured endothelial cells. Proc. Natl. Acad. Sci. USA. 1991; 88: 2384-2387. ГЛАВА 3 ГЛЮКОКОРТИКОИДЫ История применения ГК в медицине неразрывно связана с развитием ревматологии. Хорошо известно, что именно ревматологи (Phillip Hench и сотрудники из клиники Мейо) в 1949 г. впервые в мировой практике применили ГК для лечения РА и достигли поразительного клинического эффекта. Это исследование ознаменовало начало новой эры в лечении воспалительных заболеваний человека. До сих пор, несмотря на большие достижения в области фармакотерапии, ГК остаются наиболее универсальными противовоспалительными агентами, обладающими, кроме того, и выраженной иммуномодулирующей активностью (L. Axelrod, 1993). Сфера применения ГК при заболеваниях человека чрезвычайно широка и включает трансплантацию, аллергические и гематологические болезни, патологию почек, печени, кишечника, болезни глаз, кожи и др. Каждый год в США ГК используют более 5 миллионов человек. При этом наиболее широкое применение ГК нашли при лечении ревматических болезней (R. P. Kimberley, 1992). Заболевания, для лечения которых используются ГК Первичная и вторичная надпочечниковая недостаточность Ревматические заболевания (см.табл. 3.3) Заболевания почек: • гломерулонефрит • нефротический синдром Аллергические заболевания: • ангионевротический отек • крапивница • контактный и атопический дерматит • аллергический ринит • многоформная эритема Заболевания легких: • бронхиальная астма • фулминантный или диссеминированный туберкулез • аспирационная пневмония • респираторный дистресс-синдром • саркоидоз Дерматологические заболевания: • пузырчатка • буллезный герпетиформный дерматит • псориаз Гастроэнтерологические заболевания: • язвенный колит • болезнь Крона • глютеновая энтеропатия • хронический активный гепатит • алкогольный гепатит Злокачественные новообразования: • рак молочной железы • лейкозы • лимфомы • рвота, индуцированиня химиотерапией Другие заболевания: • септический шок • гиперкальциемия • пересадка органов • идиопатическая тромбоцитопеническая пурпура • гемолитическая анемия • рассеянный склероз • отек мозга В ревматологии системное применение ГК является основным (или одним из основных) подходом к лечению таких заболеваний, как гигантоклеточный артериит, ревматическая полимиалгия, ПМ/ДМ, СКВ, рецидивирующий полихондрит, смешанное заболевание соединительной ткани, системные васкулиты, эозинофильный фасциит, синдром эозинофилии/миалгии, позволяющим существенно улучшить прогноз и качество жизни больных. ГК широко используют в комплексном лечении ССД, РА и даже у больных с тяжелыми формами серонегативных спондилоартропатий и подагры. Локальное введение ГК — важный метод лечения артритов, патологии мягких тканей и связочного аппарата (L. Axelrod, 1993). Общая характеристика ГК ГК являются производными прегнана с кетоновыми группами у 3-го и 20-го и гидроксильной группой у 21-го атомов углерода и ненасыщенной связью между 4-м и 5-м атомами, синтезируются из холестерина. Термин "глюкокортикоид" подчеркивает способность этих агентов стимулировать отложение гликогена в печени и глюконеогенез. Основным ГК, образующимся у человека, является гидрокортизон (кортизол). Глюкокортикоидная активность зависит от присутствия гидроксильной группы в положении 11 стероидной молекулы и усиливается при образовании двойной связи между 1-м и 2-м атомами углерода (преднизон, преднизолон), присоединением а-метильной группы в 6-м положении (6-метилпреднизолон) или введении фтора в 9-е положение и функциональной группы в 16-е положение (триамсинолон, дексаметазон, бетаметазон). Продукция кортизола клетками коры надпочечников непосредственно регулируется АКТГ передней доли гипофиза и опосредованно — кортикотропным гормоном (кортиколиберин) гипоталамуса, который усиливает секрецию АКТГ. Регуляция синтеза ГК происходит по принципу обратной негативной (feedback) ингибиции: увеличение уровня кортизола или введение синтетических ГК вызывает уменьшение секреции АКТГ. Этот процесс носит быстрый и пролонгированный характер и приводит к ингибиции синтеза как АКТГ, так и кортиколиберина. Однако у здоровых людей стрессорные стимулы преодолевают влияние обратной ингибиции. Важными факторами, стимулирующими высвобождение АКТГ и ГК, являются провоспалительные цитокины (ИЛ-1 и ИЛ-6), в свою очередь ГК подавляют синтез этих цитокинов (E. M. Sternberg и соавт., 1992). Таким образом, опосредованное ИЛ-1 увеличение секреции ГК — компонент нормального ответа организма на стрессорные (в том числе антигенные) стимулы и этот процесс контролируется за счет обратной ингибиции ГК. Фармакодинамика Системные эффекты ГК зависят от трех основных факторов: 1) количества активного препарата, поступающего в системную циркуляцию; 2) взаимодействия ГК с соответствующими рецепторами; 3) продолжительности связывания ГК с рецепторами. Биодоступность пероральных ГК зависит от уровня их растворимости в ЖКТ и абсорбции в системную циркуляцию. Биодоступность ГК, для активации которых необходима биотрансформация в печени (например, преднизона в преднизолон), определяется уровнем активного компонента, присутствующего в циркуляции. При пероральном приеме преднизолона его биодоступность колеблется от 50 до 100%, максимальная концентрация в сыворотке достигается через 1.3-3.0 ч. Прием пищи не влияет на биодоступность ГК, за исключением препаратов со специальным кишечным покрытием, биодоступность которых существенно снижается. Системная абсорбция ГК наблюдается и при их локальном применении, она колеблется от 30 до 90%. Кожная абсорбция ГК увеличена у детей, усиливается при аппликации после мытья, на пораженную кожу или на участки тела с тонкой кожей. ГК — липофильные вещества, которые распределяются по всему организму. Примерный объем распределения преднизолона, однако, составляет 0.35-0.7 л/кг. Это связано с очень высокой способностью ГК связываться с белками сыворотки. К стероидсвязывающим белкам относятся альбумин и транскортин. При низкой концентрации ГК 80-90% связаны с белком. Но при использовании высоких доз ГК их белковосвязывающая способность снижается до 60-70% за счет насыщения транскортинсвязывающих участков и большее количество свободного препарата диффундирует в периферические ткани. Это приводит к более высокому объему распределения ГК и увеличивает риск развития побочных эффектов. Сходные процессы могут наблюдаться у больных с гипоальбуминемией, при которой рекомендуется использовать меньшие дозы препарата. Метаболизм ГК происходит в печени путем гидроксилирования и конъюгации. Менее 15% ГК выводится в неизмененном виде с мочой. При увеличении объема распределения и концентрации препарата происходит увеличение его клиренса. Однако нарушение общего клиренса заметно только при использовании высоких концентраций преднизолона (около 70 мг/день). Факторы, приводящие к снижению клиренса ГК, включают печеночную и почечную недостаточность, пожилой возраст больных (более 65 лет), сочетанное использование некоторых препаратов (кетоконазол, оральные контрацептивы и др). Увеличение клиренса наблюдается у лиц, длительно получающих ГК, страдающих гипертиреозом, а также при использовании некоторых лекарственных препаратов (фенитоин, рифампин и барбитураты и др.) ГК в основном различаются по продолжительности биологических эффектов и по соотношению между глюкокортикоидной и минералокортикоидной активностью (таблица 3.1.). Используемые в клинике ГК условно подразделяются на препараты короткого, среднего и длительного действия. Это деление основывается на продолжительности супрессии АКТГ после однократного приема препарата в дозе, эквивалентной 50 мг преднизона. Продолжительность полужизни кортизола в плазме составляет 80-115 мин., а для других ГК этот показатель составляет 0,5 ч для кортизола, 3.4-3.8 ч для преднизона, 2.2-3.5 ч для преднизолона, 1.3-3.1 ч для метилпреднизолона, 1.8-4.7 ч для дексаметазона. Различия в длительности полужизни ГК зависят от фармакокинетики препаратов, которая в свою очередь тесно связана с используемыми дозами ГК. Изменение кинетики ГК в зависимости от дозы является следствием нелинейного связывания ГК с белками плазмы; при увеличении дозы возрастает уровень не связанных с белком ГК. Связь между длительностью полужизни и биологической активностью также не является абсолютной. Например, преднизолон и дексаметазон имеют одинаковую продолжительность полужизни, но дексаметазон обладает более выраженной глюкокортикоидной активностью. На основании данных о длительности супрессии АКТГ ГК подразделяются на 3 основные группы. В первую группу входят короткоживущие ГК (кортизон, гидрокортизон, преднизон, преднизолон, метилпреднизолон), после приема которых синтез АКТГ восстанавливается через 24-36 ч. Во вторую группу включены ГК со средним по длительности периодом полужизни (триамсинолон и параметазон), которые подавляют АКТГ на 48 ч. Третью группу составляют длительноживущие ГК (дексаметазон и бетаметазон), подавляющие АКТГ более чем на 48 ч. Очевидно, что продолжительность супрессии синтеза АКТГ существенно превосходит время полужизни препаратов в плазме. Следовательно, длительность действия ГК зависит не только от времени их присутствия в циркуляции. Полагают, что ГК могут находиться внутри клетки в связи с ГК-рецепторами в течение определенного времени после их исчезновения из циркуляторного русла. Кроме того, воздействие ГК на клетки ведет к серии событий, выраженность и последствия которых прямо не зависят от биологического действия самих ГК. ГК хорошо всасываются в ЖКТ. Как уже отмечалось, в норме кортизол и его синтетические аналоги в плазме быстро связываются с кортикостероидсвязывающим белком (транскортином), который относится к фракции а-глобулинов и обеспечивает доставку ГК к клеткам. При этом связанные с транскортином ГК не обладают биологической активностью. Часть ГК (10-15%) связывается с альбумином. Таким образом, только небольшая часть ГК остается в свободной форме и обеспечивает физиологическую и фармакологическую активность ГК. У здоровых людей связывающая способность транскортина подвержена суточным колебаниям. У лиц, длительно леченных ГК, связывание ГК с транскортином снижено, а нормальные суточные колебания кортикостероидсвязывающей активности нивелируются. Следовательно, длительное лечение ГК оказывает влияние не только на эндогенную секрецию стероидов, но и на транспорт ГК в циркуляции. Существует несколько патологических состояний, которые потенциально могут негативно влиять на фармакологические свойства ГК. Так, у больных с гипоальбуминемией наблюдается увеличение частоты побочных эффектов на фоне терапии ГК, а у больных с циррозом печени отмечено нарушение конверсии преднизона в преднизолон. Развитие гипертиреоза приводит к снижению биодоступности ГК, что обусловлено нарушением абсорбции и усилением печеночного клиренса преднизолона. В то же время беременность не оказывает существенного влияния на эффективность терапии ГК и не приводит к увеличению частоты побочных эффектов. Хотя ГК проходят через плаценту, супрессии гипоталамо-гипофизарной оси и развития синдрома Кушинга у плода не наблюдается. Терапия ГК не приводит к увеличению частоты врожденных дефектов, хотя и может вызывать некоторое снижение массы плода. Предполагается, что вследствие очень низкой концентрации ГК в молоке у кормящих матерей прием этих препаратов матерями не дает серьезных нежелательных эффектов у новорожденных. Метаболизм ГК ускоряется на фоне лечения такими препаратами, как фенитоин, карбамазепин, барбитураты, рифампин, которые индуцируют активность печеночных микросомальных ферментов. Лечение этими препаратами может привести к необходимости увеличения дозы ГК, и их по возможности следует избегать. С другой стороны, кетоконазол увеличивает биодоступность больших доз преднизолона за счет подавления активности печеночных микросомальных ферментов. Кроме того, биодоступность ГК снижается на фоне лечения антацидными препаратами. Прием ГК приводит к снижению уровня салицилатов, увеличению потребности в гипогликемических и гипотензивных средствах. Для лечения ревматических заболеваний используются главным образом ГК короткого действия — преднизолон и метилпреднизолон. Длительный прием триамсинолона (полькартолон, кенакорт) нежелателен из-за более частого развития мышечной атрофии, похудания, слабости, поражения ЖКТ, а дексаметазона (дексазон) — из-за выраженного подавления функции коры надпочечников и значительной задержки жидкости (таблица 3.1.). Таблица 3.1. Характеристика глюкокортикоидных препаратов Кортизолон Преднизо- Триамсинолон лон Метилпредни- Дексаметазон золон Эквивалентная доза, мг Время полужизни, мин. ГК-активность Минералокортикоидная активность Подавление гипофиза Увеличение массы тела Мышечная атрофия Подавление функции нейтрофилов Гипертензия Экхимозы Угри, гипертрихоз Поражение ЖКТ Синдром Кушинга Психические нарушения 20 80-120 0.8 1 1 ++++ 0 ++++ ++++ +++ ++++ ++++ ++++ ++++ 5 200-210 4 0.8 4 ++ 0 — ++ ++ ++ ++ +++ +++ 4 180-240 5 0.5 5 ++++ ++++ — + ++ + + + +++ 4 200-210 5 0.5 5 + 0 + + + 0 + + + 0.75 150-270 25 0 40-50 +++ + ++ +++ ++++ +++ ++++ ++++ +++ Механизм действия ГК проявляют свою биологическую активность за счет связывания с цитоплазматическими глюкокортикоидными рецепторами, расположенными внутри клеток-мишеней, регулирующих транскрипцию нескольких генов. Полагают, что в каждой чувствительной к ГК клетке содержится от 10 до 100 стероидотвечающих генов. Гены, кодирующие синтез ГК-рецепторов, относятся к суперсемейству, в которое входят гены цитозольных рецепторов других стероидных гормонов, таких, как прогестерон, эстрогены, а также гормонов щитовидной железы, ретиноевой кислоты и витамина D (R. L. Miesfeld, 1990; P. S. Fuller, 1991). ГК-рецепторы экспрессируются почти во всех клетках, однако их плотность в разных клетках не одинакова. С помощью клонированной кДНК установлена первичная структура гена ГК-рецептора, состоящего из 800 аминокислотных остатков. Имеется один класс ГКрецепторов, не различающихся по структуре и аффинности к ГК в различных тканях. Однако минералокортикоидный рецептор также связывается с ГК с высокой аффинностью. С-терминальный участок неактивного ГК-рецептора связан с крупным белковым комплексом (примерно 300 kD), включающим две субъединицы стрессорного белка Hsp90. Кроме того, в состав этого комплекса могут входить и другие белки, такие, как иммунофилин с мол. массой 59 kD и различные ингибиторные белки. Hsp90 обеспечивает правильную сборку ГК-рецептора, способствуя приобретению конформации, оптимальной для связывания с ГК, и действуя, как чаперон для предотвращения локализации не связанного с ГК рецептора в ядре. После связывания ГК с рецептором происходит диссоциация Hsp90, позволяющая активированному ГК-рецептору очень быстро локализоваться в ядре и связаться с ДНК. Экспрессия ГК-рецепторов регулируется различными факторами на транскрипционном, трансляционном и посттрансляционном уровнях (M. Karin и соавт., 1993). Длительная стероидная терапия вызывает подавление экспрессии ГК-рецепторов в циркулирующих моноцитах и лейкоцитах. Внутри клетки ГК-рецепторы образуют димер, который связывается с участками ДНК, получившими название глюкокортикоидотвечающих элементов (glucocorticoid response elements-GRE), расположенными в промоторном участке стероидотвечающего гена. Уровень транскрипции гена зависит от того, происходит ли индукция или репрессия гена. ГК являются естественными регуляторами транскрипции генов нескольких цитокинов и других молекул, играющих важную роль в развитии воспаления (таблица 3.2.). Таблица 3.2. Влияние ГК на экспрессию генов некоторых молекул, принимающих участие в развитии воспаления (R. P. Schleimer, 1993) Влияние на экспрессию генов подавление усиление Цитокины: ИЛ -1, ИЛ-6, ИЛ-8, ФНО-a, ÈÔ-y, ГМ-КСФ Ингибитор активатора плазминогена Эластаза Вазокортин, липокортин Активатор плазминогена Нейтральные эндопептидазы Коллагеназа Рецепторы для гормонов Циклооксигеназа Синтетаза окиси азота Недавно было показано, что ГК-рецепторы взаимодействуют с различными факторами транскрипции, что может иметь важное значение для реализации их противовоспалительного эффекта (P. J. Barnes и I. Adcoch, 1993). Например, ГК являются мощными ингибиторами транскрипции гена коллагеназы, индуцированной ФНО-а и форболовым эфиром, которая регулируется фактором транскрипции — активаторным белком-1 (АБ-1). Последний является гетеродимером Fos и Jun белков, являющихся продуктами протоонкогенов c-fos и c-jun. АБ-1 образует белковый комплекс с активированными ГК-рецепторами в ядре, вызывая репрессию связывания ДНК. Полагают, что ГК могут подавлять эффекты тех цитокинов, действие которых обусловлено активацией АБ-1. Например, ФНО-а и форболовый эфир увеличивают связывание АБ-1 с ДНК, которая ингибируется ГК. Этот эффект опосредуется взаимодействием с GRE, что вызывает репрессию соответствующих цитокиновых генов. Установлено, что гены цитокинов особенно чувствительны к ингибиторному действию ГК. Например, ген ИЛ-6 имеет не менее 4 отрицательных GRE, расположенных очень близко к промоторному участку, и 1 GRE, локализующийся около участка, с которого начинается транскрипция. Кроме того, ГК усиливают разрушение иРНК таких цитокинов, как ИЛ-1, ИЛ-3 и ГМ-КСФ. ГК не только блокируют синтез цитокинов, но и могут отменять их эффекты за счет нескольких механизмов. Один из них связан с подавлением синтеза некоторых цитокиновых рецепторов, в частности ИЛ-2Р. Некоторые цитокины оказывают свое действие за счет активации факторов транскрипции, таких, как АБ-1 и NFkB, которые активируют или репрессируют мишеневые гены, в то время как ГК оказывают противоложное влияние на ГК-рецепторы. Так, ФНО-а активируют АБ-1 и NFkB, и эта активация подавляется ГК. Стимуляция Т-лимфоцитов опосредуется активацией АБ-1, что приводит к индукции нескольких генов, кодирующих ИЛ-2, ИЛ-2Р, и пролиферации клеток. Эта активация также подавляется ГК. Кроме того, ГК могут ингибировать экспрессию генов цитокинов опосредованно. Например, ЦсА (глава 10) подавляет транскрипцию гена ИЛ-2, блокируя связывание специфического фактора транскрипции, так называемого ядерного фактора активации Т-клеток (NF-AT), который реагирует с распознающей последовательностью ИЛ-2 промотора. Поскольку NF-AT образует комплекс с АБ-1, предполагается, что ГК, блокируя АБ-1, могут интерферировать с NF-AT и таким образом подавлять транскрипцию гена ИЛ-2. Описаны следующие механизмы, обеспечивающие противовоспалительный, антидеструктивный и иммунный эффекты терапии ГК: 1. Предотвращение миграции лейкоцитов в зону воспаления за счет подавления экспрессии молекул адгезии (ELAM-1 и IСАМ-1) ЭК и транскрипции генов провоспалительных цитокинов (ИЛ-1, ФНО-а, ИЛ-8). 2. Индукция синтеза липокортина — ингибитора активности фосфолипазы А2. 3. Подавление активности ЦОГ-2. 4. Негативная регуляция экспрессии генов металлопротеиназ (коллагеназа, стромелизин), которая может быть связана как с ингибицией синтеза ИЛ-1 и ФНО-а, усиливающих экспрессию генов металлопротеиназ, так и с подавлением активности фактора транскрипции (АБ-1), участвующего в регуляции экспрессии генов металлопротеиназ. ГК блокируют образование иРНК и посттрансляционную экспрессию фосфолипазы À2, играющей ключевую роль в активации простагландинового каскада. Один из механизмов антипростагландиновой активности ГК связан с регуляцией синтеза липокортина (E. Solito и соавт., 1991; N. J. Goulding и соавт., 1990). Напомним, что липокортин-1 (также называемый аннексином-1), белок с мол. массой 37 kD, является членом суперсемейства, включающего по крайней мере 10 тесно связанных белков, и обнаруживается во многих клетках, особенно лейкоцитарной и эпителиальной природы. Липокортин предотвращает мобилизацию арахидоновой кислоты из мембранных фосфолипидов путем подавления гидролиза этих фосфолипидов фосфолипипазой À2 (G. Cirino и соавт., 1990). Это в свою очередь ингибирует синтез эйкозаноидов: ПГ, простациклинов, тромбоксана и ЛТ. Интересно, что один из представителей семейства липокортинов имеет сходство с белками цитоскелета, так называемыми кальпактинами, которые, образуя комплекс с фосфолипидами, отменяют их способность выполнять роль субстрата для фосфолипазы (F. F. Davidson и соавт., 1987). Недавно было обнаружено, что липокортин-1 подавляет индуцированный ИЛ-1 хемотаксис нейтрофилов, образование супероксидных радикалов, ПГЕ2 и ЛТ (M. Perretti и соавт., 1993), то есть in vitro проявляет противовоспалительные эффекты, аналогичные тем, которые характерны для ГК (M. Perretti и соавт., 1987; R. J. Flower, 1990). Однако более универсальный механизм противовоспалительной и антидеструктивной активности ГК связан с прямым подавлением транскрипции генов ферментов, участвующих в образовании липидных медиаторов. Показано, что ГК ингибируют транскрипцию цитозольной формы фосфолипазы À2, индуцированной цитокинами, подавляют экспрессию генов цитокининдуцированной ЦОГ-2 в моноцитах (J. L. Masferrer и соавт., 1994) и синтез самой ЦОГ-2 (J-Y. Fu и соавт., 1990). Кроме того, ГК оказывают влияние на метаболизм воспалительных медиаторов. Например, брадикинин расщепляется несколькими ферментами, включая АПФ и нейтральную эндопептидазу, которые индуцируются ГК. Нейтральная эндопептидаза играет роль в расщеплении тахикинина, которая высвобождается из чувствительных нервных окончаний. Очевидно, что увеличение экспрессии нейтральной эндопептидазы может иметь значение в подавлении нейрогенного воспаления. Поскольку ЛТВ4 и ФАТ индуцируют экспрессию c-fos, c-jun и активируют связывание АБ-1 в воспалительных клетках, эффекты этих медиаторов могут быть подавлены ГК. Это относится и к окиси азота, образование которой индуцируется несколькими цитокинами, активирующими синтетазу окиси азота. ГК оказывают опосредованное (за счет подавления синтеза цитокинов) и прямое ингибирующее (на уровне транскрипции генов) влияние на экспрессию молекул адгезии, таких, как IÑÀÌ-1 и Е-селектин (B. N. Cronstein и соавт., 1992). Общими эффектами ГК-терапии являются подавление активности нейтрофилов и моноцитов, способность вызывать лимфопению и депрессия клеточных иммунологических реакций. (D. T. Boumpas и соавт., 1991; C. Christiansen и S. M. Krane, 1993). При этом В-лимфоциты более устойчивы к цитопеническому воздействию ГК, чем Т-лимфоциты, среди которых СD4+Т-лимфоциты (Т-хелперы) чувствительны к ГК в большей степени, чем СD8+Т-лимфоциты (Т-супрессоры). ГК-индуцированная лимфопения обычно носит транзиторный характер и в большей степени связана с перераспределением циркулирующих лимфоцитов в костный мозг, чем с лизисом клеток. Однако имеются данные о том, что ГК могут существенно усиливать лимфолитическое действие цитотоксических препаратов. Это объясняет причины депрессии клеточного иммунитета на фоне лечения ГК. Кроме того, в недавних исследованиях были расшифрованы дополнительные механизмы ГК-индуцированной иммуносупрессии, связанные с усилением процессов апоптоза (программированная клеточная гибель) Т- и В-лимфоцитов (B. A. Garry и соавт., 1993; M. A. Nieto и соавт., 1992), подавлением Т-клеточной активации за счет прерывания проведения активационного сигнала, опосредованного ИЛ-2Р (F. Paliogianni и соавт., 1993) и В-клеточной дифференцировки (D. Emilie и соавт., 1988). В то же время у больных с общим вариабельным иммунодефицитом ГК обладают способностью подавлять супрессорную активность лимфоцитов и тем самым вызывать восстановление гуморального иммунитета. Другая важная сторона активности ГК — модуляция экспрессии и функциональной активности Fc- и С3-рецепторов мононуклеарных фагоцитирующих клеток ретикулоэндотелиальной системы (P. G. Comber и соавт., 1989; L. Y. Pan и соавт., 1990). Примечательно, что в присутствии ИФ-у, ГК не подавляют, а усиливают экспрессию и функциональную активность Fc-ðåöåïòîðîâ in vitro и in vivo (K. C. Petroni и соавт., 1988). Имеются данные об увеличении функциональной активности Fc-ðåöåïòîðîâ у больных волчаночным нефритом на фоне пульс-терапии метилпреднизолоном (J. E. Salmon и соавт., 1989). Механизмы резистентности к ГК Резистентность к ГК наиболее часто встречается при бронхиальной астме, реже — при Других воспалительных заболеваниях. Этот тип резистентности по своей природе отличается от редкой семейной формы резистентности к ГК, которая характеризуется увеличением концентрации кортизола и АКТГ и обусловлена дефектом связывания ГК с рецептором. Развитие резистентности к ГК при воспалительных заболеваниях невозможно объяснить только фармакокинетическими особенностями ГК, например нарушением их абсорбции или быстрой элиминацией. У стероидрезистентных больных наблюдается ослабление ответа моноцитов и Тлимфоцитов на ГК, что свидетельствует о клеточной природе этого дефекта. У некоторых больных ревматическими заболеваниями с резистентностью к ГК наблюдается синтез антител к липокортину-1 (N. J. Goulding и соавт., 1990), однако эти антитела отсутствуют у стероидрезистентных больных с бронхиальной астмой. Кроме того, при РА наблюдается ослабление индуцированной ГК продукции липокортина-1 (E. F. Morand и соавт., 1994). Имеются данные о снижении экспрессии ГК-рецепторов на лимфоцитах больных PA (R. Schalaghecke и соавт., 1992), однако это не коррелирует с развитием резистентности к ГК (R. Schlaghecke и соавт., 1994). У резистентных к стероидам больных описано ослабление связывания ГК рецепторов с GRE, а также нарушение способности АБ-1 ингибировать активацию GRE. Предполагают, что это частично объясняет причины селективной резистентности к противовоспалительному действию ГК при сохранности метаболической и гомеостатических эффектов препаратов. Имеются данные о связи стероидрезистентности с дефектом в t1 домене ГК-рецепторов, который принимает участие во взаимодействии с фактором транскрипции. Клиническое применение Несмотря на то, что ГК используются при ревматических заболеваниях очень давно, по-прежнему остается немало вопросов, касающихся наиболее адекватных доз и лекарственных форм препаратов, путей введения, длительности терапии, путей предотвращения побочных эффектов. При ревматических заболеваниях широко применяются все основные варианты ГК-терапии: 1. Локальное введение ГК (внутрисуставное и др.). 2. Местное применение ГК (мази, капли, аэрозоль). 3. Системное применение ГК: • ежедневный прием ГК в низких дозах, • ежедневный прием ГК в высоких дозах, • альтернирующий режим приема ГК, • пульс-терапия ГК, • "мини-пульс" терапия ГК. • сочетанное применение ГК с другими противоревматическими препаратами (в первую очередь цитотоксиками). Основные показания к системному применению ГК и ориентировочные дозы препаратов суммированы в таблице 3.3. Таблица 3.3. Основные показания к системному назначению ГК при ревматических заболеваниях. Заболевания Показания Обычная начальная доза преднизолона (мг/день) РА Высокая воспалительная активность, не контролирующаяся НПВС; < 15 некоторые формы РА в пожилом возрасте; компонент "bridge"-тepaпии Некротизирующий васкулит 60 Плеврит/перикардит 15-60 СКВ/смешанное Артрит или умеренные системные проявления <15 заболевание соединительной ткани Нефрит 60 Люпус-ЦНС 60 Цитопении 30-60 Плеврит/перикардит 15-60 Миокардит 60 Системный васкулит 60 ПМ/ДМ 60 Синдром Шегрена Системный васкулит 15-60 Системные Гигантоклеточный артериит 60 васкулиты Узелковый периартериит 60 Гранулематоз Вегенера 60 Кожный гиперчувствительный васкулит 15-30 Ревматическая Достоверный диагноз или для подтверждения диагноза 15 полимиалгия ССД Миозит, плеврит, перикардит, артрит 30-60 Острый При наличии противопоказаний к НПВС и колхицину 30 микрокристалличес кий артрит Рефлекторная ? 60 симпатическая дистрофия Побочные эффекты 15-60 базисной терапии — соли золота, дпеницилламии и дp. В то же время существует целый ряд ревматических заболеваний или синдромов, при которых системное применение ГК нецелесообразно из-за низкой эффективности или возможности развития тяжелых осложнений. К ним относятся остеоартроз, дегенеративные поражения позвоночника, фибромиалгия, остеонекроз, спондилоартропатии, инфекционные артриты, а также феномен Рейно и склеродермический фиброз. При назначении ГК при любом ревматическом заболевании необходимо иметь в виду существование ряда общих принципов фармакотерапии этими препаратами, знание которых позволяет повысить эффективность и безопасность лечения (T. W. Behrens и J. S. Goodwin, 1989) Лечение ГК необходимо проводить только по строгим показаниям. При этом не только врач, но и больной должен быть детально информирован о всех достоинствах и недостатках ГК-терапии. В процессе лечения врач должен стремиться использовать ГК короткого действия, в оптимальной дозе и по возможности не назначать эти препараты на срок, более длительный, чем это необходимо для контролирования активности болезни. Однако при наличии явных показаний, ГК следует назначать как можно раньше, не пытаясь использовать вначале более "мягкие" методы лечения. В случае необходимости длительного применения ГК следует стремиться как можно быстрее перейти на однократный прием всей дозы в утренние часы, а затем на альтернирующий режим ГК-терапии. Подбор начальной дозы ГК, длительности терапии и темпов снижения дозы следует проводить не эмпирически, а ориентируясь на стандартизованные клинико-лабораторные параметры активности заболевания, В процессе лечения необходимо помнить о том, что супрессия оси гипоталамус—гипофиз—надпочечник сохраняется у больных, получавших ГК даже в небольшой дозе (10 мг/сут в течение 3 недель и более), длительное время (до 1 года) после отмены препарата. При развитии тяжелых интеркуррентных заболеваний или необходимости хирургических вмешательств, эти больные должны получать заместительную терапию ГК. Побочные эффекты Лечение ГК обязательно приводит к развитию тех или иных нежелательных реакций. По неясным причинам у одних больных они развиваются быстрее, чем у других. В целом частота побочных эффектов нарастает при увеличении дозы и длительности приема ГК. Иногда выраженность побочных эффектов зависит от состояния органов, на функцию которых оказывают влияние ГК. Например, больные с постменопаузальным остеопорозом более чувствительны к развитию ГК-индуцированного остеопороза. Частота и выраженность побочных реакций увеличиваются при гипоальбуминемии. Побочные эффекты ГК-терапии непосредственно связаны со спектром фармакологической активности этих препаратов и включают в первую очередь разнообразные клинические проявления, характерные для синдрома Кушинга, супрессии оси гипоталамус—гипофиз—надпочечник, подавления нормальных воспалительных и иммунологических реакций и отрицательного азотистого баланса. При этом ГК-индуцированный ятрогенный синдром Кушинга по спектру клинических проявлений отличается от спонтанного синдрома Кушинга (L. Axelrode, 1993). В частности, при спонтанном синдроме Кушинга практически не встречаются такие характерные клинические проявления ятрогенного синдрома, как доброкачественное повышение внутричерепного давления, глаукома, задняя субкапсульная катаракта, панкреатит и аваскулярный некроз, которые развиваются на фоне длительного приема больших доз ГК. В то же время артериальная гипертензия, образование угрей, нарушение менструального цикла, гирсуитизм и вирилизм, импотенция у мужчин, стрии и пурпура чаще наблюдаются при спонтанном синдроме Кушинга, а увеличение массы тела, психические нарушения, отеки и ухудшение заживления ран в одинаковой степени характерны для обеих форм синдрома. Различия связывают с тем, что при ятрогенном синдроме Кушинга наблюдается подавление синтеза АКТГ, а при спонтанном синдроме в результате гиперплазии надпочечников происходит активация синтеза этого гормона. Поэтому при ятрогенном синдроме не увеличивается секреция андрогенов и минералокортикоидов. Описаны следующие побочные эффекты ГК-терапии (по T. W. Behrens и J. S. Goodwin, 1989): 1. Очень частые: • отрицательный кальциевый баланс, ведущий к остеопорозу • усиление аппетита • тучность • ухудшение заживления раны • увеличение чувствительности к инфекциям • подавление оси гипоталамус—гипофиз—надпочечник • задержка роста у детей 2. Частые: • миопатия • аваскулярный некроз • артериальная гипертензия • истончение, ломкость кожи, стрии, пурпура • отеки, связанные с задержкой натрия и жидкости • гиперлипидемия • психические расстройства • сахарный диабет • задняя субкапсульная катаракта 3. Редкие: • глаукома • повышение внутричерепного давления • "скрытая" перфорация кишечника • пептическая язва (как правило, желудка) • гипокалиемический алкалоз • гиперосмолярная некетоновая кома • панкреатит • гирсуитизм • панникулит • • • • вторичная аменорея импотенция эпидуральный липоматоз аллергия к синтетическим стероидам Поражение ЦНС У многих больных, даже получающих низкие дозы ГК, могут развиться повышенная раздражительность и бессонница. Очень тяжелым осложнением является "стероидный психоз" (M. Kaufmann и соавт., 1982) — психиатрический синдром, как правило, развивающийся при назначении высоких доз ГК (более 30 мг/день) (P. Kershner и R. Whang-Cheng, 1989). При невозможности быстро отменить ГК для его купирования приходится применять психотропные препараты (галоперидол и др.) или соли лития, которые оказывают определенное профилактическое действие. Поражение глаз Лечение высокими дозами ГК может приводить к следующим офтальмологическим побочным эффектам: задней субкапсульной катаракте, увеличению внутриглазного давления, глаукоме и нарушению заживления ран роговицы (B. Girand, 1990). Задняя субкапсульная катаракта, обычно двусторонняя, является одним из частых побочных эффектов ГК-терапии. Полагают, что развитие катаракты в большей степени связано с кумулятивной дозой ГК. Низкие дозы могут вызывать катаракту не ранее чем через 2 года беспрерывного приема ГК (H. W. Shalka и Prchal, 1980). Чаще это осложнение развивается у детей, реже — у взрослых, страдающих РА. Поражение ЖКТ Данные о связи между приемом ГК и язвенным поражением ЖКТ противоречивы. Доказано, что у больных, получающих высокие дозы ГК, увеличивается риск развития пептических язв, желудочнокишечного кровотечения и панкреатита. С другой стороны, имеются данные о том, что прием низких доз ГК (менее 20 мг/день) не ассоциируется с развитием острых язв ЖКТ. Частота язв желудка составляет 2% в группе больных, получавших ГК, и 1% в контроле (J. Messer и соавт., 1983). Предполагается, что ГК ухудшают заживление предсуществующих язвенных поражений желудка и предрасполагают к развитию желудочных геморрагий. Таким образом, вероятность желудочно-кишечных осложнений на фоне приема ГК невелика и связана с общей дозой и длительностью терапии (Н. О. Conn, T. Poynard, 1985). Относительную редкость тяжелых гастропатий на фоне лечения ГК связывают с особенностями фармакологической активности этих веществ, блокирующих активность ЦОГ-2, а не ЦОГ-1 (глава 2). Гематологические нарушения Лечение ГК приводит к развитию лимфопении и эозинофилии. В течение первых нескольких дней лечения высокими дозами ГК наблюдается выраженное увеличение концентрации лейкоцитов. Хотя на фоне более длительного приема ГК уровень лейкоцитов постепенно возвращается к норме, в ряде случаев он остается повышенным, несмотря на снижение дозы ГК. Обычно общее количество лейкоцитов у больных, находящихся на ГК-терапии, составляет 15000-20000 мм3, изредка наблюдаются умеренный палочкоядерный сдвиг и токсическая зернистость. Синдром отмены ГК и мышвчно-скелетные нарушения Мышечно-скелетные нарушения ("стероидный псевдоревматизм") часто являются признаком синдрома отмены ГК, развивающегося при очень быстром прекращении ГК-терапии, Основные клинические проявления синдрома отмены — артралгии, миалгии, потеря аппетита, тошнота, рвота, "загруженность", лихорадка, шелушение кожи, постуральная гипотензия, депрессия (R. B. Dixon и соавт., 1980; L. Kahl и T. A. Jr Medsger, 1986). Перечисленные симптомы обычно сохраняются в течение нескольких часов или дней и быстро купируются при назначении адекватной дозы ГК. Иногда синдром отмены развивается у больных, получающих альтернирующее лечение ГК, по характеру симптомов он может напоминать клинические проявления основного заболевания. В большинстве случаев синдром отмены наблюдается у больных, принимавших преднизолон в дозе, превышающей 10 мг/день, в течение года и более (R. B. Dixon и соавт., 1980). Стероидная миопатия — синдром, основным признаком которого является проксимальная мышечная слабость (A. Askari и соавт., 1976; A. K. Affini и соавт., 1976). Обычно стероидная миопатия проявляется мышечной слабостью в нижних конечностях, но может прогрессировать, захватывая верхние конечности и дистальную мускулатуру. При морфологическом исследовании мышц выявляются атрофия мышечных волокон типа 2, а при лабораторном исследовании — только увеличение экскреции креатинина. Наиболее часто стероидная миопатия развивается у больных, получающих фторированные производные ГК (триамсинолон, дексаметазон) (D. N. Golding и T. B. Begg, 1960). В отличие от идиопатических воспалительных миопатий при лабораторном исследовании у больных стероидной миопатией не обнаруживается увеличения уровня мышечных ферментов (креатинфосфокиназы и альдолазы). Интересно, что, по данным одного исследования, выраженность стероидной миопатий при СКВ коррелирует с концентрацией сывороточной лактатдегидрогеназы (Y. Kanayama и соавт., 1981). При уменьшении дозы ГК мышечная сила восстановливается в течение 2-4 мес. Полагают, что стероидная миопатия иногда может наблюдаться и у больных, получающих ГК в низких дозах. Остеопороз, как правило, развивается у больных, принимающих ГК в высоких дозах и в течение длительного времени, реже — при лечении низкими дозами ГК (R. G. G. Russel и S. M. Krane, 1993). Однако, по данным B. P. Lukert и L. G. Raisz (1990), низкие дозы ГК (7.5 мг преднизолона в день) также увеличивают потерю кальция и риск переломов трабекулярных костей. Полагают, что все больные, получающие ГК более 1 года, должны принимать кальций и другие препараты, обладающие потенциальной антиостеопоретической активностью (эстрогены, бифосфонаты, кальцитонин и др.) Поражение кожи Длительное лечение ГК (как в высоких, так и в низких дозах) приводит к развитию разнообразных кожных нарушений, которые рассматриваются как один из наиболее частых побочных эффектов. Самые характерные — легкость травматизации кожи, ухудшение заживления раневой поверхности, образование грубых рубцов, появление стероидных угрей на лице и туловище, стрий. Последние нередко сохраняются после прекращения лечения и могут быть очень распространенными. Развитие кожных нарушений на фоне лечения ГК связано со способностью препаратов подавлять синтез коллагена фибробластами кожи (J. Uitton и соавт., 1972). Кожные побочные эффекты наиболее часто встречаются у пожилых женщин, принимающих фторированные ГК (S. Capewell и соавт., 1990). Локализованная липогипертрофия Одним из проявлений локализованной липогипертрофии является "лунообразное" лицо. Если она развивается в области средостения, то может приводить к неправильной трактовке рентгенограмм легких. Эпидуральная липогипертрофия может изредка вызывать компрессию спинного мозга и ее наличие должно быть заподозрено у всех больных с миелопатией, возникшей на фоне лечения ГК. Наиболее частые и тяжелые проявления липогипертрофии наблюдаются при локализации этого процесса в костном мозге и эпифизах костей, что приводит к повышению внутримедуллярного давления и развитию асептического (аваскулярного) некроза костной ткани или остеонекроза (D. S. Hungerford и соавт., 1989). К другим причинам остеонекроза относят подавление активности остеокластов, периартериальный или перивенозный отек, гиперкоагуляцию, жировую эмболизацию и др. (R. M. Zizic и соавт., 1985). Остеонекроз может развиваться в любых костях, но наиболее часто — в головках бедренной и плечевой кости. Обычно аваскулярный некроз наблюдается у больных, длительно получающих высокие дозы ГК (E. S. Weiner и соавт., 1989; R. M. Zizic и соавт., 1985; P. N. Sambrook и соавт., 1984), и иногда ассоциируется со стероидным диабетом. По данным R. M. Zizic и соавт. (1985), при СКВ аваскулярный некроз развивается только у больных, получавших преднизолон в дозе, превышающей 20 мг/день, но не связан с длительностью приема ГК. Однако при оценке связи между аваскулярным некрозом и лечением ГК необходимо иметь в виду, что это осложнение может иметь место в отсутствие ГК-терапии и являться осложнением основного заболевания. Метаболические и эндокринные эффекты Теоретически лечение преднизолоном или другими синтетическими ГК не должно сопровождаться выраженными минералокортикоидными и андрогенными эффектами. Предполагается, что во многих случаях развитие импотенции, нарушений менструального цикла в большей степени связано с основным заболеванием, а не с ГК-терапией. Длительное лечение ГК может индуцировать атеросклеротическое поражение сосудов, что связывают со способностью ГК вызывать нарушение липидного обмена (K. Kalbak и соавт., 1972). У больных РА, получавших ГК (средняя доза 17 мг/день) более 3 лет, отмечено снижение уровня липопротеидов высокой плотности (D. B. Jeffers и соавт., 1980). Однако, по данным других авторов, нарушение концентрации липидов при РА не связано со стероидной терапией. В исследовании K. L. G. Svenson и соавт. (1987) показано, что длительное лечение ГК в низких дозах не приводит к существенному изменению уровня липидов при исключении других факторов риска дислипопротеидемии. Длительное лечение ГК может сопровождаться повышением АД. Однако при учете исходного уровня АД и возраста больных длительная терапия ГК в низких дозах ГК (менее 10 мг преднизолона в день, в течение в среднем 2.7 года) не ассоциировалась с увеличением частоты артериальной гипертензии (S. H. D. Jackson и соавт., 1981). Подавление оси гипоталамус—гипофиз—надпочечник отмечено у больных, принимавших даже небольшие дозы ГК несколько дней. Супрессия может сохраняться в течение 1 года после отмены ГК, и ее выраженность варьирует у разных больных (E. K. Garber и соавт., 1981). Это диктует необходимость адекватной заместительной терапии в процессе стрессорных воздействий. Заместительная терапия у больных с супрессией оси гипоталамус— гипофиз—надпочечник в процессе хирургического лечения (по T. W. Behrens и J. S. Goodwin, 1989): 1. В день операции: гидрокортизон гемисукцинат 100 мг в/м каждые 6 ч в сочетании с обычной поддерживающей дозой ГК, 2. С первого по третий послеоперационный день: пункт 1; 3. Четвертый послеоперационный день: отменить гидрокортизон, при необходимости продолжить поддерживающую терапию. Альтернирующая терапия вызывает менее выраженную супрессию, чем ежедневный прием ГК, а при назначении ГК 1 раз в день развивается меньшая супрессия, чем при двукратном приеме, даже если доза преднизолона меньше 10 мг в сутки. Способны ли ГК индуцировать развитие сахарного диабета или только демаскировать предсуществующую патологию поджепудочной железы, остается не ясным. Известно, что высокие дозы ГК могут вызывать гипергликемию и даже кетоз, причем чувствительность к индуцированному ГК нарушению толерантности к глюкозе особенно высока у тучных больных пожилого возраста. Развитие стероидного диабета сопровождается увеличением концентрации инсулина в плазме, снижением потребления глюкозы всеми тканями и усилением глюконеогенеза. У больных сахарным диабетом лечение даже низкими дозами ГК может привести к ослаблению толерантности к глюкозе, что часто диктует необходимость усиления гипогликемической терапии. Наличие сахарного диабета само по себе не является противопоказанием к назначению ГК, однако больные с нарушением толерантности к глюкозе в анамнезе нуждаются в более тщательном наблюдении (R. Gunnarsson и соавт., 1980). Иммуносупрессия Противовоспалительный и иммуносупрессивный эффекты ГК не только приводят к увеличению чувствительности к инфекционным осложнениям, но и маскируют их клинические симптомы. Длительное лечение ГК создает предпосылки для развития герпетической инфекции и реактивации туберкулеза, хотя последнее встречается очень редко. Поскольку ГК обладают очень слабой жаропонижающей активностью, повышение температуры тела на фоне лечения, как правило, является признаком инфекционных осложнений. До начала ГК-терапии рекомендуется проведение реакции Манту, однако профилактическое лечение противотуберкулезными препаратами, даже при положительных кожных пробах, не показано. Более того, мета-анализ результатов нескольких контролируемых клинических исследований показал, что у больных, получавших менее 10 мг преднизолона в день или принявших суммарную дозу менее 700 мг, не отмечено увеличения частоты инфекционных осложнений (A. E. Stuck и соавт., 1989). Применение ГК при некоторых заболеваниях РА Основным показанием к лечению ГК при РА является непереносимость НПВС, в первую очередь поражение ЖКТ. Кроме того, ГК иногда рекомендуется назначать в комбинации с медленнодействующими (базисными) противоревматическими препаратами, в рамках так называемой мост-терапии ("bridge"), на время, необходимое для достижения терапевтического эффекта в процессе лечения последними (E. D. Harris и соавт., 1983; J. R. Caldwell и D. E. Furst, 1991; M. Wess, 1989). Основным ограничением при использовании терапии низкими дозами ГК у больных РА является развитие побочных эффектов, выраженность и спектр которых несколько отличаются от наблюдающихся на фоне лечения средними или высокими дозами ГК. Полагают, что длительное лечение низкими дозами ГК достоверно ассоциируется с поражениями глаз (катаракта, глаукома), провоцирует развитие стероидного диабета и поражение кожи (угри, гирсуитизм, стрии, атрофия кожи). Вероятными побочными эффектами являются остеопороз, миопатия, подавление коры надпочечников, в то время как развитие остеонекроза, поражения ЖКТ, сердечно-сосудистых и психических нарушений, склонность к инфекциям и др. не являются строго доказанными (J. R. Caldwell и D. E. Furst, 1991). Поданным исследователей группы G. S. Panayi, введение метилпреднизолона ацетата (в/м по 120 мг однократно) в начале лечения миокризином (фаза индукции) и на 4-й и 8-й неделе позволяет существенно снизить активность РА и избежать токсических побочных реакций кризотерапии (М. М. Corkill и соавт., 1990; E. H. Choy и соавт., 1993). Интересно, что введение депо-препаратов метилпреднизолона внутримышечно оказалось в этом отношении более эффективным, чем пульсовой пероральный прием высокой дозы метилпреднизолона (500 мг). В этой связи заслуживают внимания результаты E. M. Sternberg (1989), который в экспериментальных исследованиях обнаружил, что дефицит эндогенного кортизола может вносить определенный вклад в хронизацию артрита, а по данным I. C. Chikanza и G. S. Panayi (1990), сходный дефект выявляется и у больных РА. Это позволило высказать предположение о том, что постоянный прием небольших доз ГК в ряде случаев может быть более эффективным, чем пульс-терапия ГК (B. W. Lirkham и соавт., 1988). СКВ Основные показания к назначению ГК при СКВ суммированы в таблице 3.4. Таблица 3.4. Применение ГК при СКВ (по B. H. Hahn, 1993) Проявления, обычно контролирующиеся ГК: Проявления, не всегда контролирующиеся ГК: васкулит тромбоз (включая инсульт) гломерулонефрит (ХПН, дерматит мембранозный гломерулонефрит) психические нарушения полисерозит миокардит пневмонит резистентная тромбоцитопения/гемолитическая анемия гломерулонефрит гемолитическая анемия феномен Рейно тромбоцитопения органические мозговые синдромы миелопатия периферическая нейропатия "волчаночный криз" СКВ является наиболее ярким примером заболеваний, для лечения которых используется длительный пероральный прием высоких или средних доз ГК. Доза ГК варьируются от 40 до 100 мг (0.5 мг/кг/день-1,5 мг/кг/день). Вначале дневную дозу препарата делят на 3 приема, затем переходят на однократный прием препарата в утренние часы. Клинический эффект в одних случаях развивается довольно быстро (в течение 1-5 дней), в других случаях (например, у больных с нефритом) для этого требуется 3-10 нед. Длительное лечение высокими дозами ГК (более 0,5 мг/кг/день) не рекомендуется. При достижении оптимального эффекта следует незамедлительно начинать снижение дозы ГК. Каждую неделю общую дозу препарата можно уменьшать на 5-10%, если это не сопровождается обострением заболевания. У некоторых больных могут развиться признаки синдрома отмены. При отсутствии эффекта необходимо обсудить вопрос об альтернативных методах назначения ГК (пульс-терапия) или присоединении цитотоксических препаратов. Начинать лечение активной СКВ ГК в альтернирующем режиме не рекомендуется. Но переход на альтернирующий прием ГК весьма желателен при полном подавлении активности болезни в процессе перевода больного на поддерживающую дозу препарата. В процессе лечения ГК очень важно контролировать развитие интеркуррентных инфекций (опоясывающий лишай, мочевая инфекция грам-отрицательными бактериями, стафилококки, кандидоз, микобактерии, грибы, цитомегаловирус, Pnevmocystic carine), некоторые проявление которых (например, легочные инфильтраты, лихорадка, неврологические нарушения, пиурия), могут имитировать обострение СКВ. ПМ/ДМ ГК короткого действия (преднизолон, метилпреднизолон) являются единственной группой препаратов, которые доказали свою эффективность в контролируемых исследованиях, они рассматриваются как средство выбора при лечении ИВМ (P. H. PIotz и соавт., 1989). Полный или частичный ответ на ГК удается достигнуть у 75-90% больных, которым назначалась адекватная доза препарата. Ретроспективный анализ результатов лечения показывает, что у больных, получивших в начале болезни высокую дозу ГК (не менее 50 мг/сут.) статистически достоверно более высокая выживаемость, чем у больных, принимавших малые или средние дозы или вообще не леченных ГК. Минимальная терапевтически эффективная доза ГК при ПМ/ДМ составляет около 1 мг/кг как для взрослых, так и детей, причем, чем в более ранние сроки начато лечение, тем более вероятно, что оно будет эффективным (M. C. Dalakas, 1991; C. V. Oddis и T. A. Medsger, 1989). Следует иметь в виду, что улучшение у больных ПМ/ДМ на фоне лечения высокими дозами ГК развивается не так быстро, как при других ДБСТ, в среднем в промежутке между 2 и 4 мес. (P. J. Vignos, 1985). Отсутствие хотя бы минимальной динамики клинических и лабораторных показателей на фоне приема преднизолона (1 мг/кг/день) в течение 4 нед., является основанием для увеличения дозы препарата. Корректировку дозы ГК следует проводить постепенно по 0.25 мг/кг, мониторируя клиническую и лабораторную эффективность в течение 2-3 нед. При этом максимальная доза преднизолона при длительном пероральном приеме составляет 2 мг/кг в день. В случае отсутствия эффекта в течение 4 нед., следует еще раз обсудить правильность диагноза и при его подтверждении попытаться найти пути для преодоления стероидрезистентности. Уменьшение дозы ГК следует начинать лишь при достижении клинико-лабораторной ремиссии, но не раньше, чем через 4-6 нед. от начала лечения. После достижения полной клинико-лабораторной ремиссии возможна попытка перехода на альтернирующий режим ГК терапии. В большинстве случаев, даже при самом благоприятном варианте заболевания, общая продолжительность гормональной терапии составляет 2-3 года. При этом только 25% больных могут полностью отказаться от ГК, а многие вынуждены в течение очень длительного времени принимать поддерживающие дозы препарата, варьирующие от 2.5 до 40 мг/день. Альтернирующая терапия ГК Альтернирующая терапия ГК — метод лечения, заключающийся в назначении ГК короткого действия без выраженной минералокортикоидной активности (преднизон, преднизолон, метилпреднизолон) однократно утром (около 8 ч) каждые 48 ч. Целью альтернирующей терапии является уменьшение выраженности побочных эффектов ГК при сохранении терапевтической эффективности. Полагают, что возможность альтернирующей терапии ГК должна обсуждаться у всех больных, которым планируется длительное лечение ГК (более нескольких недель). Показано, что альтернирующий режим в начале болезни может быть успешно применен при язвенном колите, хронических дерматозах, миастении гравис, бронхиальной астме (J. B. Tyrrel и J. D. Baxter, 1987). Однако при системных ревматических заболеваниях такая тактика лечения, как правило, неэффективна. Основанием для разработки этого метода лечения явилось предположение о том, что противовоспалительное действие ГК длится дольше, чем нежелательные метаболические эффекты. Поэтому может существовать такой ритм приема ГК, при котором в перерыве между приемами препаратов сохраняется их противовоспалительное действие, но уменьшается риск развития побочных эффектов. Предполагается также, что интермиттирующее назначение ГК задает определенный ритм колебаний уровня кортикостероидов в крови, стимулирующий нормальный диульнарный цикл, что и может позволить предотвратить развитие синдрома Кушинга и супрессию гипоталамо-гипофизарной оси. Однако не исключается, что уменьшение выраженности побочных эффектов при альтернирующем приеме ГК связано только со снижением общей дозы препарата, что определяется особенностями биодоступности ГК. Действительно, имеются данные о том, что при пероральном приеме биодоступность преднизолона ниже при назначении суточной дозы в один прием, чем в несколько приемов. В то же время только с этой точки зрения трудно объяснить уменьшение числа побочных эффектов при сохраняющейся эффективности этого варианта ГК-терапии. Использование ГК короткого действия и 48-часовой интервал между приемом препаратов не является эмпирическим. Имеются данные о том, что 36-, 24- и 12-часовой интервалы ассоциируются с супрессией надпочечников, а 72-часовой интервал снижает терапевтическую эффективность ГК. Установлено, что альтернирующий режим ГК-терапии в определенной степени снижает выраженность клинических проявлений синдрома Кушинга (некоторые соматические и психотические реакции, артериальная гипертензия, задержка роста у детей, повышение чувствительности к инфекции и др.) и в меньшей степени подавляет функцию надпочечников. Положительные свойства альтернирующей терапии проявляются только при длительном лечении ГК. Поэтому ее не следует назначать больным, у которых планируется кратковременное использование этих препаратов, а также в начальной стадии лечения или в период обострения заболевания. Необходимо иметь в виду, что альтернирующий режим не во всех случаях позволяет предотвратить развитие синдрома Кушинга или подавление гипоталамо-гипофизарно-надпочечниковой оси. Кроме того, у многих больных проведение альтернирующей терапии затруднено из-за ухудшения самочувствия больных в день приема низкой дозы ГК. Ниже представлена одна из возможных схем перехода на альтернирующий прием ГК (по T. W. Behrens и J. S. Goodwin, 1989). ДНИ ПРЕДНИЗОЛОН, мг ДНИ ПРЕДНИЗОЛОН, мг 1-7-й 60 14-й 90 8-й 60 15-й 10 9-й 40 16-й 95 10-й 70 17-й 5 11-й 30 18-й 95 12-й 80 19-й 5 13-й 20 Вся доза преднизолона принимается в утренние часы; с 18-го дня начинается медленное снижение большей дозы препарата через каждые 5-7 дней. Пульс-терапия В начале 70-х годов появились сообщения о возможности подавления отторжения почечного трансплантата путем внутривенного введения больным очень больших доз ("мегадоз") метилпреднизолона (P. R. F. Bell и соавт., 1971; N. J. Federska и соавт., 1972). Этот метод получил название пульс-терапии. Хотя четкого определения этого термина до сих пор не существует, под ним обычно подразумевают быстрое (в течение 30-60 мин.) внутривенное введение больших доз ГК (около 1 г) 1 раз в день на протяжении 3 сут. Наиболее часто при проведении пульс-терапии используют метилпреднизолон в виде раствора гемисукцината натрия (Е. Л. Насонов и соавт., 1994). При изучении фармакокинетики было обнаружено, что уровень метилпреднизолона в плазме при внутривенном введении в течение 1 ч достигает максимума и быстро снижается в последующие 6-7 ч. Через 4 дня в периферической крови обнаруживаются только следовые количества активного метилпреднизолона, явно недостаточные для депо-эффектов. Однако при изучении метаболизма меченного изотопами метилпреднизолона было показано, что последний очень активно накапливается в различных тканях, особенно эритроцитах, некоторых висцеральных тканях и головном мозге. При этом на основании некоторых косвенных данных было высказано предположение, что метилпреднизолон обладает способностью в большей степени депонироваться в воспаленных, чем в нормальных тканях. Эти фармакологические свойства метилпреднизолона, а также минимальная минералокортикоидная активность позволяют рассматривать его как средство выбора при проведении пульс-терапии (I. Williams, 1984). Первое сообщение о пульс-терапии при ревматических болезнях обычно приписывают E. S. Cathcart и соавт., которые в 1976 г. использовали этот метод при волчаночном нефрите. Основанием для проведения пульс-терапии явилось сходство морфологических изменений в почках у больного волчаночным нефритом с картиной, наблюдаемой при отторжении почечного трансплантата. Это позволило авторам применить для лечения волчаночного нефрита протокол, ранее разработанный для больных с острым отторжением трансплантата, то есть пульс-терапию ГК. Показания к проведению пульс-терапии суммированы в таблице 3.5. Необходимо подчеркнуть, что у больных с наиболее тяжелыми формами ревматических болезней, такими, как волчаночный нефрит, ЦНС-люпус, ревматоидный васкулит, системные некротизирующие васкулиты, пульс-терапия ГК обязательно должна сочетаться с активной цитотоксической терапией, в первую очередь ЦФ, так как только комбинированное лечение позволяет реально улучшить прогноз заболевания (С. К. Соловьев и соавт., 1985). Таблица 3.5. Показания к проведению пульс-терапии в ревматологии (по J. G. Hardin и G. L. Longenecker, 1992) Заболевание Примечание СКВ без поражения почек Эффективный метод купирования большинства внепочечных проявлений: лихорадка, артралгии, серозит, миозит; гематологические нарушения, поражение ЦНС? Волчаночный нефрит При сочетании с другими методами лечения (ЦФ) Васкулиты При сочетании с другими методами лечения (цитостатики) ПМ/ДМ При опасных для жизни осложнениях; снижает частоту развития кальциноза при ювенильном дерматиомиозите РА Ревматоидный васкулит (в сочетании с цитостатиками) острое активное воспаление суставов; Анкилозирующий спондилит Тяжелое рефрактерное воспаление (S. Yoshida и соавт., 1993) "Замороженное" плечо Быстрое купирование процесса; часто (C. R. Morris и соавт., 1994) полное исчезновение симптомов после введения 1 г метилпреднизолона По данным неконтролируемых исследований на фоне пульс-терапии быстрое улучшение наблюдается у 75% больных с активным волчаночным нефритом, поражением ЦНС, пневмонитом, полисерозитом, васкулитом, тромбоцитопенией. На пульс-терапию отвечают некоторые больные, резистентные к пероральному приему высоких доз ГК (М. М. Иванова и соавт., 1983). Полагают, что одним из преимуществ пульс-терапии является возможность более быстрого перевода больного на поддерживающую дозу ГК. Предварительные результаты свидетельствуют также о том, что повторные процедуры пульс-терапии (1-3 дня в месяц) являются возможной альтернативой назначению цитостатиков. Ряд авторов наблюдали развитие на фоне пульс-терапии ремиссии у детей с ДМ без поддерживающего лечения ГК (R. M. Laxer и соавт., 1987). Однако, по данным большинства исследователей, при ПМ/ДМ у взрослых пульс-терапия дает лишь кратковременный эффект, а у больных, резистентных к стандартным дозам ГК, вообще неэффективна (C. V. Oddis и T. A. Medsger, 1988). Тем не менее иногда введение сверхвысоких доз стероидов позволяет в дальнейшем вести больных на более низких дозах ГК. Недавно появились предварительные сообщения о высокой эффективности у больных резистентной тромбоцитопенической пурпурой пульс-терапии дексаметазоном (J. C. Andersen, 1994). При рефрактерном РА пульс-терапия, как правило, позволяет достигнуть быстрого, но достаточно кратковременного подавления активности суставного процесса (M. R. Liebling, 1993; T. Danao и A. M. Segal, 1990). По данным M. Radia и D. E. Furst (1987), существенно не различается краткосрочный эффект при введении больным высоких (1000 мг внутривенно) или средних (320 мг внутривенно или внутримышечно) доз метилпреднизолона. Имеются данные о том, что проведение пульс-терапии в начале лечения препаратами золота позволяет существенно улучшить непосредственные результаты кризотерапии (в течение первых 6 мес.) и снизить выраженность инвалидизации в более отдаленные периоды болезни (38 мес. (M. Heytman и соавт., 1994). Одним из преимуществ пульс-терапии является относительно низкая частота побочных реакций, а также возможность в дальнейшем использовать более низкие дозы ГКС для поддерживающей терапии. Тем не менее на фоне пульс-терапии зарегистрирован ряд характерных побочных эффектов, перечень которых представлен ниже: 1. ЧАСТЫЕ: • гиперемия лица • изменение вкусовых ощущений • транзиторная артериальная гипертензия • транзиторная гипергликемия • невоспалительный артрит, артралгии • миалгии • сердцебиение • транзиторная задержка жидкости • остеонекроз 2. РЕДКИЕ: • некупирующаяся икота • анафилактические реакции, коллапс • неврологические расстройства (судороги, галлюцинации, головные боли, тошнота) • диссеминация инфекции\ • внезапная смерть (связана с развитием аритмий на фоне электролитных нарушений). Смертельные исходы, зарегистрированные при проведении пульс-терапии у 3 больных с пересаженной почкой, как полагают, были связаны с сочетанным приемом диуретиков и очень быстрым (менее 20 мин.) введением препарата (S. S. Subbs и R. M. Morrel, 1973). Было высказано предположение, что индуцированная ГК вазодилатация в сочетании с нарушением входа кальция в кардиомиоциты привела к резкому снижению сердечного выброса и сосудистому коллапсу, что послужило причиной смерти больных. M. E. Shipley и соавт. (1988) сообщили о двух смертельных исходах у больных РА через 6 нед. после пульс-терапии. Однако, оба пациента имели факторы риска: в одном случае — тяжелая ишемическая болезнь сердца, а в другом — системный амилоидоз. Таким образом, для снижения риска внезапной смерти пульс-терапию не следует сочетать с мочегонными препаратами, способными индуцировать электролитные нарушения. Тактика снижения дозы и отмены ГК Кратковременный прием небольших доз ГК (менее 20 мг/сут) в течение 3 нед. позволяет быстро прекратить лечение без развития синдрома отмены (E. L. Hefler и L. I. Rose, 1989). Однако у больных, длительно принимающих ГК, отмена препарата является продолжительным и трудным процессом. Одна из возможных схем отмены ГК у больных, длительно получавших эти препараты, представлена в таблице 3.6. Таблица 3.6. Схема постепенного снижения и отмены ГК (начальная доза 40 мг/сут) (по E. S. Shlom. 1992) День Доза.мг День Доза, мг А. Снижать дозу по 5 мг через день до 10 мг/день 1-й 40 7-й 40 2-й 35 8-й 20 3-й 40 9-й 40 4-й 30 10-й 15 5-й 40 11-й 40 6-й 25 12-й 10 Б. Снижать по 2.5 мг через день каждые 3 цикла до полной отмены ГК в альтернативный день 13-й 40 25-й 40 14-й 7.5 26-й 2.5 15-й 40 27-й 40 16-й 7.5 28-й 2.5 17-й 40 29-й 40 18-й 7.5 30-й 2.5 19-й 40 31-й 40 20-й 5 32-й 0 21-й 40 33-й 40 22-й 5 34-й 0 23-й 40 35-й 40 24-й 5 36-й 0 В. При стабильном состоянии больного снижать по 5 мг через день до дозы 10 мг через день 37-й 35 43-й 20 38-й 0 44-й 0 39-й 30 45-й 15 40-й 0 46-й 0 41-й 25 47-й 10 42-й 0 48-й 0 Г. Снижать по 2.5 мг через день каждые 3 цикла до достижения дозы 2.5 через день 49-й 7.5 58-й 0 50-й 0 59-й 5 51-й 7.5 60-й 0 52-й 0 61-й 2.5 53-й 7.5 62-й 0 54-й 0 63-й 2.5 55-й 5 64-й 0 56-й 0 65-й 2.5 57-й 5 66-й 0 Д. Снижать по 1 мг через день в течение нескольких недели или месяцев Е. Отменить ГК ДЕФЛАЗАКОРТ (Deflazacort) Дефлазакорт — синтетический кортикостероид, являющийся дериватом преднизолона. Рассматривается как представитель третьего поколения ГК. Присутствие оксазолина в положении С-17 приводит к снижению жирорастворимости препарата (G. G. Nathansohn и соавт., 1979). Предполагается, что эта структурная особенность обусловливает способность дефлазакорта оказывать более выраженное действие на лимфоциты, чем на другие ткани (G. Buniva и соавт., 1979). Дефлазакорт быстро адсорбируется в ЖКТ, биодоступность приближается к 70%. Пик концентрации в плазме при пероральном приеме достигается через 1-2 ч; в плазме 40% препарата находится в связи с альбумином. Метаболиты дефлазакорта выводятся главным образом с мочой, в меньшей степени с калом. Эквивалентная преднизолону терапевтическая доза препарата варьирует от 0.66: 1 до 1: 1. Предполагается, что дефлазакорт обладает более длительной и выраженной иммуномодулирующей активностью, чем преднизолон (M. Scudeletti и соавт., 1993). В клинических исследованиях было показано, что терапевтическая эффективность дефлазакорта у больных РА, ревматической полимиалгией и другими хроническими воспалительными заболеваниями сходна с таковой преднизона (R. E. S. Gray и соавт., 1991; M. Scudeletti и соавт., 1987). Основным преимуществом дефлазакорта перед преднизолоном и другими ГК является менее выраженная способность нарушать метаболизм глюкозы и кальциевый обмен (M. Scudeletti и соавт., 1993; R. E. S. Gray и соавт., 1991). Список литературы к главе "Глюкокортикоиды". 1. 2. 3. 4. Иванова М. М., Соловьев С. К., Сперанский Ф. И., Насонов Е. Л.: Внутривенное введение ударных доз метилпреднизолона (пульс-терапия) при люпус нефрите. Терапевт. архив. 1983; 7: 114-117. Насонов Е. Л., Иванова М. М., Панин Д. И.: Глюкокортикоиды в ревматологии: опыт использования Солю-Медрола. Клин. фарм. тер. 1994; 3: 46-49. Соловьев С. К., Иванова М. М., Насонов Е. Л. и др.: Комбинированное применение ударных доз 6 метилпреднизолона и циклофосфана у больных системной красной волчанкой. Терапевт, архив. 1985; 8: 7-12. Affini А. К., Bergman R. A., Harvey J. C.: Steroid myopathy. John Hopkins Med. J. 1968; 107: 158- 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 174. Andersen J. С.: Response of resistant idiopathic thrombocytopenic purpura to pulsed highdose dexamethasone therapy. N. Engl. J. Med. 1994; 330: 1560-1564. Askari A., Vignos P. J., Moskowitz R.: Steroid myopathy in connective tissue disease. Am. J. Med. 1976; 61: 485-491. Axelrod L.: Glucocortocoids. In: Textbook of rheumatology. Ed. W. N. Kelley, Å. D. Harris, S. Ruddy, C. B. Sledge. W. B. Saunders Company. Philadelphia, London, Toronto. 1993.779-796. Barnes P. J., Adcock I.: Anti-inflammatory actions of steroids: molecular mechanisms. TIPS 1993; 14: 436-441. Behrens Т. W. Goodwin J. S.: Glucocorticoids. In: Arthritis and allied conditions. A textbook of rheumatology. Ed. D. J. McCarty. Lea. Febiger. Philadelphia London. 1989: 604-621. Bell P. R. F., Briggs J. D., Calman K. C., et al.: Reversal of acute clinical and experimental organ rejection using large doses of IV prednisolone. Lancet. 1971; 1: 876-880. Boumpas D. Т., Palilogrianni F., Anastassiou E. D., Balow J. E. Glucocorticosteroid action on the immune system: molecular and cellular aspects. Clin. Exp. Rheumatol. 1991, 9, 413-423. Caldwell J. R., Furst D.: The efficacy and sefety of low-dose corticosteroids for rheumatoid arthritis. Semin. Arthritis. Rheum. 1991; 21: 1-11. Capewell S., Reynolds S., Shuttleworth D., et al.: Purpura and dermal thinning, associated with high dose inhaled corticosteroids. Brit. Med. J. 1990; 300: 1548-1551. Cathcart E. S., Idelson B. A., Scheinberg M. A., Couser W. G.: Beneficial effects of methylprednisolone "pulse" therapy in diffuse proliferative lupus nephritis. Lancet. 1976. I, 163-166. Chikanza I. C., Panayi G. S.: The interaction between the neurological and immune system in the pathogenesis of rheumatoid arthritis (abstract). Clin. Rheumatol. 1990; 90: 26-27. Choy E. H., Kingsley G. H., Corkill M. M., Panayi G. S,: Intramuscular methylprednisolone is superior to pulse oral methylprednisolone during the induction phase of chrysotherapy. Brit. J. Med. 1993; 32: 734-739. Christiansen C., Krane S. M.: Advances in corticosteroids. Adis Int. Langhorne. 1993, 70p. Cirino G., Peers S. H., Flower R. J., et al.: Human recombinant lipocortin I has acute local antiinflammatory properties in the rat edema test. Proc. Natl. Acad. Sci. USA. 1989; 86: 3428-3432. Comber P. G., Rossman M. D., Rappaport E. F., et al.: Modulation of human mononuclear phagocyte Fc-gamma II mRNA and protein. Cell. Immunol. 1989; 124: 297-307. Conn H. O., Poynard Т.: Adrenocorticosteroid administration and peptic ulcer: A critical analysis. J. Chronic Dis. 1985; 38: 457. Corkill M, M., Kirkham B. W., Chikanza I. C., Gibson Т., Panayi G. S.: Intramuscular depot methylprednisolone induction of chrysotherapy in rheumatoid arthritis: a 24 week randomized controlled trial. Brit. J. Rheumatol. 1990; 29: 274-279. Cronstein B. N., Kimmel S. C., Levin R. I., et al.: A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of the endothelial leukocyte adhesion receptor ELAM-1 and ICAM-I. Proc. Natl. Acad. Sci. USA. 1992; 89: 9991-9995. Dalakas M. C.; Polymiositis, dermatomiositis and inclusion-body myositis. New Engl. J. Med. 1991; 1487-1498. Danao Т., Segal A. M.: Pulsed suppressive treatment in rheumatoid arthritis: intravenous methylprednisolone and nitrogen mustard. J. Rheumatol. 1990; 17: 893-899. Davidson E. F., Dennis E. A., Powell M., et at.: Inhibition of phospholipase A2 by lipocortin and calpactins. An effect on binding to substrate phospholipid. J. Biol. Chem. 1987; 262: 1698-1705. Dixon R. B., Christy N. P.; On the various form of corticoste roid withdrawal syndrome. Amer. J. Med. 1980; 68: 224-230. Emilie D., Crevon M. C., Auffredou M. T., Galanaud P.: Glucocorticosteroid-dependent synergy between interleukin I and interleukin 6 for human В lymphocytes differentiation. Eur. J. Immunol. 1988: 18: 2043-2047. Flowers R. S.: Lipocortin: what is it and what does it mean? Brit. J. Rheumatol. 1992, 31, 506-508. Federska N. J., Turcotte J. C., Gikas P. W., et al.: Reversal of renal allograft rejection with intravenous methylprednisolone pulse therapy. J. Surg. Res., 1972; 12: 208-215. Fuller P. J.: The steroid receptor superfamily: mechanisms of diversity. FASEB. 1991, 5, 3092-3099. Fu J.-Y., Masferrer J. L., Seibert K., et al.: The induction and supression of prostaglandin H2 synthase (cyclooxigenase) in human monocyte. J. Biol. Chem. 1990; 265: 16737-16740. Hardin J. O., Longenecker G. L.: Handbook of drug therapy in rheumatic disease. Pharmacology and clinical aspects. Little, Brown and Company. Boston, Roronto, London. 1993: 47-76. Harris E. D., Emkey R. D., Nichols J. E., et al.: Low dose prednisone therapy in rheumatoid arthritis: a double blind study. J. Rheumatol. 1983; 10: 713-721. Heytman M., Ahern M. J., Smith M. D., Robert-Thompson P. J.: The longterm effect of pulsed 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. corticosteroids on the efficacy and toxicity of chrysotherapy in rheumatoid arthritis. J. Rheumatol. 1994; 21: 435-441. Heifer E. L., Rose L. I.: Cortocosteroids and adrenal suppression. Characteristic and avoid the problem. Drugs 1989; 38: 838-854. Hungerford D. S.: Response: the role of core decompression in the treatment of ischemic necrosis of the femoral head. Arthritis Rheum. 1989; 32: 801-806. Garber E. K., Fan P. T., Bluestone R.: Realistic guidelines of corticosteroid therapy in rheumatic disease. Semin. Arthritis Rheum. 1981; II: 231-256. Girard В.: Corticosteroids and ophtalmology. Rev. Pract. 1990; 40: 563-540. Golding D. N., Begg T. B.: Dexametason myopathy.: Brit. Med. J. 1960; 2: 1129-1130. Goulding N. J., Podgorski M. R., Hall H. D., et. al.: Autoantibodies to recombinant lipocortin-1 in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 1989, 48,843-850. Goulding N. J., Godolphin J. L., Sharland P. R., et al.: Anti-infammatory lipocortin I production by peripheral blood leukocytes in response to hydrocortisone. Lancet 1990; 335: 1416-1418. Gray R. E. S., Doherty S. M., Galloway J., et al.: A double-blind study of deflazacort and prednisone in patients with chronic inflammatory disorders. Arthritis Rheum. 1991; 34: 287-295. Gunnarsson R., Lundgren G., Magnusson G., et al.: Steroid dia betesa sign of overtreatment with steroids in the vrenal graft recipient. Scand. J. Urol. Nephrol (suppl); 54: 135-138. Kahl L., Medsger T. A. Jr: Severe arthralgias after wide fluctuation in corticosteroid dosage. J. Rheumatol. 1986; 13: 1063-1065. Kalbak К.: Incidence of arteriosclerosis in patients with rheumatoid arthritis receiving long-term corticosteroid therapy. Ann. Rheum. Dis. 1972; 31: 196-200. Kanayama Y., Shieta F., Horiguchi T. et al.: Correlation between steroid myopathy and serum lactic dehydrogenase in systemic lupus erythematosus. Arch. Int. Med. 1981; 141: 1176-1179. Karin M., Yang-Yen H.-M., Chambard J.-C., et al.: Various modes of gene regulation by nuclear receptors for steroid and thyroid hormones. Eur. J. Clin. Pharm. 1993 (suppl. 1); 45: S9-S15. Kaufmann M., Kahaner K., Peselow E. D. et al.: Steroid psychoses: case report and brief overview. J. Clin. Psychiatr. 1982; 43: 75-76. Kershner P., Wang-Cheng R.: Psychiatric side effects of steroid therapy. Psychomatics. 1989; 30: 135-139. Kimberley R. P.: Glucocorticoid therapy for rheumatic diseases. Curr. Opin. Rheumatol. 1992,4,325331. Lirkham В. W., Panayi G. S.: Duurnal periodicity of cortisol secretion, immune reactivity and disease activity In rheumatoid arthritis: implication for steroid treatment. Brit. J. Rheumatol. 1988; 28: 154-157. Jackson S. H. D., Beevers D. G., Myers K.: Does long-term low-dose corticosteroid therapy cause hypertension? Clin. Sci. 1981; 61: 381S-382S. Jefferys D. B., Lessof M. H., Mattock M. B.: Corticosteroid treatment, serum lipids and coronary artery disease. Postgrad. Med. 1980; 56: 491-493. Laxer R, M., Stein L. D., Petty R. E.: Intravenous pulse methylprednisolone treatment of juvenile dermatomyositis. Arthritis Rheum. 1987; 30: 328-334. Liebling M. R. Pulse methylprednisolone therapy in active rheumatoid arthritis. Int. Med. 1983; 4: No. 3. Lukert В. P., Raisz L. G.: Glucocorticoid-induced osteoporosis: pathogenesis and managment. Ann. Intern. Med. 1990; 112: 352-364. Masferrer J. L., Zweifer B, S., Manning P. T., et al.: Selective inhibition of inducible cyclooxygenase 2 in vitro is antiinflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. USA. 1994; 91: 3228-3232. Messer J., Reitman D., Sacks H. S. et al.: Association of adrenocorticosteroid therapy and pepticulcer disease. New Engl. J. Med. 1983; 309: 21-24. Morand E. F., Jefferiss C. M., Dixey J., et al.: Impaired glucocorticoid induction of mononuclear leukocyte lipocortin-1 in rheumatoid arthritis. Arthritis Rheum. 1994; 37: 207-211. Morris С. R., Morris A. J.: Intravenous pulse corticosteroids for the treatment of frozen shoulder. Arthritis Rheum. 1994; 37 (suppl. ): S241. Nathansohn G. G., Winters G., Testa D.: Steroid possessing nitrogen. HI. Synthesis of new highly active corticosteroid (17 alpha, 16 alpha) oxazoline steroids. J. Med. Chem. 1979; 10: 799-811. Oddis С. V., Medsger T. A.: Current management of polymyositis and dermatomyositis. Drugs, 1989; 37: 382-390. Paliogianni F., Ahuja S. S., Balow J. P., et al: Novel mechanism for inhibition of human T cells by glucocorticoids: glucocorticoids inhibit signal transduction through IL-2 receptor. J. Immunol. 1993; 252: 4081-4089. Pan L. Y., Mendel D. B., Zurlo J., Guyre P. M.: Regulation of the steady state level of Fc-gammaRI 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. mRNA by gamma-interferon and dexametasone in human monocytes, neutrophils, and U-939. J. Immunol. 1990; 145: 267-275. Perretti M., Flower R. J.: Modulation of IL-l-induced neutrophil migration by dexametasone and lipicortin 1. J. Immunol. 1993; 150: 992-999. Plotz P. H., Dalakas M. C., Left R. L., et al.: Current concepts in the idiopathic inflammatory myopathies: dermatomyositis and related disorders. Ann. Int. Med. 1989; III: 143-157. Radia M., Furst D. E.: Comparison of three pulse methylprednisolone regimens in the treatment of rheumatoid arthritis. J. Rheumatol. 1988; 15: 242-246. Russel R. G. G., Krane S. M. Glucocorticoids and bone. Brit. J. Rheumatol. 1993; 32 (Suppl. 2). Salmon J. E., Kapur S., Meryhew N. L., et al.: High-dose, pulse intravemnous methyl prednisolone enhances Fc receptor-mediated mononuclear phagocyte function in systemic lupus erythematosus. Arthritis Rheum, 1989; 32: 717-725. Sambrook P. N., Hassall J. E., York J. R.: Osteonecrosis after high dosage, short term corticosteroid therapy. J. Rheumatol. 1984; II: 514-516. Schlaghecke R., Kornely E., Wollenhaupt J., Specker C.: Glucocorticoid receptors in rheumatoid arthritis. Arthritis Rheum. 1992; 35: 740-744. Schlaghecke R., Beuscher D., Kornely E., Specker C.: Effects of glucocorticoids in rheumatoid arthritis. Arthritis Rheum. 1994; 37: 1127-1131. Schleimer R. P.: An overview of glucocorticoid anti-inflammatory actions. Eur. J. Clin. Pharmacol. 1993; 45 (suppl. 1): S3-S7. Scudeletti M., Piccardo C., Piovano P. L., et al.: Effects of a new heterocyclic glucocorticoid, deflazacort (DFC), on rheumatoid arthritis. Int. J. Immunopharmacol. 1987; 9: 133-139. Scudeletti M., Puppo F., Lanza L., et al.: Comparison of two glucocorticoid preparations (deflazaxort and prednisone) in the treatment of immune-mediated disease. Eur. J. Clin. Pharmacol. 1993; 45 (suppl. 1): S29-34. Shalka H. W., Prchal J. Т.: Effect of corticosteroids on cataract formation. Arch. Ophtalmol. 1980; 98: 1772-1777. Shipley M. E., Bacon P. A., Berry H., et al.: Pulse methylprednisolone in active early rheumatoid arthritis: A dose ranging study. Br. J. Rheumatol. 1988; 27: 211-214. Shiom E. S.: Adrenocortical dysfunction and clinical use of steroids. In. Clinical pharmacy and therapeutidc. Ed. E. T, Herfindal, D. R. Gourley, L L. Hart. Williams. Wilkins. 1992: 245-266. Solito E., Raugei G., Melli M., Parente L.: Dexamethasone induces the expression of the mRNA of lipocortin I and 2 and the release of lipocortin I and 5 in the differentiated, but not undifferentiated U-937 cells. FEBS lett. 1991; 291: 238-244. Stenberg E. M., Young W. S.. Bernardini R.,et al.: A central nervous system defect in biosynthesis of corticotropin-releasing hormone is associated with susceptibility to streptococcal cell wallinduced arthritis in Lewis rats. Proc. Nati, Acad. Sci. USA 1989: 86: 4771-4775. Sternberg E. M., Chousos G. P., Wilder R. L, Gold P. W.: The stress response and the regulation of inflammatory disease. Ann. Intern. Ned. 1992; 117: 854-866. Stuck A. E., Minder C. E., Frey F. J.: Risk of infectious complications in patients taking glucocorticosteroids. Rev. Infect. Dis. 1989; 2: 954-963. Subbs S. S., Morrel R. M.: Intravenous methylprednisolon sodium succinate adverse reaction reported in assocoation with immunosupressive therapy, Transpl. Proc. 1973; 5: 1145-1146. Svenson К. L. G., Lithell H., Hallgren R., Vesby B.: Serum lipoprotein in acute rheumatoid arthritis and other chronic inflammatory arthritides. II. Effects of antiinflammatory and disease-modifying drug treatment. Arch. In tern. Med. 1987; 147: 1917-1920. Tyrrel J. B., Baxter J. D. Glucocorticoid therapy. In: Endocrinology and methabolism. Ed. Feling P., Baxter J. D., Broadus A. E., Frohman L. A. New York, McGraw-Hill, 1987, p. 787. Uitto J., Teir H.,Mustakallio K. K.: Corticosteroid-induced inhibition of the biosynthesis of human skin collagen. Biochim Pharmacol. 1972; 21: 2161-2167. Weiner E. S.,Abeles M.: Aseptic necrosis and glucocortiticosteroids in systemic lupus erythematosus: a reavalution. J. Rheumatol. 1989; 16: 604-608. Vignos P. J.: Polymiositis and dermatomyositis. Landmarks in rheumatol. 1985; 3: 1012. Weiss M.: Corticosteroids in rheumatoid arthritis. Semin. Arthritis. Rheum., 1989; 19: 9-21. Wenner M. H: The use of steroidal agents via intermitent pulse administration. In.: Therapeutic controversies in the rheumatic diseases. Ed. R. F. Willkens, S. L. Dahl. Grune and Stratton Inc. Orlando, New York, London. 1987. 93-132. Williams 1.: A survey of pulsed intravenous methylprednisolon In the treatment of rheumatoid diseases. Geriart. Med. Today. 1984, 3, 97-108. Yoshida S., Matai Y., Hattori H., et al.: A case of HLA-B27 negative ankylosing spondilitis treated with methylprednisolone pulse therapy. J. Rheumatol. 1993; 20: 1805-1806. Zizic R. M., Marcoux C., Hungerford D. S., et al.: Corticosteroid therapy associated with ischemic necrosis of bone in systemic lupus erythematosus. Am. J. Med. 1985; 790: 596-604. ГЛАВА 4 ПРЕПАРАТЫ ЗОЛОТА Препараты золота используются в лечении больных РА с 1929 г. и рассматриваются как одно из наиболее эффективных средств терапии этого заболевания (D. A. Gordon, 1989). К препаратам золота относят несколько лекарственных форм. В России широко применяется отечественный препарат кризанол, представляющий собой масляную суспензию кальцийауротиопропанолсульфата, а за рубежом наиболее популярен миокризин (myochrysine) — натриевая соль ауротиояблочной кислоты. Оба препарата предназначены для внутримышечного введения, являются водорастворимыми и серосодержащими веществами. Кризанол содержит 33.5% металлического золота, а миокризин — 50%. Существует препарат золота для перорального приема — ауранофин, представляющий собой триэтилфосфорное соединение с содержанием золота 25%. Особенностью ауранофина является низкая абсорбция (25% от принятой дозы), мономерная форма, жирорастворимость, слабое взаимодействие с сульфгидрильными группами. Фармакологические свойства После парентерального введения золото наиболее активно накапливается в почках, надпочечниках и ретикулоэндотелиальной системе. Внутри клеток золото локализуется в ядре, митохондриях и лизосомальной фракции. У больных РА золото обнаруживается во многих тканях, в наиболее высокой концентрации — в костном мозге (26%), печени (24%), коже (19%), костях (18%), реже — в других органах (N. L. Gottlieb и соавт., 1972). В синовиальной жидкости уровень золота составляет примерно 50% от сывороточного. Однако в процессе кризотерапии золото преимущественно локализуется в синовиальной мембране, причем более активно в воспаленном, чем в невоспаленном синовиуме. Фармакокинетика препаратов золота изучена главным образом на примере миокризина. Установлено, что после однократного введения 50 мг препарата его уровень в сыворотке достигает максимума — 700 мкг/дл — через 2 ч., а период полувыведения препарата равен 7 дням. Через 6-8 нед. после еженедельного введения концентрация золота в сыворотке стабилизируется на уровне 300400 мкг/дл. При введении 50 мг в 3-4 нед. в качестве поддерживающей дозы концентрация золота в сыворотке составляет 75-125 мг/дл. После приема ауранофина (6 мг/день) его уровень в сыворотке постепенно нарастает в течение 3 мес., а затем выходит на плато. Около 40% внутримышечно введенного миокризина быстро выводится из организма, 70% — с мочой, 30% — с каловыми массами. При приеме ауранофина адсорбируется примерно 25% золота. Выведение ауранофина более полное, чем парентеральных препаратов: 95% с каловыми массами, 5% с мочой. Степень задержки золота в организме зависит от ряда факторов, в том числе от дозы, частоты и лекарственной формы. У больных, получающих еженедельные инъекции, в организме задерживается 60%, а при ежемесячном введении — только 20%. При приеме ауранофина задерживается менее 30% препарата. В настоящее время возник интерес к изучению фармакологической активности сульфгидрильных компонентов препаратов золота. Показано, что при еженедельном введении миокризина его меркаптосукцинатный компонент быстро исчезает из плазмы, 60% обнаруживается в моче на следующий день после введения препарата. У экспериментальных животных меркаптосукцинат обнаруживается в почках, печени, селезенке, легких, мышцах и коже (E. Jellum и E. Munthe, 1982). При изучении фармакокинетических параметров было установлено, что клиническая эффективность кризотерапии не коррелирует с уровнем золота в цельной крови. Однако, по данным атомно-абсорбционной спектрометрии, развитие токсических реакций ассоциируется с увеличением уровня свободного золота по отношению к общему уровню золота (M. Heath и соавт., 1988). Механизм действия Несмотря на длительную историю применения препаратов золота в ревматологии, механизмы, обеспечивающие его терапевтическую эффективность, остаются не ясными. Между тем фармакологические эффекты препаратов золота, зарегистрированные in vitro и in vivo, весьма многообразны. Фармакологические эффекты препаратов золота • Подавление роста микроорганизмов • Подавление митоген-индуцированного синтеза IgM РФ (N. J, 0lsen, H. E. Jasin, 1988) • Подавление митоген/антиген-индуцированной пролиферации лимфоцитов (P. E. Lipsky, M. Ziff, 1977; D. E. Grisword и ñîàâò., 1985) • Уменьшение инфильтрации синовиальной ткани Т- и В-лимфоцитами (M. Rooney и соавт., 1989) • Стимуляция (низкие дозы золота) или ингибиция (высокие дозы) синтеза ИЛ-1 макрофагами (V. A. Danis и соавт., 1990) • Ингибиция ИЛ-1-стимулированной пролиферации фибробластов (T. Matsubara и M. Ziff, 1987) 119 • Усиление синтеза растворимого антагониста ИЛ-1 (M. Shingu и соавт., 1995) • Подавление хемотаксиса нейтрофилов, фагоцитоза, деактивация синглетного кислорода (Р. Но и соавт., 1978; A. G. Mowat, 1978; J. Jesop и соавт., 1973; M. Hart и соавт., 1985; E. J. Corey и ñîàâò., 1987) • Гипогаммаглобулинемия с селективным дефицитом IgA и IgG (F. Guillemin и соавт., 1987) • Уменьшение экспрессии молекул адгезии (ECLAM-1, VCAM-1 и Е-селектин) (M. Corkill и соавт., 1991) • Подавление пролиферации ЭК и неоваскуляризации (R. Saura и соавт., 1994) • Снижение активности протеолитических ферментов (N. Penneys и соавт., 1974) • Ингибиция миелопоэза (J. A. Hamilton, 1993) • Инактивация C1-êîìïîíåíòà комплемента Показано, что моновалентные препараты золота in vitro подавляют активность аденилатциклазы в мембранах нестимулированных и стимулированных форсколином Т- и В-лимфоцитов, что свидетельствует о возможной роли цАМФ в реализации иммуномодулирующих эффектов кризотерапии (M. B. Lazarevic и соавт., 1992). Полагают, что не только сама молекула золота, но и солевой компонент некоторых препаратов может обладать противовоспалительными свойствами. Известно, что миокризин быстро диссоциирует в организме, после чего золото связывается с сульфгидрильными группами белков, а свободная натриевая соль ауротиояблочной кислоты (меркаптосукцинат) частично накапливается в тканях (E. Jellum и соавт., 1980). Отсутствие корреляции между концентрацией золота в крови и терапевтической эффективностью препарата позволило высказать предположение о том, что сам меркаптосукцинат может обеспечивать противовоспалительную активность миокризина (S. R. Rudge и соавт., 1984; E. Jellum и соавт., 1982). По данным P. M. Newman и соавт. (1994), меркаптосукцинат имитирует некоторые эффекты миокризина, в частности проявляет способность подавлять экспрессию молекул адгезии ЭК. Предварительные результаты свидетельствовали об эффективности меркаптосукцината при PA (E. Munthe и E. Jellum, 1983), хотя в дальнейшем другие авторы не подтвердили эти данные (S. R. Rudge и соавт., 1988). Полагают, что механизм действия как миокризина, так и меркаптосукцината на экспрессию молекул адгезии связан с их способностью увеличивать уровень цАМФ в ЭК. Препараты золота в очень низких концентрациях in vitro снижают число миелоидных костномозговых колоний. Это может приводить к уменьшению образования зрелых моноцитов и гранулоцитов и тем самым к снижению их поступления в воспаленный сустав (J. A. Hamilton и N. Williams, 1985; 1987). Клиническое применение и тактика лечения Показания к назначению препаратов золота суммированы в таблице 4.1. Таблица 4.1. Применение солей золота для лечения ревматических заболеваний (по D. A. Gordon. 1989; 1993) Заболевание Примечание Активный, прогрессирующий РА, включая Лечение контролирует активность и рентгенологическое синдром Фелти и синдром Шегрена прогрессирование; эффективность подтверждена в многочисленных контролируемых исследованиях Ювенильный РА (полиартикулярный) Эффективность подтверждена в контролируемых исследованиях Псориатический артрит Умеренно превосходит НПВС Дискоидная красная волчанка (ауранофин) Отдельные наблюдения Лечение кризанолом начинают с одной или нескольких пробных инъекций небольших доз (8.5-17 мг металлического золота, то есть 0.5-1,0 мл 5% раствора кризанола с интервалом 7 дней). Затем переходят на однократное еженедельное введение 2 мл 5% или 1 мл 10% кризанола в течение 7-8 мес. При хорошей переносимости и достижении клинического эффекта интервал между инъекциями удлиняют до 2 нед. и более. Поддерживающее лечение кризанолом с введением 34 мг золота 1 раз в 3-4 нед. может проводиться очень длительно. Если в период поддерживающей терапии наблюдается обострение РА, больному назначают более частые инъекции (В. А. Насонова и Я. А. Сигидин, 1985). Лечение миокризином начинают с введения 25 мг препарата, а затем дозу увеличивают до 50 мг в неделю. После достижения ремиссии продолжают лечение препаратом в дозе 25-50 мг в 2-4 нед. Ауранофин обычно назначают в дозе 3 мг 2 раза в день, хотя некоторым больным для достижения клинического эффекта требуется доза 9 мг/день, а другим — только 3 мг/день. Развитие диареи и желудочных спазмов является основным фактором, ограничивающим возможность увеличения дозы препарата. При ювенильном РА ауранофин назначают из расчета 0.1-0.2 мг/кг в день. Как уже отмечалось, эффективность солей золота при РА была продемонстрирована более 50 лет назад. Соли золота были первым препаратом, тестированным в контролируемых исследованиях (T. N. Faser, 1945). Многочисленные данные литературы, свидетельствующие об эффективности кризотерапии при РА в сравнении с плацебо и другими базисными противоревматическими препаратами, суммированы D. A. Gordon (1993). Имеются данные о том, что длительное лечение парентеральными препаратами золота приводит к увеличению продолжительности жизни больных PA (K. Lehtinin и H. Isomaki, 1991). Основным показанием к назначению солей золота является достоверный прогрессирующий полиартикулярный РА. Многие ревматологи по прежнему предпочитают начинать лечение РА базисными препаратами именно с солей золота. При этом имеются данные об эффективности кризотерапии как у больных с ранним неэрозивным артритом, так и у больных в развернутой стадии заболевания с тяжелым поражением суставов, а также ревматоидным моно- или олигоартритом (А. Б. Шехтер и соавт., 1990; D. A. Gordon, 1993). Лечение солями золота не показано в поздней стадии заболевания при отсутствии признаков активного синовита. Эффективность кризотерапии не зависит от пола и возраста больных, наличия и характера внесуставных проявлений, степени увеличения СОЭ и присутствия РФ. При синдроме Фелти лечение препаратами золота позволяет в ряде случаев уменьшить выраженность нейтропении и спленомегалии (A. M. Dilon, 1986), а у больных РА с хронической анемией способствует положительной динамике показателей красной крови. Не отмечено ухудшения переносимости препаратов золота у больных РА с синдромом Шегрена (M. H. Gordon и соавт., 1975). Не рекомендуется назначать соли золота больным с протеинурией и дерматитом, что в первую очередь связано с трудностями мониторирования побочных эффектов кризотерапии. Эффективность ауранофина изучена хуже, чем парентеральных препаратов золота. В целом, по данным большинства исследователей, при РА ауранофин уступает парентеральным препаратам золота и другим базисным противоревматическим препаратам по эффективности, но несколько превосходит их по переносимости (Г. А. Слободина и соавт., 1988; P. Clark и соавт., 1989; D. T. Felson и соавт., 1990). В двойном слепом контролируемом исследовании, проведенном в США и России, было показано,, что ауранофин относительно безопасный препарат при ювенильном РА, однако его эффективность по сравнению с плацебо была статистически недостоверной (E. H. Giannini и соавт., 1990). Выявлена более высокая эффективность парентеральных препаратов золота по сравнению с ауранофином у больных псориатическим артритом (J. Palit и соавт., 1990). Появились наблюдения об успешном применении ауранофина при тяжелой дискоидной красной волчанке (K. Dalzier и соавт., 1986). Доза препарата составила 3 мг 2 раза в день, а продолжительность лечения — 1 год. У 4 из 23 больных развилась полная ремиссия, а у 25—частичное улучшение. Побочные эффекты (таблица 4.2.) На фоне парентерального введения солей золота побочные реакции в среднем наблюдаются у трети больных с колебаниями от 5 до 80% (D. A. Gordon, 1993). При этом частота и спектр побочных эффектов парентеральных препаратов и ауранофина существенно различаются, Например, поражение слизистых и некоторые другие тяжелые осложнения, наблюдаемые при введении внутримышечном введении препаратов, реже встречаются при приеме ауранофина (J. R. Ward и соавт., 1983). Прервать лечение из-за токсических реакций вынуждены около 30% больных, леченных парентеральными препаратами золотом, и только 5%, принимающих ауранофин. Имеются данные об иммуногенетической предрасположенности к развитию некоторых токсических реакций на фоне кризотерапии. Протеинурия и тромбоцитопения чаще наблюдаются у носителей HLA-DR3: относительный риск протеинурии составляет 32, тромбоцитопении — 9. Таблица 4.2. Побочные эффекты кризотерапии (по D. A. Gordon, 1993) Побочный эффект Парентеральные Ауранофин препараты золота Поражение кожи и слизистых оболочек: +++ +++ • дерматит/стоматит +++ +++ • зуд ++ + • алопеция ++ + • крапивница ++ + • трофические изменения ногтей ++ + хризиазис/пигментация фотосенсибилизация ++ + Нарушение костномозгового кроветворения: ++ ++ • лейкопения • тромбоцитопения • эозинофилия • лимфоцитопения • апластическая анемия Нарушение функции почек: • протеинурия • гематурия • нефротический синдром • почечная недостаточность Поражение ЖКТ: • энтероколит Поражение печени: • некроз гепатоцитов • холестатическая желтуха Поражение поджелудочной железы: • панкреатит Поражение легких: • легочная инфильтрация Поражение нервной системы: • полинейропатия • энцефалопатия Постинъекционные реакции: • нитроидные (вазомоторные) • анафилактические • миалгия/артралгия Поражение глаз: • хризиазис роговицы и хрусталика • конъюнктивит • ирит/язвы роговицы Другие: • металлический привкус во рту • головные боли ++ +++ + ++ ++ ++ ++ + +++ + ++ ++ ++ ++ ++ + ++ +++ ++ ++ + + ++ ++ ++ ++ ++ ++ ++ ++ + ++ +++ ++ + ++ +++ ++ ++ ++ + + +++ ++ +++ ++ Примечание: +++ — частые; ++ — редкие; + — очень редкие Поражение кожи и слизистых оболочек Дерматит и стоматит относятся к числу самых частых побочных эффектов кризотерапии — 6080% от их общего количества. Дерматит может проявляться разнообразными кожными высыпаниями, в большинстве случаев сопровождается кожным зудом, длится до 2 мес., может сочетаться с другими токсическими проявлениями: трофическим изменением ногтей, эозинофилией, протеинурией. Поражение кожи и слизистых иногда ассоциируется с синтезом IgE антител к вводимым препаратам золота (J. Betza и соавт., 1983). Развитие дерматита является основанием для отмены препарата до полного исчезновения всех кожных проявлений. В последующем допускается попытка возобновления лечения, но в очень низких дозах с постепенным их увеличением. Интересно, что у некоторых больных развивается контактная аллергия на золотые украшения (I. P. Wicks и соавт., 1988). Редким кожным осложнением кризотерапии является изменение окраски кожи (серый или голубой цвет) в сочетании с гиперпигментацией, которое получило название "хризиазис". Имеются данные о том, что развитие кожно-слизистых осложнений ассоциируется с присутствием в сыворотках антител к Ro-àíòèãåíó (M. Tishler и соавт., 1994). Поражение почек Поражение почек может проявляться транзиторной протеинурией, микроскопической гематурией и нефротическим синдромом (C. L. Hall и соавт., 1987). Частота протеинурии нарастает при увеличении дозы препарата (A. Cats, 1976), но иногда она исчезает, несмотря на продолжение лечения. В некоторых случаях повторное назначение препаратов золота, но в меньших дозах, после прекращения лечения в связи с развитием протеинурии позволяет избежать рецидивов протеинурии (A. V. Klinkhoff и A. Teufel, 1994). Тяжелым ранним осложнением кризотерапии является нефротический синдром, который ассоциируется с носительством HLA-B8/DR3 (C. L. Hall, 1989). Нефротический синдром редко приводит к почечной недостаточности и обычно заканчивается выздоровлением после отмены препаратов. Поражение почек крайне редко развивается на фоне приема ауранофина, что, несомненно, является достоинством этого препарата. Гематологические нарушения К гематологическим осложнениям кризотерапии относят эозинофилию, лейкопению, агранулоцитоз, тромбоцитопению, анемию, панцитопению, апластическую анемию. Иногда повышение уровня эозинофилов коррелирует с увеличением концентрации сывороточного IgE. Тромбоцитопения является редким (1-3%), но потенциально смертельным осложнением кризотерапии (J. D. Adachi и соавт., 1987), иногда развивается через много месяцев после прекращения лечения и ассоциируется с носительством HLA-DR3. Механизмы тромбоцитопении связывают как с костномозговой супрессией, так и с синтезом антител к тромбоцитам. Необходимо иметь в виду, что если кризотерапия вызывает тромбоцитопению, то она, как правило, развивается и на фоне применения других базисных противоревматических препаратов, включая ауранофин (M. J. Wijnands и соавт., 1990; F. V. Cicuttini и соавт., 1988). В отличие от тромбоцитопении нейтропению и лимфопению не относят к числу серьезных побочных эффектов кризотерапии. Самым тяжелым осложнением лечения препаратами золота является панцитопения, связанная с аплазией костного мозга, часто заканчивающаяся смертельным исходом (A. Yan и P. Davis, 1990). Иногда лечение препаратами золота приводит к генерализованной гипогаммаглобулинемии, вплоть до синдрома комбинированного иммунодефицита взрослых, сопровождающегося тяжелыми инфекционными осложнениями (B. G. A. Stuckey и соавт., 1986; J. C. Olson и соавт., 1986; D. O. Hascard и D. Macfarlane, 1988), Поражение легких Легочные осложнения кризотерапии проявляются непродуктивным кашлем, одышкой, плевральными болями. При рентгенологическом исследовании выявляются инфильтраты, а при функциональном исследовании — изменения, характерные для рестриктивного поражения легких ("золотое" легкое). Поражение легких, как правило, развивается у больных, получивших суммарно не менее 500 мг препарата золота, и купируется при их отмене и назначении ГК. Поражение ЖКТ У некоторых больных, преимущественно женщин среднего возраста, на фоне парентерального введения небольших доз препарата развивается тяжелый энтероколит, нередко приводящий к летальному исходу (A. G. Fam и соавт., 1986). Клинически это осложнение проявляется болями в животе, диареей, тошнотой, рвотой, лихорадкой. Диарея существенно чаще развивается при лечении ауранофином, чем парентеральными препаратами золота. У 5% больных, леченных ауранофином, развитие диареи является основанием для прекращения лечения. Холестатическая желтуха с гипергаммаглобулинемией, повышением уровня трансаминаз и щелочной фосфатазы, часто сопровождающаяся эозинофилией, — хорошо известное осложнение лечения препаратами золота, как правило, развивающееся в ранние сроки терапии и связанное с реакцией гиперчувствительности (J. Edelman и соавт., 1983). Редким осложнением кризотерапии является панкреатит (A. D. Eisemann и соавт., 1989). Поражение ЦHC Поражение ЦНС может протекать по типу синдрома Гийена— Барре, энцефалопатии или пареза черепно-лицевых нервов (A. G. Fam и соавт., 1984). На фоне лечения ауранофином описано развитие энцефалопатии и моторной невропатии. (P. Gambari и соавт., 1984). Поражение глаз Частота отложения золота в роговице и хрусталике четко связана с кумулятивной дозой. Постинъекционные реакции Нередко наблюдаются постинъекционные реакции вазомоторного (нитроидного) типа, проявляющиеся слабостью, головокружением, тошнотой, рвотой, покраснением лица, снижением АД. Особенно часто они встречаются у лиц, получающих на фоне внутримышечного введения препаратов золота ингибиторы АПФ (L. A. Healey и Backes, 1989). Невазомоторные реакции характеризуются болями и отеком суставов, миалгиями, которые появляются через 6-24 ч после введения препаратов. В целом развитие постинъекционных реакций не является основанием для прерывания лечения. Столь широкий спектр токсических реакций на фоне парентерального лечения препаратами золота диктует необходимость тщательного клинико-лабораторного наблюдения за больными. Рекомендации по наблюдению за больными в процессе лечения препаратами золота: 1. 2. 3. 4. Тщательный сбор анамнестических данных о лекарственной аллергии, поражении кожи, почек. Полный общий анализ крови, мочи, биохимический профиль. Повторный общий анализ крови и мочи каждые 1-3 нед. Прерывание лечения при появлении перечисленных выше клинических симптомов или лабораторных нарушений, таких, как эозинофилия, лейкопения, анемия, гематурия, протеинурия. 5. Учитывая возможность вазомоторных реакций, следует вводить препарат в положении больного лежа, не назначать ингибиторы АПФ. 6. Не назначать препараты беременным и кормящим женщинам. Лечение токсических осложнений кризотерапии Большинство побочных реакций купируются при отмене препарата в течение нескольких недель. Дерматит контролируется антигистаминными препаратами, стероидными мазями, при генерализованном дерматите помогают ГК. Умеренные или высокие дозы ГК (20-60 мг/день) в большинстве случаев оказываются эффективными при нефротическом синдроме, тромбоцитопении, в меньшей степени — при энтероколите, поражении легких, гематологических нарушениях. Больным, которые не реагируют на ГК, целесообразно назначать хелирующие препараты (D. A. Gordon, 1993). Имеются данные о возможности коррекции гематологических нарушений на фоне лечения солями золота с помощью рекомбинантного ГМ-КСФ (глава 15). Недавно для лечения этого осложнения стали использовать пересадку костного мозга. Список литературы К главе "Препараты золота" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Насонова В. А., Сигидин Я. А. Патогенетическая терапия ревматических болезней, М. Медицина. 1985. стр. 94-99. Слободина Г. А., Каневская М. З., Крель А. А., Чичасова Н. В., Покрышкин В. И.: Возможности перорального препарата золота (ридауры) в лечении ревматоидного артрита по данным проспективного наблюдения. Терапевт, архив 1988; 12: 80-84. Шехтер А. Б., Слободина Г. А., Крель А. А., Каневская М. З., Чичасова Н. В.: Сравнительная оценка эффективности и переносимости препаратов золота и Д-пеницилламина у больных ревматоидным артритом. II. Влияние на морфологические признаки ревматоидного синовита. Ревматология 1990; 2; 23-29. Adachi J. D., et al.: Gold Induced thrombocytopenia: 12 cases and review of the literature. Semin. Arthritis Rheum. 1987; 16: 288-293. Bretza J., Wells I., Novey H. S.: Association of IgE antibodies to sodium aurothiomalate and adverse reactions to chrysotherapy for rheumatoid arthritis. Amer. J. Med. 1983; 74: 945. Champion G. D., Graham G. G., Ziegler J. B.: The gold complexes. Bailliere's Clin. Rheumatol. 1990; 4: 491-534. Clark P., Tugwell P., Bennett K., Bombardier C.: Meta-analysis in injectable gold In rheumatoid arthritis. J. Rheumatol. 1989; 16: 442. Corey E. J., Mehrotta M. M., Khan A. U.: Antiarthritic gold compaunds effectively quench electronically excited singlet oxygen. Science 1987; 236: 68. Corkill M. M., Kirkman B. W., Haskard D. O., et al.: Gold treatment of rheumatoid arthritis decreases synovial expression of the endothelial leukocyte adhesion receptor ELAM-1. J. Rheumatol. 1991; 18: 1453-1460. Dalziel К., Going G., Cartwright P. H., et al.: Treatment of chronic discoid lupus erythe-matosus with oral gold compaund (auranofin). Brit. J. Dermatol. 1986; 115: 211-216. Danis V. A., Kulesz A. J., Nelson D. S., Brook P. M.: The efffect of gold sodium thiomalate and auranofin on lipopolysaccharide-induced interleukin-I production by monocytes in vitro: variation in healthy subjects and patients with arthritis. Clin. Exp. Immunol. 1990; 79: 335-340. Dilon A. M., Luthra H. S., Conn D. L. et al.: Parental gold therapy in the Felty syndrome. Experience with 20 patients. Medicine (Baltimore)1986; 65: 107. Gordon M. H., Tiger L. H., Ehlich C. E.: Gold reactions are not more common in Sjogren's syndrome. Ann. Intern. Med. 1986; 82: 47. 14. Gordon D. A.: Gold compaunds in the rheumatic disease. In: Textbook of rheumatology. Ed. Kelly W. N., Harris E. D., Ruddy S., Sirdge C. B. W. B. Saunders Comp. Philadelphia London Toronto. 1993: 743-759. 15. Felson D. T., Anderson J. J., Meenan R. F.: The comparative efficacy and toxicity of second-line drugs in rheumatoid arthritis. Arthritis Rheum. 1990; 33: 1449. 16. Hart Ì.: Mechanisms of action of disease modifying antirheumatic drugs. J. Rheumatol. 1992 (suppl. 32); 19: 100-103. 17. Hall C. L., et al.: The natural course of gold nephropathy: long term study of 21 patients. Br. Med. J. 1987; 295: 745-748. 18. Hamilton J. A., Williams N.: In vitro inhibition of myelopoiesis by gold salts and D-peni-cillamine. J. Rheumatol. 1985; 12: 892-896. 19. Hamilton J. A., Williams N.: Elects of auronofin and other antirheumatic drugs on human myelopoiesis. J. Rheumatol. 1987; 14: 216-220. 20. Hamilton J. A.: Rheumatoid arthritis: opposite actions of haemopoietic growth factors and slowacting anti-rheumatic drugs. Lancet 1993; 342: 536-539. 21. Healey L. A., Blackes M. B.: Nitritoid reactions and angiotensin-converting-enzyme inhibitors. J. Rheumatol. 1989; 321: 763. 22. Heath M. J.: Measurement of "free" gold in patients receiving disodium aurothiomalate and the association of high free to total gold levels with toxicity. Ann. Rheum. Dis. 1988; 47: 18 23. Klinkhof A. V., Teufel A.: Reinstitution of intramuscular gold after gold induced proteinuria. Arthritis Rheum. 1994; 37 (suppl.): S-255. 24. Kawakami A., Eguchi K., Migita H., et al.: Inhibitory effects of gold sodium thiomalate on the prolipheration and interferon-gamma induced HLA-DR expression in human endothelial cells. J. Rheumatol. 1990: 17: 430-435. 25. Lazarevic M. B., Van K., Swedler W. I., et al.: Effect of gold compounds on the activity of adenylyl cyclase In human lymphocyte membrane. Arthritis Rheum. 1992; 35: 857-864. 26. Lihtinin K., Isomaki H.: Intramuscular gold therapy is associated with long term survival in patients with rheumatoid arthritis. J. Rheumatol. 1991; 18: 524-529. 27. Lipsky P. E., Ziff Ì.: Inhibition of antigen-and mitogen-induced human lymphocyte prolipheration by gold compaunds. J. Clin. Invest. 1977; 59: 455-466. 28. Matsubara Т., Ziff Ì.: Inhibition of human endothelial cell proliferation by gold compaunds. J. Clin. Invest. 1987; 79: 1440-1446. 29. Munthe E., Jellum E.: Is gold nessesarry in so called chrysotherapy? J. Rheumatol. 1983 (suppl.); 51: 46-51. 30. Newman P. M., To S. S. T., Robinson B. G., et al.: Effect of gold sodium thiomalate and its thiomalate component on the in vitro expression of endothelial cell adhesion molecules. J. Clin. Invest. 1994; 94: 1864-1871. 31. Palit J., Hill J., Capell H. A., et al.: A multi-centre double-blind comparison of auranofine, intramuscular gold thiomalate and placebo in patients with psoriatic arthritis. Br. J.Rheumatol. 1990; 29: 280. 32. Saura R., Matsubara Т., Mizuno K.: Inhibition of neovascularisation in vivo by gold compounds. Rheumatol. Int. 1994; 14: 1-7. 33. Scosey J. L. Gold compaunds. In: Arthritis and allied conditions. A Textbook of rheumatology. Ed. D. J. McCarty. Lea/Febiger. Philadelphia, London. 1989, p. 544-555. 34. Scheinberg M. A., Santos L. M., Finkelstein A. E.: The effect of auranofin and sodium aurothiomalate on peripheral blood monocytes. J. Rheumatol. 1982; 9: 366-369. 35. Shingu S., Fujikawa Y., Wada Т., et al.: Increased IL-I receptor antagonist (IL-1ra) production and decreased IL-I beta/IL-1ra ratio in mononuclear cells from rheumatoid arthritis patients. Brit. J. Rheumatol. 1995; 34: 24-30. 36. Tishler Ì., Golbrut B., Shoenfeld Y., Yaron M.: Anti-Ro (SSA) antibodies in patients with rheumatoid arthritis- a possible marker for gold induced side effects. J. Rheumatol. 1994; 21: 10401042. 37. Van A., Davis P.: Gold induced marrow supression: a review of 10 cases. J. Rheumatol. 1990; 17: 47-51 ГЛАВА 5 D-ПЕНИЦИЛЛАМИН И ДРУГИЕ ТИОЛСОДЕРЖАЩИЕ ВЕЩЕСТВА С БЛИЗКОЙ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ 5.1. D-ПЕНИЦИЛЛАМИН Пеницилламин (3,3-диметилцистеин) — трифункциональная аминокислота, содержащая карбоксильные, амино- и сульфгидрильные группы и являющаяся аналогом естественной аминокислоты цистеина. Наличие асимметрически расположенного атома углерода позволяет пеницилламину существовать в виде D- и L-èçîìåðîâ. Пеницилламин, получаемый при контролируемом гидролизе пенициллина, существует только в форме D-èçîìåðà, который в настоящее время и используется в клинической практике (L. A. Jaffe, 1989). Являясь водорастворимым веществом, D-ïåíèöèëëàìèí хорошо адсорбируется в верхних отделах ЖКТ, выделяется с мочой в виде окисленных метаболитов, обладает способностью длительно задерживаться в тканях после прекращения лечения. Механизм действия 1. Хелирование тяжелых металлов (медь, цинк, золото, ртуть) и участие в реакции сульфгидрилсульфидного обмена растворимых в воде активных сульфгидрильных групп D-ïåíèöèëëàìèíà (предполагается, что этот механизм определяет способность D-ïåíèöèëëàìèíà снижать содержание меди при болезни Вильсона). 2. Нарушение образования витамина В6, играющего роль в функционировании иммунной системы, за счет тиазолидинового связывания D-ïåíèöèëëàìèíà с пиридоксинфосфатом. 3. Нарушение перекрестного связывания молекул коллагена и увеличение содержания водорастворимого коллагена в связи со взаимодействием D-ïåíèöèëëàìèíà с альдегидными группами коллагена. 4. Нарушение образования IgM РФ (полимерной молекулой, отдельные субъединицы которой связаны S-S мостиками) за счет межцепочечного обмена сульфгидрильных (SH) групп молекулы D-ïåíèöèëëàìèíà и дисульфидных (S-S) связей различных макромолекул. Механизмы, определяющие фармакологическую активность D-ïåíèöèëëàìèíà именно при ревматических заболеваниях, до конца не ясны. Примечательно, что в отличие от других базисных противоревматических препаратов D-ïåíèöèëëàìèí не подавляет развитие поражения суставов при экспериментальном моделировании артрита. Однако на фоне лечения больных и in vitro описаны многообразные иммунологические и противовоспалительные эффекты, имеющие непосредственное отношение к иммунопатологическим механизмам развития ревматических заболеваний: 1. селективная ингибиция активности CD4+T-ëèìôoöèòîâ (Т-хелперы); подавление синтеза ИФ-у и ИЛ-2 CD4+T лимфоцитами; 2. подавление синтеза РФ, образования циркулирующих иммунных комплексов и диссоциация РФ-содержащих иммунных комплексов; 3. антипролиферативное действие на фибробласты; 4. подавление пролиферации эндотелия и неоваскуляризации; 5. ускорение дисмутации супероксидов; 6. подавление миелопоэза. D-ïåíèöèëëàìèí не подавляет функциональную активность макрофагов, В-лимфоцитов и CD8-Tcynpeccopныx клеток in vitro, но селективно ингибирует активность CD4+T-xeлперных лимфоцитов. (P. E. Lipsky и M. Ziff, 1980). По данным M. Walters и соавт. (1987), лечение D-ïåíèöèëëàìèíîì вызывает существенное снижение содержания CD4+ Т-лимфоцитов в синовиальной ткани больных РА. На фоне лечения D-ïåíèöèëëàìèíîì обычно наблюдается снижение уровня иммунных комплексов и IgG и IgM РФ в сыворотке и синовиальной жидкости (I. A. Jaffe, 1975). В опытах in vitro было показано, что Dïåíèöèëëàìèíà обладает способностью подавлять пролиферацию фибробластов человека (T. Matsubara и соавт., 1988). Полагают, что этот механизм обусловливает способность D-ïåíèöèëëàìèíà влиять на образование ревматоидного паннуса и неконтролируемую продукцию коллагена при системной склеродермии. Кроме того, D-ïåíèöèëëàìèí ингибирует пролиферацию эндотелиальных клеток и неоваскуляризацию (M. Tsukasa и соавт., 1989). По мнению J. A. Hamilton (1993), один из возможных механизмов действия D-ïåíèöèëëàìèíà (как и других базисных противоревматических препаратов) связан с подавлением миелопоэза, что приводит к медленному снижению числа зрелых моноцитов и гранулоцитов, мигрирующих в полость воспаленного сустава. Клиническое применение и тактика лечения D-ïåíèöèëëàìèí используется в лечении активного РА (серопозитивного и серонегативного), в том числе и с разнообразными системными проявлениями (васкулит, синдром Фелти, амилоидоз, ревматоидное поражение легких), палиндромного ревматизма (E. C. Huskisson, 1976), некоторых форм ювенильного артрита и ССД. По данным большинства исследователей, D-ïåíèöèëëàìèí не эффективен при центральной форме анкилозирующего спондилита и при других HLA-Â27-íåãàòèâных спондилоартропатиях (H. A. Bird и A. Dixon, 1977; М. М. Steven и соавт., 1985). В начале лечения рекомендуется назначать препарат 1 раз в день по 125-250 мг за 1-2 ч до завтрака, а при дробном приеме вторую дозу D-ïåíèöèëëàìèíà следует принимать за 2-3 ч до ужина. Это связано с тем, что пища существенно снижает абсорбцию и биодоступность препарата (D. Perett, 1981). D-ïåíèöèëëàìèí назначают после еды только в том случае, если прием до еды вызывает развитие желудочно-кишечных симптомов. Через 8 нед. дозу препарата увеличивают на 125-250 мг/день. Полагают, что 8 нед. — оптимальное время для оценки клинической эффективности лечения Dïåíèöèëëàìèíîì. Увеличение дозы не на 250 мг/день, а на 125 мг/день особенно показано при появлении тошноты, рвоты, потере аппетита и других признаков токсичности. В том случае, если дневная доза Dïåíèöèëëàìèíà достигает 1 г/день, ее делят на 2 приема. Следует стремиться использовать не фиксированную дозу препарата, а подбирать оптимальную — в зависимости от клинической эффективности. Показано назначение витамина В6 (50-100 мг/день) и поливитаминных добавок, особенно при нарушении питания больных. Хотя клинические признаки дефицита витамина Â6 развиваются крайне редко, описаны больные с периферической нейропатией, купировавшейся только введением витамина Â6 (K. D. Pool и соавт., 1981). Недавно появилось сообщение о том, что при РА, интермиттирующий прием D-ïåíèöèëëàìèíà (3 раза в неделю через день) позволяет существенно снизить частоту побочных реакций при сохранении клинической эффективности (M. Hakoda и соавт., 1994). РА D-ïåíèöèëëàìèí относится к числу наиболее эффективных и токсичных препаратов, использующихся для лечения РА. В серии контролируемых исследований установлено, что при РА Dïåíèöèëëàìèí по эффективности не уступает солям золота и азатиоприну (Multicenter trial group, 1973; D. T. Felson и соавт., 1990). Оптимальная терапевтическая доза D-ïåíèöèëëàìèíà варьирует в очень широких пределах. Хотя небольшие дозы D-ïåíèöèëëàìèíà (в пределах 500 мг/день) значительно лучше переносятся и во многих случаях позволяют эффективно контролировать активность заболевания, некоторые исследователи полагают, что истинная модификация течения РА, судя по замедлению рентгенологического прогрессирования, достигается только при приеме препарата в дозе 1 г/день и более. Тем не менее в настоящее время в клинической практике используется лечение низкими дозами препарата. При лечении D-ïåíèöèëëàìèíîì требуется не менее 16 нед. от начала лечения для первичной оценки эффективности терапии. В этот период, который нередко удлиняется до 1-2 лет, больные должны продолжать прием "симптоматических" препаратов. Положительная динамика лабораторных показателей наблюдается не ранее чем через 6-9 мес. от начала терапии. Рентгенологическое улучшение, проявляющееся уменьшением числа эрозий в суставах, становится заметным не ранее чем через 2 года и более (I. A. Jaffe, 1989). Нередко в процессе лечения наблюдается обострение, иногда заканчивающееся спонтанной ремиссией, в других случаях требуется увеличение дозы препарата или перевод больного на двукратный прием дневной дозы. Обычно в процессе лечения частота обострений снижается и клиническое улучшение становится более стойким. Иногда обострение артрита является проявлением индуцированного D-ïåíèöèëëàìèíîì волчаночноподобного синдрома, для купирования которого необходима отмена препарата. Данные об оптимальной продолжительности лечения противоречивы. По данным M. Ahern и соавт. (1984), прекращение лечения в 80% случаев приводит к обострению РА. ССД Возможность использования D-ïåíèöèëëàìèíà при ССД определяется следующими факторами: 1. способностью препарата блокировать альдегидные группы, участвующие во внутримолекулярном и межмолекулярном перекрестном связывании зрелого коллагена, что приводит к увеличению количества растворимых форм и ускоряет обмен коллагена (M. E. Nimmi, 1979); 2. иммуносупрессивной активностью; 3. подавлением образования кислородных радикалов. D-ïåíèöèëëàìèí — препарат, наиболее широко использующийся для лечения ССД (M. I. V. Jayson и соавт., 1977; М. М. Иванова и соавт., 1977). D-ïåíèöèëëàìèí существенно уменьшает плотность кожи, развитие суставных контрактур, увеличивает выживаемость, снижает частоту поражения почек (V. D. Steen и соавт., 1982) и подавляет прогрессирование интерстициального легочного фиброза (T. A. Medsger, 1987; V. D. Steen и соавт., 1985; L. S. DeClerck и соавт., 1987). Однако около 25% больных ССД вынуждены прервать лечение из-за развития побочных реакций (V. D. Steen и соавт., 1987). Частоту побочных реакций можно снизить при очень медленном увеличении дозы препарата. Начинать лечение следует с дозы 250 мг/день, постепенно (в течение нескольких месяцев) увеличивая ее до 750-1000 мг/день. Лекарственные взаимодействия Поскольку D-ïåíèöèëëàìèí обладает очень высокой способностью реагировать с различными молекулами, оценка лекарственных взаимодействий имеет очень большое значение в процессе лечения этим препаратом. Поэтому промежуток между приемом D-ïåíèöèëëàìèíà и других лекарственных средств должен быть не менее 2 ч. D-ïåíèöèëëàìèí не следует назначать в сочетании с пероральными препаратами железа, поскольку отмена последних может привести к быстрому увеличению нефротоксичности фиксированной дозы Dпеницилламина (J. A. L. Harkness и D. R. Blake, 1982). При одновременном приеме D-ïåíèöèëëàìèíà и гидроксихлорохина наблюдается уменьшение терапевтической эффективности обоих препаратов (T. W. Bunch и соавт., 1984). Не следует принимать большие дозы аскорбиновой кислоты, которые вызывают инактивацию D-ïåíèöèëëàìèíà. В то же время взаимодействия между D-ïåíèöèëëàìèíîì и НПВС не обнаружено. D-ïåíèöèëëàìèí не следует назначать во время беременности, а ее развитие на фоне лечения является основанием для отмены препарата. Аллергические реакции на пенициллин или соли золота не являются противопоказанием к лечению D-ïåíèöèëëàìèíîì. Отсутствие эффекта от кризотерапии не может служить предиктором неэффективности D-ïåíèöèëëàìèíà. С особой осторожностью следует назначать D-ïåíèöèëëàìèí больным с нарушением функции почек, развитие которого является относительным противопоказанием к использованию этого препарата. Побочные эффекты (таблица 5.1) Основным фактором, ограничивающим использование D-ïåíèöèëëàìèíà в ревматологии, является высокая частота побочных эффектов. Некоторые из них имеют зависимый от дозы характер и купируются при кратковременном прерывании лечения или при уменьшении дозы препарата. Другие побочные эффекты связаны с идиосинкразией и не зависят от дозы (I. A. Jaffe, 1989). Большая часть побочных эффектов развивается в первые 18 мес. лечения, реже — в другие периоды терапии. Таблица 5.1. Побочные эффекты, возникающие на фоне лечения D-ïåøöèëëàìèíîì Частые, нетяжелые (не требуют отмены Частые, тяжелые (требуют прекращения лечения): препарата): • Тромбоцитопения • Понижение вкусовой чувствительности • Лейкопения • Дерматит • Протеинурия/нефротический синдром • Стоматит • Тошнота • Потеря аппетита Редкие, тяжелые: • Апластическая анемия • Аутоиммунные синдромы (миастения, пузырчатка, СКВ, синдром Гудпасчера, полимнозит, синдром Шегрена?) В процессе лечения необходимо тщательное наблюдение за больными, включающее клинический осмотр, исследование крови (в том числе определение количества тромбоцитов) и мочи в первые несколько месяцев лечения каждые 2 нед., а затем не реже одного раза в мес. Существует несколько факторов, предрасполагающих к развитию токсических реакций. К ним относятся: 1) носительство HLA-B8/DR3 (нефропатия); 2) нарушение способности к сульфоксидации (расщеплению карбоксицистеина), которое чаще, чем в норме, наблюдается при РА, что позволяет объяснить худшую переносимость D-ïåíèöèëëàìèíà при РА, чем при болезни Вильсона; 3) сочетание РА с синдромом Шегрена и продукция антител к SSA/Ro-àíòèãåíó; 4) синтез антител к D-пеницилламину. Кожный зуд и разнообразные виды кожной сыпи, появляющиеся в различные сроки лечения, являются самыми частыми побочными эффектами лечения D-ïåíèöèëëàìèíîì. Небольшой кожный зуд, кожная сыпь, афтозные язвы и стоматит в начале лечения часто проходит при кратковременной отмене, уменьшении дозы D-ïåíèöèëëàìèíà или назначении антигистаминных препаратов. Позднее поражение кожи носит более устойчивый характер и часто требует прерывания лечения. Язвы в ротовой полости обычно появляются в первые 6 мес., полностью исчезают после отмены D-ïåíèöèëëàìèíà, однако в половине случаев рецидивируют при повторном назначении препарата даже в минимальной дозе. Тем не менее у больных с кожными побочными эффектами, если лечение эффективно, допускается по крайней мере одна попытка повторного назначения D-ïåíèöèëëàìèíà. У ряда больных описано развитие тяжелого поражения кожи, так называемая Dïåíèöèëëàìèíèíäóöèðîâàííàÿ пузырчатка, требующая окончательной отмены препарата и назначения ГК. Обычно этот тип токсичности развивается не ранее чем через 2 мес. от начала лечения. Поражение ЖКТ Желудочно-кишечные расстройства, как правило, не очень тяжелые, проявляются потерей аппетита, тошнотой, обычно проходят самостоятельно в течение 2-3 мес., несмотря на продолжение лечения. Для уменьшения этих проявлений рекомендуется принимать препарат с едой. Тяжелое поражение печени наблюдается редко (B. D. Macleod и T. B. Kinsella, 1979; J. Rosenbaum и соавт., 1980). Гематологические нарушения Гематологические нарушения относятся к числу наиболее тяжелых побочных эффектов Dïåíèöèëëàìèíà, проявляются развитием лейкопении, тромбоцитопении и апластической анемии. Количество лейкоцитов менее 3000/мм3 и тромбоцитов менее 100000/мм3 является основанием для прекращения лечения. В некоторых случаях развитие тромбоцитопении и нейтропении связано с дозой препарата. В этих случаях возможна попытка возобновить лечение, тщательно титруя дозу Dпеницилламина. Лекарственная апластическая анемия, связанная с идиосинкразией к D-ïåíèöèëëàìèíó, может возникнуть в любой период болезни и не зависит от дозы. Поскольку тромбоцитопения, агранулоцитоз и/или апластическая анемия могут развиться в интервале между проведением лабораторных тестов, следует информировать больных о необходимости самостоятельного прерывания лечения в случае появления следующих симптомов: кровоточивости, геморрагий, лихорадки, боли в горле, гриппоподобных симптомов, При развитии зависимой от дозы цитопении нормализация гематологических показателей происходит в течение недели, однако идиосинкразическая аплазия может персистировать в течение нескольких месяцев. Как и при других апластических состояниях, у больных индуцированной Dïåíèöèëëàìèíîì аплазией не рекомендуется проведение глюкокортикоидной терапии. Поражение почек Нефропатия мембранозного типа, связанная с отложением циркулирующих иммунных комплексов, проявляется бессимптомной протеинурией или признаками нефротического синдрома. Полагают, что основанием для отмены D-ïåíèöèëëàìèíà является суточная протеинурия, превышающая 2 г (C. L. Hall и R. Tighe, 1989). Микроскопическая гематурия, как правило, доброкачественная и не может служить основанием для прекращения лечения (D. Barraclough и соавт., 1981). Описано развитие синдрома Гудпасчера, потребовавшее гемодиализа и иммуносупрессивной терапии (T. E. Gavaghan и соавт., 1981). Поражение легких Поражение легких относится к числу редких осложнений лечения D-ïåíèöèëëàìèíîì. Развитие кровохарканья позволяет заподозрить индуцированный D-ïåíèöèëëàìèíîì синдром Гудпасчера. Описано развитие фиброзирующего альвеолита, связь которого с лечением подтверждается быстрым исчезновением клинических и рентгенологических изменений после отмены препарата (C. J. Eastmond, 1976). Поражение нервной системы Нервно-мышечные расстройства иногда могут проявляться развитием миастении гравис (R. C. Bucknall и соавт., 1975) и ПМ/ДМ (К. Таkahashi и соавт., 1986), клинически не отличимых от идиопатических форм этих заболеваний. Отмена препарата приводит к исчезновению клинических признаков данных осложнений. Аутоиммунные нарушения Уникальной особенностью D-ïåíèöèëëàìèíà является его способность индуцировать развитие ряда аутоиммунных нарушений, в число которых входят наряду с перечисленными выше формами нервномышечной патологии пузырчатка, синдром Гудпасчера, волчаночноподобный синдром (A. Chalmers и соавт., 1982) и, вероятно, синдром Шегрена и хронический тиреоидит. У некоторых больных в сыворотке обнаруживают антитела к инсулину и инсулиновым рецепторам, ассоциирующиеся с развитием гипогликемии (E. A. Benson и соавт., 1985). 5.2. БУЦИЛЛАМИН (Bucillamine) Буцилламин-[N-(2-меркапто-2-метилпропионил)-L-цистеин) — новый базисный антиревматический препарат (Y. Shiokawa и соавт:, 1985). Синтезирован Santen Pharmaceutical Company (Osaka, Japan). Является тиолсодержащим веществом, отличающимся от D-ïåíèöèëëàìèíà присутствием двух свободных сульфгидрильных групп, благодаря которым образует внутримолекулярный дисульфид (SA-981) между двумя сульфгидрильными группами. (S. Hirohata и P. E. Lipsky, 1993). В отличие от D-ïåíèöèëëàìèíà способен предотвращать развитие экспериментального артрита, индуцированного коллагеном типа II у крыс (M. Hayashi и соавт., 1991). Это свидетельствует о том, что по механизму действия он отличается от D-ïåíèöèëëàìèíà. Полагают, что иммуносупрессивный эффект буцилламина определяется двумя механизмами: 1. образованием активного метаболита внутримолекулярного дисульфида между двумя сульфгидрильными группами, обладающего способностью непосредственно подавлять функциональную активность В-лимфоцитов (S. Hirohata и P. E. Lipsky, 1993; 1994); 2. пеницилламиноподобным эффектом интактной молекулы, обладающей способностью подавлять активность Т-лимфоцитов (S. Hirihata и P. E. Lipsky, 1994). Недавно было показано, что в отличие от D-ïåíèцилламина, который оказывает супрессорное действие на Т-лимфоциты за счет образования перекиси водорода, буцилламин специфически ингибирует метаболизм клеток в поздней G1- и S-ôàçàõ клеточного цикла (S. Hirohata и соавт., 1994). Эффективность буцилламина при РА была подтверждена в нескольких клинических испытаниях (С. Аbе, 1985; S. Kashiwazaki и Y. Shiokawa, 1987). 5.3. ПИРИТИНОЛ (Pyritinol) Пиритинол (Recuperol) — дисульфид 5-тиопиридоксина. Используется главным образом для лечения сенильной депрессии. Поскольку он является структурным аналогом D-ïåíèöèëëàìèíà, то был использован как базисный противоревматический препарат (J. P. Camus и соавт., 1981) По данным открытых неконтролируемых испытаний (более 400 больных) пиритинол (300 мг 2 раза в день) по эффективности несколько уступает солям золота и D-ïåíèöèëëàìèíó, но превосходит их по переносимости (J. P. Camus и соавт., 1981). Результаты многоцентрового двойного слепого контролируемого испытания пиритинола в сравнении с ауранофином свидетельствуют не только о более высокой эффективности пиритинола, но и о более высокой частоте побочных эффектов (E. M. Lemmel и соавт., 1993). Однако в целом частота побочных реакций на фоне лечения пиритинолом (особенно тяжелых гематологических осложнений) существенно ниже, чем на фоне лечения D-ïåíèöèëламином. 5.4. ТИОПРОНИН (Thiopronin, alpha-mercaptopropionylglycine) Тиопронин — препарат, близкий к D-ïåíèöèëëàìèíó, используемый для лечения болезни Вильсона. В открытом исследовании, проведенном G. Pasero и M. L. Ciompi (1979), было показано, что тиопронин в дозе до 1 г/сут. вызвал клиническое улучшение у 19 из 32 больных РА в сроки от 4 до 18 мес. Кроме того, оказалось, что при лечении тиопронином частота и выраженность побочных эффектов такие же, как и при лечении D-ïåíèöèëëàмином. По данным B. Amor и соавт. (1979), назначавших препарат в максимальной дозе 1500 мг/день 32 больным РА, клинико-лабораторное улучшение, о котором судили по таким параметрам, как длительность утренней скованности, суставной индекс, сила сжатия кисти, отек суставов, СОЭ, отмечено у 23 больных. Основными побочными эффектами были кожная сыпь, протеинурия, стоматит. Сходные результаты получены в процессе контролируемого многоцентрового исследования (G. Pasero и соавт., 1982). Интересно, что только у некоторых больных с побочными эффектами, при приеме D-ïåíèöèëëàìèíà отмечено их развитие в процессе лечения тиопронином. По данным G. F. Ferraccioli и соавт., (1989), эффективность и токсичность тиопронина такая же, как и препаратов золота. Имеются данные о том, что на фоне лечения тиопронином у больных РА отмечается не только уменьшение титров РФ, но и снижение функциональной аффинности РФ в сыворотке и синовиальной жидкости, сочетающееся с восстановлением сиализации IgG. Кроме того, в сыворотке и синовиальной жидкости наблюдалось достоверное снижение концентрации растворимых ИЛ-2Р (P. Youinou и соавт., 1992). Это может свидетельствовать о влиянии тиопронина на Т-лимфоциты, приводящем к нормализации процессов гликозилирования. Имеются данные о том, что нарушение гликозилирования является важным фактором патогенеза PA (T. W. Rademacher и соавт., 1988). Недавно J. Sany и соавт. (1994) в результате 24-недельного контролируемого сравнительного исследования получили данные о сходной клинической эффективности МТ и тиопронина при РА. 5.5. КАПТОПРИЛ Каптоприл (êàïîòåí-Bristol-Myers-Squibb) относится к группе ингибиторов АПФ и рассматривается как один из наиболее эффективных сосудорасширяющих препаратов. Наличие тиоловой (SH) группы обусловливает определенное сходство каптоприла с пеницилламином. Имеются следующие доказательства противовоспалительной и иммунной активности каптоприла: 1. Лечение каптоприлом оказывает благоприятное влияние на течение иммунопатологического процесса у мышей линии MRL lpr/lpr, проявляющееся в снижении АД, уменьшении лимфоидной гиперплазии, выраженности кожного васкулита, увеличении продолжительности жизни (H. Herlitz и соавт., 1988). 2. Имеются данные об участии АПФ и ангиотензина II (АII) в развитии воспалительных и иммунных реакций. АПФ синтезируется макрофагами и периферическими моноцитами больных РА. В сыворотке и синовиальной жидкости обнаружено повышение концентрации АПФ, коррелирующее с увеличением СОЭ и уровня СРБ. Выявлено синхронное увеличение синтеза ИЛ-1b и АПФ моноцитами больных РА (A. E. Goto и соавт., 1990). 3. В экспериментальных исследованиях in vitro было обнаружено, что All стимулирует активность СD8+Т-лимфоцитов, проявляющих супрессорную активность. В свою очередь каптоприл обладает способностью подавлять пролиферативный ответ лимфоцитов на растительные митогены (обсуждается роль простагландинзависимых механизмов) и ингибировать первичный антительный ответ Т-лимфоцитов путем активации моноцитов и супрессорных Т-лимфоцитов (S. F. Delpraissy и соавт., 1984). 4. В опытах in vitro и in vivo было показано, что каптоприл ингибирует синтез ЛТВ4 нейтрофилами (K. Shindo и соавт., 1994). Клиническая эффективность Имеется несколько сообщений о противовоспалительном действии каптоприла при РА (В. А. Насонова и соавт., 1992; I. A. Jaffe, 1983; M. Martin и соавт., 1982; C. L. Mertel и соавт., 1989). В. А. Насонова и соавт. (1992) использовали каптоприл в дозе 150 мг/сут. для лечения 10 больных РА с синдромом артериальной гипертензии. Получены данные о том, что каптоприл не только нормализует АД, но и проявляет очевидное противовоспалительное действие, сходное с таковым других базисных противоревматических препаратов. Значимых взаимодействий между каптоприлом и различными НПВС не выявлено. Список литературы К главе "D-пеницилламин и тиолсодержащие вещества с близкой фармакологической активностью" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. Гусева Н. Г. Системная склеродермия. Медицина. М. 1993. 267 стр. Насонова В. А., Саморядова О. С., Масенко В. П., Балабанова P. M., Насонов Е. Л.: Эффективность капотена при лечении ревматоидного артрита с артериальной гипертензией: анализ гипотензивного и противовоспалительного действия. Терапевт, архив. 1992; 8: 68-73. Abe С.: Clinical evaluation of immunomodulators. Int. J. Immunother. 1985; 1: 7-10. Bird H. A., Dixon A.: Failure of d-penicillamine to affect peripheral joint involvment in ankylosing spondylitis or HLA B27 associated arthropathy. Ann. Rheum. Dis. 1977; 36: 289. Camus J. P., Courset J., Prior A., Bergevin H.: Pyrithioxin and thiopronin: a new penicillamine-like drugs in rheumatoid arthritis. J. Rheumatol. 1982; 8(suppl. 7): 175-177. DeClerck L. S., Dequeker J., Francx L., Demedts M.: D-penicillamine therapy and intestitial lung disease in scleroderma. Arthritis Rheum. 1987; 30: 643-650. Delpraissy S. F., Balavoine Y. F., Wallon C., Dorment J.: Captoril and immune regulation. Kidney Int., 1984; 25: 923-925. Ferracioli G. F., Salaffi F., Nervetti A., Manganelli P.: Long-term outcome with gold thiosulphate and tiopronin in 200 rheumatoid patients. Clin. Exp. Rheumatol. 1989; 7: 577-581. Goto A. E., Fujisawa M., Yamacio A., et al.: Spontaneous release of angiotensin converting enzyme and interleukin I beta from peripheral blood monocyte from patients with rheumatoid arthritis under a serum free conditions. Ann. Rheum. Dis., 1990; 49: 172-176. Hakoda M., Taniguchi A., Kamatani N., et al.: Intermittent treatment with D-penicillamine is effective in lower doses and with fewer adverse effects in patients with rheumatoid arthritis. J. Rheumatol. 1994: 21: 1637-1641. Hamilton J. A.: Rheumatoid arthritis: opposite actions of haemopoietic growth factors and slowacting drugs. Lancet 1993; 342: 536-539. Harkness J. A. L., Blake D. R,: Penicillamine nephropathy and iron. Lancet 1982; 2: 1368-1369. Hayashi M., Matsunaga К., Okahara A., Mita S.: Effect of bucullamine (SA96) on type II collagen inducred arthritis in rats. J. Rheumatol. 1991; 18: 691-695. Herlitz H., Tarkowski A., Svalander C., et al.: Beneficial effect of captopril on systemic lupus erythematosus-like disease in MRL lpr/lpr mice. Int. Arch. Allergy. 1988; 85: 272-277. Hirohata S., Lipsky P. E.: Regulation of В cell function by Bucillamine, a novel diseasemodifying antirheumatic drug. Clin. Immunol. Immunopathol. 1993; 66: 43-51. Hirohata S., Lipsky P. E.: Comparative inhibitory effects of bucillamine and D-penicillamine on the function of human В cells and T cells. Arthritis Rheum. 1994; 37: 942-950. Hirohata S., Yanagida Т., Lipsky P. E.: Analysis the mechanism of action of bucillamine, a novel antirheumatic drug on human В cell function. Arthritis Rheum. 19994; 37 (suppl.): S340. Huskisson E. C.: Treatment of palindromic rheumatism with D-penicillamine. Br. Med. J. 1976; II: 979-980. Kashiwazaki S., Shiokawa Y.: Bucillamine: a new immunomodulator. Int. J. Immunother. 1987; 3: 1-6. 18. Jaffe J. A.: Penicillamine treatment of rheumatoid arthritis: Effect on immune complexes. Ann. N. Y. Acad. Sci. 1975; 256: 330-337. 19. Jaffe I. A., Thyol compaunds with penicillamine-like activity and possible mode of action in rheumatoid arthritis. Anti-Rheumatic drugs. Ed. E. S. Huskisson, 1984. p555-558. 20. Jaffe I. A.: Angoitensin converting enzyme inhibitors in rheumatoid arthritis. Arthritis Rheum., 19841 27: 7: 840. 21. Jaffe I. A. Penicillamine. In: Arthritis and allied conditions. A textbook of rheumatology. Ed. D. J. McCarty. Lea/Febiger. Philadelphia, London. 1989: 593-603. 22. Jayson M. I. V., Lovell C., Black C. M.: Penicillamine therapy in systemic sclerosis. Proc. R. Soc. Med. 1977; 70: 82-88. 23. Joyce D. A.: D-penicillamine. Bailleres Clinical Rheumatol. 1990; 4: 553-574. 24. Lemmel E.-M., for the European Multicentre Study Group. Comparison of pyritinol and auranofin in the rheumatoid arthritis. Brit. J. Rheumatol., 1993; 32: 375-382. 25. Lipsky P. E., Ziff M.: Inhibition of human helper T cell function in vitro by d-penicillamine and CuS04. J. Rheumatol. 1980; 15: 1069. 26. Lyle W. H. Penicillamine. In: Anti-rheumatic drugs. Ed. E. C. Huskisson. Praeger. 1983: 521-554. 27. Martin M. F. R., McKenna F., Bird H. A., et al.: Captopril. A new treatment for rheumatoid arthritis. Lancet 1984; 2: 1325. 28. Medsger T. A. Jr.: D-penicillamine treatment of lung involvement in patients with systemic sclerosis, (scleroderma). Arhritis Rheum. 1987; 30: 832-834. 29. Mertel C. L., Camus J. P., Prier A., et al.: Captopril in rheumatoid arthritis. Cong. Rheumatol. Paris., 1989; 1389. 30. Nimni M. E.: Penicillamine and collagen metabolism. Scand. J. Rheumatol. 1979; 8: 71-78. 31. Pasero G., Ciompi M. L.: Thypronine therapy in rheumatoid arthritis. Arthritis Rheum. 1979; 22; 803-804. 32. Pasero G., Pellegrini P., Ambanelli U., et al.: Controlled multicentric trial of tiopronin and Dpenicilliamine for rheumatoid arthritis. Arthritis Rheum. 1982; 25: 923-929. 33. Perett D.: The metabolism and pharmacology of D-penicillamine. J. Rheumatol. 1981; 8 (suppl. 7): 41-50. 34. Pool K. D., Feit H., Kirkpatrick J.; Penicillamine-induced neuropathy In rheumatoid arthritis. Ann. In tern. Med. 1981; 95: 457-458. 35. Rashiwazaki S., Shiokawa Y.: Bucillamine: a new immunomodulator. Int. J. Immunother. 1987: 3: 1-6. 36. Steen V. D., Medsger T. A., Rodnan G. P.: D-penicillamine therapy in progressive systemic sclerosis (scleroderma). Ann. Intern, Med. 1882; 97: 652-658. 37. Steen V. D. et. al.: The effect of D-penicillamine on pulmonary findings in systemic sclerosis. Arthritis Rheum. 1985; 28: 882-888. 38. Steven M. M., Morrison M., Sturrock R. D.: Penicillamine in ankylosing spondilitis. J. Rheumatol. 1985; 12: 735. 39. Sany J., Jorqensen C., Ash L., et al.: A Randomized double blind controlled study (RCS) of methotrexate (MTX) and tiopronin in rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S-81. 40. Shindo K., Baker J. R., Munafo D. A., Bigby T, D.: Captopril inhibits neutrophil synthesis of leukotriene B4 in vitro and in vivo. J. Immunol. 1994; 153: 5750-5795. 41. Shiokawa Y., Abe C., Warabi S., et al,: Clinical evaluation of SA96 on rheumatoid arthritis. Jpn. J. Inflamm. 1985; 5: 333-343. 42. Youinou P., Keusch J.,Casburn-Budd R., et al.: Tiopronin-induced reduction of rheumatoid factor functional affinity and asialyted IgG in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 1992; 10: 333-338 ГЛАВА 6 МЕТОТРЕКСАТ Метотрексат (МТ) относится к группе антиметаболитов, по структуре близок к фолиевой кислоте. Фолиевая (птиролглутаминовая) кислота состоит из птеридиновых групп, связанных с парааминобензойной кислотой, соединенной с остатками глутаминовой кислоты. МТ отличается от фолиевой кислоты заменой аминогруппы на карбоксильную группу в 4-м положении птеридиновой молекулы и добавлением метиловой группы в 10-м положении 4-аминобензойной кислоты. Фармакологические свойства После перорального приема 10-25 мг МТ уровень абсорбции препарата колеблется в очень широких пределах —от 25 до 100%, в среднем составляя 60-70%, а биодоступность варьирует от 28 до 94% (D. Oguey и соавт., 1992). У одного и того же больного на фоне достаточно длительного лечения уровень абсорбции МТ при увеличении дозы от 7.5 до 25 мг не меняется. Максимальная концентрация препарата в крови достигается через 2-4 ч. Прием МТ вместе с пищей замедляет время, необходимое для достижения максимальной концентрации в сыворотке, примерно на 30 мин., но уровень его абсорбции и биодоступность не меняются (D. Oguey и соавт., 1992; G. D. KozIovski и соавт., 1992). Следовательно, больные могут принимать МТ вместе с едой (G. Kozlovski и соавт., 1992). Элиминация МТ происходит преимущественно в почках за счет клубочковой фильтрации и канальцевой секреции. Время полужизни препарата колеблется от 2 до 6 ч. Развитие почечной недостаточности приводит к замедлению экскреции препарата и увеличению его токсичности. Несмотря на достаточно быстрое выведение МТ из крови, его полиглутамированные метаболиты обнаруживаются внутриклеточно в течение не менее чем 7 дней после однократного приема. При РА особенно высокая концентрация МТ выявляется в синовиальной мембране, что частично позволяет объяснить его высокую эффективность при РА (C. Bologna и соавт., 1993). Механизм действия Возможные механизмы действия метотрексата при ревматических заболеваниях: 1. подавление пролиферации мононуклеарных клеток и синтеза антител (РФ) В-лимфо-цитами; 2. подавление образования ЛТВ4; 3. снижение синтеза ИЛ-1; 4. подавление пролиферации ЭК и фибробластов; 5. подавление функциональной активности нейтрофилов; 6. подавление активности протеолитических ферментов в суставной полости; 7. усиление синтеза растворимых антагонистов IL-1 и растворимых ФНОР. В организме человека фолиевая кислота расщепляется ферментом дегидрофолатредуктазой с образованием метаболически активных продуктов: дигидрофолиевой и тетрагидрофолиевой кислот, которые принимают участие в конверсии гомоцистеина в метионин, образовании пуринов и тимидилата, необходимых для синтеза ДНК. Одним из основных фармакологических эффектов МТ является связывание и инак-тивация дегидрофолатредуктазы. Кроме того, в клетке МТ подвергается полиглутамилированию с образованием дериватов, которым в настоящее время придают очень важное значение в реализации биологической активности МТ. Например, в клетках опухоли молочной железы 55% МТ подвергается полиглутамированию в течение 24 ч. (J. Jolivet и соавт., 1982). Эти метаболиты в отличие от "нативного" МТ оказывают ингибиторное действие не только на дегидрофолатредукта-зу, но и на "дистальные" фолатзависимые ферменты, включая тимиди-латсинтетазу, фосфорибозиламиноамидазолкарбоксамид трансформи-лазу (АИКАР), а возможно, и трифункциональные ферменты (10-фор-милтетрагидрофолатсинтетазу, циклогидролазу, дегидрогеназу), также принимающие участие в синтезе ДНК. Таким образом, терапевтические эффекты и токсичность МТ определяются фолатзависимыми механизмами. Однако они зависят от степени ингибиции не только дегидрофолатредуктазы, но и других ферментов, участвующих в биотрансформации фолиевой кислоты. Предполагают, что именно этим объясняется терапевтическая эффективность низких доз МТ, недостаточных для полной ингибиции активности дегидрофолатредуктазы. Действительно, подавление активности дегидрофолатредуктазы, ведущее к снижению синтеза ДНК, наблюдается главным образом при назначении сверхвысоких доз МТ онкологическим больным и составляет основу антипролиферативного действия препарата. При использовании низких доз МТ ингибиция дегидрофолатредуктазы, вероятно, имеет существенно меньшее значение, а клиническая эффективность связана с действием его глутаминированных производных. Поскольку метаболизм МТ завершается в течение 24 ч., большая часть принимаемого 1 раз в неделю МТ находится внутри клеток в форме полиглутаминированных дериватов. Последние особенно активны в отношении ингибиции тимидилатсинтетазы и особенно АИКАР, рибозидный компонент которой обладает способностью инги-бировать аденозиндезаминазу и S-àäåíîçèë-ãîìîöèñòåèí гидролазу (J. E. Baggot и соавт., 1993). Примечательно, что активность этих ферментов нарушена у больных с синдромом тяжелого комбинированного иммунодефицита, связанного с генетическим дефектом аденозиндеза-миназы. Представляется весьма вероятным, что на фоне лечения низкими дозами МТ именно накопление метаболитов АИКАР может быть одной из причин противовоспалительного и мягкого иммуносупрессив-ного действия препарата. В недавнем экспериментальном исследовании B. N. Cronstein и соавт. (1993) были получены новые доказательства того, что противовоспалительный эффект МТ не зависит от его антипролиферативного действия и связан со свойством препарата вызывать внутриклеточное накопление АИКАР, что в свою очередь ведет к высвобождению аденозина, проявляющего антивоспалительную активность. На модели экспериментального воспаления у мышей было показано, что противовоспалительное действие МТ ассоциируется с увеличением концентрации АИКАР в спленоцитах и с продукцией аденозина. При этом селективный антагонист аденозиновых рецепторов типа 2 (3,7 метил-1-пропаргилксантин) полностью устранял противовоспалительное действие МТ. По мнению авторов, эти данные позволяют рассматривать МТ, по крайней мере при использовании его в низких дозах, не как иммуносупрессивное средство, а как новый тип НПВС, противовоспалительный эффект которого связан со стимуляцией высвобождения аденозина в очаге воспаления. По данным экспериментальных исследований, МТ подавляет пролиферацию ЭК (B. Haraoui и соавт., 1991) и рост синовиальных фибробластов (S. Hirata и соавт., 1989; F. A. Meyer и соавт., 1993), что также связывают со способностью препарата стимулировать высвобождение аденозина из ЭК и фибробластов (B. N. Cronstein и соавт., 1992; R. Wolf и соавт., 1992). Примечательно, что индуцированное МТ высвобождение аденозина вызывает образование многоядерных гигантских клеток, принимающих участие в образовании множественных ревматоидноподобных узелков — характерного осложнения лечения МТ (J. Merrill и соавт., 1993). У больных РА на фоне лечения низкими дозами МТ отмечается подавление спонтанного синтеза IgM и IgM РФ нестимулированными и стимулированными митогеном лаконоса мононуклеарными клетками, снижение концентрации иммуноглобулинов (P. A. Anderson и соавт., 1985), титров IgM РФ и IgA РФ (N. Olsen и соавт.. 1987; G. S. Alarcon и соавт., 1990) и IgA РФ, коррелирующее с клиническим улучшением. При этом спонтанный синтез IgM РФ снижается очень быстро, уже через 24-48 ч. после однократного приема препарата. На фоне лечения МТ не отмечается подавления пролиферативного ответа лимфоцитов, оцениваемого по включению тимидина (C. Johnston и соавт., 1988). Однако использование тимидиновой метки не позволяет определить истинное состояние лимфоцитов, поскольку МТ ингибирует активность тимидилатсинтетазы. Кроме того, in vitro высокие дозы МТ блокируют клеточный цикл в G2-фазе, не влияя на включение тимидина. По данным N. J. Olsen и L.A. Murray (1989), МТ подавляет пролиферацию мононуклеарных клеток в тесте, в котором вместо тимидина использовали дезоксиуридин. При интерпретации этих результатов необходимо учитывать тот факт, что МТ, ингибируя конверсию дезоксиуридиновой кислоты в тимидилат, блокирует включение не только тимидина, но и дезоксиуридина. По данным P. Martinez-Osuma и соавт. (1993), у больных РА, леченных МТ, не наблюдается изменения пролиферативного ответа лимфоцитов по данным метода, основанного на определении активности тимидилатсинтетазы (включение ЗН-дезок-сиуридина). По мнению авторов, эти результаты, а также то, что лей-коворин не снижает эффективность МТ, свидетельствуют том, что лечебный эффект низких доз МТ не связан с иммунодепрессивным действием. При низкой концентрации фолатов в культуре пролифератив-ный ответ лимфоцитов больных РА, леченных МТ, ниже, чем в норме, и у больных РА, не леченных МТ (R. J. Hine и соавт., 1992). Число В-, CD4+-, СD8+-лимфоцитов, соотношение СD4+Т-лимфоцитов и СD8+Т-лимфоцитов существенно не меняется (R. Segal и соавт., 1990; J. M. Kramer и соавт., 1993), но при проспективном анализе результатов отмечена тенденция к увеличению абсолютного количества СD4+Т-лимфоцитов (M. E. Weinblatt и соавт., 1975) и усиление активности ЕК-клеток. Предполагается, что, поскольку ЕК-клетки обладают способностью ингибировать дифференцировку В-лимфоцитов, это является одним из возможных механизмов, определяющих ингибирую-щее влияние МТ на синтез аутоантител, в первую очередь РФ. В клинических исследованиях было показано, что при использовании МТ клиническое улучшение развивается значительно быстрее, чем при лечении другими базисными препаратами, в том числе цитостати-ческими, имеет четкую зависимость от дозы, быстро исчезает после отмены препарата и коррелирует со снижением концентрации острофа-зовых белков, но не СОЭ (N. Songsiridej и D. E. Furst, 1990). Предполагается, что противовоспалительное действие МТ связано с ингибицией синтеза ЛТВ4 (J. L. LeRoux и соавт., 1992; R. I. Speling и соавт., 1990), с подавлением образования супероксидных радикалов (I. M. M. Laurindo и соавт., 1993), провоспалительных цитокинов и протеолитических ферментов. По данным J. L. LeRoux и соавт. (1992), однократный прием 10 мг МТ больными РА вызывает снижение синтеза LTB4 на 32%. В исследовании R. I. Speling и соавт. (1990) было показано, что на следующие сутки после приема МТ наблюдается снижение активации лейкоцитов кальциевым ионофором. Подавление функции нейтрофилов может быть также связано со способностью МТ стимулировать высвобождение аденозина из клеток соединительной ткани (B. N. Cronstein и соавт., 1991). Экспериментальные данные D. Goddard и соавт. (1993) говорят о том, что один из возможных противовоспалительных механизмов действия МТ связан с подавлением активности фосфолипазы А2. Имеются многочисленные данные, свидетельствующие о влиянии МТ на синтез провоспалительных и иммунорегуляторных цитокинов. Установлено, что МТ снижает синтез ИЛ-1 макрофагами при адъю-вантном артрите у крыс (К. М. Соnnоlу и соавт., 1988) и подавляет индуцированное ИЛ-1 обострение коллагенового артрита у мышей, В опытах in vitro было показано, что МТ подавляет биологическую активность ИЛ-1 (L. C. Miller и соавт., 1988; R. Segal и соавт., 1990). Предполагается, что этот эффект препарата может быть обусловлен или связыванием МТ с ИЛ-1 b вследствие структурной гомологии последнего с дегидрофолатредуктазой (L. C. Miller и соавт., 1988), или антифо-латными эффектами МТ (R. Segal и соавт., 1990). Кроме того, in vitro МТ подавляет биоактивность ИЛ-6, не влияя на ФНО-а (R. Segal и соавт., 1990). Длительное лечение низкими дозами МТ приводит к снижению синтеза ИЛ-1 периферическими мононуклеарными клетками (D. M. Chang и соавт., 1992), концентрации ИЛ-1, ИЛ-6 и ИЛ-8 в сыворотках больных PA (J. M. Kremer и соавт., 1993) и концентрации ИЛ-1-b в синовиальной жидкости (R. Thomas и соавт., 1993). P. Barrera и соавт. (1993) при изучении в динамике уровня растворимых рецепторов для ФНО (р55 и р75), ИЛ-2 рецепторов (ИЛ-2Р) и ИЛ-6 в сыворотках больных РА обнаружили, что МТ в отличие от A3 существенно снижает уровень ÈË2Ð, рФНОр55 и ИЛ-6, не влияя на концентрацию ФНО-a и ð75 ФНО-Р. Интересно, что МТ увеличивает синтез ИЛ-2 как in vitro, так и in vivo (S. L. Aaron, 1989; J. M. Kremer, 1994). Этот эффект МТ связывают с его способностью подавлять синтез полиаминов, гиперпродукция которых вызывает дефицит синтеза ИЛ-2 при РА (E. Flescher и соавт., 1989). Усиление синтеза ИЛ-2 на фоне лечения МТ было зарегистрирована и у крыс с экспериментальным артритом, индуцированным мембраной стрептококка (S. C. Ridge и соавт., 1986). Установлено также, что не только длительный, но и однократный прием МТ вызывает снижение уровня острофазовых белков (R. Segal и соавт., 1989), ингибирует хемотаксис нейтрофилов (J. W. O'Callaghan и соавт., 1988), образование ЛТВ4 (R. I. Sperling и соавт., 1990; J. L. Leroux и соавт., 1992) и синтез ИЛ-1-b мононуклеарными клетками периферической крови больных PA (P. Barrera и соавт., 1994). Другой точкой приложения МТ является продукция протеолитических ферментов. Обнаружено, что на фоне лечения МТ достоверно снижается уровень экспрессии гена коллагеназы в биоптатах синовиальной оболочки, в то время как экспрессия генов тканевого ингибитора металлопротеиназа и стромелизина не меняется (G. S. Firestein и соавт., 1994). МТ также не оказывал влияния на уровень иРНК коллагеназы и тканевого ингибитора металлопротеиназ после стимуляции ИЛ-1. Предполагается, что поскольку именно стромелизин играет основную роль в разрушении протеогликана хряща, эти результаты в определенной степени позволяют объяснить причины прогрессирования сужения межсуставных щелей у больных, леченных МТ, несмотря на достоверное уменьшение числа новых костных эрозий. В то же время, по данным T. Murul и соавт. (1993), МТ в терапевтических концентрациях зависимым от дозы образом подавляет высвобождение коллагеназы и стромелизина из хондроцитов. Клиническое применение В настоящее время МТ рассматривается как один из эффективных препаратов, используемых не только в ревматологии, но и в лечении широкого круга заболеваний внутренних органов: Ревматические заболевания: • РА • синдром Фелти • псориатический артрит • синдром Рейтера • ювенильный артрит • болезнь Стилла у взрослых • ПМ/ДМ • ССД • системные васкулиты (граиулематоз Вегенера, болезнь Такаясу, гигантоклеточныйартериит, гангренозная пиодермия, синдром Когана) • СКВ • множественный ретикулогистиоцитоз Другие: • склерозирующий холангит • первичный билиарный цирроз • воспалительные заболевания кишечника • псориаз • саркоидоз • бронхиальная астма Тактика лечения и клиническое применение: При ревматических заболеваниях МТ назначают 1 раз в неделю (перорально или парентерально), поскольку более частый прием препарата, как правило, ассоциируется с развитием острых и хронических токсических реакций. В связи с возможной непереносимостью одномо-ментного приема больших доз препарата рекомендуется назначать его дробно, с 12-часовым интервалом, в утренние и вечерние часы. Начальная доза МТ в большинстве случаев составляет 7.5 мг/нед, а у лиц пожилого возраста — 5 мг/нед. Эффект оценивается через 4-8 нед., и в случае его отсутствия при нормальной переносимости дозу МТ постепенно увеличивают по 2.5 мг в неделю. При этом суммарная недельная доза при пероральном приеме не должна превышать 25 мг. Это связано, как с возможностью развития токсических реакций, так и с ухудшением всасывания более высоких доз препарата в ЖКТ. При повышении дозы МТ оценка токсичности проводится через 6 дней после приема препарата, а при достижении кумулятивной дозы 1500 мг показана биопсия печени. Парентеральное введение МТ используется в случае отсутствия эффекта от перорального приема или при развитии токсических реакций со стороны ЖКТ. При этом необходимо иметь в виду, что отсутствие эффекта при пероральном приеме МТ в ряде случаев связано не столько с неэффективностью самого препарата, сколько с низкой абсорбцией в ЖКТ, не позволяющей достичь оптимальной концентрации вещества в крови. Отмена МТ, как правило, приводит к обострению заболевания в сроки между 3-й и 4-й неделями. Хотя негативное влияние терапии МТ на заживление ран и развитие постоперационных инфекционных осложнений не доказано, рекомендуется отменять препарат за неделю до предполагаемого оперативного вмешательства и не назначать его в течение 2 нед. после операций. Основные подходы, направленные на предотвращение развития побочных эффектов, уменьшение токсичности МТ и методы наблюдения за больными суммированы в таблице 6.1. Таблица 6.1. Подходы к предотвращению побочных эффектов и уменьшению токсичности МТ Абсолютные противопоказания: Относительные противопоказания: • алкоголизм • потребление алкоголя • заболевания печени • ожирение • беременность • сахарный диабет • ожидаемая беременность • умеренная почечная недостаточность (снижение дозы) • тяжелое поражение легких • цитопения в анамнезе • тяжелая почечная недостаточность • опухоли в анамнезе • панцитопения • язвенная болезнь • злокачественные новообразования • антикоагулянтная терапия • ВИЧ-инфекция Исследования до лечения: Сообщать о следующих симптомах: • общий анализ крови • кашель • печеночные пробы • желтуха • рентгеноскопия грудной клетки • тошнота, рвота, диарея • исследование почек • инфекция • исследование легких? • боли в ротовой полости • антитела к ВИЧ • маркеры гепатита В и С Мониторинг: Профилактика побочных реакций: • полный анализ крови (каждые 3-4 нед.) • фолиевая кислота или лейковорин • печеночные пробы (каждые 6-8 нед.) Важной особенностью. МТ является его способность взаимодействовать с многими лекарственными препаратами. Например, абсорбция препарата существенно снижается при совместном приеме с некоторыми антибиотиками. Урикозурический препарат пробеницид снижает клиренс МТ на 60% и увеличивает концентрацию препарата в плазме до 400%. Совместное применение МТ и триметоприм/сульфаметоксазола, являющегося, как и МТ, антагонистом фолатов, также может вести к нарастанию частоты побочных реакций. Особое значение имеет взаимодействие МТ и НПВС, поскольку такая комбинация наиболее часто используется при лечении различных ревматических заболеваний. В литературе описано около 50 больных, у которых при сочетанном приеме МТ и НПВС развились тяжелые токсические реакции. Особенно часто токсические реакции регистрируются при использовании комбинации МТ и салицилатов (C. Stewart и соавт., 1991). При сочетанием введении МТ и салицилатов лабораторным животным у них развивается тяжелая лейкопения и снижается выживаемость. По данным M. Zuik и M. A. Mandel (1975), у 7 из 176 больных развилась тяжелая цитопения при лечении МТ в высоких дозах. 6 из них получали салицилаты. Салицилат натрия на 26-42% снижает клиренс МТ, что связано с ингибицией его секреции в почках, в то время как напроксен (1 г/день) не дает подобного эффекта (C. Stewart и соавт., 1991). У больных РА, получающих аспирин, отмечается снижение клиренса МТ, главным образом за счет не связанного с белком МТ. Однако вытеснение МТ НПВС из связывания с белком в большинстве случаев не имеет существенного значения для реализации терапевтического эффекта и развития побочных реакций на фоне лечения МТ, так как не связанная с белком часть вещества составляет 50%. Все эти результаты свидетельствуют о том, что на фоне лечения больных МТ следует избегать назначения салицилатов и использовать НПВС короткого действия. Некоторые авторы рекомендуют в день приема МТ заменять НПВС ГК в низких дозах (R. Rao, 1994). Имеются данные о достоверном снижении выраженности большинства побочных эффектов МТ (за исключением цитопении и поражения легких) на фоне приема фолиевой кислоты в дозе 5-50 мг/день (S. L. Morgan и соавт., 1993; S. L. Morgan и соавт., 1994). Наряду с фолиевой кислотой рекомендуется использовать лейковорин, представляющий собой синтетическую форму редуцированного метаболически активного коэнзима фолата (5-формил-тетрагидрофолиевая кислота), для последующего расщепления которого необходимость в дигидрофолат редуктазе отсутствует. Препарат специально создан для преодоления метаболического блока, возникающего при введении высоких доз МТ, и используется как антидот при токсических реакциях на МТ и профилактически для снижения частоты и выраженности побочных эффектов лечения высокими дозами препарата. В исследованиях некоторых авторов было показано, что лейковорин снижает эффективность МТ (D. A. Joyce и соавт., 1991; M. Tishler и соавт., 1988). Однако результаты многоцентрового двойного слепого контролируемого исследования показали, что прием лейковорина существенно уменьшает частоту побочных эффектов МТ без уменьшения его терапевтической активности. Важно, что для получения оптимальных результатов необходимо назначать лейковорин через 24 ч. после приема МТ в дозе более низкой, чем доза МТ. (J. B. Shiroky и соавт., 1993). РА Внедрение в клиническую практику терапии низкими дозами МТ является важнейшим достижением в лечении РА. Обсуждается вопрос о возможности лечения МТ раннего PA (D. E. Furst, 1993). Эффективность МТ при РА подтверждена во многих контролируемых исследованиях, результаты которых обобщены в серии недавно опубликованных обзоров (R. Rau, 1994; A. Schnabel и W. L. Gross, 1994, M. E. Weinblatt, 1993) Эффективность МТ в дозе 7.5-25 мг/нед в сравнении с плацебо доказана в серии клинических испытаниях. Клинический эффект на уровне 50% отмечен у 90% больных, он сочетался со снижением потребности в ГК; уменьшение СОЭ наблюдалось у 80% пациентов. При этом в большинстве случаев лечение МТ проводилось больным активным РА, не отвечающим на другие базисные препараты. Действие препарата начинает проявляться весьма быстро — в сроки между 4-й и 8-й неделями и достигает максимума к 6-му месяцу. Однако полная ремиссия развивается довольно редко, а при отмене препарата часто возникает обострение. Важным достоинством МТ является возможность его приема в течение длительного времени без уменьшения активности и развития побочных эффектов. Установлено, что 46-75% больных продолжают принимать препарат через 5-7.5 года от момента назначения (J. M. Kremer и C. T. Phelps, 1992). Это значительно больше, чем при лечении другими базисными противоревматическими препаратами. По данным M. E. Weinblatt и соавт. (1993), в 36-недельном контролируемом исследовании у 281 больного РА МТ (7.5 мг с постепенным увеличением дозы до 15 мг/нед) превосходил ауранофин (6-9 мг/день) по всем параметрам оценки клинической эффективности и отличался лучшей переносимостью. Особенно важными представляются данные о том, что в группе больных, получавших МТ, но не ауранофин, наблюдалось замедление рентгенологического прогрессирования деструктивного процесса в суставах, судя по количеству эрозий и выраженности сужения межсуставных щелей. Однако Н. Wiliams и соавт. (1992) не выявили достоверных различий в эффективности МТ и ауранофина, хотя частота прерывания лечения была выше у больных, получавших ауранофин. При сравнении результатов лечения МТ и парентерально вводимыми препаратами золота обнаружена их сходная эффективность, но лучшая переносимость МТ. По данным R. Rau, проведшего сравнительное исследование МТ и препарата золота у 174 больных в течение 13 мес., существенное улучшение наблюдалось более чем у 50% больных в обеих группах, но переносимость МТ оценена выше. Недавно было проведено 34-недельное многоцентровое контролируемое исследование эффективности и токсичности МТ (7.5-15 мг/нед) и ЦсА (2.5-5.0 мг/кг/день) в сравнении с плацебо у 261 больного рефрактерным РА. Оба препарата вызывали статистически достоверное улучшение большинства клинических параметров по сравнению с плацебо, но лечение МТ было более эффективным, чем ЦсА (S. Cohen и соавт., 1993). Результаты 24-недельного двойного слепого контролируемого исследования (J. Sany и соавт., 1993), в которое вошел 61 больной активным РА, свидетельствуют о сходной эффективности МТ и тиопринина. M. E. Weinblatt и соавт. (1994) представили результаты 5-летнего наблюдения за 123 больными РА, леченными МТ. В целом лечение было эффективным у 64% пациентов, 36% больных прекратили прием препарата. По данным F. Salaffi и соавт. (1995) вероятность продолжения лечения МТ больных РА составляет 80.3% через 12 мес., 74.5% через 24 мес. и 70.5% через 35 мес. При этом у всех больных, которым было проведено 36-недельное лечение МТ, отмечено существенное клиническое улучшение. В целом побочные реакции отмечены у 80% больных, но только у 15.6% они явились причиной прерывания лечения. Только у 8% больных лечение было прекращено из-за неэффективности. Все эти данные подтверждают достаточную эффективность и приемлемую токсичность МТ при РА. При этом именно токсические реакции, а не отсутствие эффекта являются основной причиной прекращения лечения МТ (G. S. AIarcon и соавт., 1989; J. Sany и соавт., 1991). Имеются данные об успешном лечении МТ больных с синдромом Фелти (L. S. Allen и G. Groff, 1986; J. J. Fiechtner и соавт., 1989). Данные о влиянии МТ на рентгенологическое прогрессирование деструктивного процесса при РА, как, впрочем, и других базисных противоревматических препаратов, противоречивы. Мета-анализ опубликованных к 1992 г. результатов не позволил четко установить преимущества МТ перед другими базисными противоревматическими препаратами в отношении рентгенологического прогрессирования эрозивного процесса (G. S. Alarcon и соавт., 1992). По оценке R. Rau (1994), обобщившего результаты 6 исследований, включавших 161 больного РА, наблюдавшихся в течение 24-90 мес., МТ замедляет рентгенологическое прогрессирование у тех больных, у которых удается подавить общую воспалительную активность болезни. В серии исследований изучена возможность комбинированной терапии РА МТ в сочетании с другими базисными препаратами. R. F. Wilkens и соавт. (1992) провели двойное слепое контролируемое исследование A3, МТ и их комбинации. Применение МТ в сочетании с A3 или только МТ было достоверно эффективнее, чем только A3. Вызывает интерес тот факт, что комбинация МТ и A3 не приводила к нарастанию частоты побочных реакций, которая в целом была ниже, чем при монотерапии A3 в течение первых 6 нед. лечения. Причины этого феномена не ясны, поскольку представляется маловероятным, что МТ способен предохранять от азатиоприновой токсичности. Возможно, что речь идет о чисто субъективных факторах, связанных с тем, что хороший противовоспалительный эффект комбинации МТ и A3 как бы маскирует некоторые субъективные отрицательные эффекты, связанные с приемом A3. H. Williams и соавт. (1992) провели 48-недельное проспективное двойное слепое контролируемое испытание МТ (7.5 мг/нед), ауранофина (6 мг/день) и их комбинации у 335 больных РА. Существенных различий по клинической эффективности между сравниваемыми группами не зарегистрировано, однако у больных, получавших только ауранофин, эффект от лечения проявлялся более медленно, чем у больных, принимавших МТ или МТ в сочетании с ауранофином. Важно отметить, что частота побочных эффектов лишь незначительно нарастала на фоне комбинации двух препаратов, а частота отмен из-за неэффективности была самой высокой у больных, находившихся на монотерапии ауранофином. По данным R. Rau (1994), проведшего открытое испытание МТ и комбинации МТ и парентерально вводимого золота у 271 больного РА, по эффективности и переносимости сравниваемые группы не различались. Однако больные, получавшие комбинированное лечение, изначально имели более тяжелые проявления РА. Другой перспективной комбинацией является сочетание МТ и сульфасалазина, который, как и МТ, обладает антифолатной активностью. C. Haagsma и соавт. (1993) провели сравнительное исследование эффективности МТ (7.5 мг/нед с постепенным увеличением до 15 мг/нед) и сульфасалазина (2000 мг/день) и только МТ у 40 больных активным РА, резистентных к сульфасалазину в течение 6 мес. Установлена более высокая эффективность комбинированной терапии по сравнению с монотерапией МТ при сходной токсичности. Аналогичные данные получены E. F. Morand и соавт. (1993), продемонстрировавшими отсутствие нарастания частоты побочных эффектов на фоне комбинированной терапии МТ и сульфасалазином и возможность более длительного лечения. Однако, несмотря на положительные результаты комбинированной терапии МТ и сульфсалазином при РА, эффективность и безопасность этой комбинации требует дальнейшего изучения. Это в первую очередь связано с тем, что оба препарата обладают антифолатной активностью, что потенциально существенно увеличивает риск развития гематологических нарушений и других осложнений, связанных с дефицитом фолатов (S. L. Morgan и соавт., 1993). Описано развитие панцитопении, тромбоцитопении, кожной сыпи и стоматита у больных псориазом и РА, получавших комбинированное лечение МТ и триметопримом/сульфаметоксазолом (последний также обладает антифолатной активностью) (M. H. Thomas и соавт., 1986). С другой стороны, антифолатные эффекты описаны у некоторых НПВС, например ибупрофена, однако лечение НПВС и МТ не ассоциируется с существенным увеличением риска токсических реакций. Недавно была проведена серия исследований, посвященных оценке эффективности комбинированной терапии МТ и антималярийными препаратами. Интерес к этой комбинации возник после исследований J. F. Fries и соавт. (1990), которые обнаружили, что при сочетанием назначении МТ и гидроксихлорохина достоверно снижается частота гепатотоксических побочных эффектов МТ. По мнению авторов, это явление связано с гепатопротективным действием аминохинолиновых препаратов и их способностью вызывать стабилизацию лизосомальных ферментов. Однако, по данным Seideman и соавт. (1994), у больных РА, леченных МТ, прием аминохинолиновых препаратов вызывает снижение уровня МТ в плазме. Этот эффект связывают со способностью аминохинолиновых препаратов уменьшать биодоступность МТ. Но в недавнем контролируемом исследовании, выполненном D. O. Clegg и соавт. (1993), были получены данные об определенных преимуществах комбинированного использования МТ и гидроксихлорохина по сравнению с монотерапией этими препаратами. В частности, было показано, что гидроксихлорохин пролонгирует продолжительность ремиссии после отмены МТ и сокращает частоту и длительность обострений. В подавляющем большинстве исследований, посвященных оценке эффективности МТ, доза препарата не превышала 15 мг/нед. В то же время, по данным ряда авторов, клиническая эффективность МТ имеет зависимый от дозы характер (D. E. Furst и соавт., 1989; S. Gabriel и соавт., 1990; R. N. Thompson и соавт., 1984). Однако увеличение дозы МТ ассоциируется с нарастанием частоты побочных эффектов (D. E. Furst и соавт., 1989; R. N. Thompson и соавт., 1990). По данным A. Schnabel и соавт. (1994) использование МТ в начальной дозе 25 мг/нед приводит к увеличению частоты желудочнокишечных побочных эффектов и вызывает более выраженное повышение концентрации печеночных ферментов по сравнению с начальной дозой 15 мг/нед. Появились сообщения о возможности интенсификации лечения МТ у больных РА, рефракторных к стандартным дозам препарата. H. Bauer и соавт. (1993) применили у 40 таких больных метод лечения, основанный на "болюсном" внутривенном введении МТ в дозе 40 мг/нед, которое продолжалось — 4-8 нед. У 30 больных достигнуты хороший клинический эффект и снижение лабораторных параметров воспалительной активности, а развития тяжелых токсических реакций, которые потребовали бы прерывания лечения, не отмечено, Примечательно, что достигнутое улучшение эффективно поддерживалось ранее не эффективными низкими дозами перорального МТ. Имеются данные об использовании МТ при болезни Стилла у взрослых (T. Fujii и соавт., 1994). Препарат в начальной дозе 5 мг/нед с последующим увеличением до оптимальной (5-20 мг/нед) назначали 13 больным с обострением заболевания. У 6 пациентов достигнута полная ремиссия, а у остальных — существенное улучшение в сроки между 3-12-й неделями лечения. В целом по группе отмечена положительная динамика лабораторных показателей (СОЭ, лейкоциты, СРБ и др.). У 5 больных удалось существенно снизить дозу преднизолона. Эти результаты свидетельствуют об определенной эффективности МТ при лечении болезни Стилла у взрослых. Появилось сообщение об успешном применении МТ в виде внутрисуставных инъекций у больных с синовитом, резистентным к ГК (H. Durk и соавт., 1994). Внутрь суставов МТ вводили в дозе 7.5-10 мг, курс лечения составил 6-12 инъекций. У всех больных отмечен очень хороший клинический эффект, а у 5 — развилась длительная ремиссия моноартрита. Ювенильный артрит МТ зарекомендовал себя как эффективное средство для лечения тяжелого ювенильного артрита, не отвечающего на НПВС. В группе больных, состоящих из 49 человек с полиартикулярным ювенильным артритом, леченных низкими дозами МТ, у 45% достигнута ремиссия артрита в течение 13.6 мес. (E. H. Giannini и соавт., 1992; С. А. Walace и соавт., 1993). У некоторых детей эффект отмечался при увеличении дозы МТ до 0.82-1.1 мг/кг/нед. (C. A. Walace и соавт., 1992). Имеются данные о том, что лечение низкими дозами МТ приводит к замедлению прогрессирования рентгенологических изменений в суставах (L. Harel и соавт., 1993). Низкие дозы МТ не вызывают развития тяжелых побочных эффектов, по крайней мере в течение 5 лет наблюдения (L. D. Graham и соавт., 1992). СКВ Первое сообщение о лечении СКВ МТ появилось еще в 1965 г., когда Р. А. Miescher и D. Riethmuller использовали этот препарат у 10 больных в дозе 2.5 мг/день или в виде "пульсов" по 50 мг внутривенно 1 раз в неделю. В целом отмечен хороший эффект в отношении большинства клинических проявлений болезни, но высокая частота лейкопении не позволяла проводить терапию достаточно длительное время. Вскоре М. А. Swanson и R. S. Scwartz (1967) применили внутривенное введение МТ (50-75 мг/день 5-7 раз в течение 6 нед.) у 3 больных СКВ. В 2 случаях была достигнута частичная, а в 1 — полная ремиссия. Однако, по данным DuBois, у 7 больных СКВ МТ в дозе 7.5 мг/день в течение 6 нед. был неэффективен. R. J. Rothenberg и соавт. в 1988 г. применили МТ (7.5 мг/нед) у 10 больных активной СКВ, проявляющейся артритом, кожной сыпью, серозитом, поражением почек, миозитом, судорожными приступами; доза преднизолона во всех случаях была больше 20 мг/день. За 13 мес. лечения клинический эффект был достигнут в 7 случаях, а у 3 больных препарат был отменен из-за развития побочных эффектов. W. S. Wilke и соавт. (1991) проанализировали результаты лечения 17 больных СКВ, получавших МТ в средней недельной дозе 8.47±1,72 мг. Установлено, что лечение МТ оказывает положительное влияние на такие проявления СКВ, как артрит, сыпь, алопеция, плеврит, слабость, более чем у половины больных, позволяет снизить дозу ГК. Однако на фоне лечения отмечалась высокая частота побочных эффектов (71%), особенно цитопении, изъязвления слизистой полости рта, алопеции. Их было трудно мониторировать, поскольку аналогичные проявления могут быть обусловлены активностью самой СКВ. По данным C. M. Wise и соавт. (1994), при СКВ МТ более эффективен в отношении артрита, поражения кожи, миозита, чем поражения ЦНС и серозита. B. A. E. Walz LeBlanc и соавт. (1994) продемонстрировали благоприятное действие МТ (доза 7.5-20.0 мг/нед) у 5 больных СКВ, у 2 из которых превалировало поражение суставов, а у 3 поражение почек. По данным S. Shatten и соавт. (1992), существенное улучшение на фоне лечения МТ отмечено у 12 из 17 больных СКВ или с перекрестным синдромом. D. Galarza и соавт. (1992) наблюдали 10 больных активным волчаночным нефритом, плохо контролирующимся ЦФ и преднизолоном, у которых наблюдалась существенная положительная динамика на фоне лечения МТ в дозе 7.5-15.0 мг/нед. J. R. Davidson и соавт. (1987) также сообщили об успешном применении МТ у 1 больной СКВ, резистентной к ГК, A3, ЦФ и плазмаферезу. Таким образом, создается впечатление, что МТ может быть с успехом использован у определенной части больных СКВ, но из-за возможности развития тяжелых побочных реакций лечение следует проводить под строгим клиническим и лабораторным контролем. ССД Недавно Р. Barrera и соавт. (1994) представили результаты двойного слепого контролируемого исследования МТ у 23 больных ССД. После 24 нед. приема МТ клиническое улучшение, оцениваемое по общему кожному счету и визуальной аналоговой шкале, отмечено у 9 больных (у 8 — получающих МТ и у 1—в контрольной группе). J. S. Seibord и соавт. (1994) провели открытое испытание эффективности МТ у 20 больных с ранней диффузной формой ССД; доза МТ составила 7.5-15 мг/нед, а продолжительность лечения — 1 год. Установлено, что у 61% больных достоверно улучшился кожный счет, за это время не наблюдалось вовлечения в процесс других висцеральных органов. ПМ/ДМ Эффективность МТ при ПМ/ДМ колеблется от 50 до 75% и не зависит от того, назначают ли его перорально или внутривенно (P. H. Plotz и соавт., 1989; J. N. Whitaker и M. J. Penals, 1984). Внутримышечное введение МТ не рекомендуется, так как это может индуцировать повышение концентрации КФК и тем самым затруднить оценку эффективности лечения. Доза МТ при пероральном приеме варьирует от 7.5 до 25-30 мг/нед. Лечение следует начинать с небольшой дозы, постепенно увеличивая ее (по 0.25 мг/нед) до оптимальной. У больных, плохо переносящих пероральный прием, следует назначать МТ внутривенно. Лечение начинают с дозы 0.2 мг/кг в неделю, затем постепенно увеличивают дозу препарата по 0.2 мг/кг через каждые 7 дней. Снижение дозы МТ должно проводится постепенно, под тщательным контролем клинических показателей и уровня КФК. Существует две схемы отмены МТ — по 1/4 от еженедельной дозы или путем увеличения интервалов между приемом препарата, в начале до 2 нед., затем до 4 нед. Имеются данные об определенных преимуществах МТ по сравнению с A3, которые заключаются в первую очередь в более быстром развитии стероидсберегающего эффекта. Это может иметь немаловажное значение у больных, длительно принимающих высокие дозы ГК, или при быстро прогрессирующем ПМ/ДМ. Гранулематоз Вегенера (ГВ) G. S. Hoffman и др. (1992) провели исследование эффективности МТ у 39 больных ГВ без быстропрогрессирующего нефрита и тяжелого поражения легких. Тяжесть ГВ определялась активным гломерулонефритом, прогрессирующим поражением легких и ЛОР-органов. Еженедельное введение МТ в средней дозе 20 мг в сочетании с ГК вызвало улучшение у 29 (76%) больных, а у 69% — достигнута ремиссия. Только у 2 пациентов после отмены препарата развилось обострение. По данным K. Handrock и соавт. (1994), низкие дозы МТ (0.3 мг/кг внутривенно 1 раз в неделю) во многих случаях позволяют эффективно поддерживать индуцированную ЦФ ремиссию у больных ГВ, но не могут использоваться в качестве основного метода лечения у пациентов с тяжелыми проявлениями заболевания. Болезнь Такаясу Открытое проспективное исследование эффективности МТ (средняя доза 17.1 мг/нед) в сочетании с небольшими дозами ГК проведено у 18 больных, 16 из которых наблюдались около 3 лет. Ремиссия достигнута у 81% пациентов; у 7 больных с обострением, возникшим после отмены препарата, повторное назначение МТ вновь позволило индуцировать ремиссию. У 9 больных ремиссия была достаточно длительной —от 4 до 34 мес. (в среднем 18 мес.). Таким образом, предотвращение прогрессирования болезни Такаясу на фоне применения МТ достигнуто у 50% больных (A. S. Fauci, 1994). Гигантоклеточный артериит В открытом испытании у II больных гигантоклеточным артериитом было показано, что МТ оказывает благоприятное влияние на течение заболевания, в первую очередь позволяет быстрее снизить дозу ГК и поддерживать более длительную ремиссию (C. Hernandez-Garcia и соавт., 1994). Синдром Когана Появилось сообщение об использовании МТ для лечения синдрома Когана — редкой формы некротизирующего васкулита (B. Richardson, 1994), при этом удалось снизить дозу ГК. Имеются данные об эффективности МТ у больных с остеоартикулярными и мышечными проявлениями саркоидоза, не отвечающих на лечение колхицином и ГК (O. Kave и соавт., 1993). Побочные эффекты Несмотря на то что побочные эффекты отмечаются примерно у 50% больных, получающих низкие дозы МТ, в целом этот препарат переносится лучше, чем другие базисные противоревматические средства. Побочные эффекты МТ суммированы в таблице 6.2. Таблица 6.2: Побочные эффекты лечения низкими дозами МТ Частые, обычно нетяжелые: Частые, потенциально тяжелые: • тошнота, рвота боли в животе диарея • тромбоцитопения • повышение уровня печеночных ферментов • лейкопения • макроцитоз • панцитопения • стоматит • зуд/дерматит • головные боли, ослабление памяти • постдозовые реакции (артралгии, миалгии, недомогание, усталость) Редкие, потенциально летальные: • легочная инфильтрация и гипоксемия • цирроз печени • острая печеночная недостаточность Примечание. Тяжелые побочные эффекты чаще развиваются у лиц пожилого возраста. Поражение ЖКТ Тошнота и рвота обычно развиваются через 1-8 дней после приема препарата и длятся 1-3 дня. Для уменьшения выраженности этих явлений следует снизить дозу МТ или перевести больных на парентеральный прием. Рекомендуется также назначение симптоматических средств, например метаклопрамида. Поскольку МТ замедляет рубцевание язв желудка и двенадцатиперстной кишки, язвенное поражение является относительным противопоказанием к назначению МТ. Поражение кожи и слизистых оболочек Частым осложнением лечения является стоматит, из-за которого препарат отменяют в 6% случаев. Изредка развивается алопеция или кожный васкулит. Имеются данные о возможности использования полоскания рта раствором аллопуринола для лечения индуцированных МТ изъязвлений слизистой полости рта (C. Montecucco и соавт., 1994). Раствор готовится непосредственно перед употреблением следующим образом; 300 мг аллопуринола растворяют в 60 мл дистиллированной воды (финальная концентрация аллопуринола 5 мг/мл). Этого количества достаточно для 2-4 полосканий рта, которые рекомендуется повторять 2-4 раза в течение 3 дней каждую неделю. Продолжительность каждого полоскания составляет около 1 мин. Гематологические нарушения Тяжелые гематологические нарушения развиваются редко (Аl-Awadhil и соавт., 1993), что связано с низким уровнем накопления МТ в костномозговых клетках. Риск гематологических осложнений повышается при почечной недостаточности, дефиците фолиевой кислоты и при сочетанием использовании МТ с салицилатами и препаратами, обладающими антифолатной активностью, особенно триметоприма/сульфаметоксазолом. В то же время комбинированная терапия МТ и другим препаратом с антифолатной активностью — сульфасалазином повышает эффективность лечения и не ассоциируется с нарастанием частоты гематологической токсичности (M. Nissar и соавт., 1994). Поражение легких Очень редким осложнением лечения МТ является пневмонит (1-8%), обусловленный развитием аллергической реакции на препарат. Клинически это осложнение проявляется одышкой, сухим кашлем, развитием цианоза; при рентгенологическом исследовании выявляется двусторонняя интерстициальная инфильтрация в базальных отделах легких, а при трансбронхиальной биопсии — признаки гиперчувствительного пневмонита (R. F. Willkens, 1990; P. Barrera и соавт., 1994). Поскольку сходные симптомы могут быть связаны с ревматоидным поражением легких или быть следствием инфекции, диагностика этого осложнения представляет большие сложности. Развитие пневмонита на фоне лечения МТ описано почти исключительно у больных РА. Метотрексатный пневмонит является потенциально смертельным осложнением, однако в большинстве случаев отмена препарата и назначение ГК приостанавливает прогрессирование пневмонита. Поражение печени Транзиторное увеличение уровня трансаминаз, нарастающее при увеличении дозы МТ, — нередкий побочный эффект МТ. Как правило, концентрация трансаминаз достигает максимума через 4-5 дней после приема препарата и сохраняется в течение 1-2 нед. Дву-трехкратное повышение уровня трансаминаз не является основанием для отмены МТ, а более существенное увеличение концентрации ферментов свидетельствует о необходимости снижения дозы или прерывания лечения. Данные, касающиеся опасности развития фиброза и цирроза печени на фоне лечения МТ, противоречивы. Полагают, что поражение печени чаще наблюдается при лечении МТ больных псориазом, чем РА (Q. E. White-O'Keefe и соавт., 1991; S. K. Gilbert и соавт., 1990). По данным R. E. Wilkens и соавт. (1990), фиброз печени обнаруживается почти у трети больных, получающих МТ более 2 лет, но цирроз печени развивается редко. При электронной микроскопии признаки печеночного фиброза выявляются у большинства больных (Q. E. Whiting-O'Keefe и соавт., 1991). В процессе длительного наблюдения 15 больных у 5 — при электронной микроскопии (более чувствительный метод для выявления печеночного фиброза, чем световая микроскопия) отмечено увеличение количества коллагена в печени на 2-й год приема МТ. Однако необходимо иметь в виду, что у больных РА, не леченных МТ; также обнаруживаются признаки фиброза, хотя и менее выраженные, чем на фоне лечения МТ. W. J. Shergy и соавт. (1988) выявили прогрессирование печеночного фиброза у 14 из 61 пациента по данным серийных биопсий. У 2 больных РА описано развитие острой обратимой печеночной недостаточности (D. O. Clegg и соавт., 1989). В исследовании C. A. Philips и соавт. (1992) развитие тяжелого фиброза и цирроза было констатировано у 3 (2.2%) из 134 больных РА, получавших МТ в дозе 7.5-15 мг/нед в течение 2-4 лет. Анализ данных литературы и собственных данных, касающихся поражения печени на фоне лечения МТ, проведенный Q. E. White-O'Keefe, позволил сделать следующие выводы. Поражение печени прогрессирует почти у трети больных, что связано с кумулятивной дозой МТ, причем прием каждого последующего грамма МТ в целом соответствует 7% вероятности прогрессирования печеночного фиброза на один гистологический класс. При этом у больных, принимавших алкоголь в количестве более 100 г/нед., этот показатель увеличивался до 18%, а у принимавших менее 100 г/нед. — снижался до 4.5%. Больные, страдавшие псориазом, чаще имели более серьезное поражение печени и более высокий уровень прогрессирования, чем больные РА. Инфекционные осложнения Лечение низкими дозами МТ может сопровождаться увеличением чувствительности к инфекционным осложнениям. Имеются единичные клинические наблюдения, свидетельствующие о возможности развития необычных тяжелых грибковых и вирусных инфекций, таких, как Nocardia asteroides (L. Gruberg и соавт., 1991), легочный аспергиллез и токсоплазмоз (D. E. Furst и соавт., 1990), герпес (M. S. Antonelli и соавт., 1991), криптококкоз (M. Alt-Smith и соавт., 1987), пневмоцистная пневмония (J. B. Shiroky и соавт., 1991; R. L. Leff и соавт., 1990). Развитие инфекции является основанием для отмены МТ. Показано, что увеличение чувствительности к инфекции на фоне лечения МТ чаще наблюдается при РА, чем при псориазе. По данным S. L. Briges и соавт. (1991), среди 38 больных РА, которым производились ортопедические операции на суставах, частота инфекционных послеоперационных осложнений была достоверно выше у больных, получавших МТ, чем у пациентов, не получавших его. Однако в ретроспективном исследовании R. S. Perhala и соавт. (1991) было показано, что лечение МТ не приводило к нарастанию частоты локальных инфекционных осложнений у оперированных больных. Несмотря на то что полиглутамированные дериваты МТ длительно сохраняются в клетках, предполагается, что отмена МТ за 4-6 нед. до операции позволяет снизить опасность развития инфекционных послеоперационных осложнений. Другие осложнения Описано развитие так называемых постдозовых реакций, проявляющихся болями в суставах и мышцах, общим недомоганием, изредка лихорадкой, которые возникают через 24 ч после приема МТ и продолжаются в среднем 96 ч. (J. T. Halla и J. G. Hardin, 1993). По данным этих авторов, это осложнение является второй по частоте причиной прерывания лечения после желудочно-кишечных расстройств. У некоторых больных, особенно у лиц пожилого возраста с нарушением функции почек, наблюдаются неврологические расстройства, проявляющиеся головными болями, нарушением памяти, светобоязнью. Эти симптомы иногда могут быть выражены в такой степени, что приводят к прерыванию лечения. Редким побочным эффектом, ранее описанным только у больных с онкологическими заболеваниями, получающими очень высокие дозы препарата, является остеопатия, характеризующаяся болями в костях, остеопорозом, спонтанными переломами (S. J. Preston и соавт., 1994). При РА на фоне лечения МТ описано развитие своеобразного феномена, проявляющегося быстрым образованием подкожных узелков (нодулез), отличающихся от ревматоидных меньшим размером и нетипичной локализацией, но сходных с ними по морфологическим и иммуноморфологическим признакам (P. J. S. M. Kerstens и соавт., 1992). Существенного нарастания частоты онкопатологии на фоне лечения МТ не отмечено (A. Nyfos и соавт., 1983). 10-ДИАЗАМИНОПТЕРИН (10-DEAZAAMINOPTERIN) 10-ДАМ — новый аналог фолатов, используемый для лечения РА. По данным G. Alarcon и соавт. (1992), эффективность и токсичность 10-ДАМ сходны с МТ. Список литературы К главе "Метотрексат" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. Alarcon G. S., Tracy I. C., Blackburn W. D.: Methotraxate in rheumatoid arthritis. Toxic effects as the major factor in limiting long-term treatment. Arthritis Rheum., 1989; 32: 671-676. Alarcon G. S., Schrohenloher R. E., Bartolucci A. A. et al: Supression of rheumatoid factor production by metotrexate in patients with rheumatoid arthritis: Evidence for different influences of therapy and clinical status on IgM and IgA rheumatoid factor expression. Arthritis Rheum., 1990; 33: 1156. Alarcon G. S., Castaneda 0., Ferrandiz M., et al.: Efficacy and safety of 10-deazaaminopterin in the treatment of rheumatoid arthritis. Arthritis Rheum., 1992; 35: 1318-1321. Alarcon G. S., Lopez-Mendez A., Walter J., et al.: Radiographic evidence of disease progression im methotrexate treated and non methotrexate disease modifying antirheumatic drugs tested rheumatoid arthritis patients: a meta-analysis. J. Rheumatol, 1992; 19: 1868-1873. Al-Awadhil, Dale P., McKendry R J. R.: Pancytopenia associated with low dose methotrexate therapy. A regional survey. J. Rheumatol. 1993; 20: 1121-1125. Allen L. S., Groff G.: Treatment of Felty's syndrome with low dose oral methotrexate. Arthritis Rheum. 1986; 29: 902-905. Altz-Smith M., Kendal L. G., Jr. Stamm A. M.: Cryptococcus associated with low-dose methotrexate for arthritis. Am. J. Med. 1987; 83: 179-181. Andersen P. A., West S. G., O'Dell J. R., et al. Weekly pulse methotrexate in rheumatoid arthritis. Ann. Inter. Med., 1985; 103: 489-496. Antonelli M. S., Moreland L. W., Brick J. E.: Herpes zoster in patients with RA treated with weekly, low dose methotrexate. Am. J. Med. 1991; 90: 295-298. Baggott J. E., Morgan S. L., Ha T. S., et al: Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem J. 1992; 282: 197-203. Baggott J. E., Morgan S. L., Ha T.-S., et al. Antifolates in rheumatoid arthritis: a hypotetical mechanism of action. Clin. Exp. Rheumatol. 1993; II (Suppl. 8): S101-S105. Barrera P., Boerbooms A. M., Janssen E. M, met al. Circulating soluble tumor necrosis factor receptors, interleukin-2 receptors, tumor necrosis factor alpha, and interleukin-6 levels in rheumatoid arthritis. Longitudinal evaluation during methotrexate and azathioprine therapy. Arhtritis Rheum., 193; 36: 1070-1079. Ваггега P., Van der Hoogen F. H. J., Boerbooms A. M. Th., et al. Metotrexate-placebo controlled trial in systemic sclerosis: effects in circulating concentrations of bioactive interleukin-6. Brit. J. Rheumatol., 1994 (Sulll. 1); 33: 114 (abst.). Barrera P., Laan F. J. M., Van Riel P. L. C. M., et al.: Methotrexate-related pulmonary complications in rheumatoid arthritis. Ann. Rheum. Dis. 1994; 53: 434-439. Barrera P., Boerbooms A. M. Th., Demacker P. N. M., et al.: Circulating concentrations and production of cytokines and soluble receptors in rheumatoid arthritis patients: effects of a single dose methotrexate, Br. J. Rheumatol 1994; 33: 1017-1024. Bauer H., Breitbart A., Brado B., Pezzutto A.: Intravenous intensification of MTX-therapy in severe rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S232. Bologna C., Edno L., Anava J, M., et al.: Methotrexate (MXT) concentration in synovial membrane (SM) of rheumatoid arthritis (RA) patients. Arthritis Rheum. 1993; 36 (suppl.): S80. Bjorkman D. J., Boschert M., Tolman K., Clegg D., Ward J. R.: The effect of long-term metotrexate therapy on hepatic fibrosis in rheumatoid arthritis. Arthritis Rheum., 1993; 36: 1697.1701. Bridges S. L, Jr. Lopez-Mendez A., Han K.-H., et al.: Should methotrexate be discontinued before elective orthopedic surgery in patients with RA. J. Rheumatol., 1991; 19: 984-988. Buchbinder R., Hall S., Sambrook P. N., et al.: A life table review of 587 patients treated in community practice. J. Rheumatol. 1993; 20: 639-644. Chang D. M., Weinblatt M. E., Schur P. H.: The effects of methotrexate on interleukin-I in patients with rheumatoid arthritis. J. Rheumatol. 1992: 19: 1678-1682. Clegg D. O., Furst D. E., T9: Tolman K. G. et al.: Acute rever siblehepatic failure associated with methotraxate treatment of RA. J. Rheumatol. 1989; 16: 1123-1126. 23. Clegg D. O., and HCQ/MTX study group.; Combination hydroxychloroquine and methotrexate in the treatment of rheumatoid arthritis. Arthritis Rheum 1993; 36 (suppl.): S-53. 24. Connolly K. M, Stecher V. J., Danis E., et al.: Alteration of interleukin-I production and the acute phase response following medication of adjuvant arthritic rats with cyclosporin-A or methotrexate. Int. J. Pharmacol., 1988; 10: 717-728. 25. Cohen S., Rutstein J., Luggen M., et al. Comparison of the safety and efficacy of cyclosporin and methotrexat in refractory rheumatoid arthritis: a randomized, multicentered, placebo-controlled trial. Arthritis Rheum. 1993: 36 (suppl.): S-56. 26. Cronstein B. N., Eberle M. A., Gruber H. E., et al.: Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl. Acad. Sci. USA 1992; 88: 2441-2445. 27. Davidson J. R., Grazino P. M., Rothenburg R. J.: Methotrexate therapy for severe systemic lupus erythematosus. Arthritis Rheum. 1987; 30: 1195-1196. 28. Durk H., Koffler 1., Saal J. D.: Intraarticular methotrexate (MTX) therapy in corticosteroid resistant monoarthritis. Artritis Rheum. 1994; 37 (suppl.) S-252. 29. Fauci A. S. Treatment of glucocorticoid-resistent or relapsing Takayasu's arteritis with metotrexate. Arthritis Rheum. 1994; 37: 578-582. 30. Fietcher J. J., Miller D. R., Starkebaum G.: Reversal of neutropenia with methotrexate treatment in patients with Felty's syndrome: correletion of response with neutrophil-reac-tive IgG. Arthritis Rheum. 1989; 32: 194-201. 31. Firestein G. S., Paine M. M., Boyle D. L. Mechanism of metotrexate action in rheumatoid arthritis. Selective decrease in synovial collagenase gene expression. Arthritis Rhe um., 1994; 37: 193-200. 32. Fries J. F., Singh G., Lenert L., Furst D. E.: Aspirin, hydroxychloroquin, and hepatic enzyme abnormalities with methotrexate in rheumatoid arthritis. Arthritis Rheum., 1990; 33: 1611-1619. 33. Fujii Т., Akizuki M., Matsumara M., et al.: Efficacy of methotrexate therapy in patients with adult Still's disease. Arthritis. Rheum. 1994; 37 (suppl.): S-417. 34. Furst D. E., Erickson N., Cluete L., et al.: Adverse experience with methotrexate during 176 weeks of a long-term prospective trial in RA. J. Rheumatol. 1990: 17: 1628-1635. 35. Furst D. E., Koehnke R., Burmeister L. F., et al.: Increasing methotrexate effect with increasing dose in the treatment or resistant rheumatoid arthritis. J. Rheumatol. 1989; 16: 313-320. 36. Furst D. E.: Should methotrexate be used to treat early rheumatoid arthritis. Semin. Arthritis Rheum. 1993; 6 (suppl. 2): 39-43. 37. Gabriel S., Regan E., O'Fallon M., et al.: Treatment of rheumatoid arthritis with higher dose intravenous methotrexate. J. Rheumatol. 1990; 17: 460-465. 38. Galarza D., Esquivel J., Villarreal M., et al.: Methotrexate in lupus nephritis: an uncontrolled study, preliminary results. Arthritis Rheum. 1992; 34 (suppl.): S187. 39. Gilbert S. C., Klintman G., Menter A., et al.: Methotrexate induced cirrhosis requiring liver transplantation in three patients with psoriasis. Arch. Int. Med. 1990; 150: 889-891. 40. Goddard D., Gutilla R., Clark M., Bomalaski J. L: Methotrexate inhibits eicosanoid biosynthesis in the rat air pouch through diminished phospholipase A2 activity. Arthritis Rheum. 1994; 137 (suppl,): S-253. 41. Grospham J., Weinblatt M. E.: Methotrexate: mechanism of action, pharmacokinetics, clinical indications, and toxicity. Curr. Opin. Rheumatol., 1991; 3: 363-368. 42. Gruberg L., Thaler M., Rozenman J. et al.: Nocardia asteroides infection complicating rheumatoid arthritis. J. Rheumatol, 1991; 18: 459-461. 43. Haagsma С., van Reid P., de Rooil D. J., van der Putte L.; Combination therapy In RA: sulphasalazine and methotrexate. Arthritis Rheum. 1993; 36 (suppl.): S-53. 44. Halla J. T., Hardin J. G.: Post-dosing reactions to methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S-80. 45. Handrock K., Reinhold-Keller E., Duncker G., et al.: Beneficial effects of low-dose methotrexate in Wegener's granulomatosis. Arhtritis Rheum. 1994: 37 (suppl.): S-353. 46. Haraoui B., Pelletier J. P., Cloutier J. M., et al.: Synovial membrane histology and immunopathology in rheumatoid arthritis and osteoarthritis. In vivo effects of antirheumatic drugs. Arthritis Rheum., 1991: 34: 153-163. 47. Harel L., Wagner-Weiner L., Poznanski A. K., et al. Effects of methotrexate on radiologic progression in juvenile rheumatoid arthritis. Arthritis Rheum., 1993; 10: 1370-1374. 48. Herman R. A., Veng-Pedersren P., Hoffman J., et. al. Pharmako kinetics of low-dose metotrexate in rheumatoid arthritis patients. J. Pharm. Sci., 1989; 78: 165-171. 49. Hernandes-Garcia С., Soraino С., Morando C., et al.: Methotrexate treatment in the management of giant cell arteritis. Scand. J. Rhgeumatol. 1994; 23: 295-298. 50. Hine R. J., Everson M. P., Hardin J. M., et al.: Methotrexate therapy in rheumatoid arthritis diminished lectin-induced mononuclear cell prolipheration. Rheumatol. Int., 1990; 10: 165-169. 51. Hirata S., Matsubara Т., Saura R., et al.: Inhibition of in vitro vascular endothelial cell prolipheration and in vivo neovascularization by low-dose methotrexate. Arthritis Rheum., 1989; 32: 1065-1073. 52. Hoffman G. S., Leavitt R. Y., Kerr G. S., Fauci A. S.: The treatment of Wegeners's granulomatosis with glucocorticoids and metotrexate. Arthritis Rheum, ml992; 35: 1322-1329. 53. Hoffman G., Leavitt R., Kerr G., Rotterm M., Sneller M. C., Johnston C., Russel A. S., Aaron S.: The effect of in vivo and in vitro methotrexate on lymphocyte prolipheration as measured by uptake of tritiayed thymidine and tritiated guanosine. Clin. Exp. Rheumatol., 1988; 6: 391-393. 54. Jolivet J., Schilsky R. L., Bailey B. D., et al.: Synthesis, retention and biological activity of methotrexate polyglutamates in cultured human brest cancer cells. J. Clin. Invest., 1982; 70: 351360. 55. Joyce D. A., Wikk R. K., Hoffman D. M, et al.: Exacerbation of rheumatoid arthritis in patients treated with methotrexate after administration of folinic acid. Ann. Rheum. Dis.: 1991; 50: 913-914. 56. Kaye 0., Kahn M. F., Palazzo E., Bourgeois P.: Low dose methotrexate in musculoskeletal manifestations of sarcoidosis. Arthritis Rheum. 1993; 36 (suppl.): S-203. 57. Kerstens P. J. S. M., Boerbooms A. M. T. H., Jeurissen M. E. C., et al.: Accelerated nodulosis during low dose metotrexate therapy for rheumatoid arthritis. An analysis of ten cases. J. Rheumatol., 1992; 19: 867-871. 58. Kozlovski G., De Vito J. M., Kisicki J. C., Johnson J. B.: The effect of food on the absorbtion of metotrexate sodium tablets in healthy volunteres. Arthritis Rheum., 1992; 35: 761-764. 59. Kremer J. M., Phelps C. T. Long-term prospective study of the use of methotrexate in the treatment of rheumatoid arthritis. Update after a mean of 90 months. Arthritis Rhe um., 1992; 35: 138-145. 60. Kremer J. M., Petrillo G. F., Lawrence D. A.: Methotrexate (MTX) induces significant changes in IL-1, IL-2. IL-6 and IL-8, but not lymphocyte markers in patients with rheumatoid arthritis. Arthritis Rheum. 1993: 36: S-77. 61. Kremer J. M.: The mechanism of action of methotrexate in rheumatoid arthritis: the search continues. J. Rheumatol. 1994; 21: 1-5. 62. Laurindo I. M. M., Mello S. B. V., de Falco V., et al.: Action of methotrexate on superoxide production (abstract). Rev. Esp. Rheumatol. 1993; 20 (suppl. 1): 6 ( 12). 63. Leff R. L, Case J. P., McKenzie R.: Rheumatoid arthritis, methotrexate therapy and pneumocystic pneumonoa. Ann. Int. Med. 1990; 1123: 716. 64. LeRoux J. L. Damon M., Chavis C., et al.: Effects of a single dose of metotrexate on 5-and 12lipoxygenase products in patients with rheumatoid arthritis. J Rheumatol., 1992; 19: 863-866. 65. Martinez-Osuna P., Zwolinska J. B., Sikes D. H., et al.: Lack of immunosupressive effect of lowdose oral methotrexate on lymphocytes in rheumatoid arthritis. Clin. Exp. Rheuma tol. 1993; II: 249254. 66. Marul Т., Matsubara Т., Mizuno К., Itoh H.: Inhibition of metalloproteinase activity derived from chondrocytes by MT. Arthritis Rheum. 1993; 36 (suppl.): S-79 67. Merrill J., Lahita R. G., Shen C., Cronstein B.: Rheumatoid nodulosis in patients treated with methotrexate: adenosin, acting primary at A2 receptors promote CR3-mediated formation of multinucleated giant cells (MGC) Arthritis Rheum. 1993: 36 (suppl): S-79. 68. Meyer F. A., Yaron I., Mashiah V., Yaron M.: Methotrexate inhibits proliferation but not interleukin I stimulated secretory activities of cultured human synovial fibroblasts. J. Rheumatol. 1993; 20: 238242. 69. Miescher P. A., Riethmuller D.: Diagnosis and treatment of systemic lupus erythematosus. Semin. Hematol. 1965; 2: 1-28. 70. Miller L. C., Cohen E. E., Orencole S. F. et al.: lnterleukin-1 beta is structurally related to dihydrofolate reductase: effects of methotrexate (abstract). Cytokine Res. 1988; 7: 49. 71. Montecucco C., Rossi S., Porta C.: Allopurinol mouthwasher in methotrexate-induced stomatitis. Arthritis Rheum. 1994; 37: 777-778. 72. Morand E. F., Axtens R. S. K., Littejohn G. O.: Combination methotrexate and sulphasala-sine therapy in patients with rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S-53. 73. Morgan S. L, Baggot J. T., Vaughn W. H., et al.: 5mg or 50 mg/week of folic acid supplementation does not alter the efficacy of methotrexate treated rheumatoid arthritis patients. Arthritis Rheum. 1992; 35 (suppl. 9): S343. 74. Morgan S. L., Alarcon G. S., Krumdieck C. L.: Folic acid supplementatrion during methotrexate therapy: It makes sense. J. Rheumatol. 1993; 20: 1121-1125. 75. Morgan S. L., Baggott J. E., Alarcon G. S.: Methotrexate and sulfasalazine combination therapy: is it worth the risk. Arthritis Rheum. 1993; 36: 281-282. 76. Morgan S. L. et al.: Supplementation with folic acid during methotrexate therapy for rheumatoid arthritis: a double-blind placebo controlled trial. Ann. Int. Med., 1994; 121: 833-841. Nissar M., Carlisle L., Amas R. S.: Methotrexate and sulphasalazine combination therapy in rheumatoid arthritis. Br. J. Rheumatol. 1994; 7: 651-654. 77. Nyfors A., Jensen H.: Frequency of malignant neoplasms in 248 long term methotrexate treated psoriasis. Dermatologica 1983; 167: 260-261. 78. O'Callaghan J. W., Forrest M., Brooks P. M.: Inhibition of neutrophil chemotaxis in rheumatoid arthritis patients. Rheumatol. Int. 1988; 8: 41-45. 79. Oguey D., Kolliker F., Gerber N. J., Reichen J.: Effect of food on the bioavalability of low-dose metotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 1992; 35: 611-614. 80. Olsen N. J., Callahan L. F., Pincus T. Immunologic studies of rheumatoid arthritis patients treated with metotrexate. Arthritis Rheum., 1987; 30: 481-488. 81. Olsen N. J., Murray L. M.: Antiprolipherative effects of methotrexate on peripheral blood mononuclear cells. Arthritis Rheum. 1989; 32: 378-385. 82. Paters L. J., Olsen N. J.: Mechanisms of action of metotrexate. Bull. Rheum. Dis., 1991; 41: 5-7. 83. Perhala R. S., Wlike W. S., Clough J. D., et al.: Local infectious complications following large replacement in RA patients treated with methotrexate versus those not treated with methotrexate. Arthritis Rheum. 1991; 34: 146-152. 84. Philips C. A., Cera P. J., Mangan T. F., et al.: Clinical liver disease in patients with RA taking methotrexate. J. Rheumatol. 1992; 19: 229-233. 85. Plotz P. H., Dalakas M., Leff R. L., Love L. A., Miller F. W., Cronin M. E.: Current concepts in the idiopathic inflammatory myopathies: dermatomyositis and related disorders. Ann. Intern. Med. 1989; III: 143- 157. 86. Preston S. J., Diamond Т., Scott A., Laurent M. R.: Methotrexate osteopathy in rheumatic disease. Ann. Rheum. Dis. 1994; II: 13-16. 87. Rau R.: Metotrexate therapy in rheumatoid arthritis. Rheumatol. in Europe 1994; 23: 60-65. 88. Richardson В.: Methotrexare therapy for hearing loss in Cogan's syndrome. Arthritis Rheum. 1994; 37: 1559-1561. 89. Rothenburg R. J., Graziano F. M., Grandone J. T., et al,: The use of methotrexate in steroid resistant systemic lupus erythematosus. Arthritis Rheum. 1988; 31: 612-615. 90. Salaffi F., Carotti M., Cervini C.: A prospective study of the long-term efficacy and toxic-ity of lowdose methotrexate in rheumatoid arthritis. Clin. Exp. Rheumatol. 1995; 13: 23-28. 91. Sany J., Anaya J. M., Lussiez V., et al.: Treatment of rheumatoid arthritis with methotrexate: a prospective open-term study of 191 case. J. Rheumatol. 1991; 18: 1323-1327. 92. Schnabel A., Gross W. L.: Low-dose methotrexate in rheumatic disease-efficacy, side effects, risk factors for side effects. Semin. Arthritis Rheum., 1994; 23: 310-327. 93. Schnabel A., Reinhold-Keller E., Willmann V., Gross W. L.: Tolerabillity of metotrexate starting with 15 to 25 mg/week for rheumatoid arthritis. Rheum. Int. 1994; 14: 33-38. 94. Seibord J. R., McCloskey D. A., Furst D. E.: Pilot trial of methotrexate (MTX) in treatment of early diffuse scleroderma. Arthritis Rheum. 1994; 37 (suppl.): S-262. 95. Seideman P., Albertioni R., Beck O., et. al. Chloroquine reduced the bioavailability of metotrexate in patients with rheumatoid arthritis. Arthritis Rheum., 1994, 37: 830-833. 96. Segal R., Caspi D., Tisher M., et al.: Short term effects of low dose methotrexate on the acute phase reaction in rheumatoid arthritis. J. Rheumatol. 1989; 16: 14-18. 97. Segal R., Mozes E., Yaron M., Tartakowsky B.: The effects of methotrexate on the production and activity of interleukin 1. Arthritis Rheum. 1990; 32: 370-377. 98. Segal R., Yaron M., Tartakovsky B.: Methotrexate: mechanism of action in rheumatoid arthritis. Semin. Arthritis Rheum., 1990; 20: 190-200. 99. Segal R., Yaron M., Tartakowsky B.: Rescue of interleukin-I activity by leukovorin following inhibition by methotrexate in a murine in vitro system. Arthritis Rheum. 1990; 33: 1745-1749. 100. Shatten S., Asherson R. A., Hughes G. R. V.: Methotrexate (MTX) therapy for systemic lupus erythematosus (SLE) and SLE overlap syndrome. Arthritis Rheum. 1992; 34 (suppl.): S187. 101. Shergy W. J., Polisson P. P., Cadwell D. S. et al.: Methotrexate associated hepatotoxicity: Retrospective analysis of 210 patients with RA. Am. J. Med. 1988; 85: 771-777. 102. Shiroky J. B., Frost A., Skelton J. D. et al.: Complicatins of immunosupession associated with weekly low dose methotrexate. J. Rheumatol. 1991; 18: 1172-1175. 103. Shiroky J. B., Neville C., Esdaile J. M. et al. Low-dose metotrexate with leukovorin (folinic acid) in the managemeht of rheumatoid arthritis: results of a multicentrer random-ized, double-blind, placebo control trial. Arthritis Rheum., 1993; 36: 795-803. 104. Singh R. R., Malaviya A. N., Pandey J. N., Guleiria J. S. Fatal interaction between metotrexate and naproxen (letter). Lancet 1986; i: 1390. 105. Songsiridej N., Furst D. E.: Methotrexate — The rapidly acting drug. Clin. Rheumatol, 1990; 4: 575594. 106. Sperling R. I., Benincaso A. I., Anderson R. J., et al.: Acute and chronic suppression of leukotriene B4 synthesis ex vivo in neutrophils from patients with rheumatoid arthritis beginning treatment with methotrexate. Arthritis Rheum. 1992; 35: 376-384. 107. Stewart C. F., Fleming R. A., German B. F., et al.: Aspirin alters metotrexate disposition in rheumatoid arthritis patients. Arthritis Rheum., 1991; 34: 1514-1520. 108. Thomas R., Carrol G. J.: Reduction of leukocyte and interleukin-I beta concentrations in the synovial fluid of rheumatoid arthritis patients treated with methotrexate. Arthritis Rheum. 1993; 36: 12441252. 109. Thomas M. H., Gutterman L. A.: Methotrexate toxicity in patient receiving trimethoprimsulphamethoxazole. J. Rheumatol. 1986: 13: 440-441. 110. Thompson P. M., Watts C., Edelman J., et al.: A controlled two-centre trial of parental methotrexate therapy for refractory rheumatoid arthritis. J. Rheumatol. 1984; II: 760-763. 111. Tishler M., Caspi D., Fishel B., Yaron M.: The effects of leucovorin (folinic acid) on methotrexate therapy in rheumatoid arthritis. 1988; 31: 906-908. 112. Walz Le Blanc B. A. E., Dagenais P., Urowitz M. B., Gladman D. D.: Methotrexate in systemic lupus erythematosus. J. Rheumatol. 1994; 21: 836-838. 113. Weinblatt M. E., Polisson R., Blotner S. D., et al.: The effect of drug therapy on radiographic progression of rheumatoid arthritis. Arthritis Rheum., 1993; 36: 613-619. 114. Weinblatt M. E., Kaplan H., Germain B. F., et al.: Methotrexate in rheumatoid arthritis. A five-year prospective multicenter study. Arthritis Rheum. 1994; 37: 1492-1498. 115. Whitaker, J. N., Pinals M. S.: Dermatomyositis/polymyositis. Current therapy in allergy and immunology 1984; Ed. L. M. Lichtenstein, A. S. Fauci. B. C. Decker. Inc. Philadelphia, Toronto, pl 22-125. 116. White-O'Keefe Q. E., Fye K. H., Sack C. D,: Metotrexate and histologic hepatic abnormalities: a meta-analysis. Am. J. Med., 1991; 90: 711-716. 117. White-O'Keefe Q. E., Fye K. H., Sack K. D.: Liver biopsy and methotrexate use: See no evil...? Am. J. Gastroenterol. 1991; 70: 711-716. 118. Wilke W. S., Krall P. L., Scheetz R. J., et al.: Metotrexate for systemic lupus erythematosus: a retrospective analysis of 17 unselected patients. Clin. Exp, Rheumatol., 1991; 9: 581-587. 119. Williams H. J., Willkens R. F., Samuelson C. O., et al.: Comparison of low-dose oral pulse metotrexate and placebo in the treatment of rheumatoid arthritis: a controled trial. Arthritis Rheum., 1985; 28: 721-730. 120. Williams H., Ward J. R., Reading J. C., et al.: Comparison of auranofin, metotrexate, and the combination of both in the treatment of rheumatoid arthritis. Arthritis Rheum., 1992; 35: 259-269. 121. Wilkens R. F., Leonard P. A., Clegg D. D. et al.: Liver histology in patients receving low dose pulse methotrexate for the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 1990; 49: 591-593. 122. Willkens R. F., Urowitz M. B., Stablein D. M., et al.: Comparison of azathioprine, metotrexate, and the combination of both in the treatment of rheumatoid arthritis. Arthritis Rheum. 1992; 35: 849-856. 123. Wise C. M., Vuyyru S., Roberts W. N.: Methotrexate in systemic lupus and lupus-like illness- a review of 31 patients. Arthritis Rheum. 1994; 37 (suppl.): S-1467. Wolff R., Asako H., Granger D. N.: Adenosine mediated mithotrexate-induced attenuation of leukocyte adherence and emigration in postcappillary venules. Arthritis Rheum., 1992; 35: S-35. ГЛАВА 7 АНТИМАЛЯРИЙНЫЕ (АМИНОХИНОЛИНОВЫЕ) ПРЕПАРАТЫ Вещества с антималярийными свойствами, впервые полученные из коры произрастающего в Перу хинного дерева, применяются в медицине в течение многих лет. Их активные компоненты, хинин и цинхонин, были изолированы Pelletier и Caventau в 1820 г. В 1894 г. английский врач J. P. Payne впервые выявил благоприятный клинический эффект хинина при красной волчанке у 18-летнего юноши и связал его со способностью хинина влиять на "сосудистые нарушения". В ревматологии наиболее часто используют 2 синтетических 4-аминохинолиновых аналога: хлорохин дифосфат (делагил) и гидроксихлорохин сульфат (плаквенил; Sanofi Winthrop Pharm.). Эти вещества имеют сходную структуру и отличаются заменой этиловой группы в боковой цепи молекулы хлорохина на гидроксиэтиловую группу в молекуле гидроксихлорохина. Полагают, что оба препарата обладают примерно одинаковой клинической эффективностью, однако, по данным токсикологических исследований, хлорохин примерно в 2-3 раза токсичнее, чем гидроксихлорохин (E. W. McChesney, 1983). Фармакологические свойства После перорального приема оба вещества очень быстро абсорбируются в ЖКТ; большая их часть экскретируется с мочой в неизмененном виде, меньшая часть (примерно 1/3) подвергается метаболизму с образованием продуктов деградации боковых цепей аминоэтильных групп. Период полужизни хлорохина колеблется от 3.5 до 12 дней, стабильный уровень в плазме достигается после 2-5 нед. постоянного приема препарата в терапевтической дозе. Концентрация препаратов в ткани выше, чем в плазме. Механизм действия Механизмы, определяющие терапевтическую эффективность аминохинолиновых препаратов, остаются неясными. Лишь в последние годы была проведена серия исследований, результаты которых свидетельствуют о том, что антималярийные препараты дают более широкий спектр терапевтических эффектов, чем считалось до сих пор, а также имеют своеобразные молекулярные механизмы фармакологической активности (R. I. Fox, 1993), которые отличаются от механизмов действия других противоревматических препаратов. Анализ имеющихся в настоящее время фактов свидетельствует о том, что аминохинолиновые препараты в терапевтических дозах могут давать клинически значимые противовоспалительный, иммуномодулирующий, фотопротективный, антиоксидантный, антимикробный, антипролиферативный, антиагрегантный, гиполипидемический, гипогликемический и анальгетический эффекты. Кроме того, недавно было обнаружено, что аминохинолиновые производные, по крайней мере in vitro, обладают способностью восстанавливать чувствительность опухолевых клеток к цитотоксическим химиотерапевтическим препаратам (J. Zamora и W. Beck, 1986). R. I. Fox и H.-O. Kang (1993) высказали предположение о том, что аминохинолиновые производные, являясь слабыми основаниями при нейтральном рН, могут диффундировать к кислым вакуолям цитоплазмы макрофагов, где протонируются и вызывают повышение рН цитоплазмы. Это в свою очередь приводит к нарушению процесса расщепления и презентации аутоантигенов макрофагами СD4+Т-лимфоцитам (глава 2). Кроме того антималярийные препараты в терапевтических концентрациях проявляют многообразные эффекты в отношении клеточных иммунных реакций и синтеза цитокинов (таблица 7.1), обладают способностью стабилизировать мембраны лизосом, подавлять хемотаксис лейкоцитов, образование супероксидных радикалов (N. P. Hurst и соавт., 1986; Y. Miyachi и соавт., 1986) и снижать синтез фибронектина и фибриногена. Таблица 7.1. Иммунные эффекты антималярийных препаратов 1. Подавление процессинга и презентации антигенов (R. I. Fox и H-I Kang, 1993) 2. Ингибиция пролиферативного ответа лимфоцитов при стимуляции митогенами, активности ЕКклеток (C. M. Ausiello и соавт., 1986; G. S. Panayi и соавт., 1973; S. Salmeron и P. E. Lipsky, 1983) 3. Подавление синтеза ИЛ-1, ФНО (S. Picot, 1985) макрофагами и ИЛ-1 индуцированного синтеза ПГЕ2 синовиоцитами (D. G. Baker и соавт., 1984) 4. Нарушение экспрессии ИЛ-2 Рецепторов (A. Ferrante и соавт., 1986) 5. Ингибиция индуцированного ИЛ-1 разрушения хряща (K. D. Rainsford, 1985) 6. Посттранскрипциоиная блокада синтеза ИЛ-1-а моноцитами и ИЛ-6 моноцитами и Т-клетками, стимулированными ËÏÑ, при отсутствии влияния на синтез ИЛ-2, ИЛ-4 и ИФ-y (К. Sperber и соавт., 1993) 7. Подавление синтеза ИФ-у активированными Т-лимфоцитами (R. B. M. Landewe и соавт., 1992) 8. Снижение уровня ИЛ-б, CD8, рИЛ-2Р в сыворотках больных СКВ (D, J. Wallace и соавт., 1993) Антиагрегационное действие Имеются данные о способности антималярийных препаратов подавлять агрегацию и адгезию тромбоцитов, не влияя на время свертывания крови. Лечение этими препаратами снижает частоту послеоперационных эмболий у больных, подвергнутых ортопедическим операциям (J. R. London, 1988). У больных СКВ, леченных гидроксихлорохином, частота тромбоэмболий существенно ниже, чем у больных СКВ, которые не принимали этот препарат (D. J. Walace, 1987), Недавно выявлен профилактический эффект гидроксихлорохина в отношении развития тромботических осложнений у больных СКВ с АФС (M. Petri и соавт., 1994; D. J. Wallace и соавт., 1993). Анальгетическая активность Недавно были получены данные об анальгетической активности гидроксихлорохина. По данным C. D. Middleton и соавт. (1995), прием гидроксихлорохина в дозе 400 мг 2 раза в день ассоциируется с очень быстрым увеличением болевого порога, который оценивали с помощью долориметрии. По мнению авторов, эти результаты соответствуют ранее полученным данным о более быстрой динамике болевого счета и болезненности суставов на фоне лечения гидроксихлорохином при РА (P. Clark и соавт., 1993) и волчаночном артрите (H. J. Williams и соавт., 1994) по сравнению с изменением других клинических показателей. Кроме того, по данным тех же авторов, гидроксихлорохин эффективно повышает болевой порог у больных СКВ с синдромом вторичной полимиалгии (G. D. Middleton и соавт., 1994). Полагают, что анальгетический эффект аминохинолиновых производных может быть связан с их основным механизмом действия, а именно с повышением рН цитоплазмы в различных клетках, что потенциально может вести к изменению активности нейротрансмиттеров, регулирующих болевые реакции. Лекарственная резистентность Новый аспект фармакологической активности антималярийных препаратов связан с возможным влиянием на механизмы, определяющие множественную лекарственную резистентность, нередко развивающуюся у больных со злокачественными новообразованиями, ранее чувствительных к противоопухолевой цитотоксической терапии. Установлено, что один из механизмов устойчивости к химиотерапии у онкологических больных связан с экспрессией на биомембране опухолевых клеток 170000 kD гликопротеина, который функционирует как насос, вымывающий препараты из клеток (B. A. Chamber и W. Wilson, 1991). Развитие устойчивости к ранее эффективным препаратам (в том числе цитотоксическим) нередко наблюдается в процессе лечения больных с ревматическими заболеваниями, особенно РА. В связи с этим представляют интерес данные о способности хинина (M. Lehnert и соавт., 1991) и аминохинолиновых производных (J. Zamora и W. Beck, 1986) in vitro ингибировать насосную функцию этого гликопротеина и, таким образом, способствовать преодолению лекарственной резистентости. Эти данные могут иметь очень важное значение для разработки методов комбинированной цитотоксической терапии РА, а возможно, и других воспалительных ревматических заболеваний. Липидный обмен Антималярийные препараты оказывают гиполипидемическое действие in vitro и in vivo. Предполагается, что этот эффект связан с подавлением протеолиза ЛНП-рецепторов или увеличением их экспрессии за счет ингибиции гидролиза интернализованных эфиров холестерина (A. C. Benyen и соавт., 1986; А. С. Веnуеn и соавт., 1986; J. L. Goldstein и соавт., 1975). По данным клинических исследований при СКВ лечение аминохинолиновыми препаратами приводит к снижению на 35-54% уровня ЛНП, триглицеридов, липопротеинов высокой плотности, липопротеинов очень низкой плотности, аполипопротеина CIII (H. N. Hokdis и соавт., 1993) и общего холестерина (M. Petri и соавт., 1994). Клиническое применение Начиная с 50-х годов, антималярийные препараты с успехом применяются для лечения многих воспалительных ревматических заболеваний (таблица 7.2.). Таблица 7.2. Использование антималярийных препаратов в ревматологии Заболевание Комментарий Дискоидная волчанка СКВ Ремиссия или улучшение у 60-90% больных Лечение умеренно выраженных обострений; антитромботический эффект (АФС); гиполипидемический эффект РА Эффект через 6 мес. и более; не влияют на рентгенологическое прогрессирование; уменьшают гепатотоксичность МТ; важный компонент комбинированной базисной терапии Серонегативные спондилоартрoпатии Могут быть эффективны при псориатическом артрите (иногда вызывают обострение кожного процесса) и анкилозирующем спондилите (влияют только на периферический артрит) Палиндромный ревматизм Доказана эффективность в неконтролируемых исследованиях Эозинофильный фасциит Доказана эффективность в неконтролируемых исследованиях Ювенильный ДМ Положительный эффект в отношении поражения кожи; Синдром Шегрена Эрозивный остеоартрит стероидсберегающее действие Нормализация лабораторных показателей; клиническая эффективность не доказана Положительный эффект при неэффективности НПВС Дискоидная волчанка и СКВ Антималярийные препараты в течение многих лет широко используются для лечения дискоидной волчанки и СКВ (В. А. Насонова и Я. А. Сигидин, 1985; М. М. Иванова, 1994). При дискоидной волчанке их эффективность колеблется от 60 до 90%, в то время как спонтанные ремиссии развиваются не более чем у 15% больных. Однако для купирования кожного синдрома часто приходится применять очень высокие дозы препарата (до 1200 мг/день), что существенно увеличивает возможность развития токсических реакций. Кроме того, после отмены препаратов нередко развивается обострение. При СКВ положительный эффект в отношении отдельных клинических проявлений заболевания был подтвержден в серии открытых и контролируемых исследований, главным образом у больных с минимальной или умеренной активностью патологического процесса (J. M. Esdale, 1993). Первое обширное ретроспективное исследование эффективности антималярийных препаратов, включающее 165 больных, было проведено R. D. Rudnick и соавт. (1975), которые показали, что назначение высоких доз хлорохина (1500 мг/день) приводит к достоверному снижению выраженности конституциональных симптомов, поражения кожи и ассоциируется с уменьшением частоты обострений заболевания. По данным Канадской группы по изучению эффективности гидроксихлорохина (1991), прием препарата (средняя доза 272 мг/день) в течение 37 мес. приводит к уменьшению частоты обострений: их вероятность была в 2.5 раза выше у больных, получавших плацебо, чем у пациентов, принимающих гидроксихлорохин. Результаты многоцентрового 48-недельного испытания свидетельствуют об эффективности гидроксихлорохина (200 мг 2 раза в день) при волчаночном артрите, по крайней мере в отношении выраженности субъективных симптомов, в то время как влияние препарата на другие проявления СКВ было менее очевидным (H. J. Williams и соавт, 1994). В настоящее время общепринятым является мнение, что антималярийные препараты не играют существенной роли в лечении больных тяжелой СКВ, хотя и не исключается их положительное действие на некоторые проявления болезни при сочетании с другими препаратами. Действительно, не хватает страниц 174-177 Побочные эффекты Результаты мета-анализа 71 клиническего испытания свидетельствуют о том, что антималярийные препараты значительно менее токсичны, чем другие базисные противоревматические средства, включая метотрексат (D. T. Felson и соавт., 1990). Частота побочных эффектов на фоне лечения варьирует в очень широких пределах и зависит в первую очередь от дозы препарата. Например, побочные эффекты встречаются у больных РА, получающих 400 мг гидроксихлорохина, в 3 раза чаще, чем у больных РА, получающих 200 мг этого препарата. Желудочно-кишечные осложнения Желудочно-кишечные расстройства напоминают те, которые возникают на фоне лечения НПВС, однако антималярийные препараты не вызывают желудочно-кишечных кровотечений. Тошнота и потеря аппетита обычно наблюдаются в начале лечения. Такие симптомы, как диарея, спазмы желудка, вздутие живота, более часто встречаются на фоне лечения антималярийными, чем другими противоревматическими препаратами. Гидроксихлорохин менее токсичен для ЖКТ, чем хлорохин. Поражение кожи Наиболее частым побочным эффектом, приводящим к прерыванию лечения, является кожная сыпь, которая возникает с одинаковой частотой на фоне как гидроксихлорохина, так и хлорохина. Поражение кожи может проявляться лихеноидными и макупапулярными высыпаниями и крапивницей. Очень редко наблюдаются эксфолиативный дерматит, нарушение пигментации кожи и волос, алопеция. Неврологические нарушения Неврологические нарушения обычно умеренные, быстро исчезают при снижении дозы препаратов и поэтому редко имеют клиническое значение. Описано развитие головных болей, головокружения, бессонницы, обратимого миастенического синдрома, очень редко — тяжелой миопатии, проявляющейся проксимальной мышечной слабостью на фоне нормальной концентрации КФК или кардиомиопатии. Офтальмологические осложнения Наиболее грозным побочным эффектом антималярийных препаратов является поражение глаз, которое может быть трех типов: 1. дефекты аккомодации или конвергенции, приводящие к нарушению зрения, затруднениям при быстром изменении фокусного расстояния и диплопии, которые связаны с центральными эффектами или параличом глазных мышц; 2. отложение препаратов в роговице, обычно не приводящее к тяжелым нарушениям зрения и быстро исчезающее после прерывания лечения или даже несмотря на продолжение лечения; 3. токсическое поражение сетчатки, являющееся наиболее тяжелым осложнением, нередко приводящим к прогрессирующему ухудшению зрения. К симптомам, характерным для антималярийной ретинопатии, относятся центральная скотома, сужение периферических полей зрения, позднее общее ухудшение зрения. Поражение сетчатки может быть двух типов: премакулопатия, обратимая при отмене препаратов, и истинная ретинопатия, редко купирующаяся при прерывании лечения. Интересно, что случаев прогрессирования премакулопатии в ретинопатию не характерно. При этом необходимо подчеркнуть, что гидроксихлорохин реже вызывает развитие ретинопатии, чем хлорохин, причем в подавляющем большинстве случаев больные получали дозы препаратов, значительно более высокие, чем рекомендуемые. В таблице 7.4. суммированы рекомендации, которые необходимо соблюдать для предотвращения возможности развития ретинопатии. Таблица 7.4. Рекомендации по офтальмологической безопасности лечения антималярийными препаратами (R. Rynes, 1993) Суточная доза: Гидроксихлорохин Хлорохин 400 мг/день (6.5 мг/кг) 200 мг/день (4 мг/кг) Офтальмологический контроль: Частота Протокол осмотра больного До лечения, затем каждые 3 мес. Задать вопрос о зрительных расстройствах. Исследование глазного дна (пигментация). Исследование полей зрения. Самоконтроль Хотя использование антималярийных препаратов не рекомендуется на фоне беременности (описано рождение детей с дефектами слуха), имеются сообщения об успешных родах у женщин, длительное время получавших эти препараты (A. L. Parke, 1993). Таким образом, использование антималярийных препаратов по-прежнему рассматривается как эффективный (основной или вспомогательный) подход к лечению различных воспалительных ревматических заболеваний. Новые сведения, касающиеся механизмов действия и клинических эффектов этих препаратов, позволяют существенно расширить спектр их применения в ревматологии, включив в перечень показаний профилактику тромбозов, токсических эффектов на фоне лечения МТ и коррекцию нарушений липидного и углеводного обмена, особенно у больных, получающих длительную глюкокортикоидную терапию. Место антималярийных препаратов в комплексном лечении РА, особенности при развитии резистентности к базисной терапии требует дальнейшего изучения. Список литературы к главе "Антималярийные препараты" 1. 2. 3. 4. 5. 6. 7. 8. Насонова В. А.. Сигидин Я. А.: Патогенетическая терапия ревматических заболеваний. Москва. Медицина. 1985. 286 с. Иванова М. М.: Системная красная волчанка. В книге: : Диффузные болезни соединительной ткани. Руководство для врачей. Авторы: Сигидин А. Я., Гусева Н. Г., Иванова М. М. Москва, Медицина. 1994: 231-300. Ausiello С. М., Barbieri P., Spagnoli G. C., et al.: In vitro effects of chloroquine treatment on spontaneous and interferon-induced natural killer activities in rheumatoid arthritis. Clin. Exp. Rheumatol. 1986; 1: 255259. Baker D. G., Baumgarten D. F., Dwyer J. P.: Chloroquine inhibits the production of mononuclear cell factor by inhibition of lectin binding. Arthritis Rheum. 1984; 27: 888-896. Benyen A. C., van der Moolen A. J., Geelen M. J. H.: Inhibition of hepatic cholesterol synthesis by chloroquine. Lipids 1981; 16: 472-474. Benyen A. C.: Can chloroquine be of value in the treatment of hypercholesterolemia? Artery 1986; 13: 340351. The Canadian Hydroxychloroquin study group. A randomized study of the effect of withdrawing hydroxichloroquin sulfate in systemic lupus erythmato-sus. New Engl. J. Med., 1991; 324: 150-154. Chamber Â. À., Wilson W.: Reversal of multidrug resistance. J. Clin. Oncol. 1991; 9: 4-6. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. Clark P., Casas E., Tugwell P., et al.: Hydroxichlorochine compared with placebo in rheumatoid arthritis. A randomized controlled trial. Ann. Intern. Med., 1993; 119: 1067-1071. Clegg D. 0., and HCQ/MT study group: Combination hydroxychloroquine and methotrexate in the treatment of rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S53. Combe B., Guttierez M., Anaya J., Sany J.: Possible efficacy of hydroxichloroquine on accelerated nodulosis during methotrexate therapy of rheumatoid arthritis. J. Rheuma tol. 1993; 20: 755-756. Epstein W. V.: Efficacy of standart slow-acting antirheumatic drug. Semin. Arthritis Rheum. 1994; 23 (suppl. 2): S32-S38. Esdale J. M. The efficacy of antimalarias in systemic lupus erythematosus. Lupus 1993; 2 (suppl.): S-2-S-8. Felson D. T., Anderson J. J., Meenan R. F.: The comparative efficacy and toxicity of second-line drugs in rheumatoid arthritis. Arthritis Rheum., 1990; 33: 1. Felson D. T., Anderson J. J., Meenan R. F.: Use of short-term efficacy/toxicity tradeoffs to select secondline drugs in rheumatoid arthritis. Arthritis Rheum. 1992; 35: 1117-1125. Ferrante A., Rpwan-Kelly B., Seow W. K., Thong Y. H.: Depression of human polymorphonuclear leucocyte function by anti-malarial drugs. Immunol., 1986; 58: 125-130. Fox R. I., Chan E., Benton L, et al.: Treatment of primary Sjodren's syndrome with hydroxychloroquine. Amer. J. Med., 1988; 85: 62. Fox R. I., Kang H-I.: Mechanism of action of antimalarial drugs: inhibition of antigen processing and presentation. Lupus 1993; 2 (suppl.): S-9-S-12. Fox R. I.: Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin. Arthritis Rheum. 1993; 23 (suppl. 1): 82-91. Fries J. F., Singh G., Lenert L., Furst D. E.: Aspirin, hydroxychloroquine, and hepatic enzyme abnormalities with methotrexate in rheumatoid arthritis. Arthritis Rheum. 1990; 33: 1611-1619. Goldstein J. L., Brunschede G. Y., Brown M. S.: Inhibition of the proteolytic degeneration of low density lipoprotein in human fibroblasts by chloroquine, con A and triton WR 1339. J. Biol. Chem. 1975; 250: 7854-7862. Hokdis H. N., Quismorio Jr. F. P., Wichman E., Blankenhom D. H.: The lipid, lipoprotein, and apolipoprotein effects of hydroxychloroquine in patients with systemic lupus erythe matosus. J. Rheumatol. 1993; 20: 661-665. Hurst N. P., French J. K., Bell A. L. et al.: Differential effects of mepacrine, chloroquine and hydroxychloroquine on superoxide anion generation, phospholipid methylation and arachi-donic acid release by human blood monocyte. Biochem. Oharmavol. 1986; 35: 3083-3089. Kruize A. A., Hene R. J., Kallenberg C. G. M., et al.: Hydroxychloroquin treatment for primary Sjogren's syndrome: a two year double blind crossover trial. Ann. Rheum. Dis. 1993; 52: 360-364. Landewe R. B. M., Dijkmans B. A. C., van der Woude F. J., et al.: Long term cyclosporin in patients with rheumatoid arthritis. Arthritis Rheum. 1994; 37 (suppl.): S-361 Lehnert M., Dalton W. S., Roe D., et al.: Synergistic induction of verapamil and quinine of p-glycoproteinmediated multidrug resistance in human myeloma cell line model. Blood 1991; 77: 348-357. London J. R.: Hydroxychloroqiine and postoperative thromboembolism after total hip replacement. Amer. J. Med.: 1988; 85 (suppl. A): 57-51. Masksymowych W., Russel A. S.: Antimalarials in rheumatology: efficacy and safety. Semin. Arthritis Rheum., 1987; 16: 206-221. McChesney E. W.: Animal toxicity and pharmakokinetics of hydroxychloroquine sulphate. Amer. J. Med. 1983; 75: 11-18. McLachlan A. J., Tett S. E., Cutler D. J., Day P. O.: Bioavailability of hydroxychloroquine tablets in patients with rheumatoid arthritis. Brit. J. Rheumatol. 1994; 33: 235-239. Middleton G. D., McFarlin J. E., Lipsky P. E.: The prevalence and clinical impact of fibromyalgia in systemic lupus erythematosus. Arthritis Rheum. 1994; 37: 1181-1188. Middleton G. D., McFarlin J. E., Lipsky P. E.: Hydroxychloroquine and pain threshold. Arthritis Rheum. 1995; 38: 445-447. Miyachi Y., Yoshioka A., Imamura S., Niwa Y.: Antioxidant action of antimalarias. Ann. Rheum. Dis. 1986: 45: 244-248. Panayi G. S., Neill W. A., Duthie J. J. R., McCormick J. N.: Action of chloroquine phosphate in rheumatoid arthritis. 1. Immunosupressive effect. Ann. Rheum. Dis. 1973; 32: 316-318. Parke A. L.: Antimalarial drug, pregnancy and lactation. Lupus 1993; 2 (suppl. 1): S21-S23. Petri M., Lakatta C., Magder L., Goldman D. W.: Effect of prednisolone and hydroxychloroquine on coronary artery disease risk factors in systemic lupus erythematosus Am. J. Med. 1994; 96: 254-259. Petri M., Ferman D., Goldman D.: Hypoglycemic effect of hydroxychloroquin in systemic lupus erythematosus. Arthtitis Rheum. 1994; 37: R-24 (abst. 21). Picot S.: Chloroquine inhibits tumor necrosis factor production by human macrophages in vitro. J. Infect. Dis. 1991; 164: 830-831. Rainsford K. D.: Effects of anti-inflammatory drugs on catabolin-induced cartilage destructiun in vitro. J. Tissuie React. 1985; 7: 123-126. 40. Robertson C. R., Rice J. R., Alien N. B.: Treatment of erosive osteoarthritis with hydroxychloroquine. Arthritis Rheum. 1993; 36 (suppl.): S-167. 41. Rudnick R. D., Greshman G. E., Rothfield N. F.: The efficacy of antimalarias in systemic lupus erythematosus. J. Rheumatol., 1975; 2: 323-330. 42. Rynes R.: Antimalarial drugs. In: Textbook of rheumatology. Ed.: W. N. Kelley, E. D. Harris, S. Ruddy, C. B. Sledge. W. B. Saunders company. Philadelphia, London, Toronto. 1993: 731-742. 43. Salmeron S., Lipsky P. E.: Immunosupressive properties of antimalarias. Amer. J. Med., 1983; 75 (suppl): 19-22. 44. Seideman P., Albertioni R., Beck 0., et al.: Chloroquine reduced the bioavaliability of metotraxate in patients with rheumatoid arthritis. Arthritis Rheum. 1994; 37: 830-833. 45. Sperber К., Quraishi H., Kalb Ò. Í., et al.: Selective regulation of cytokine secretion by hydroxychloroquine: inhibition of interleukin I alpha (IL-I alpha) and interleukin 6 in human monocyte and T cell. J. Rheumatol., 1993; 20: 803-808. 46. Walace D. J.: Does hydroxychloroquine sulphate prevent clot formation in systemic lupus erythematosus? Arthritis Rheum. 1987; 30: 1435-1436. 47. Wallace D. J., Linker-Israeli M., Metzger A. L., Stecher V. J.: The relevance of antimalarial therapy with regard thrombosis, hypercholesterinemla and cytokines in SLE. Lupus 1993; 2 (suppl.): S-13-S-15. 48. Williams H. J., Egger M. J., Singer J. Z., et al.: Comparison of hydroxychloroquine and placebo in the treatment of the arthropathy of mild systemic lupus erythematosus. J. Rheumatol. 1994; 21: 1457-1462. 49. Yousef W., Yan A., Russel A.: Palindromic rheumatism: a response to chloroquine. J. Rheumatol., 1991; 18: 1. 50. Zamora J., Beck W.: Chloroquine enhancement of anticancer drug cytotoxicity in multiple drug resistent human leukemic cells. Biochim. Pharmacol. 1986; 35: 4303. ГЛАВА 8 ЦИТОТОКСИЧЕСКИЕ ПРЕПАРАТЫ Цитотоксические препараты отличаются от других лекарственных средств способностью вызывать необратимое повреждение клеток. Несмотря на то что цитотоксики, как правило, обладают и иммуносупрессивной активностью, термин "цитотоксик" не является синонимом термина "иммунодепрессант". Иммуносупрессивной активностью обладает целый ряд фармакологических препаратов (например, ГК), не являющихся цитотоксиками. Цитотоксические препараты имеют общие механизмы действия на иммуновоспалительные процессы, связанные с элиминацией или подавлением функциональной активности сенсибилизированных и несенсибилизированных лимфоцидных клеток (A. S. Fauci и R. Young, 1993). Для лечения ревматических заболеваний используются цитотоксические препараты 3 основных классов: алкилирующие агенты (циклофосфамид, хлорамбуцил), пуриновые аналоги (азатиоприн) и антагонисты фолиевой кислоты (метотрексат). Как уже отмечалось, последний в низких дозах не обладает явной цитотоксической активностью; его характеристика представлена в главе 6. 8.1. Алкилирующие агенты ЦФ и ХБ являются производными азотистого иприта (nitroge mustard). Нативные молекулы этих веществ не обладают какой-либо биологической активностью; образование активных метаболитов происходит в печени за счет окисления в гладком эндоплазматическом ретикулуме. Активные формы обоих препаратов имеют 2 полифункциональные хлорэтиловые группы, образующие реактивные ионы, посредством которых вещества связываются с сульфгидрильными, амино-, фосфатными, гидроксильными и карбоксильными группами различных молекул. Эта реакция определяет способность алкилирующих агентов вызывать перекрестное связывание ДНК, РНК и некоторых белков. Например, перекрестное связывание двух цепей молекулы ДНК происходит между близлежащими парами гуаниновых оснований, что приводит к нарушению репликации и трансляции ДНК и гибели клетки. 8.1.1. Циклофосфамид Фармакологические свойства ЦФ хорошо адсорбируется в ЖКТ, обладает минимальной белково-связывающей способностью. Активные и неактивные метаболиты ЦФ элиминируются почками. Период полувыведения препарата составляет около 7 ч, пик концентрации в сыворотке достигается в течение 1 ч после введения. Нарушение функции почек может привести к нарастанию иммуносупрессивной и токсической активности препарата. Механизм действия Активные метаболиты ЦФ дают общий эффект на все быстро делящиеся клетки, особенно находящиеся в S-ôàçå клеточного цикла. Одним из важных метаболитов ЦФ — акролеин, образование которого является причиной токсического повреждения мочевого пузыря. ЦФ обладает способностью влиять на различные этапы клеточного и гуморального иммунного ответа ( A. S. Fauci и K. R. Young, 1993). Он вызывает: 1) абсолютную Т- и В-лимфопению с преимущественной элиминацией В-лимфоцитов; 2) подавление бласттрансформации лимфоцитов в ответ на антигенные, но не митогенные стимулы; 3) подавление синтеза антител и кожной замедленной гиперчувствительности; 4) снижение уровня иммуноглобулинов, развитие гипогаммглобулинемии; 5) подавление функциональной активности В-лимфоцитов in vitro. Однако, наряду с иммуносупрессией, описано иммуностимулирующее действие ЦФ, связанное, как полагают, с разной чувствительностью Т- и Влимфоцитов к воздействию препарата. Эффекты ЦФ на систему иммунитета в определенной степени зависят от особенностей терапии. Например, имеются данные о том, что длительный постоянный прием низких доз ЦФ в большей степени вызывает депрессию клеточного иммунитета, а интермиттирующее введение высоких доз ассоциируется в первую очередь с подавлением гуморального иммунитета. В недавних экспериментальных исследованиях, выполненных на трансгенных мышах, по спонтанно развивающимся аутоиммунным заболеваниям было показано, что ЦФ в неодинаковой степени влияет на различные субпопуляции Тлимфоцитов, контролирующих синтез антител и аутоантител. Установлено, что ЦФ в большей степени подавляет Тh2-засисимые, чем Тh1-зависимые иммунные реакции (глава 2), что объясняет причины более выраженного подавления синтеза аутоантител на фоне лечения ЦФ аутоиммунных заболеваний (S. J. Schulman и D. Lo, 1994). Клиническое применение ЦФ широко применяется в лечении различных ревматических заболеваний: 1. СКВ: гломерулонефриты, тромбоцитопения, пневмонит, цереброваскулит, миозит 2. Системные васкулиты: гранулематоз Вегенера, узелковый периартериит, болезнь Такаясу, синдром Чарга—Строc, эссенциальная смешанная криоглобулинемия, болезнь Бехчета, геморрагический васкулит, ревматоидный васкулит 3. PA 4. ПМ/ДМ 5. Синдром Гудпасчера 6. ССД Существуют 2 принципиальные схемы лечения ЦФ: пероральный прием в дозе 1-2 мг/кг/день и болюсное интермиттирующее внутривенное введение высоких доз (пульс-терапия) препарата (500-1000 мг/м2) в течение первых 3-6 мес. ежемесячно, а затем 1 раз в 3 мес. в течение 2 лет и более. При обеих схемах лечения стремятся поддерживать уровень лейкоцитов у больных в пределах 4000мм3. Обычно лечение ЦФ (за исключением РА) сочетают с назначением умеренных или высоких доз ГК, включая пульс-терапию (глава 3). Превалирует мнение, что обе схемы лечения примерно одинаково эффективны, но на фоне интермиттирующего внутривенного введения частота токсических реакций меньше, чем при постоянном пероральном приеме (H. A. Austin и соавт., 1986), однако последнее доказано только при волчаночном нефрите, В то же время имеются данные о том, что у больных гранулематозом Вегенера пульс-терапия и пероральный прием ЦФ в равной степени эффективны только в отношении ближайших результатов, но длительную ремиссию удается достигнуть только при длительном пероральном ежедневном приеме препарата (G. S. Hoffman и соавт., 1991). Таким образом, пульс-терапия и длительный прием низких доз ЦФ имеют разный терапевтический профиль (T. R. Cupps, 1991). По мнению T. R. Cupps (1990), в некоторых случаях пероральный прием низких доз ЦФ имеет преимущества перед интермиттирующим введением высоких доз. Например, в фазу индукции риск супрессии костного мозга выше у больных, леченных с помощью пульс-терапии, чем у больных, получающих низкие дозы ЦФ. Поскольку истинное изменение количества лейкоцитов в периферической крови после пульс-терапии становится очевидным через 10-20 дней, дозу ЦФ можно модифицировать только через месяц, в то время как при ежедневном приеме препарата дозу ЦФ можно подбирать на основе постоянного мониторинга уровня лейкоцитов в периферической крови и изменения функции почек. По мнению автора, опасность токсических реакций в ранние сроки лечения высокими дозами ЦФ особенно велика у больных с нарушениями функции многих органов, быстрым прогрессированием почечной недостаточности, ишемией кишечника, а также у больных, получающих высокие дозы ГК. В процессе лечения ЦФ крайне необходимо тщательно контролировать лабораторные показатели. В начале лечения общий анализ крови, определение уровня тромбоцитов и мочевого осадка следует проводить каждые 7-14 дней, а при стабилизации процесса и дозы препарата — каждые 2-3 мес. (P. J. Clements и Davis J., 1986). СКВ Эффективность ЦФ при тяжелой СКВ доказана в серии открытых и контролируемых исследований (D. T. Boumpas и соавт., 1990, 1992; W. J. McCune и D. T. Fox, 1989). По данным длительных наблюдений (10 лет и более) частота развития почечной недостаточности и степень прогрессирования волчаночного нефрита достоверно ниже у больных, получавших комбинированное лечение ЦФ и ГК, чем у больных, леченных только ГК (A. D. Steinberg и S. Steinberg, 1991). Так, ХПН развилась у 75% больных, получавших только преднизолон, в то время как в группе леченных ЦФ прогрессирование в ХПН наблюдалось в 10% случаев. Однако в отличие от ГК ЦФ плохо контролирует многие внепочечные проявления СКВ, которые обычно сопутствуют активности заболевания. Поэтому в подавляющем большинстве случаев ЦФ применяют вместе с ГК. Имеется несколько сообщений об эффективности внутривенного введения ЦФ (в высоких или низких дозах) при тромбоцитопении, резистентной к ГК, и спленоэктомии (D. T. Boumpas и соавт., 1990; B. A. Roach и G. J. Hutchison, 1993), системном васкулите (T. J. Liang и соавт., 1988), интерстициальном поражении легких (A. Eiser и H. M. Shanies, 1994), тяжелых нейропсихических проявлениях заболевания (D. T. Boumpas и соавт., 1991), резистентном к ГК миозите (D. Kono и соавт., 1990). Согласно рекомендациям B. Hahn, ежемесячное внутривенное лечение ЦФ в максимально переносимой дозе (без тошноты и выраженной лейкопении) следует продолжать до тех пор, пока не будет достигнут очевидный клинический и лабораторный эффект, а затем увеличивать интервал между введениями препарата до 4-6, 8, 12 нед., а затем продолжить лечение в течение 2 лет. При плохой переносимости целесообразно заменить ЦФ на AÇ. По данным F. A. Houssiau и соавт. (1991), достаточно эффективным методом лечения (по крайней мере в отношении краткосрочного прогноза) больных с тяжелым волчаночным нефритом является еженедельное внутривенное введение ЦФ в низких дозах (500 мг) в течение 2-4 нед. в сочетании с преднизолоном в умеренных дозах (0.5 мг/кг/день). Преимуществом этого метода лечения является низкая частота инфекционных осложнений и возможность быстрого уменьшения дозы ГК. Системные васкулиты ЦФ является эффективным средством лечения гранулематоза Вегенера (A. S. Fauci и соавт., 1983; G. S. Hoffman и соавт., 1991.), узелкового периартериита и синдрома Чарга—Стросс (C. C. Chow и соавт., 1989; L. Guillevin и соавт., 1991; S. DeVita и соавт., 1991; W. J. McCune и A. W. Friedman, 1992). РА В нескольких открытых и контролируемых исследованиях была доказана эффективность ЦФ (1.5 мг/кг/день) при PA (M. B. Yunus, 1988). Однако доза ЦФ, подавляющая образование эрозий, довольно высока (150 мг/день) и часто ассоциируется с развитием побочных реакций. Максимальный эффект достигается к 16-й неделе лечения. По клинической эффективности при РА ЦФ не уступает AÇ и несколько превосходит парентерально вводимые препараты золота. Интермиттирующая пульс-терапия ЦФ рассматривается как наиболее эффективный метод лечения системного ревматоидного васкулита (D. G. L. Scott и Р. А. Васоn, 1984). ССД ЦФ в дозе 2.0-2.5 мг/кг/день перорально в сочетании с низкими дозами преднизолона вызывает достоверное улучшение функционального состояния легких у больных ССД с легочным фиброзом (A. Akesson и соавт., 1994; R. M. Silver и соавт., 1993). ПМ/ДМ По данным M. E. Cronin и соавт. (1989), болюсное введение 7 больным ЦФ (0.75-1.375 г/м2 в мес.) в сочетании с ГК-терапией сопровождалось клиническим улучшением только в 1 случае, у 3 больных развились тяжелые осложнения (1 больной погиб от сердечной недостаточности). В то же время S. Bombardieri и соавт. (1989) достигли определенного клинического эффекта у всех 10 больных с резистентным к ГК ПМ/ДМ на фоне лечения ЦФ в дозе 500 мг в 3 нед. Имеются единичные наблюдения, свидетельствующие об эффективности перорального приема ЦФ как при ДМ у детей, так и у взрослых. Побочные эффекты Основные побочные эффекты ЦФ представлены в таблице 8.1. Таблица 8.1. Основные побочные эффекты ЦФ Потенциально обратимые: Подавление костномозгового кроветворения: лейкопения тромбоцитопения панцитопения Поражение мочевого пузыря Поражение ЖКТ: тошнота/рвота Потенциально необратимые: карциногенез бесплодие геморрагический цистит инфекционные осложнения кардиотоксические эффекты интерстициальный легочный фиброз диарея боли в животе Алопеция печеночный некроз Поражение мочевого ПУЗЫРЯ Наиболее частым осложнением, возникающим на фоне лечения ЦФ, является геморрагический цистит, развитие которого описано почти у 30% больных. Частота геморрагического цистита несколько меньше на фоне парентерального, чем перорального введения ЦФ. Хотя геморрагический цистит относится к числу обратимых осложнений, в ряде случаев он предшествует развитию фиброза и даже рака мочевого пузыря. Для профилактики геморрагического цистита рекомендуется прием препарата месна (C. R. deVries и E. S. Freiha, 1990). Месна, выпускаемая компанией Бристоль-Майер-Сквиб (США), является детоксицирующим агентом, снижающим опасность развития геморрагического цистита, индуцируемого ЦФ. Активный компонент месна — синтетическое сульфгидрильное вещество, обозначаемое как 2-меркаптоэтансульфонат. Месна выпускается в форме стерильного раствора, содержащего 100 мг/мл месна и 0.025 мг/мл эдетата (рН 6.6-8.5). После внутривенного введения месна очень быстро окисляется в свой основной метаболит месна дисульфид (димесна), который элиминируется почками. В почках месна дисульфид редуцируется до свободных тиоловых групп (месна), которые обладают способностью химически реагировать с уротоксическими метаболитами ЦФ — акролеином и 4-гидрокси-ЦФ. Месна вводится внутривенно в количестве, составляющем 20% от дозы ЦФ (объем/объем), до и через 4 и 8 ч после приема ЦФ. Общая доза месна составляет 60% от дозы ЦФ. Примерная схема использования месна на фоне лечения ЦФ представлена в таблице 8.2. Таблица 8.2. Схема использования месна на фоне лечения ЦФ Препарат До приема ЦФ После приема ЦФ Через 4 ч Через 8 ч ЦФ 1г/м2 Месна 240 мг/м2 240 мг/м2 240 мг/м2 Примечание: Месна вводится в любом растворе (физиологический раствор или 5% глюкоза) в концентрации 20мг/мл. Расчет площади тела по Костеффу: S=4P+7/P+90, где S — площадь тела, м2, Р — масса тела, кг. Развитие тяжелого цистита является абсолютным показанием к отмене ЦФ. У больных с умеренным циститом можно попытаться продолжить лечение меньшими дозами препарата под тщательным клиническим и инструментальным (цистоскопия) контролем. Гематологические нарушения и инфекционные осложнения Хотя на фоне лечения ЦФ наблюдается подавление всех элементов костномозгового кроветворения, развитие нейтропении является наиболее частым осложнением, лимитирующим возможность продолжения лечения. Длительный прием ЦФ дает кумулятивный эффект в отношении гематологических нарушений. Поэтому на фоне лечения необходимо часто контролировать содержание лейкоцитов в периферической крови. Описаны больные с персистирующей гипогаммаглобулинемией. Этот побочный эффект может быть очень опасным, особенно при сочетании с нейтропенией, так как способствует развитию инфекционных осложнений. Необходимо иметь в виду, что увеличение чувствительности к инфекции наблюдается и у больных с нормальной концентрацией нейтрофилов и иммуноглобулинов. Инфекционные осложнения являются показанием к прерыванию лечения. Злокачественные новообразования У больных, получавших ЦФ, наблюдается увеличение частоты злокачественных новообразований. В литературе описано около 100 больных СКВ, у которых развилась неходжкинская лимфома, и 13 случаев лимфогранулематоза (R. Bhalla и соавт., 1993). Недавно было обнаружено, что у больных СКВ уже в течение первых 4 лет от начала лечения ЦФ умеренно нарастает частота рака влагалища и шейки матки (B. Pryor и соавт., 1993). Желудочно-кишечные расстройства. Одним из наиболее частых токсических проявлений лечения ЦФ является тошнота, нередко делающая невозможным проведение пульс-терапии. Для профилактики тошноты рекомендуется внутривенное введение метоклопрамида в дозе 1-3 мг/кг (в 100 мл 0.9% хлористого натрия) за 15 мин. до начала инфузии ЦФ (максимальная доза метоклопрамида составляет 10 мг/кг) в сочетании с седативными средствами (диазепам 5-10 мг). Другие осложнения Алопеция чаще развивается на фоне приема высоких доз препарата, прерывание лечения ведет к нормализации роста волос. Олигоспермия или азоспермия у мужчин и олигоменорея или аменорея у женщин — почти обязательный побочный эффект лечения ЦФ. Имеются данные о том, что проведение пульс-терапии ЦФ во время менструации позволяет снизить риск стерилизации (D. T. Boumpas и соавт., 1993). Описаны развитие легочного фиброза и кардиотоксические эффекты. В целом риск таких токсических реакций, как бесплодие, азоспермия, аменорея, возрастает с увеличением продолжительности лечения, дозы препарата и возраста больных. У мужчин, получивших ЦФ в суммарной дозе более 18 г, как правило, развивается необратимая азоспермия. 8.1.2 Хлорамбуцил (ХБ) Хлорамбуцил, так же как и ЦФ, является бифункциональным (то есть имеет 2 хлорэтиловых радикала) алкилирующим агентом. ХБ назначают перорально, поскольку из-за быстрого гидролиза при внутривенном введении препарат имеет низкую стабильность. При пероральном приеме ХБ очень быстро абсорбируется, концентрация его в плазме достигает пика через 1 ч, элиминируется он в течение 1,5 ч. Основным метаболитом ХБ является фенилуксусный иприт, обладающий высокой алкилирующей активностью. Общепринятая терапевтическая доза ХБ составляет 0.1-0.2 мк/кг/день. ХБ реже используется для лечения ревматических заболеваний, чем ЦФ, в первую очередь из-за потенциально онкогенного эффекта. В целом показания к назначению ХБ те же, что и для ЦФ (G. W. Cannon и J. R. Ward, 1989). Оба препарата, вероятно, обладают сходными клинической эффективностью и спектром побочных эффектов, за исключением геморрагического цистита. Поэтому ХБ может быть показан больным, эффективно леченным ЦФ, но у которых развился геморрагический цистит (A. S. Steinberg, 1993). В отдельных клинических наблюдениях обнаружена особая эффективность ХБ при рефракторном ДМ (P. A. Sinoway и J. P. Callen, 1993), болезни Бехчета (E. L. Matteson и J. D. O'Duffy, 1992), амилоидозе, болезни Стилла. Побочные эффекты По сравнению с ЦФ на фоне лечения ХБ чаще развиваются тромбоцитопения, необратимая супрессия костномозгового кроветворения и злокачественные новообразования (частота зависит от длительности приема и суммарной дозы), особенно острый миелолейкоз (A. S. Steinberg, 1993). 8.2. Азатиоприн Фармакологические свойства Существует два основных пуриновых аналога — 6-меркаптопурин и азатиоприн (AÇ), однако в клинической практике в настоящее время используется только последний. 6-меркаптопурин является аналогом гипоксантина, в котором 6-ОН радикал заменен на тиоловую группу. В свою очередь AÇ представляет молекулу, отличающуюся от 6-меркаптопурина включением имидазольного кольца в S-ïîëîæåíèå. По сравнению с 6-меркаптопурином АЗ лучше абсорбируется при пероральном приеме и обладает большей длительностью действия. В организме человека AÇ метаболизируется в эритроцитах и печени с образованием биологически активных молекул (6-тиогуаниновой и 6-тиоинозиновой кислот) и экскретируется почками. В настоящее время в клинической практике используется главным образом AÇ. По механизму действия AÇ относится к классу веществ, получивших название "антиметаболиты". Он обладает способностью инкорпорировать как "ложное основание" в молекулу ДНК и, таким образом, нарушать репликацию ДНК. AÇ рассматривается как фаза-специфический препарат, поражающий клетки в определенной фазе роста, преимущественно в S-ôàçå. В более высоких дозах AÇ нарушает синтез РНК и белка в GI- и G2фазах. В отличие от алкилирующих агентов AÇ обладает не цитотоксической, а цитостатической активностью. Механизм действия AÇ вызывает периферическую Т- и В-лимфопению, в высоких дозах уменьшает уровень Т-хелперов, при длительном приеме снижает синтез антител. Однако, поскольку Т-супрессоры особенно чувствительны к действию AÇ, на фоне приема низких доз препарата синтез антител может несколько увеличиваться. Характерным действием AÇ является подавление активности ЕК-клеток и К-клеток, принимающих участие в развитии соответственно естественной и антителозависимой клеточной цитотоксичности. Клиническое применение Для исключения острой реакции гиперчувствительности к AÇ лечение следует начинать с пробной дозы 25-50 мг/день в течение первой недели. Затем дозу увеличивают на 0.5мг/кг/день каждые 4-8 нед. Оптимальная доза составляет 1-3 мг/кг/день. В начале лечения необходимо регулярно (каждые 2 нед.) выполнять общий анализ крови (с определением количества тромбоцитов), а при достижении стабильной дозы лабораторный контроль проводится один раз в 6-8 нед. Необходимо иметь в виду, что эффект AÇ начинает проявляться не ранее, чем через 5-12 мес. от начала терапии. Доза AÇ должна быть существенно снижена (на 50-75%) у больных, получающих аллопуринол или имеющих почечную недостаточность. РА Эффективность AÇ в дозе 1.25-3.0 мг/кг/день при РА подтверждена в серии контролируемых исследований, результаты которых обобщены в обзоре R. J. R. McKendry (1991). В целом клиническая эффективность AÇ при РА сопоставима с таковой ЦФ, парентерально вводимых препаратов золота, Dïåíèöèëëàìèíà и антималярийных препаратов. Предполагается, что при РА AÇ следует назначать больным пожилого возраста с вариантом начала, напоминающим ревматическую полимиалгию (R. Luqmani и соавт., 1994), когда необходим стероидсберегающий эффект. Комбинированная терапия AÇ и преднизолоном предотвращает прогрессирование сосудистого процесса у больных РА с васкулитом (A. H. Herkens и соавт., 1991). СКВ При СКВ, по данным краткосрочных наблюдении (1-2 года), существенных различий по клинической эффективности между группами больных, получавших только ГК или ГК в сочетании с AÇ не наблюдается (B. H. Hahn и соавт., 1975; J. V. Donadio и соавт., 1972). Но при оценке результатов лечения через 5-15 лет обнаружено, что комбинированная терапия имеет определенные преимущества, заключающиеся в замедлении прогрессирования поражения почек, уменьшении количества обострений и возможности использования более низкой поддерживающей дозы ГК (E. M. Ginzler и соавт., 1980). Однако у больных, получающих АЗ, достоверно нарастает частота различных побочных эффектов, включающих инфекционные осложнения (особенно опоясывающий лишай), недостаточность яичников, лейкопению, повреждение печени, увеличение чувствительности к опухолям (J. H. Klippel, 1990). ПМ/ДМ При ПМ/ДМ на обычно используемую дозу AÇ (2-3 мг/кг/день) отвечают около трети больных, резистентных к ГК, а стероидсберегающее действие отмечается в половине случаев, что несколько хуже, чем при лечении МТ. Максимальный клинико-лабораторный эффект при лечении AÇ проявляется только через 6-9 мес. Поддерживающая доза препарата — 50 мг/день. Результаты небольших контролируемых исследований свидетельствуют об эффективности A3 при псориатическом артрите, синдроме Рейтера, болезни Бехчета (R. J. R. McKendry, 1991; A. A. Calin, 1986; H. Yazici и соавт., 1990). Побочные эффекты Побочные эффекты, возникающие на фоне лечения AÇ суммированы в таблице 8.3. Таблица 8.3. Побочные эффекты, возникающие на фоне длительного приема A3 Частота, % Тактика ведения больных Цитопения (преимущественно нейтропения, До 30 Снижение дозы или отмена, динамический реже тромбоцитопения или и то и другое) контроль анализов крови Гепатотоксичность Редко Отмена Инфекционные осложнения 1-3 Отмена Желудочно-кишечные нарушения (тошнота, До 16 Прием с пищей и дробными дозами рвота, понос) Панкреатит Редко Отмена Стоматит Редко Снижение дозы Облысение Редко Снижение дозы или отмена Онкологические заболевания Редко Отмена Побочные эффекты AÇ во многом зависят от дозы препарата. Наиболее часто отмечаются тошнота и рвота, выраженность которых можно снизить при приеме препарата с пищей и дробными дозами. Гепатотоксичность встречается не более чем у 1% больных и, вероятно, связана с развитием аллергической реакции на препарат. Тем не менее печеночные показатели рекомендуется контролировать не реже чем 1 раз в 3 мес. Лейкопения, развивающаяся в начале лечения, также, как полагают, связана с гиперчувствительностью к препарату. Однако в большинстве случаев развитие цитопении отражает подавление костномозгового кроветворения и является зависимым от дозы феноменом. Общая частота цитопении может достигать 30%. При развитии цитопении препарат отменяют и лечение в последующем можно возобновить меньшими дозами. Наиболее частым инфекционным осложнением является герпетическая инфекция, развитие которой не всегда коррелируют с выраженностью нейтропении. Сообщается о больном РА, у которого на фоне лечения AÇ отмечено развитие ускоренного нодулеза (P. Langevitz и соавт., 1991). Ранее это своеобразное осложнение было описано только у больных, леченных МТ (глава 6). Потенциально наиболее тяжелым осложнением лечения AÇ является развитие злокачественных новообразований, хотя относительный риск возникновения опухолей у больных РА, леченных AÇ, невысок и составляет 1.3, за исключением лимфомы, для которой этот показатель достигает 10 (A. J. Silman и соавт., 1988). Имеются данные о существовании генетически детерминированной предрасположенности к развитию токсических реакций на фоне лечения AÇ (L. Lennard и соавт., 1989). Противопоказанием к применению AÇ является беременность, наличие злокачественных новобразований, относительным противопоказанием — поражение печени. 8.3. Цитозин-арабинозид Цитозин-арабинозид (цитарабин) оказывает мощное иммунодепрессивное действие, затрагивающее как Т-, так и В-клеточный иммунный ответ (R. O. Gordon и соавт., 1969; M. M. Beyer и соавт., 1975; D. E. Griswold и соавт., 1972; M. S. Mitchell и соавт., 1969). Кроме того, препарат быстро метаболизируется и в небольших дозах обладает умеренной токсичностью в отношении печени, почек, мочевого пузыря и легких (P. J. Burke и соавт., 1969). Показано, что цитарабин эффективно купирует экспериментальный артрит (E. M. Glenn и соавт., 1968). Эти данные, а также клиническое наблюдение развития суставной ремиссии у больного, страдающего РА и получавшего цитарабин по поводу острого миелолейкоза (R. Roubenoff и соавт., 1987), создали предпосылки для его применения в ревматологии, в первую очередь у больных, резистентных к стандартной терапии. G. E. Bayllis и соавт. (1992) использовали цитарабин для лечения 9 больных РА. 6 пациентам препарат назначали в дозе 2 мг/кг внутривенно, 3 больным — подкожно; продолжительность лечения составила 5 дней. Клинический эффект зарегистрирован у 5 больных. Побочные эффекты в виде тошноты, купирующейся при уменьшении дозы, и транзиторной лейкопении отмечены также у 5 больных. R. Yung и соавт. (1994) сообщили о применении цитозин-арабинозида (2мг/кг/день подкожно в течение 5 дней по одному курсу в мес.) у 2 больных СКВ, основным клиническим проявлением которой было тяжелое поражение кожи, не поддающееся лечению преднизолоном, антималярийными и цитотоксическими препаратами. В обоих случаях отмечена положительная динамика кожного синдрома в первые 2 нед. лечения при отсутствии побочных эффектов. Список литературы к главе "Цитотоксические препараты" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Akesson A., Scheja А., Lundin A., Wollheim F. A.: Improved pulmonary function in systemic sclerosis after treatment with cyclophosphamide. Arthritis Rheum. 1994; 37: 729-735. Austin H. A., Klippel J. H., Balow J. E., et al.: Therapy of lupus nephritis. Controlled trial of prednisolon and cytotoxic drugs. New Engl. J. Med. 1989; 313: 614-619. Baylliss G. E., Royer G. R., Juozevicius J. L., et al. Cytarabine therapy for rheumatoid arthritis. Clin. Exp. Rheumatol., 1992; 10: 420-421. Bhalla R., Ajmani H. S., Kim W. W., et al.: Systemic lupus erythematosus and Hodgkin's lymphoma. J. Rheumatol. 1993; 20: 1316-1320. Bombardieri S., Hughes G. R. V., Nerri R., Del Bravo P., Del Bono I. Cyclophosphamide in severe polymiositis. Lancet 1989; 1: 1138-1139. Boumpas D. T., Baretz S., Klippek J. H., Balow J. E.: Intermitent cyclophasphamide for the treatment of autoimmune thrombocytopenia in systemic lupus erythematosus. Ann. Int. Med. 1990; 112: 674-677. Boumpas D. T., Yamada H., Patronas N. J., et al.: Pulse cyclophosphamide for severe neu-ropsychiatric lupus. Q. J. Med. 1991; 296: 975-984. Boumpas T. D., Austin H. A., Vaughn E. M., et al.: Controlled trial of pulse methylpred-nisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. New Engl. J. Med. 1992; 296: 975-984. Boumpas D. T., Austin H. A., Vaughan E. M., et al.: Risk for sustained amenorrhea in patients with systemic lupus erythematosus received intermittent pulse cyclophosphamide therapy. Ann. Int. Med. 1993; 119: 366-369. Calin A. A.: A placebo controlled cross-over study of azathioprine in Reiter's syndrome. Ann. Rheum. Dis. 1986; 45: 653-655. Cannon G. W., Ward J. R.: Cytotoxic drugs and sulfasalazine. In: Arthritis and allied conditions. A textbook of rheumatology. Ed. D. J. McCarty. Lea/Febiger. 1989: 563-592. Chow C. C., Ki E. K. M., MacMoune L. A. I. F.: Allergic granulomatosis and angiltis (Churg-Strauss syndrome): response to "pulse" intravenous cyclophosphamide. Ann. Rheum. Dis. 1989; 48: 605-674. Clements P. J., Davis J. Cytotoxic drugs: their application to the rheumatic disease. Semin. Arthritis Rheum. 1986; 4: 231-254. 14. Cronin M. E., Miller F. W., Hics J. E., et al: The failure of intravenous cyclophosphamide therapy in refractery idiopathic inflammatory myopathy. J. Rheumatol. 1989; 16: 1225-1228. 15. Cupps T. R.: Cyclophosphamide: to pulse or not to pulse?. Amer. J. Med. 1990; 89: 399-402. 16. DeVita S., Neil R., Bombardieri S.: Cyclophosphamide pulses in the treatment of rheumatic disease: an update. Clin. Exp. Rheumatol. 1991; 9: 179-193. 17. DeVries C. R., Freiha E. S.: Hemorrhagic cystitis: a review. J. Urol., 1990; 143: 1. 18. Eiser A. R., Shanies H. M.: Treatment of lupus interstitial lung disease with intravenous cyclophosphamide. Arthritis Rheum., 1994; 37: 428-431. 19. Fauci A. S., Haynes B. F., Katz P., Wolff S. M.: Wegener's granulomatosis: prospective clinical trial and therapeutic experience with 85 patients for 21 years. Ann. Int. Med. 1983; 98: 76-85. 20. Fauci A. S., Young K. R.: Immunoregulatory agents. In. Textbook of Rheumatology. Wd. W. N. Kelly, E. D. Harris, S. Ruddy, C. B. Sledge. W. B. Saunders Coimpany. Philadelphia, London, Toronto, Montreal, Sydney, Tokyo. 1993: 797821. 21. Herkens A. H., Westedt M. L., Breedveld F. C.: Prednisone plus azathyoprine treatment in rheumatoid arthritis complicated by vasculitis. Arch. Int. Med. 1991; 151: 2249-2254. 22. Hoffman G. S., Leavitt R. Y., Fleischer T. A., et al.: Treatment of Wegener's granulomatosis with intermittent high-dose Intravenous cyclophosphamide. Am. J. Med. 1990: 89: 403-410. 23. Houssiau F. A., D'Cruz D. P., Haga H.-J., Hughes G. R. V.: Short course of weekly low-dose intravenous pulse cyclophosphamide in the treatment of lupus nephritis: a preliminary study. Lupus 1991; 1: 31-36. 24. Glenn E. M. Inhibition of adjuvant-induced polyartritis with cytarabin. Proc., Soc., Exp. Biol. Med., 1968; 70: 860-865. 25. Guillevin L., Jarrousse B., Lok C., et al.: Longterm follow-up after treatment of polyarthritis nodosa and Churg-Strauss angiitis with comparison of steroids, plasma exchange and cyclophosphamide to steroids and plasma exchange: a prospective ran-domizes trial of 71 patients. J. Rheumatol. 1991; 18: 567-574. 26. Kono D., Klasnman D., Gilbert R. C.: Successful IV pulse cuclophosphamide in refractory PM in 3 patients with SLE. J. Rheumatol. 1990; 17: 982-983. 27. Lennard L., Van Loon J. A., Welnshilboum R. M.: Pharmacogenetics of acute azathyoprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharm. Ther. 1989; 46: 146-154. 28. Liang T. J.: Gastrointestinal vasculitis and pneumatosisintestinalis due to systemic lupusa erythematosus: successful treatment with pulse intravenous cyclophasphamide. Amer. J. Med. ; 8-5: 555-558. 29. McCune W. J., Fox F. D.: Intravenous cyclophosphamide therapy of severe SLE. Rheum. Dis. Clin. Nort. Amer. 1989; 15: 455477. 30. McCune W. J., Friedman A. W.: Immunosupressive drug therapy for rheumatic disease. Curr. Opin. Rheumatol. 1992; 4: 321. 31. McKendry R. J. R. Purine analogs. In: Second line agents in the treatment in rheumatic diseases. Eds. Dixon J., Furst D. E. New York: Marcel Dekker, 1991. 32. Pryor B., Bologna S., Ernst C., et al.: Risk of malignancy in cyclophosmamide-treated SLE patients. Arthritis Rheum. 1993; 36 (suppl.): S91. 33. Roach B. A., Hutchinson G. J. Treatment of refractory systemic lupus erythematosus-asso-ciated thrombocytopenia with intermittent low-dose intravenous cyclophosphamide. Arthritis Rheum., 1993; 36: 682-684. 34. Roubernoff R., Jones R. J., Karp J. E. . Stevens M. B.: Remission of rheumatoid arthritis with the succesful treatment of acute myelogenous leukemia with cytosine arabi-noside. Arthritis Rheum., 1987; 30: 11871190. 35. Schulman S. J., Lo D.: Cyclophosphamide treatment induces shift production of a Th1-type cytokine pattern in an in vitro transgenic model of autoimmune disease. Arthritis Rheum., 1994; 37 (Suppl.): S165. 36. Scott D. G. L., Bacon P. A.: Intravenous cyclophosphamide plus methylprednosolone in the treatment of systemic rheumatoid vasculitis, Amer. J. Med. 1984; 76: 377-384. 37. Silman A. J., Petrie J., Hazleman B., Evans S. J.: Lymphoprolipherative cancer and other malignancy in patients with rheumatoid arthritis treated with azathioprine: twenty year follow up study. Ann. Rheum. Dis.: 1988; 47: 988-992. 38. Silver R. M., Warrick J. H., Kinsella M. B., et al.: Cyclophosphamide and low-dose pred-nisolone therapy in patients with systemic sclerosis (scleroderma) with intestinal lung disease. J. Rheumatol. 1993; 20: 838844. 39. Steinberg A. D., Steinberg S.: Long-term preservation of renal function in patients with lupus nephritis receiving treatment that include cyclophosphamide versus those treated with prednisolone alone. Arthritis Rheum. 1991; 34: 945. 40. Steinberg A. D.: Chlorambucil in the treatment of patients with immune-mediated rheumatic disease. Arthritis Rheum., 1993; 36: 325-328. 41. Synoway P. A., Callen J. P.: Chlorambucile. An effective corticosteroid sparing agent for patients with recalcitrant dermatomyositis. Arthritis Rheum. 1993; 36: 319-324. 42. Yazici H., Pazali H., Barnes C., et al.: A controlled trial of azathyoprine in Beheet's disease. New Engl. J. Med. 1990; 322: 281-285. 43. Yunus M. B. Investigational therapy in rheumatoid arthritis. Semin. Arthritis Rheum. 1988; 17: 163-184. 44. Yung R., McCune W. J., Richardson B. C.: Cytosine arabinoside therapy for cutaneous systemic lupus erythematosus. Arthritis Rheum., 1994 (Suppl.); 37: R16 (abst.). ГЛАВА 9 СУЛЬФАСАЛАЗИН Сульфасалазин представляет собой конъюгат 5-аминосалициловой кислоты и сульфапиридина, обладающих соответственно противовоспалительной и антимикробной активностями; является единственным базисным препаратом, специально разработанным для лечения РА. Фармакологические свойства Сульфасалазин нерастворим в воде и поэтому практически не всасывается в желудке и тонкой кишке. Большая часть препарата достигает толстой кишки, где под влиянием кишечных бактерий происходит его расщепление на исходные составляющие: 5-аминосалициловую кислоту и сульфапиридин. Сульфасалазин обладает очень высокой способностью связываться с белками, экскретируется с мочой и желчью, а также подвергается печеночной рециркуляции. Пик концентрации в плазме достигается через 3-5 ч, период полувыведения составляет 6-17 ч, 30% высвобожденной из сульфасалазина 5-аминосалициловой кислоты экскретируется с мочой в виде N-àöåòèëèðîâàííîãî продукта, 50% — выделяется с каловыми массами. Сульфапиридин появляется в плазме через 4-6 ч, интенсивно метаболизируется в печени и затем экскретируется с мочой. Механизм действия Механизм противоревматической активности сульфасалазина до конца не ясен. В ряде клинических и экспериментальных исследований были продемонстрированы многочисленные системные и локальные противовоспалительные и иммунные эффекты препарата (таблица 9.1). Таблица 9.1. Механизмы действия сульфасалазина • Антипростагландиновая активность, подавление 5-липоксигеназного пути метаболизма арахидоновой кислоты (C. L. Hawkey и соавт., 1983; O. H. Nielsen и соавт., 1987) • Подавление образования супероксидных радикалов и гипохлорной кислоты активированными нейтрофилами (О. Агиота и соавт., 1987; J. Williams и соавт., 1989; T. Pullar и соавт., 1987)) • Ингибиция хемотаксиса нейтрофилов (J. M. Rhode и соавт., 1981). • Подавление митогениндуцированной бласттрансформации лимфоцитов in vltro (P. Sheldon и соавт., 1988: S. Cormer и H. Jasin, 1988) и in vivo (коррелирует с клинической эффективностью) (A. Samanta и соавт., 1992) • Подавление активации В-лимфоцитов и синтеза IgM РФ (S. Comer и H. Jasin, 1988; F.Imal и соавт., 1991) • Локальная иммуномодулирующая активность в кишечнике (M. Fujiwara и соавт., 1990) • Антифолатное действие (C. L. Baum и соавт., 1981) • Подавление естественной цитотоксичности (M. N. Aparicio-Pages и соавт., 1990) • Подавление синтеза ИЛ-1-подобных субстанций моноцитами человека (L. Remving и соавт., 1990); подавление синтеза ИЛ-1 b, ИЛ-6 и ФНО-а лейкоцитами человека, стимулированными ЛПС (A. Gronberg и соавт., 1994) • Подавление продукции ИЛ-2 Т-лимфоцитами (G. Calin и соавт., 1994) Имеются данные о важной роли воспалительных и иммунопатологических процессов в кишечнике и нарушений кишечной флоры в развитии РА и серонегативных артритов (A. W. Segal и соавт., 1986; I. Bjarnason и соавт., 1984; P. J. Rooney и соавт., 1986). Предполагается, что один из механизмов действия сульфасалазина при артритах связан с локальным противовоспалительным, иммуномодулирующим и антимикробным действием. По мнению P. Sheldon и соавт. (1988), эффект сульфасалазина реализуется в первую очередь за счет воздействия на перемещение лимфоцитов в лимфоидную ткань тонкой кишки. Действительно, хотя сульфасалазин обладает способностью подавлять митогенстимулированную пролиферацию Т- и В-лимфоцитов, этот эффект достигается только при использовании супратерапевтических доз препарата (P. J. Sheldon и соавт., 1988). Показано, что высокая концентрация сульфасалазина, обнаруживаемая в тонкой кишке, подавляет активность как В-, так и Т-лимфоцитов; в то время как сывороточный уровень сульфасалазина значительно ниже, и в такой концентрации он оказывает действие только на В-лимфоциты (D. P. M. Symmons и соавт., 1988; S. S. Comer, H. E. Jasin, 1988; F.Imai и соавт., 1991). По данным L. Kanerud и соавт. (1994), у больных РА в процессе лечения сульфасалазином отмечается снижение числа СDЗ+Т-лимфоцитов и Т-лимфоцитов, экспрессирующих у/б ТКР в биоптатах тонкой кишки, что отражает локальный иммуномодулирующий эффект препарата. У здоровых добровольцев, иммунизированных инактивированной противогриппозной вакциной, на фоне приема сульфасалазина развивается супрессия системного и кишечного гуморального иммуногенспецифического иммунного ответа. Это коррелировало со способностью сульфасалазина ингибировать индуцированное вакцинацией увеличение концентрации ИФ-у в плазме. (L. Klareskog и соавт., 1994). В опытах in vitro было показано, что сульфасалазин зависимым от дозы образом подавляет продукцию провоспалительных цитокинов (ИЛ-1 b, ИЛ-6 и ФНО-а) периферическими мононуклеарными клетками человека, стимулированными ЛПС (A. Gronberg и соавт., 1994). Важные результаты получены при изучении механизмов антипростагландиновой активности сульфасалазина. По данным G. Calin (1994), сульфасалазин, в отличие от НПВС и ГК не обладает способностью ингибировать активность двух ключевых ферментов, участвующих в образовании ПГ, — ЦОГ-1 и ЦОГ-2. Это свидетельствует о том, что антипростагландиновые эффекты сульфасалазина связаны с ингибицией других ферментных систем арахидонового каскада или подавлением клеточных стимулов, ведущих к активации этих ферментов. A. Calin и соавт. (1994) недавно обнаружили, что сульфасалазин подавляет синтез ИЛ-2 Т-лимфоцитами, стимулированными фитогемагглютинином. Интересно, что основные метаболиты сульфасалазина (сульфапиридин и 5аминосалициловая кислота) не проявляли подобной иммуномодулирующей активности. Совсем недавно C. Maple и соавт. (1995) обнаружили, что лечение сульфасалазином больных активным РА сопровождается выраженным снижением концентрации в сыворотке растворимых молекул адгезии (Е-селектина и Р-селектина), что можут указывать на существование еще одной точки приложения активности препарата. Клиническое применение и тактика лечения В таблице 9.2. суммированы данные о применении сульфасалазина при лечении ревматических болезней. Таблица 9.2. Применение сульфасалазина в ревматологии Заболевание Комментарий РА Эффект достигается через 4 нед. и позже и быстро исчезает после отмены препарата Анкилозирующий спондилоартрит/ HLA-B27 Эффект достигается через 2-3 мес.; лечение показано ассоциированные реактивные артриты (включая больным с периферическим артритом и высокой ВИЧ-ассоциированный синдром Рейтера) лабораторной активностью. Препарат рекомендуется назначать всем недавно заболевшим пациентам Псориатический артрит Эффективен при симметричном артрите и артрите, ассоциирующемся со спондилитом, не вызывает обострения поражения кожи (M. Farr и соавт.,1990) Генерализованная морфеа Отдельные клинические наблюдения Рецидивирующий передний увеит Предварительное сообщение (A. Breilbart и соавт., 1993) Обычно используемая доза у взрослых: 2 г (1.5-3 г; 40 мг/кг/день) по 1 г 2 раза в день с едой. Для уменьшения риска развития побочных эффектов рекомендуется следующая схема приема: 1-я неделя — 500 мг; 2-я неделя — 1000 мг; 3-я неделя — 1500 мг; 4-я неделя — 2000 мг. Общий анализ крови: первые 3 мес. 1 раз в 2-4 нед., затем каждые 3 мес. При появлении болей в горле, язв во рту, лихорадки, выраженной слабости препарат необходимо немедленно самостоятельно отменить. РА При РА эффективность сульфасалазина достоверно превосходит таковую у плацебо и не уступает эффективности кризотерапии, лечения D-ïåíèöèëëàìèíà, антималярийных препаратов и МТ (D. Porter и соавт., 1992). По переносимости сульфасалазин приближается к аминохинолиновым производным и превосходит ауранофин, парентеральное золото и D-ïåíèöèëëàìèí (D. Porter и соавт., 1992). По сравнению с другими базисными противоревматическими препаратами (например, D-ïåíèöèëëàìèíîì) эффект сульфасалазина проявляется существенно быстрее, уже через 1-2 мес. лечения (M. Farr и соавт., 1994). В недавних исследованиях было показано, что лечение сульфасалазином, но не другими базисными противоревматическими препаратами (соли золота, D-ïåíèöèëëàìèí, плаквенил), уменьшает выраженность скрытого кровотечения из кишечника и воспалительных изменений в кишечной стенке (J. Hayllar и соавт., 1994), иногда возникающих на фоне хронического приема НПВС (глава 2). В последние годы широко изучается возможность комбинированной терапии сульфасалазином и МТ (глава 6). У больных РА, активность заболевания у которых не контролируется МТ, добавление сульфасалазина в стандартной дозе приводит к достоверному клиническому улучшению в сроки от 3 до 6 мес. (G. C. Liang и соавт., 1994; C. J. Haasma и соавт., 1994). Анкилозирующий спондилоартрит Оценке эффективности сульфасалазина при анкилозирующем спондилите посвящена серия контролируемых исследований, результаты которых представлены в обзоре J. T. Gran и G. Husby (1992). По данным большинства авторов, на фоне лечения сульфасалазином отмечается достоверно снижение СОЭ, концентрации С-реактивного белка и IgA, что свидетельствует о способности препарата подавлять активность воспалительного процесса. Однако результаты, касающиеся влияния сульфасалазина на клинические проявления анкилозирующего спондилита, более противоречивы. Мета-анализ материалов 5 контролируемых исследований свидетельствует о следующих благоприятных клинических эффектах сульфасалазина: снижение продолжительности утренней скованности на 28.2%, уменьшение выраженности утренней скованности на 30.6%, снижение интенсивности болей на 26.7%, уменьшение СОЭ на 9.2%, снижение концентрации IgA на 11.7%, общее улучшение состояния больных на 7.1%. Эти результаты говорят об эффективности (по крайней мере краткосрочной) сульфасалазина при анкилозирующем спондилите, который наиболее очевиден при периферическом артрите. Примечательно, что развитие периферического артрита при анкилозирующем спондилите часто ассоциируется с воспалительными изменениями в тонкой кишке, выявляемыми при илеоколоноскопии (H. Mielants и E. M. Veys, 1990). Учитывая высокую эффективность сульфасалазина при язвенном колите и болезни Крона, можно полагать, что подавление кишечного воспаления является одним из основных механизмов действия сульфасалазина при анкилозирующем спондилоартрите. По мнению J. T. Gran и G. Husby (1992), сульфсалазин показан всем больным с недавно развившимся анкилозирующим спондилитом, а также больным с высокой лабораторной активностью болезни. У пациентов с поражением преимущественно позвоночника и крестцовоподвздошных сочленений без признаков лабораторной активности лечение сульфасалазином нецелесообразно. Имеются данные о высокой эффективности сульфасалазина при синдроме Рейтера, развившемся у больных СПИДом (P. Youssel и соавт., 1992) Сообщают также об определенном положительном действии сульфасалазина у больных с редким вариантом склеродермии — генерализованной морфеа (D. B. Crzarnecki и E. H. Taft, 1982; Z. Stava и M. Kobokova, 1977). Побочные эффекты (таблица 9.3.). Таблица 9.3. Побочные эффекты сульфасалазина Редкие, но тяжелые: Частые, но нетяжелые: апластическая анемия агранулоцитоз Желудочно-кишечные: фиброзирующий альвеолит тошнота, рвота, потеря аппетита, реакция гиперчувствительности боли в животе, диспепсия и т.д. необратимое поражение ЦНС или нейромышечной системы Неврологические: поражение почек головная боль, лихорадка, поражение печени головокружение и тд. Редкие: Поражение кожи: сыпь (экзематозная) Поражение печени: увеличение уровня ферментов Гематологические: лейкопения, гемолиз, метгемоглобинемия Иммунологические: продукция АНФ, волчаночноподобный синдром Сульфасалазин широко используется в клинической практике для лечения воспалительных заболеваний кишечника с 1950 г. На фоне длительного приема высоких доз препарата у больных язвенным колитом зарегистрирован ряд очень тяжелых побочных эффектов со смертельным исходом. Однако при РА используются существенно меньшие дозы препарата, и тяжелых побочных реакций, которые приводили бы к смерти больных, незарегистрировано (R. Amos и соавт., 1986). По обобщенным данным (D. R. Porter и H. A. Capell, 1990), у 58% больных, леченных сульфасалазином, наблюдались те или иные побочные эффекты, но только у 20-25% пациентов это явилось основанием для отмены препарата. Таким образом, сульфасалазин, наряду с антималярийными препаратами и ауранофином, рассматривается как один из наиболее хорошо переносимых противоревматических препаратов (D. Furst, 1990). Наиболее часто побочные реакции развиваются в первые 2-3 мес. лечения (R. S. Amos и соавт., 1986), некоторые из них зависят от дозы препарата и чаще отмечаются у лиц с медленным ацетилированием. Нередко побочные эффекты со стороны ЖКТ и ЦНС исчезают при уменьшении дозы препарата. По данным метаанализа, гематологические побочные эффекты и поражение печени чаще встречаются при РА, чем при язвенном колите и серонегативных спондилоартропатиях (M. J. H. Wijnands и соавт., 1993). Аллергическая сыпь наблюдается у 5% больных, частота нейтропении колеблется от 1 до 5%, но тяжелое поражение печени и гематологические нарушения развиваются крайне редко (D. R. Portrer и H. A. Capell, 1990). Хотя лейкопения, как правило, развивается в первые 24 нед. приема сульфасалазина, описаны случаи поздних цитопений, вплоть до агранулоцитоза. Это диктует необходимость тщательного гематологического контроля на протяжении всего лечения. Поражение кожи также встречается у 1-5% больных, наиболее часто в виде кожного зуда, макулопапулярной и генерализованной сыпи, реже — крапивницы. Описано развитие алопеции, сывороточной болезни, синдрома Лайелла. Очень редко наблюдаются изменения печеночных проб и развитие острого гепатита с лихорадкой, кожной сыпью и лимфаденопатией. Известны единичные случаи эозинофильного пневмонита, фиброзирующего альвеолита (O. Boyd и соавт., 1990). К крайне редким побочным эффектам сульфасалазина относят психические нарушения, проксимальную мышечную слабость, гипогаммаглобулинемию, волчаночноподобный синдром (E. M. Walker и J. E. Carty, 1994; A. R. Siam и M. Hammouden, 1993; H. Caulier и соавт., 1994). Список литературы к главе "Сульфасалазин" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. Amos R., Pullar Т., Capell Н.: Sulphasalazine for rheumatoid arthritis: toxicity in 744 patients monitored tor one two 11 years. Br. J. Med. 1986; 293: 420. Aparicio-Pages M. N., Verspaget H. W., Hafkenscheid J. C. M. et al.: Inhibition of cell mediated cytotoxicity by sulphasalazine: effect of in vitro treatment with 5-aminosalicylic acid and sulphasalazine on in vitro natural killer cell activity. Gut 1990; 31: 1031-1032. Aruoma 0., Wasil М., Halliwell В., et al.: The scavendger of oxidants by sulphasalazine and its metabolite: a possible contribution to their anti-inflammatory effects?. Biochem. Pharmacol. 1987; 36: 3739. Baum C. L, Seihub J., Rosenberg I. H.: Antifolate actions of sulfasalazine on Intact lymphocytes. J. Lab. Clin. Med. 1981; 97: 779-784. Bjarnason 1., So A., Levi A. J., et al.: Interstinal permeability and Inflammation in rheumatoid arthritis: effects of non-steroidal anti-inflammatory drugs. Lancet 1984; 2: 1171-1174. Breilbart A., Bauer H., Krastel H., et al.: Sulfasalazine in reccurent anterior uveitis.: a new strategy. Arthritis Rheum. 1993; 36 (suppl.): S-225. Boyd 0., Gibbs A. R., Smith A. P.: Fibrosing alveolitis due to sulphasalazine in a patient with rheumatoid arthritis. Brit. J. Rheumatol. 1990; 29: 222-224. Calin G.: Sulfasalazine does not inhibit cyclooxygenases I and 2. Arthritis Rheum. 1994; 37 (suppl.): S-383. Calin G., Nyman A.-K., Gronberg A.: Effects of sulfasalazine on cytokine production by mitogenstimulated human T cells. Arthritis Rheum. 1994; 37 (suppl.): S-383. Caulier M., Dromer C., Andrien V., et al.: Sulfasalazine induced lupus in rheumatoid arthritis. J. Rheumatol. 1994; 21: Cormer S. S., Jasin H. E.: In vitro immunomodulatory effects of sulfasalazine and its metabolites. J. Rheumatol. 1988; 15: 580-586. Czarnecki D. B., Taft E. H.: Generalized Morphea succesfully treated with salazopyrin. Acta Dermatovenerol. 1982; 62; 81-82. Farr M., Johnson A., Jones P. J., et al.: Sulfasalazine (SASP) — a rapidly acting drug in rheumatoid arthritis. A comparative study with penicilamine. Arthritis Rheum., 1994; 37 (Suppl.): S256. Fujiwara M., Mitsusi K., Ishida J., Yamamoto 1.: The effect of salazosulfapyridine on the in vitro antibody production in murine spleen cells. Immunopharmacology 1990; 19: 15-21. Gran J., Husby G.: Ankylosing spondilltis. Current drug treatment. Drugs 1992; 44: 586-603. Griffits I. D., Kane S. P.: Sulphasalazine induced lupus syndrome in ulcerative colitis. Brit. Med. J. 1977; ii: 1188-1189. Gronberg A., Isaksson P., Smedegard G.: Inhibitory effect of sulfasalazine on production of IL-I beta, IL-6 and TNF-alpha. Arthritis Rheum. 1994; 37 (suppl.): S-383. Haasma C. J., Van Riel C. M., De Grooij D. J. R. A., et al.: Combination of methotrexate and sulphasalasine vs methoirexate alone: a randomized open clinical trial in rheumatoid arthritis patients resistant to sulphasalazine therapy. Brit. J. Rheumatol. 1994; 33: 1049-1055. Hawkey C. L., Truelove S. C.: Inhibition of prostaglandin synthesis in human rectal mucosa. Gut 1983; 24: 213-217. Hayllar J., Smith T., Macpherson A., et al.: Nonsteroidal antiinflammatory drug-induced small intestinal inflammation and blood loss. Effects of sulfasalazine and other disease-modifying antirheumatic drugs. Arthritis Rheum. 1994; 37: 1146-1150. Imai F., Suzuki T., Ishibashi Т., Dohi Y.: Effect of sulphasalazine on В cells. Clin. Exp. Rheumatol. 1991; 9: 259-264. Imai F., Suzuki Т., Ashibashi T., et al.: Effect of sulfasalazine on В cell hyperreactivity in patients with rheumatoid arthritis. J. Rheumatol. 1994; 21: 612-615. Kanerud L., Scheynius A., Hafstrom I.: Evidence of a local intestinal immunomodulatory effect of sulfasalazine in rheumatoid arthritis. Arthritis Rheum. 1994; 37: 1138-1145. Liang G. C., Lessard J., Forks G.: Addition of sulfasalazine to rheumatoid arthritis (RA) patients (pts) inadequaetely controlled with methotrexate. Arthritis Rheum., 1994; 37: R-14 (abst. 15). Maple С., Kirk G., Veale D., Belch J. J. R.: Reduction in cell adhesion molecules levels following sulphasalazine in patients with rheumatoid arthritis. Brit. J. Rheumatol. 1995; 34 (suppl. 1): 98. Mielants H., Veys E. M.: The gut in the spondiloarthropathies. J. Rheumatol., 1990; 17: 7-10. Nielsen O. H., Bukhave К., Elmgreen J., Ahnfelt-Ronne I.: Inhibition of 5-lipoxygenase pathway of arachidonic acid metabolism in human neutrophils by sulphasalazine and 5-aminosalicylic acid. Dig. Dis. Sci. 1987; 6: 577-582. Nissila M., Lehtinen К., Leirisalo-Repo M., et al.: Sulphasalazine in the treatment of ankylosing spondilitis: a twenty-six-week, placebo-controlled clinical trial. Arthritis Rheum., 1988; 31: I II 1-1116. Porter D. R., Capell H. A.: The use of sulphasalazine as a disease modifying antirheumatic drug. Bailliere Clinical. Rheumatol. Ed. P. Brooks. London. 1990. Pullar T., Zoma A., Cappel A., et al.: Alteration of thiol and superoxide dismutase status in rheumatoid arthritis treated with sulphasalazine. Brit. Med. J. 1987; 26: 202. 31. Revming L., Andersen B.: Salicylazosulfapyridine (Salazopyrin) effect on endotoxin-induced production of interleukin-I like factor from human monocyte in vitro. Scand. J. Rheumatol. 1990; 19: 11-16. 32. Rhodes J. M., Bartholomew T. C., Jewell D. P.: Inhibition of leucocyte motility by drugs used in ulcerative colitis. Gut 1981; 22: 642-647. 33. Rooney P. J., Jenkins R. T., Smith K. M., Coates G.: III Indium-labelled polymorphonuclear leucocyte scans in rheumatoid arthritis: an importance clinical cause of false positive results. Br. J. Rheumatol. 1986; 25: 167-170. 34. Samanta A., Webb C., Grindulis K. A. et al.: Sulphasalazine therapy in rheumatoid arthritis: qualitative changes in lymphocytes and correlation with clinical response. Brit. J. Rheumatol. 1992; 31: 259-263. 35. Segal A. W., Isenberg D. A., Hajirousou V., et al.: Preliminary evidence for gut involvment in the pathogenesis of rheumatoid arthritis? Br. J. Rheumatol. 1986; 25: 162-166. 36. Sheldon P., Webb C., Grindulis K. A.: Effect of sulphasalazine and its metabolities on mitogen induced transformation of lymphocytes: clues to its clinical actions? Br. J. Rheumatol. 1988; 27: 344-349. 37. Siam A. R. M., Hammonden M.: Sulfasalazine induced systemic lupus erythematosus in patients with rheumatoid arthritis. J. Rheumatol. 1993; 20: 207. 38. Stava Z., Kobikova M.: Salazopyrin in the treatment of scleroderma. Br. J. Dermatol. 1977; 96: 541-544. 39. Symmons D. P. M., Salmon M., Farr M., Bacon P. A.: Sulfasalazine treatment and lympho-cyte function in patients with rheumatoid arthritis. J. Rheumatol. 1988; 15: 575-579. 40. Walker E. M., Carty J. E.: Sulphasalazin-induced systemic lupus erythematosus In patients with erosive arthritis. Brit. J. Rheumatol. 1994; 33: 175-176. 41. Wijnands M. J. H., Van T'Hof M. A., Van de Putte L. B. A., Van Riel P. L. C.: Rheumatoid arthritis: a risk factor for sulphasalazine toxicity? A meta-analysis. Brit. J. Rheumatol. 1993; 32: 313-318. 42. Williams J. Hallet M.: Effect of sulphasalazine, 5-amino-salicilic acid. on toxic oxygen metabolite production by neutrophils. Gut 1989; 30: 1581. 43. Youssel P., Bertouch J. V., Jines P. D. Succesful treatment of human immunodeficiency virus-associated Reiter's syndrome with sulfasalazine. Arthritis Rheum., 1992; 35: 723-734. ГЛАВА 10 ЦИКЛОСПОРИН А И ДРУГИЕ ПРЕПАРАТЫ С БЛИЗКИМИ МЕХАНИЗМАМИ ДЕЙСТВИЯ 10.1. Циклоспорин А Циклоспорин — нейтральный, липофильный, циклический эндекапептид, впервые изолирован в 1970 г. из двух штаммов грибков Tolypocladium inflatum Gams и Cylindrocarpon lucidium в процессе разработки новых противогрибковых препаратов. При изучении биологических свойств веществ, которые синтезировались этими грибками, было установлено, что некоторые из них, обозначенные как циклоспорины А, С и G, обладают не противогрибковой, а иммунологической активностью. К 1976 г. были накоплены многочисленные экспериментальные данные, свидетельствующие о выраженных специфических иммуносупрессивных эффектах циклоспорина А (ЦсА). Это позволило, начиная с 1978 г., применять его, в клинической практике сначала в трансплантологии, а затем при широком спектре заболеваний человека, развитие которых связано с нарушениями в системе иммунитета (B. D. Kahan, 1989). В настоящее время ЦсА известен под названием "Сандиммун" (фирма "Сандоз", Швейцария). Фармакологические свойства ЦсА (таблица 10.1) Таблица 10.1. Основные фармакокинетические параметры ЦсА при применении у человека ( "Сандиммун". Практическое руководство "Sandoг") Время, необходимое для достижения максимальной концентрации 2-4 ч Пероральная биодоступность 10-57% Связывание с белками плазмы >90% Связывание с эритроцитами около 0% Степень метаболизма около 99% Время полувыведения 10-27 ч Основной путь выделения Желчь Примечание: в связи с вариабельностью всасывания рекомендуется мониторировать концентрацию ЦсА в сыворотке (или цельной крови) с помощью радиоиммунологического метода. Механизм действия Биохимические механизмы действия ЦсА на клетки изучены еще не полностью и в настоящее время являются предметом интенсивного исследования (B. D. Kahan, 1989). Вначале полагали, что ЦсА проявляет свою биологическую активность за счет связывания со специфическими мембранными рецепторами, имеющими отношение к пролактиновым рецепторам. Однако в настоящее время установлено, что ЦсА благодаря своим липофильным свойствам обладает способностью к диффузии в цитоплазму через клеточную мембрану, где связывается со специфическими белками с мол. массой 17 kD, получившими название циклофилинов (S. L. Сhriеbег, 1991; N. H. Sigal и F. J. Dumont, 1992). Последние проявляют активность, идентичную таковой фермента пептидил-пропил цис-транс изомеразы (ППИ) (G. Ficher и соавт., 1989). ППИ — семейство ферментов, которые также известны как ротамазы, они играют важную роль в реализации функциональной активности многих клеток. В частности, эти ферменты обладают способностью изменять конформацию пролиновых остатков, а следовательно, регулировать укладку белка в процессе белкового синтеза. Таким образом, блокированием ЦсА ППИ можно частично объяснить влияние препарата на транскрипцию генов цитокинов. Однако широкое распространение циклофилинов и ППИ не только в лимфоцитах, но и в различных клетках, не обладающих иммунной активностью, позволяет объяснить некоторые токсические эффекты препарата, но не причины специфического влияния ЦсА на синтез цитокинов. Кроме того, при изучении аналогов ЦсА было показано, что, хотя связывание с циклофилином является необходимым условием для развития иммунных эффектов, прямая корреляция между циклофилинсвязывающей активностью и иммуносупрессивным действием отсутствует. Недавно было установлено, что ЦсА влияет на функциональную активность нескольких ядерных белков (NF-AT, АР-3, NFkB), принимающих участие в регуляции транскрипции генов, в том числе гена ИЛ-2. При этом NF-AT (nuclear factor of activated T lymphocytes) локализуется преимущественно в Т-клетках, что позволяет частично объяснить причины влияния ЦсА именно на эту субпопуляцию лимфоцитов. Примечательно, что иммунные эффекты ряда недавно открытых иммуномодуляторов, структурно не связанных с ЦсА (FK-506, рапамицин), также опосредуются через взаимодействие с некоторыми ядерными белками. Некоторые из них, так называемые FKñâÿçûâàþùèå белки, также проявляют ротамазную активность и наряду с циклофилинами составляют семейство высокоаффинных связывающих белков, получивших название иммунофилинов, или макрофилинов (S. L. Chrieber, 1991). В настоящее время клонировано 4 циклоспоринсвязывающих белка и 4 FK-506-ñâÿçûâàþùèõ белка и установлено их присутствие практически во всех тканях. Имеются данные о том, что ЦсА и FK-506 связываются с каталитической субъединицей серин/треонин фосфатазы (кальцинейрин), которая функционирует как Са2+ и кальмодулинзависимый комплекс. Это подтверждает мнение о том, что в основе действия этих препаратов может лежать подавление кальцийзависимой активации и пролиферации Тлимфоцитов. Наконец, совсем недавно появились сообщения о том, что циклофилины могут не только выступать в роли внутриклеточных иммунорегуляторных белков, но и секретироваться различными клетками, включая макрофаги, и проявлять цитокиноподобную активность. При этом оказалось, что циклофилины имеют определенное функциональное и структурное сходство с ИЛ-8 (глава 1), Специфическое связывание ЦсА с ИЛ8 может являться одним из возможных механизмов противовоспалительного действия этого препарата. Основными клетками-мишенями для ЦсА являются CD4+T-(хелперы) лимфоциты, активация которых лежит в основе развития иммунного ответа. При этом высокая эффективность и низкая токсичность ЦсА определяются его способностью селективно блокировать ранние этапы кальцийзависимой Т-клеточной активации, опосредуемой ТКР-комплексом, и тем самым прерывать процесс трансдукции активационного сигнала, не влияя на более поздние этапы дифференцировки клеток. Установлено, что ЦсА селективно ингибирует экспрессию генов (с-myc, srs), принимающих участие в ранней активации Т-лимфоцитов, и транскрипцию иРНК некоторых цитокинов, в том числе ИЛ-2, ИЛ-3, ИЛ-4, ИФ-у. Важная точка приложения ЦсА — частичное блокирование экспрессии мембранных ИЛ-2 рецепторов на Т-лимфоцитах. Все это вместе взятое приводит к замедлению пролиферации CD4+ Т-лимфоцитов, опосредуемому паракринными и аутокринными эффектами этих цитокинов. Подавление синтеза цитокинов активированными CD4+ Тлимфоцитами в свою очередь ведет к супрессии цитокинзависимой пролиферации цитотоксических Тлимфоцитов и оказывает опосредованное влияние на функциональную активность других клеток иммунной системы, принимающих участие в реализации ее функциональной активности: В-лимфоцитов, мононуклеарных фагоцитирующих клеток и других АПК, тучных клеток, эозинофилов, ЕК-клеток (A. W. Thompson и J. I. Duncan, 1989). Более того имеются данные, свидетельствующие о способности ЦсА напрямую подавлять активацию В-лимфоцитов, ингибировать хемотаксис мононуклеарных фагоцитов, синтез ФНО-а и в меньшей степени ИЛ-1 и, наконец, подавлять экспрессию антигенов класса II ГКГ на мембранах АПК. Последнее, вероятно, в большей степени связано с влиянием ЦсА на синтез ИФ-у, ИЛ-4 и ФНО, чем с прямым действием препарата на клеточные мембраны (A. W. Thompson, 1992). Тактика лечения Предполагаeтся, что схема применения ЦсА, недавно разработанная для псориаза, подходит и для лечения других аутоиммунных заболеваний. Рекомендации по лечению больных ЦсА (по G. S. Panayi и P. Tugwell, 1994): 1. Препарат не следует назначать больным с нарушенной функцией почек, тяжелой артериальной гипертензией, инфекционными заболеваниями и злокачественными новообразованиями. 2. Перед началом лечения необходимо провести детальное клиническое и лабораторное обследование: определение уровня билирубина, печеночных ферментов, калия и магния, мочевой кислоты, липидного профиля в сыворотке, общий анализ мочи. 3. Начинать лечение следует с дозы не более Змг/кг/день, в 2 приема. 4. Затем дозу увеличивают до оптимальной по 0.5-1.0мг/кг/день в зависимости от эффективности (оценивается через 6 — 12 нед.) и переносимости. Максимальная доза не должна превышать 5мг/кг день. 5. Оценивать АД и уровень сывороточного креатинина (установить базовый уровень по данным не менее чем 2 определений до лечения) каждые 2 нед. в первые 3 мес. терапии, затем каждые 4 нед. 6. При увеличении уровня креатинина более чем на 30% уменьшить дозу препарата на 0.5-1.0 мг/кг/день в течение 1 мес. 7. При снижении уровня креатинина на 30% продолжить лечение препаратом. При сохранении 30% увеличения концентрации креатинина прекратить лечение. При снижении содержания креатинина на 10% по сравнению с базальным уровнем возобновить лечение. 8. По возможности следует избегать сочетания с препаратами, влияющими на концентрацию ЦсА в крови. Установлено, что наиболее низкая концентрация ЦсА в крови, при которой наблюдается терапевтический эффект препарата, составляет 100 нг/л, а нефротоксическая концентрация — 250 нг/л. Промежуток между этими концентрациями препарата в плазме крови определяется как "терапевтическое окно"; при этом оптимальное содержание ЦсА в плазме составляет около 200 нг/л. В то же время значительные индивидуальные колебания фармакологической чувствительности к ЦсА и его взаимодействие с различными лекарственными препаратами иногда затрудняют ведение больных. Среди препаратов, которые широко используются при лечении ревматических заболеваний, клинически значимые лекарственные взаимодействия могут наблюдаться при приеме ГК (особенно в высоких дозах), антагонистов кальция (дилтиазем, верапамил), метаклопрамида, эритромицина, назначение которых может приводить к увеличению концентрации ЦсА в крови. На фоне лечения ЦсА следует избегать приема препаратов, которые могут вызывать нарушение функции почек, таких, как гентамицин и некоторые другие антибиотики, блокаторы Н2-гистаминовых рецепторов, триметоприм/сульфаметаксозол. Клиническое применение РА В экспериментальных исследованиях было показано, что ЦсА подавляет развитие экспериментального коллагенового артрита (B. Henderson и соавт., 1984) и особенно эффективен при комбинации с МТ. Эти экспериментальные данные были подтверждены в многочисленных контролируемых исследованиях, в которых было убедительно доказано, что ЦсА является весьма эффективным препаратом для лечения РА, не только вызывающим симптоматическое улучшение, но и оказывающим базисное противоревматическое действие (С. Б. Усова, 1993; M. Dougados и H. Torley, 1993; G. Wells и P. Tugwell, 1993). Примечательно, что клиническая эффективность ЦсА была продемонстрирована у больных с активным, длительно текущим РА, нередко рефракторным к другим базисным противоревматическим препаратам. Суммарный анализ полученных результатов позволил выявить положительное влияние ЦсА на следующие показатели, использующиеся для оценки эффективности лечения РА: выраженность боли, оцениваемая как по визуальной аналоговой шкале, так и по суставному индексу; функциональная способность суставов (индекс Lee и модифицированный опросник состояния здровья); воспалительная активность, в том числе уменьшение продолжительности утренней скованности и выраженности синовита, снижение концентрации острофазовых белков (при этом изменения СОЭ и титров РФ; уменьшение дозы ГК; замедление прогрессирования суставной деструкции по данным рентгенологического исследования. Результаты мета-анализа эффективности и токсичности ЦсА при РА свидетельствуют о том, что по динамике этих параметров ЦсА сопоставим с другими базисными противоревматическими препаратами (M. Dougados, и H. Torley, 1993). При оценке эффективности ЦсА необходимо иметь в виду гетерогенность РА в отношении как клинических проявлений, так и иммунопатологических механизмов, лежащих в основе развития болезни. Некоторые предварительные результаты говорят об особенно высокой эффективности препарата у больных с определенным спектром иммунологических нарушений, таких, как кожная анергия, низкое соотношение СD4+Т-лимфоцитов и CD8+ Тлимфоцитов в периферической крови, увеличение уровня ЕК-клеток и снижение концентрации клеток, экспрессирующих ИЛ-2Р (D. E. Yocum и соавт., 1990; B. T. Walsh и соавт., 1992). Это свидетельствует о принципиальной возможности прогнозирования клинической эффективности ЦсА при РА. Результаты контролируемых исследований эффективности и токсичности ЦсА при РА были обобщены недавно H. Torley и D. Yocum (1994). Установлено, что развитие осложнений на фоне лечения не зависит от дозы ЦсА (если она не превышает 5.5 мг/кг/день), сочетанного приема НПВС и ГК, но нарастает при длительном использовании препарата (более 52 нед.) и у больных старше 65 лет. По данным R. B. M. Landewe и соавт. (1994), длительное лечение низкими дозами ЦсА больных РА не сопровождается более выраженным прогрессированием почечного фиброза, чем это наблюдается у больных, не леченных ЦсА, несмотря на биохимические признаки нарушений почечной функции. Поскольку, по данным литературы, СD4+Т-лимфоциты играют особенно важную роль на ранних этапах ревматоидного синовита, представляет интерес исследование возможности использования ЦсА при раннем PA. R. B. M. Landewe и соавт. (1994) провели 24-недельное двойное слепое .контролируемое исследование эффективности ЦсА (начальная доза 2.5 мг/кг/день, поддерживающая доза 3.6 мг/кг/день) и гидроксихлорохина (начальная доза 300 мг/день, поддерживающая доза 100 мг/день) у 44 больных с ранним РА (продолжительность болезни меньше 2 лет). Установлено, что оба препарата обладают сходными эффективностью и нефротоксичностью. Интересно, что клинический эффект ЦсА не коррелировал с изменением СОЭ. Имеются данные об эффективности ЦсА при синдроме Фелти, причем клиническое улучшение и нормализация числа нейтрофилов ассоциировались со снижением титров РФ и исчезновением антинейтрофильных антител (J. Camps и соавт., 1991). Серия исследований посвящена изучению возможности комбинации ЦсА с другими базисными противоревматическими препаратами. Полученные результаты, хотя и носят предварительный характер, свидетельствуют о том, что сочетанное применение ЦсА с МТ и ЦсА с солями золота позволяет более эффективно контролировать патологический процесс при рефракторном PA (W. Bensen и соавт., 1994). По данным многоцентрового двойного слепого контролируемого исследования (P. Tugwell и соавт., 1994), включение ЦсА в схему лечения больных РА, не полностью "отвечающих" на МТ, позволяет повысить эффективность лечения в отношении подавления признаков суставного воспаления без нарастания частоты токсических реакций. Имеются данные о синергической активности ЦсА и антималярийных препаратов при PA (R. B. M. Landewe и соавт., 1992). Болезнь Бехчета По данным K. Masuda и соавт. (1989), ЦсА в начальной дозе 5 мг/кг/день с последующим снижением до 2 мг/кг/день эфффективнее колхицина в отношении подавления прогрессирования увеита при болезни Бехчета. A. I. Binder и соавт. (1987) применили ЦсА для лечения увеита у 12 больных болезнью Бехчета. Во всех случаях зарегистрировано улучшение признаков воспаления глаз и системных проявлений, однако отмена препарата сопровождалась обострением болезни. У большинства пациентов развились побочные эффекты, однако это, вероятно, было связано с тем, что в исследовании использовались дозы препарата, более высокие (10 мг/кг/день), чем принято в настоящее время. ПМ/ДМ Обнадеживающие результаты получены при использовании ЦсА для лечения ДМ/ПМ. J. Heckman и соавт. (1989) применяли ЦсА (2.5-7.5 мг/кг/день) при рефракторном к ГК ювенильном ДМ и зарегистрировали хороший клинический эффект при отсутствии осложнений. K. Danko и соавт. (1991) использовали ЦсА в дозе 5 мг/кг/день для лечения 10 больных с рефракторным к ГК и цитостатикам дерматомиозитом. Определенный клинический эффект был зарегистрирован уже в течение первой недели лечения, уровень сывороточных ферментов нормализовался к 3-му месяцу. На фоне поддерживающей терапии ЦсА в дозе 2-3.5 мг/кг/день у большинства больных удалось отменить ГК или существенно снизить их дозу. Обращает на себя внимание отсутствие тяжелых побочных эффектов, которые потребовали бы отмены препарата. Имеется интересное наблюдение о достижении ремиссии на фоне сочетанной терапии IVIg и ЦсА (5мг/кг/день) у больных ПМ, рефракторных к предшествующей кортикостероидной и иммуносупрессивной терапии (C. K. Saadeh и соавт., 1993). Высокая эффективность ЦсА при ПМ/ДМ может быть связана с несколькими обстоятельствами. Известно, что развитие патологического процесса при воспалительных миопатиях сопровождается выраженной инфильтрацией периферической мускулатуры как активированными СD4+Т-лимфоцитами, так и СD8+Тлимфоцитами, экспрессирующими DR-àíòèãåí, а также ЕК-клетками. При этом в наибольшем количестве СD8+Т-лимфоциты располагаются вокруг миофибрилл, экспрессирующих антигены класса I ГКГ. В целом морфологическая характеристика мышечного инфильтрата в определенной степени напоминает картину, наблюдаемую при отторжении трансплантата. Таким образом, механизмы действия ЦсА при ИВМ, могут быть сходными с таковыми при подавлении реакций трансплантационного иммунитета. ЦсА обладает способностью подавлять миоцитолиз цитотоксическими лимфоцитами за счет ингибиции высвобождения цитотоксических субстанций: перфорина и сериновой эстеразы В (P. Cherin и соавт., 1993). Полагают, что эффект ЦсА реализуется на уровне ингибиции транскрипции генов, кодирующих синтез этих цитотоксических факторов. ССД Имеется несколько сообщений о лечении ЦсА больных ССД. В 1987 г. T. Appelboom и соавт. описали больную быстропрогрессирующей ССД с яркими системными проявлениями (перикардит, легочный фиброз, артрит), которые не контролировались ГК. Назначение ЦсА привело к быстрому улучшению и позволило контролировать заболевание в течение 1 года наблюдения. Позже H. Zacharie и соавт. (1990) исследовали эффективность ЦсА (начальная доза 3.5-7.5 мг/кг/день, поддерживающая доза 1.5-5.0 мг/кг/день) у 10 больных ССД. Отмечено определенное положительное влияние препарата на выраженность кожного синдрома, дигитальные язвы, однако в целом эффект лечения был незначителен. Другие авторы также обнаружили положительное влияние ЦсА только на кожные проявления заболевания. (C. Frances и соавт., 1988; M. L. Russel и соавт., 1988). В исследовании Н. Gisslinger и соавт. (1991) ЦсА в дозе 5 мг/кг/день был назначен 8 больным тяжелой ССД с последующей коррекцией дозы до уровня, поддерживающего концентрацию препарата в пределах 300-500 нг/мл. Через 6-12 мес. лечения у 7 больных уменьшились кожные проявления, у 4 — увеличилось насыщение артериальной крови кислородом, у 4 — уменьшились признаки легочной гипертензии, у 3 — увеличилась амплитуда сокращения пищевода. Прогрессирование заболевания констатировано только в 1 случае. Побочные эффекты были выражены умеренно и носили обратимый характер; не отмечено отрицательного влияния лечения на состояние функции почек. По данным P. Clements и соавт. (1990), проводивших 48-недельное открытое исследование ЦсА при ССД, препарат достоверно подавлял выраженность кожного синдрома (снижал кожный счет более чем на 35%), однако не оказаывал существенного влияния на поражение легких и сердца. По мнению авторов, при назначении ЦсА больным ССД необходим очень тщательный их подбор (исключение пациентов с АГ), контроль даже минимальных изменений концентрации креатинина и использование вначале небольших доз, в пределах 2.5 мг/кг/день, с последующим медленным увеличением до оптимальных при удовлетворительной переносимости препарата. Кроме того, необходимо иметь в виду, что данные некоторых экспериментальных и клинических исследований свидетельствуют о потенциальной возможности негативного влияния ЦсА на сосудистую стенку. В опытах in vitro было показано, что ЦсА стимулирует синтез эндотелина в культуре эндотелиальных клеток (стр. ). Сообщается о развитии синдрома Рейно на фоне лечения ЦсА у больных с трансплантированной почкой. СКВ При исследовании эффективности ЦсА у экспериментальных моделей СКВ установлено, что лечение приводит к увеличению продолжительности жизни гибридных линий мышей NZB/NZW (FI) и MRL-lpr/lpr, у которых спонтанно развивающаяся аутоиммунная патология напоминает СКВ (D. I. Israel-Biet и соавт., 1983; M. G. Jones и соавт., 1985). Интересно, что у мышей NZB/NZW (FI) отмечена связь между подавлением выраженности гломерулонефрита и снижением уровня антител к ДНК, в то время как у мышей MRL-lpr/lpr увеличение продолжительности жизни не ассоциировалось со снижением титров этих антител (J. D. Mountz и соавт., 1987). M. Blank и соавт. (1992) исследовали эффективность ЦсА на другой модели волчанки у мышей, основанной на иммунизации моноклональными анти-ДНК антителами, экспрессирующими "патогенный" идиотип 16/6. В этой работе было установлено, что при назначении в ранний период после иммунизации ЦсА более эффективно подавляет практически все клинические и иммунологаческие проявления болезни, чем при назначении в более поздний период. Результаты клинических исследований также свидетельствуют об уменьшении протеинурии у больных волчаночным нефритом на фоне лечения ЦсА (H. Favre и соавт., 1989). Кроме того, у 8 пациентов с очень хорошим клиническим эффектом наблюдалось снижение уровня антител к ДНК. Побочных эффектов, требующих отмены ЦсА, не отмечено. G. Feutren и соавт. (1987) выявили стероидсберегающий эффект ЦсА у 8 из 13 больных стероидрезистентной СКВ, у которых до назначения ЦсА были тяжелые проявления СКВ (цереброваскулит, легочная гипертензия, плевроперикардит). Однако иммунологические показатели активности болезни не улучшились. M. Tokuda и соавт. (1994) использовали ЦсА в начальной дозе Змг/кг/день у 11 больных СКВ. На фоне 20-недельной терапии выявлено достоверное снижение индекса активности СКВ (SLEDAI) у 80% больных, снижение титров АНФ у 8 больных и антител к ДНК у 5 больных, 10-летний опыт лечения ЦсА 21 больного СКВ суммирован R. Bambauer и соавт. (1992). Положительный клинический эффект зарегистрирован в 52% случаев, что, однако, по мнению авторов, не дает оснований судить о влиянии лечения на прогноз болезни. Имеется сообщение об успешных родах у 2 больных СКВ на фоне лечения ЦсА (M. M. Hussen и соавт., 1993). F. Dimmacco и соавт. (1994) обнаружили стероидсберегающее действие низких доз ЦсА у пациентов с СКВ при отсутствии побочных эффектов на фоне комбинированной терапии. СШ. В двойном слепом контролируемом исследовании, проведенном с участием 20 больных в течение 6 мес., было установлено, что ЦсА в дозе 5 мг/кг/день не влияет на основные клинические и лабораторные проявления заболевания, но несколько замедляет прогрессирование гистопатологических проявлений в слюнных железах. При более длительном применении ЦсА в той же дозе в течение последующих 6 мес. было отмечено лишь некоторое уменьшение проявлений ксеростомии на фоне явного ухудшения гистологических изменений в малых слюнных железах (A. A. Drosos и соавт., 1986). Эти результаты свидетельствуют о низкой эффективности небольших доз ЦсА в отношении стоматологических проявлений СШ. В то же время имеются данные о благоприятном эффекте ЦсА в отношении офтальмологических проявлений болезни, особенно при назначении его в ранней стадии заболевания (P. M. Leuenberger и P. A. Miesher, 1992). Место ЦсА в лечении СШ, несомненно, требует дальнейшего уточнения, тем более, что при иммуноморфологическом исследовании пораженных органов выявляется выраженная лимфоидная инфильтрация преимущественно СD4+Тлимфоцитами, экспрессирующими DR-àêòèâàöèîííûé маркер. Предварительные результаты указывают на возможность купирования ЦсА некоторых проявлений синдрома эозинофилии-миалгии, развитие которого связывают с приемом L-òðèïòîôàíà (D. J. Claw и соавт., 1991). На фоне приема препарата в дозе 5мг/кг/день в течение 11.2 нед. 8 больными у 3 из них отмечено уменьшение выраженности миалгий, а у 4— проявлений фасциита. При этом эффекта в отношении других проявлений синдрома — мышечной слабости, одышки, поражения ЦНС — не обнаружено. Недавно появилось сообщение об успешном применении ЦсА (5ìã/êã/äåíü) в сочетании с ГК у 2 больных ГВ (F. Gremmel и соавт., 1988). Следует подчеркнуть, что оба пациента не реагировали на лечение высокими дозами ЦФ и у обоих были признаки почечной недостаточности. Создается впечатление, что ЦсА в ряде случаев может быть альтернативой ЦФ, который, хотя и обладает высокой клинической эффективностью при ГВ, нередко вызывает инфекционные осложнения. Необходимо обратить внимание на то, что в основе развития некоторых характерных морфологических нарушений при ГВ, в частности гранулем, лежат иммунные реакции, связанные с активацией СD4+Т-лимфоцитов и нейтрофилов, что также подтверждает патогенетическую обоснованность применения ЦсА при этой форме системных васкулитов. Описаны случаи успешного применения ЦсА при рецидивирующем полихондрите (K. L. G. Svenson и соавт., 1984; R. Priori и соавт., 1993), синдроме Вебера—Крисчена, гангренозной пиодермии (R. K. Curley и соавт., 1985; M. L. Magid и соавт., 1989; G. Elgart и соавт., 1991; J. R. Hughes и соавт., 1994). Побочные эффекты По сравнению с другими иммуносупрессивными препаратами ЦсА в целом дает меньше как непосредственных, так и отдаленных побочных эффектов, в первую очередь в отношении развития инфекционных осложнений и злокачественных новообразований (P. T. Kurki, 1992). В то же время на фоне лечения ЦсА наблюдается развитие некоторых специфических осложнений, наиболее тяжелым из которых является поражение почек. Поражение почек ЦсА проявляет так называемую функциональную нефротоксичность, приводящую к увеличению концентрации креатинина и мочевины. Она связана с вазоконстрикцией афферентных артериол клубочков, ведущей к снижению почечного кровотока, четко коррелирует с дозой препарата и обычно не сопровождается явными морфологическими нарушениями. У больных аутоиммунными заболеваниями, получающих ЦсА в дозе не более 5 мг/кг/день, нарушение функции почек, как правило, обратимо. Однако иногда при гистологическом исследовании почек обнаруживается так называемая изометрическая вакуолизация проксимальных канальцев, а при длительном лечении (6-12 мес.) — интерстициальный фиброз и/или васкулопатия, затрагивающая мелкие артерии. Эти изменения также обычно частично обратимы, но в ряде случаев ведут к развитию хронической почечной недостаточности. Артериальная гипертензия ЦсА вызывает зависимое от дозы повышение диастолического АД в среднем на 2-3 мм рт.ст. при дозе 2.5 мг/кг/день и на 5 мм рт.ст. при дозе 5 мг/кг/день. АГ развивается у 5-15% больных, ее частота не существенно различается у больных, получавших ЦсА по 2-3 мг/кг/день и по 5 мг/кг/день. Обычно АД повышается в течение первых недель лечения ЦсА, чаще у больных, страдающих заболеваниями, которые сами предрасполагают к развитию АГ. После отмены препарата АД, как правило, быстро нормализуется. Хотя ЦсА и не рекомендуется назначать больным с длительной неконтролируемой АГ, ее развитие не является абсолютным противопоказанием к использованию ЦсА и отмене препарата, так как АД может нормализоваться на фоне гипотензивной терапии и при снижении дозы ЦсА. Препаратами выбора при лечении АГ являются антагонисты кальция, такие, как нифедипин или исрадипин, которые в отличие от дилтиазема и верапамила существенно не влияют на фармакокинетику ЦсА. Злокачественные новообразования При пересадке почки частота лимфопролиферативных заболеваний у больных, леченных ЦсА, составляет 0.4%, в то время как частота других типов злокачественных новообразований (например, аденокарцином) не увеличивается. Среди 3700 больных различными аутоиммунными заболеваниями, получавших ЦсА, развитие лимфом описано в 5 случаях (3 случая, вероятно, не связаны с ЦсА). Таким образом, частота лимфопролиферативных опухолей при аутоиммунных заболеваниях, леченных ЦсА, очень низкая и колеблется от 0.14 (5 случаев) до 0.05 (3 случая). Имеются данные о том, что отмена ЦсА приводит к обратному развитию опухоли. Другие побочные реакции К другим побочным реакциям относят гипертрихоз, гиперплазию слизистой полости рта, парестезии, тремор, желудочно-кишечные расстройства, умеренную гипербилирубинемию, анемию. Обычно они развиваются через несколько дней после начала лечения ЦсА и затем исчезают, несмотря на продолжение лечения или после отмены ЦсА. Инфекционные осложнения развиваются очень редко. Частота инфекций у больных, длительно леченных ЦсА, не отличается от таковой в группе пациентов, получавших плацебо. Не связанные с поражением почек побочные реакции являются причиной отмены ЦсА у 3-4% больных псориазом и с нефротическим синдромом и у 21%— PA, главным образом в связи желудочно-кишечными расстройствами. 10.2. FK-506 FK-506 (Tacrolimus; Fujisawa Pharm.) — природный иммуносупрессивный агент, относящийся к макролидным антибиотикам, изолированный из грибка Streptomyces tsukubaensis, имеющий следующую брутто-формулу: С44Н69NО12хН2О (мол. масса 822 kD). Механизм действия Хотя FK-506 отличается по структуре от ЦсА и связывается с другим типом цитоплазматических лигандов (белок с мол, массой 12 kD, относящийся к семейству иммунофилинов), оба препарата проявляют сходную иммуносупрессивную активность, связанную с подавлением кальцийзависимой активации транскрипции генов цитокинов. Более того, FK-506 проявляет свою активность в значительно меньших (примерно в 10 раз) концентрациях, чем ЦсА. При введении FK-506 вместе с ЦсА наблюдается аддитивный эффект (N. H. Sigal и F. J. Dumont, 1992), В опытах in vitro было показано, что FK-506 подавляет секрецию или экспрессию иРНК различных цитокинов, таких, как ИЛ-2, ИЛ-4, ИФ-у Т-лимфоцитами (J. Anderson и соавт., 1992; M. J. Tocci и соавт., 1989) и ИЛ-1 макрофагами (N. Keicho и соавт., 1991), а также ИЛ-6 синовиоцитами, полученными от больных PA (E. Sugiyama и соавт. 1994). Препарат предотвращает развитие артрита, индуцированного коллагеном типа II у крыс и мышей (C. Arita и соaвт. 1990; N.Inamura и соавт., 1988), и других экспериментально индуцированных аутоиммунных заболеваний (сахарный диабет, увеит и аллергический энцефалит) (Macleod А. М. и Thompson A. W., 1991). Клиническое применение В клинических исследованиях было показано, что благодаря способности концентрироваться в печени при ее пересадке препарат обладает более высокой терапевтической активностью и меньшей токсичностью, чем ЦсА. Отдельные клинические наблюдения свидетельствуют об эффективности FK-506 при стероидрезистентном нефротическом синдроме, связанном с очаговым гломерулосклерозом, тяжелом псориазе, увеите, болезни Бехчета, синдроме Когана, инсулинзависимом сахарном диабете. V. Aldo и соавт. (1994) использовали FK-506 для лечения 7 больных псориатическим артритом, резистентным к МТ. В 5 случаях отмечено клиническое улучшение как кожного, так и суставного процессов. FK-506 в дозе 5-24 мг/день недавно был применен для лечения 5 больных ПМ/ДМ, резистентным к ГК и комбинированной цитотоксической терапии (C. V. Oddis и соавт., 1994). Продолжительность курса составила в среднем 12.8 мес. У всех больных достигнут хороший клинический эффект, ассоциирующийся с нормализацией уровня КФК и возможностью снизить дозу ГК в среднем с 20 до 4 мг/сут. У 3 из 4 больных с антителами к Jo-1 наблюдалось исчезновение признаков артрита. Побочные эффекты FK-506 такие же, как и у ЦсА, однако нефротоксичность и способность вызывать гипергликемию выражена у FK-506 в меньшей степени, чем у ЦсА. 10.3. Рапамицин Рапамицин (Sirolimus; Wyeth-Ayerst Res.) — макролид, изолированный Streptomyces hygroscopicus (S. N. Sehgal и ñîàâò., 1975), подавляющий отторжение трансплантата. Имеет следующую брутто-формулу: C57H79NO73 Механизм действия Рапамицин по структуре напоминает FK-506 и связывается с тем же самым внутриклеточным белком с мол. массой 12 kD, относящимся к классу иммунофилинов. Несмотря на это, препараты имеют разный механизм действия. В отличие от FK-506 рапамицин подавляет не кальций-зависимые, а посттрансляционные этапы активации Т-лимфоцитов в GI-фазе клеточного цикла (N. H. Sigal и F. J. Dumont, 1992) и в отличие от ЦсА и FK-506 не оказывает действия на транскрипцию и секрецию цитокинов, а подавляет зависимую от цитокинов или факторов роста активацию Т-хелперов (В. Е. Вrier и соавт., 1990; D. J. Henderson и соавт., 1991), а также рост и дифференцировку некоторых клеток. Рапамицин является антагонистом FK-506 в отношении иммуносупрессивного действия и проявляет синергическую активность с ЦсА. Имеются данные о том, что рапамицин более эффективно ингибирует пролиферацию тимоцитов и лимфоцитов, чем ЦсА, влияя на те стадии Т-клеточной активации, которые резистентны к ЦсА (B. D. Kahan и соавт., 1991). Предполагается также, что возможной точкой приложения рапамицина является ингибиция фосфорилирования тирозиновых остатков p34cdc2 киназы и p70S6 киназы в S-фазе клеточного цикла (W. G. Morice и соавт., 1993; C. J. Kuo и соавт., 1992). Рапамицин подавляет развитие аутоиммунного процесса при экспериментальном аллергическом энцефаломиелите у крыс (модель рассеянного склероза) (R. B. Martel и соавт., 1977) и у NOD-ìûøåé (модель сахарного диабета) (W. L. Baeder и соавт., 1992). Кроме того, рапамицин значительно снижает интенсивность клинических и лабораторных проявлений у мышей MRL/MpJ/lpr/lpr (модель СКВ) (L. Warner и соавт., 1994). Имеются данные о том, что рапамицин способен подавлять развитие коллагенового артрита, усиленного введением бактериального суперантигена (J. Zimmerman и соавт., 1994). Рапамицин может модулировать Т- клеточные, макрофагальные и В-клеточные иммунные реакции, в том числе ингибирует Т-клеточную активацию и пролиферацию, уменьшает количество "double negative" Т-клеток, восстанавливает синтез ИЛ-2, ингибирует синтез IgG В-лимфоцитами и продукцию ИЛ-1. Недавно было показано, что рапамацин подавляет активацию преимущественно Тh2-клеток (N. Ferraresso и соавт., 1994). Эти данные свидетельствуют о целесообразности испытания препарата при лечении аутоиммунных ревматических заболеваний. Список литературы к главе "Циклоспорин А, FК-506, рапамицин" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. Усова С. Б.: Циклоспорин А в терапии ревматоидного артрита. Клиническая ревматология, 1993; No. 1: 44-48. Aldo V., Londino Ir., Santoro D., et al.: FK 506 in the treatment of psoriasis and psoriatic arthritis. Arthritis Rheum., 1994; 37 (Suppl.): S207. Anderson J., Nagy S., Groth C.-G. et al.: Effect of FK506 and cyclosporine A on cytokine production studied in vitro at a single-cell level. Immunology 1992; 75: 136-142. Appelboom Т., Itzkowitch D.: Cyclosporine in succesful control of rapidly progresstve scle-roderma. Am. J. Med. 1987: 82: 866-867. Arita С., Hotokebuchi Т., Miyahara H., et al.: Inhibition by FK506 of established colla-gen-induced arthritis in rats. Clin. Exp. Immunopathol. 1990; 82: 456-461. Baeder W. L, Sredy J., Sehgal S. N. et al.: Rapamycin prevens the onset of insulin-dependent diabetes mellitus (IDDM) In NOD mice. Clin. Exp. Immunol. 1992; 89: 174-178. Brahn E., Peacock D. J., Banquerico M. L.: Supression of collagen-induced arthritis by combination cyclosporin A and methotrexate therapy. Arthritis Rheum. 1981; 34: 1282-1288. Baumbauer R., Schiel R., Keller H. E.: Cyclosporin A in systemic lupus erythematosus. Lupus 1992; 1: 169 (abst). Bensen W., Tugwell P., Roberts R., et al.: Combination therapy of cyclosporin with methotrexate and gold in rheumatoid arthritis (2 pilot study). Arthritis Rheum. 1994; 37 (suppl.): S-335. Binder A. I., Grahem E. M., Sanders M. D., et. al.: Cyclosporin A in the treatment of severe Behcet's uveitis. Br. J. Rheumatol. 1987; 26: 285-291. Blank M., Ben-Bassat M., Shoenfeld Y.: The effect of cyclosporin on early and late stages of experimental lupus. Arthritis Rheum. 1992; 35: 1350-1355. Brier B. E., Mattila P., Standaert R. F. et al.: Two distinct signal transmission pathways in T-lymphocytes are inhibited by complex formed between an immunophilin and either FK-506 or rapamycin. Proc. Natl. Acad. Sci USA., 1990: 87: 9231-9235. Camps J., Sangro B., Garcia N. et al.: Felty's syndrome: response to cyclosporin A with disappearence of neutrophil auto-antibodies (letter). Arthritis Rheum. 1991; 34; 253-255. Cherin P., Crevon M.-C., Hauw J.-J., et. al. Mechanisms of lysis by cytotoxic cells in inflammatory myopathies: a study of perforin and serine esterase gene expression by in situ hybridisation and protein TIA-I expression by immunohistochemistry, from muscular biopsies. Arthritis Rheum. 1993; 36 (suppl.): S84. Chrieber S. L.: Chemistry and biology of the immunophillins and their immunosupressive ligands. Science 1991; 251: 283-287. Claw D. J., Alloway J. A., Read C., et. al.: The use of cyclosporin in the eosinophillia myalgia syndrome (EMS). Arthritis Rheum. 1991; 34 (suppl): R29. Clements P. J., Lachenbruch P. A., Sters M., et. al.: Cyclosporine in systemic sclerosis. Arthritis Rheum. 1993; 36: 75-83. Curkey R. K., MacFarlane A. W., Vickers C. F. H.; Pyoderma gangrenosa treated with cyclosporin A. Br. J. Dermatol. 1985; 113: 601-604. Dammacco F., Riza R., Ferraccioli G. F., et al.: Efficacy and tolerability of cyclosporin A in association with corticosteroids vs corticosteroids alone in systemic lupus erythematosus. Arthritis Rheum. 1994: 37 (suppl.): S-407. Danko K., Szegedi G. : Cyclosporin A treatment of dermatomyositis. Arthritis Rheum. 1991; 34: 933-934. Dougados M., Torley H.: Efficacy of cyclosporin in rheumatoid arthritis; woldwide experience. Brit. J. Rheumatol. 1993; 32 (Suppl. 1): 57-59. Drosos A. A., Scopouli F. N., Costopoulos J. S., et. al.: Cyclosporin A (CyA) in primary Sjogren's syndrome: a double blind study Ann. Rheum. Dis. 1986; 45: 732-735. Elgart G., Stover P., Larson K. et al.: Treatment of pyoderma gangrenosum with cyclosporin: result in seven patients. J. Amer. Acad. Derm. 1991; 24: 83-86. Favre H., Miesher P. A., Huang Y. P., et. al.: Cyclosporin in the treatment in the lupus nephritis. Amer. J. Nephrol 1989; 9: 57-60. Feutren D., Querin S., Noel L. H., et al. Effects of cyclosporin in severe systemic lupus erythematosus. J. Pediatr., 1987; III: 1063-1068. Ferraresso N., Tian L., Ghobrial R. et al.: Rapamicine Inhibits production of cytotoxic but not noncytotoxic 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. antibodies and preferentially activate Th2 cell that inducible long-term survival of heart allograft reaction. J. Immunol. 1994; 153: 3307-3318. Frances C., Brauchet M. C., Bletry 0. et. al.: Skin collagen from scleroderma patients before and after cyclosporin treatment Clin. exp. dermatol. 1988; 13: 1-3. Ficher G. Wittmann L. B., Lang K. et. al. Cyclophilin and peptidyl-propyl cis-trans isomerase are probably identical proteins. Nature, 1989; 337: 467-478. Gisslinger H., Burghuber O. C., Stacher G., et. al.: Efficacy of cyclosporin A in systemic sclerosis. Clin. exp. rheumatol. 1991; 1991; 9: 303-309. Henderson В., Staines N. A., Burrai 1., Сох J. H.: The anti-arthritis and immunosuppres-sive effects of cyclosporin on arthritis induced in the rat by type II collagen. Clin. Exp. Immunol. 1984; 57: 51-56. Henderson D. J., Naya I., Bundick R. V., et al.: Comparison of the effects of FK-506, cyclosporin A and rapamycin on IL-2 production. Immunol. 1991; 73: 316-323. Hughes J. R., Smith E., Higgins E. M. et al.: Pyoderma ganrenosum in a patient with rheumatoid arthritis responding to treatment with cyclosporin A, Brit. J. Rheumatol. 1994; 33: 680-681. Hussen M. M., Mooij J. M. V., Roujouleh H.: Cyclosporine in the treatment of lupus nephritis including two patients treated during pregnancy. Clin. Nephrol, 1993; 40: 160-163. Israel-Biet D. I., Noel L. H., Bach M. A.: Marked reduction of DNA antibodies production and glomerulopathy in thymulin (FTS-Zn) or cyclosporin-A treated (NZBxNZW)FI mice. Clin. exp. immunol. 1983; 54: 359-365. Inamura N., Hashomoto M., Nakahara K., et al.: Immunosupressive effect of FK-506 on collagen-induced arthritis in rats. Clin. Innunol. Immunopathol. 1988; 46: 82-90. Jones M. G., Harris G.: Prolongation of life in female NZB/NZW (Fl) mice by cyclosporin A. Clin. exp. immunol, 1985; 59: 1-9. Kahan В. D., Chang J. Y., Sehgal S. N.: Preclinical evaluation of a new potent immuno-suppressive agent, rapamycin. Transplantation 1991; 52: 185-191. Kahan B. D.: Cyclosporin. New Engl. J. Med. 1989; 321: 1725-1738. Keicho N., Sawada S., Kitamura K. et al.: Effects of immunosuppressant, FK506, on inter-leukin I alpha production by human macrophages and a macrophage-like cell line, U973. Cell. Immunol 1991; 132: 285294. Kuo C. J., Chung J., Fiorentino D. V. et al.: Rapamycine selectively inhibits interleukin 2 activatioin of p70S6 kinase. J. Exp. Med. 1992; 358: 70-79. Kurki P. Т.: Safety aspects of the long term cyclosporin A therapy. Scand. J. Rheumatol. 1992; 21 (Suppl. 95): 35-38. Landewe R. B. M., Miltenburg A. M. M., Breedveld F. C. et al.: Cyclosporine and chloroquine synergistically inhibit the interferon gamma production by CD4 positive and CD8 positive T-cell clones derived from a patient with rheumatoid arthritis. J. Rheumatol., 1992; 19: 1353-1357. Landewe R. B. M., Coel T. H. S., Rijthoven A. W. A. M. et al.: A randomized, double-blind, 24-week controlled study of low-dose cyclosporine versus chloroquine for early rheumatoid arthritis. Arthritis Rheum., 1994; 37: 637-643. Landewe R. B. M., Dijkmans B. A. C., van der Woude F. J., et al.: Long-term low-dose cyclosporin in patients with rheumatoid arthritis. Arthritis Rheum, 1994; 37 (suppl.): S-361. Leuenberger P. M., Miescher P. A.: Sjogren's syndrome: treatment with cyclosporin A (CsA). J. Autoimmun. 1992; 5: A51. Macleod A. M. Thompson A. W. FK-506: an immunosupressant for the 19890s? Lancet 1991; 337: 25-27. Magid M. L., Gold M. H.: Treatment of recalcitrant pyoderma gangrenosa with cyclosporin. J. Amer Acad. Dermatol. 1989; 20: 293-294. Martel R. R., Klicius J., Galet S.: Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can. J. Physiol. Pharmacol. 1977; 55: 48-51. Masuda К., Urayma A., Kogune M.: Double-masked trial of cyclosporin versus colchicin and long-term open study of cyclosporin in Bechcet's disease. . Lancet, 1989; 1: 1093-1095. Morice W. G., Brunn G. J., Wiederrechts G., et al.: Rapamycine-induced inhibition of p34cdc2 kinase activation is associated with g l/S-phase growth arrest in T lym-phocytes. Biol. Chem. 1993; 268: 37. Mountz J. D., Smith H. R., Wilder J., Reeves P., Steinberg A. D.: CS-A therapy in MRL-lpr/lpr mice: amelioration of immunopathology despite autoantibody production. J. Immunol. 1987: 138: 157-163. Oddis C. V., Caroll P., Abu-Elmagd K., et al.: FK 506 in the treatment of polimiositis. Arthritis Rheum., 1994 (Suppl.); 37: R19. Panayi G. S., Tugwell P.: The use of cyclosporin A in rheumatoid arthritis: conclusions of an international review. Brit. J. Rheumatol., 1994; 33: 967-969. Priori R., Paroli M. P., Luan F. L., et al.: Cyolosporin A in the treatment of relapsing poly-chondritis with severe reccurent eye involvment. Brit. J. Rheumatol. 1993; 32: 352. Russel M. L., Schachter R. K.: Cyclosporin treatment of systemic sclerosis. Arthritis Rheum. 1988; 31 (suppl. 4): S51. Saadeh O. K., Bridges W., Burwick F. et. al.: Induction of remission of dermatomiositis (DM) in an adult 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. and a child by the combination of oral cyclosporin (CsA) and high dose intravenous gamma globulin (IVIG) after failure to respond to steroids and other immunosupressive agents. Arthritis Rheum. 1993; 36 (suppl.): S90. Sehgal S. N., Baker H., Vezina C.: Rapamycin (AY-22, 989), a new antifungal antibiotic. J. Antibiot. 1975; 28: 727-232. Sigal N. H., Dumont F. J.: Cyclosporin A, FK-506, and rapamycin: pharmacologic probes of lymphocyte signal transduction. Ann. Rev. Immunol., 1992; 10: 519-560. Sugiama E., Suzuki H., Turu I. S. et al.: FK506, an immunosuppressant, partially inhibits interleukin 6 production by adherent rheumatoid synovial cells. J. Rheumatol. 1994; 21: 1597-1601. Svenson K. L. G., Hoirndahl R., Klareskos L. et. al.: Cyclosporin A threatment in a case of relapsing polychodritis. Scand J. Rheumatol. 1984; 13: 329-333. Thompson A. W.: The effects of cyclosporin A on non-T-cell components of the immune system. J. Autoimmunity. 1992; 5 (Suppl. A): 167-176. Thompson A. W., Duncan J. I.: The influence of cyclosporin A on T cell activation, cytokine gene expression and cell mediated immunity. In Cyclosporin. Mode of action and clinical application. A. W. Thompson, ed. Kluwer Academic Publishers, London, pp. 50-81,1989. Tokuda M., Kurata N., Mizoguchi A., Inoh M. et al.: Effect of low dose cyclosporin A on systemic lupus erythematosus disease activity. Arthritis Rheum., 1994; 37: 551-558. Torley H., Yocum D.: Effects of dose and treatment duration on adverse experiences with cyclosporin in RA: analysis of North American trial. Arthritis Rheum. 1994; 37 (suppl.): S-334. Tugwell P., Pincus Т., Yocum D. et al.: A multi-center, double-blind, randomozed trial of low-dose cyclosporin and placebo therapy in combination with methotrexate in patients with severe rheumatoid arthritis. Arthritis Rheum. 1994; 37 (suppl.): S-361. Walsh B. T., Seaver N., Bennett R., et al.: Immunologic profile of rheumatoid arthritis patients treated with cyclosporin A and methotrexate: results from double-blind placebo-controled study. Arthritis Rheum., 1992: 35: S106. Warner L. M., Adams L. M., Sehgal S. N.: Rapamycin prolongs survival and arrests patho-physlologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 1994; 37: 289-297. Wells G., Tugwell P.: Cyclosporin A in rheumatoid arthritis: overrewiew of efficacy. Brit. J. Rheumatol. 1993; 32 (Suppl. 1): 51-56. Yocum D. E., Wilder R. L., Dougherty et. al.: Immunologic parameters of response in patients with rheumatoid arhritis treated with cyclosporin A. Arthritis rheum. 1990: 33: 1310-1316. Zacharie H., Zacharie E.: Cyclosporin A treatment of systemic sclerosis. Brit. J. Dermatol, 1990; 5: 677681. Zimmerman J., Ochalski S., Qian Xiang et al.: Therapeutic effects of Sirolimus (Rapamycin) on superantigen-facilated collagen-induced arthritis. Arthritis Rheum. 1994: 37 (suppl.): S-398. ГЛАВА 11 ТЕНИДАП Тенидап (Pfizer Central Research, Groton, CT), который в настоящее время зарегистрирован под торговым названием ENABLEX, проходит III фазу клинических испытаний при РА в ряде стран Западной Европы и США. Тенидап представляет собой натриевую соль [5-хло-3.2-дигидро-3 (гидрокси-2-тиенилметилен)-2-оксо1Н-индол-1 карбоксимида], обладает оригинальной химической структурой, отличающейся от таковой других известных противовоспалительных препаратов (I. G. Otterness и соавт., 1989). Механизм действия Вначале препарат разрабатывался как ингибитор 2 ключевых ферментов арахидонового каскада: ЦОГ и 5-липоксигеназы; эти эффекты тенидапа были подтверждены в серии экспериментальных и клинических исследований (T. J. Carty и соавт., 1988; H. Blackburn и соавт., 1990; 1991; E. Moilanen и соавт., 1988; D. M. Smith и соавт., 1990). Однако очень скоро было показано, что тенидап обладает более широким спектром фармакологической активности, не связанным с влиянием на образование простагландинов и лейкотриенов. Важной особенностью тенидапа является его способность подавлять синтез ряда провоспалительных цитокинов, что существенно отличает его от известных НПВС и сближает с базисными противоревматическими средствами и ГК. Одним из наиболее существенных эффектов тенидапа in vitro является ингибиция синтеза ИЛ-1 мононуклеарными клетками человека и экспериментальных животных при стимуляции различными индукторами (B. McDonald и соавт., 1988; I. G. Otterness и соавт., 1991), а также ИЛ-6 и ФНО (J. D. Sipe и соавт., 1992; J. Martel-Belletier и соавт., 1990). Кроме того, тенидап зависимым от дозы образом ингибирует секрецию ИЛ-6 и экспрессию HLA-DR на клетках моноцитарной линии ТНР-1 при стимуляции ФНО-a/ИФ-y (S, Golding и соавт., 1993) и подавляет синтез острофазовых белков клетками гепатомы (Нер3В) при стимуляции последних ИЛ-6/ИЛ-1-b и продукцию ИЛ-6/ИЛ-1-b моноцитами, индуцированную ГМ-МФ КСФ (C. J. Pazoles и соавт., 1993). По данным A. M. M. Miltenburg и соавт. (1994), тенидап ингибирует Т-клеточную пролиферацию, индуцированную анти-СD3, и ИЛ-2 и подавляет уровень иРНК ФНО-а и ИФ-у. С точки зрения механизмов терапевтической эффективности особый интерес представляют данные о способности тенидапа блокировать ИЛ-1-зависимое повреждение суставной и костной ткани, в том числе деградацию хряща (J. T. Dingle и соавт., 1993), резорбцию костной ткани (A. M. Al-Humidan и соавт., 1993), потерю протеогликана культивируемыми клетками хряща (W. D. Blackburn и соавт., 1993), а также стимулировать синтез протеогликана хондроцитами (W. D. Blackburn и соавт., 1993). Тенидап в отличие от НПВС сильно ингибирует высвобождение активной коллагеназы из нейтрофилов за счет взаимодействия с медиаторами активации латентной формы фермента — гипохлорной кислотой и супероксидными радикалами (W. D. Blackburn и соавт., 1991) и в отличие от НПВС ингибирует адгезию лейкоцитов к эндотелиальным клеткам (U. Kyan-Aung и соавт., 1993). В других исследованиях было показано, что на фоне лечения тенидапом отмечается снижение уровня иРНК стромелизина и в меньшей степени коллагеназы и тканевого ингибитора металлопротеиназ (ТИМ) в синовиальной ткани больных PA (J.-P. Pelleitier и соавт., 1993; B. H. Litman и соавт., 1994). По мнению авторов, этот очень важный, с точки зрения патогенеза тканевой деструкции эффект тенидапа обусловлен подавлением синтеза ИЛ-1 и ФНО, регулирующих продукцию металлопротеиназ и ТИМ на претрансляционном уровне. О способности тенидапа подавлять активность металлопротеиназ свидетельствуют данные, полученные на модели экспериментального артрита у собак (J. C. Fernandes и соавт., 1994). Оказалось, что терапевтические концентрации тенидапа эффективно уменьшают образование остеофитов, зону поражения хрящевой ткани и гистологические признаки синовиального воспаления, что ассоциируется со снижением активности коллагеназы и стромелизина. При этом диклофенак не проявлял подобной активности. Эти данные говорят о потенциальной эффективности тенидапа при остеоартрозе у человека. Следует отметить, что все перечисленные выше эффекты тенидапа, продемонстрированные в опытах in vitro, проявлялись в очень низких (терапевтических) концентрациях и не были связаны с общим подавлением синтеза белка и снижением жизнеспособности клеток. Противовоспалительная активность тенидапа была продемонстрирована на нескольких моделях экспериментального артрита. По данным W. Van den Berg и соавт. (1989), тенидап зависимым от дозы образом подавляет клеточную инфильтрацию пораженного сустава и предотвращает деструкцию хондроцитов на модели антигениндуцированного артрита. При адъювантном артрите у крыс тенидап (как и дексаметазон, но не ЦФ) подавляет развитие острого и хронического воспаления, что коррелирует со снижением уровня СРБ (I. G. Ouerness и соавт., 1991). L. D. Loose и соавт. (1993) обобщили результаты 4 независимых исследований, в которых сравнивалось влияние тенидапа (40-120 мг/день), напроксена (1000 мг/день) и плацебо на концентрацию СРБ у больных РА. Длительность испытания варьировала от 2 до 24 недель. Во всех 4 исследованиях отмечено снижение концентрации СРБ у больных, леченных тенидапом, статистически более значимое по сравнению с больными, получавшими напроксен и плацебо. Этот эффект был заметен уже через неделю от начала приема препарата и сохранялся 6 мес. Через 24 нед. снижение уровня СРБ коррелировало с уменьшением концентрации другого ОФБ, сывороточного амилоидного белка А (САБ) и СОЭ. Эти результаты подтверждают точку зрения об особенностях фармакологической активности тенидапа. Ранее было показано, что снижение концентрации ОФБ наблюдается только при использовании ГК или базисных противоревматических препаратов, но не НПВС (C. Menkes, 1993). Установлено, что нормализация концентрации СРБ ассоциируется с уменьшением темпов прогрессирования суставной деструкции, снижением частоты эрозий в суставах и клиническим улучшением (M. A. Van Leeuwen и соавт., 1993). По данным J. D. Sipe и соавт. (1992), тенидап в отличие от НПВС обладает способностью подавлять продукцию ИЛ-6 периферическими моноцитами человека, который, как уже отмечалось, является основным цитокином, индуцирующим синтез ОФБ. Сходные результаты получены B. H. Littman и соавт. (1993), которые в 12-недельном испытании сравнивали действие тенидапа (120 мг/день) и пироксикама (20 мг/день) на уровень СРБ, ИЛ-6 в сыворотках больных РА. Оказалось, что прием тенидапа, но не пироксикама, приводит к снижению уровня СРБ, САБ, ИЛ-6 и нормализации СОЭ. Аналогичная динамика концентрации СРБ, САБ, ИЛ-6 и СОЭ на фоне 24-недельного курса тенидапом выявлена A. R. Kraska и соавт. (1993). Примечательно, что близкий эффект был достигнут в группе сравнения — у больных, леченных пироксикамом в сочетании с плаквенилом, но не у больных, получавших только пироксикам. Молекулярные механизмы действия тенидапа на клетку, приводящие к ингибиции синтеза цитокинов, являются предметом интенсивных исследований. Установлено, что тенидап, являясь слабой органической кислотой, обладает способностью диффундировать через биомембрану и вызывать быстро развивающееся и длительное снижение рН цитоплазмы (pHi) и подавление транспорта анионов (вход CL- и обмен CL-/HC03-) в различных клетках (C. A. Gabel и соавт., 1993; D. McNiff и соавт., 1994). Известно, что закисление цитоплазмы и другие изменения ионного гомеостаза играют важную роль в регуляции синтеза цитокинов (ФНО и ГМ-КСФ и др.). Показано, что подавление анионного транспорта тенидапом приводит к ингибиции АТФиндуцированного высвобождения ИЛ-1 b из макрофагов, стимулированных ЛПС (R. Laliberte и соавт., 1994). Предполагается, что эти эффекты тенидап может проявлять не во всех тканях, а только в условиях определенного микроокружения, особенно при низкой концентрации белка, рН и бикарбоната. Именно эти условия могут создаваться в зоне развития воспаления. Таким образом, полученные данные свидетельствуют O ТОм, что тенидап дает некоторые иммунные эффекты, нехарактерные для большинства НПВС и напоминающие действие базисных противоревматических препаратов. Клинические испытания В настоящее время проведены 11/111 фазы клинических испытаний тенидапа, в которые было включено уже более 3000 больных (F. Breedveld, 1994). В двойном слепом контролируемом 8-недельном исследовании (L. D. Loose и соавт., 1993), в которое вошло 427 больных РА, было показано, что тенидап достоверно превосходит плацебо по стандартным параметрам клинической эффективности противоревматических препаратов. При этом эффект тенидапа зависел от дозы препарата и был достоверно выше при приеме 120 мг/сут, чем 40 и 80 мг/сут. Эта доза, как полагают, является оптимальной, поскольку существенное нарастание клинической эффективности не сопровождалось увеличением частоты и тяжести побочных эффектов. Примечательно, что клинический эффект тенидапа коррелировал с достоверным снижением концентрации СРБ. D. S. Kirby и соавт. (1993) провели сравнительное исследование тенидапа (40-80-120 мг/сут) и напроксена (500-750-1000 мг/сут) у 486 больных РА. Оказалось, что тенидап достоверно превосходит напроксен по влиянию на большинство клинических параметров через 24-52 нед. от начала лечения. Более того, было отмечено, что снижение концентрации СРБ, САБ, ИЛ-2Р и IgM РФ в сыворотке и СОЭ было более существенным в группе больных, леченных тенидапом. В коротком 2-недельном испытании, проведенном L. D. Loose и соавт. (1993), был подтвержден выраженный антипростагландиновый эффект тенидапа при РА. Установлено, что тенидап в большей степени, чем напроксен, уменьшает число лейкоцитов в синовиальной жидкости коленного сустава, а также подавляет синтез ПГЕ2 периферическими лейкоцитами, стимулированными ионофором клетками синовиальной жидкости и снижает эндогенную продукцию этих ПГ в синовиальной жидкости. В отличие от тенидапа напроксен обладал способностью влиять только на синтез ÏÃÅ2 периферическими лейкоцитами. Оказалось также, что перечисленные выше эффекты тенидапа на синтез ПГ коррелируют со снижением концентрации СРБ. Результаты многоцентрового рандомизированного двойного слепого контролируемого исследования, в котором сравнивали клиническую эффективность тенидапа (120 мг/сут) и диклофенака (150 мг/сут) у 393 больных РА, суммированы G. Wylie (1993). Через 24 нед. от начала лечения тенидап достоверно превосходил диклофенак по клинической эффективности (по оценке врача и больного), влиянию на число воспаленных суставов, силу сжатия кисти, продолжительность утренней скованности и некоторым другим параметрам, а также влиянию на СОЭ и концентрацию СРБ и САП. Особый интерес представляют результаты еще 2 двойных слепых контролируемых исследований, посвященных оценке эффективности монотерапии тенидапом и лечения с помощью комбинации диклофенака и ауранофина (M. R. G. Leeming и соавт., 1993) и пироксикама и плаквенила (B. H. Littman и соавт., 1993). В первом исследовании 374 больным с ранним активным РА назначали тенидап (120 мг/сут) или ауранофин (6 мг/сут) в сочетании с диклофенаком (150 мг/сут). Предварительный анализ полученных результатов (через 9 мес. лечения, которое планируется продлить до 2 лет) показал, что тенидап в качестве монотерапии по клинической эффективности не уступает, а по переносимости и влиянию на ОФБ превосходит комбинацию диклофенака и ауранофина. Во втором исследовании, в которое вошло 367 больных с РА, было продемонстрировано, что тенидап (120 мг/сут) превосходит пироксикам (20 мг/сут) и не уступает комбинации пироксикама и плаквенила (400 мг/сут) по влиянию на клиническую активность болезни. Важно также подчеркнуть, что положительная динамика острофазовых показателей (СРБ, САБ), уровня ИЛ-6 и СОЭ наблюдалась только на фоне лечения тенидапом и комбинированной терапии, но не при использовании пироксикама. Получены первые данные о применении тенидапа у больных ревматической полимиалгией (B. H. Littman и соавт., 1994). Проведено 15-недельное многоцентровое двойное слепое контролируемое исследование, результаты которого свидетельствуют о том, что добавление тенидапа к стандартному протоколу лечения больных ревматической полимиалгией (10 мг/день преднизолона) позволяет более эффективно контролировать клинические проявления заболевания. Важно, что в группе пациентов, леченных тенидапом, 45% больных смогли полностью прекратить прием ГК, в то время как в контрольной группе — только 6%. Таким образом, тенидап оказывает очевидное стероидсберегающее действие. Побочные эффекты К положительным свойствам тенидапа относится не только его высокая эффективность, но и низкая токсичность. Данные мета-анализа результатов всех проведенных клинических испытаний свидетельствуют о том, что частота побочных эффектов на фоне лечения тенидапом не выше, чем при использовании НПВС, и существенно ниже, чем при комбинированной терапии НПВС и ауранофином и плаквенилом. Единственным частым побочным эффектом препарата является умеренная, не прогрессирующая и полностью обратимая канальцевая протеинурия. По данным G. Wylie и соавт. (1993), протеинурия в пределах 250мг/сут развилась у 50% больных в сроки между 12-й и 24-й неделями лечения, и лишь у 5% больных она превышала 750 мг/сут. Другие, более редкие побочные эффекты тенидапа суммированы в таблице 11.1. Таблица 11.1. Частота побочных эффектов тенидапа Побочные эффекты частота > 1% частота > 0.1% Желудочно-кишечные: Желудочно-кишечные: тошнота, рвота, запоры, диарея, диспепсия, боли в животе, язвы ЖКТ Общие: недомогание, астения, лихорадка Неврологические: шум в ушах, сонливость Поражение мочеполового тракта: изменение окраски мочи, спермы Кожные: алопеция, сыпь, зуд Сердечно-сосудистые: периферические отеки стоматит, сухость в рту, энтерит, гингивит, глоссит, увеличение аппетита Сердечно-сосудистые: боли в грудной клетке, гипертензия, сердцебиение, коллапс Общие: озноб, фотосенсибилизация Поражение мочеполового тракта: дизурия, гематурия, никтурия, полиурия Изменение репродуктивной системы: меноррагии, другие нарушения менструального цикла, импотенция Мышечно-скелетные: боли в пояснице, артралгии, миалгии, судороги Неврологические: амнезия, депрессия и др. Легочные: одышка, кашель и др. Кожные: сухость кожи, крапивница, изменения ногтей Список литературы К главе "Тенидап" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. Al-Humidan А. М., Leeming M. R. G., Russel R. G. R.: Tenidap inhibits bone resorbtion induced by PTH, 1, 25 Vit D3, IL-I, TNF and PGE2 In vitro by mechanism independent of inhibition of prostaglandin synthesis. Arthritis Rheum., 1991; 34: S192. Blackburn W. D., Heck L., Loose L. D.: Inhibition of PMN degranulation and synovial fluid lipoxygenase (LO) and cyclooxygenase (CO) avtivity by by tenidap [5-chloro-2, 3-dihydго-2-охо-3 (2thienólcarbonól)indole-l-carboxamide]. Arhritis rheum, 1990; 33 (Suppl 5): R13. Blackburn W., Loose L. D., Heck L. W., Chathman W. W.: Tenidap, in contrast to several available nonsteroldal antiinflammatory drugs, potentially inhibits the release of activated neutrophil collagenase. Arthritis Rheum., 1991: 34: 211-216. Blackburn W. D., Loose L. D., Eskra J. D., Carty T. J.: Inhibition of 5-lipoxygenase product formation and polymorphonuclear cell degradation by tenidap sodium in patients with rheumatoid artritis. Arthritis Rheum., 1991; 34: 204-216. Blackburn W. D., Loose L. D., Chathan W. W.: Tenidap enhances basal chondrosyte proteo-glycan synthesis and reverses IL-I induced effect. Clin. Rheumatol., 1994; 13: 352. Breedveld F.: Tenidap: a novel cytokine-modulating antirheumatic drug for the treatment of rheumatoid arthritis. Scand. J. Rheumatol. 1994; 3 (suppl. 100): 31-44. Dingle J. T., Leeming M. R. G., Martindale J. J.: Effect of tenidap on cartilage integrity in vitro. Ann. Rheum. Dis., 1993; 52: 292-299. Carty T. J., Showell H. J., Loose L. D., Kadin S. B.: Inhibition of both 5-lipoxygenase (5-LO) and cyclooxygenase (CO) pathways of arachidonic acid metabolism by CP-66, 248, a novel anti-inflammatory compaund. Arthritis Rheum. 1988; 31 (suppl): 89. Gabel C. A., McNoff P., Perregaux D., et. al.: Tenidap inhibits anion transport in vitro. Arthritis Rheum., 1993; 36 (Suppl. 9): S109. Golding S., Young S. P., Emery P.: Modulation of cytokine-mediated interleukln-6 (IL-6) synthesis by tenidap. Brit. J. Rheumatol., 1993; 32: 166. Fernandes J. C., Martel-Pelletier J., Otterness I. G., et al.: Effects of tenidap on canine experimental osteoarthritis (OA): morphological analysis and metalloprotease activity. Arthritis Rheum. 1994; 37 (suppl.): S-362. Kirby D. S., Loose L. D., Weiner E. S. et. al.: Tenidap versus naproxen in the treatment of rheumatoid arthritis patients. Arhritis Rheum., 1993: 36 (Suppl. 9): SI 12. Kraska A. R., Wilhelm F. E., Kirby D. S. et. al.: Tenidap versus piroxicam plus hydroxy-chloroquine in rheumatoid arthritis: a 24-week analysis. Arthritis Rheum., 1993'36 (Suppl. 9): S57. Kyan-Aung U., Lee Т. Н., Haskard D. O.: The inhibitory effect of tenidap on leukocyte-endothelial cell adhesion. J. Rheumatol., 1993; 20: 10141019. Laliberte R., Perregaux D., Svensson L. et. al.: Tenidap modulates cytoplasmatic pH and inhibits anion transport in vitro. II. Inhibition of IL-I beta production from ATP-treated monocytes and macrophages. J. Immunol., 1994; 153: 2168-2179. Leeming M. R. G. and The Tenidap Early RA Study Group.: A double-blind randomised comparison of tenidap versus auranofin plus diclofenac in early rheumatoid arthritis. Arthritis Rheum., 1993; 36 (Suppl. 1): SIII. 17. Littman B. H., Drurry C. E., Stewart C. R.: Acute phase protein and plasma IL-6 in patients with rheumatoid arthritis: effect of tenidap and piroxicam in a 12-week double-blind crossover study. Arthritis Rheum., 1993; 36 (Suppl. 9): SI 12. 18. Litman B. H., Drury C. E.: In vitro reduction of RA synovial tissue metalloproteinase mRNA levels by tenidap. Arthritis Rheum. 1994; 37 (suppl.): S-420. 19. Littman B. H., Drury C. E., Bjarnason D., et al.: Steroid sparing activity of tenidap in polymialgia rheumatica. Arthritis Rheum. 1994; 37 (suppl.): S-354. 20. Loose L. D., Sipe J. D., Kirby D. S. et. al.: Reduction of acute-phase proteins with tenidap sodium, a cytokine-modulating anti-rheumatic drug. Brit. J. Rheumatol., 1993 (suppl. 3); 3: 19-25. 21. Loose L. D., Wilhelm F. E., Kraska A. R. et. al.: Double-blind, placebo-controlled dose-response study of tenidap in rheumatoid arthritis. Arthritis Rheum., 1993; 36 (Suppl. 9): S166. 22. Loose L. D., Weiner E. S.. Kibry D. S. et. al.: Tenidap versus naproxen versus placebo in rheumatoid arthritis: a 2-week study in patients with knee effusion. Arthritis Rheum., 1993; 36 (Suppl. 9). 23. Martel-Pelletier J., Ounissi H., Coutier J. M., Pelletier J.-P.: Tenidap effectively reduced cytokine synthesis and expression by human rheumatoid arthritis synovium. Clin. Rheumatol., 1994; 13: 352. 24. McDonald В., Loose L., Rosenwasser L. J.: The influence of a novel arachidonate inhibitor, CP-66, 248, on the production and activity of human monocyte IL-I. Arthritis Rheum., 1988; 33 (Suppl. 4): S17. 25. McNiff P., Svensson L., Pazoles C. J., Gabel C. A.: Tenidap modulates cytoplasmatic pH and inhibits anion transport in vitro. 1. Mechanism and evidence of functional significance J. Immunol. 1994; 153: 2180-2193. 26. Menkes C.-J.: Effects of disease-modifying anti-rheumatic drugs, steroids and non-steroidal antiinflammatory drugs on acute-phase proteins in rheumatoid arthritis. Brit. J. Rheumatol., 1993. (Suppl. 3); 32: 14-18. 27. Miltenburg A. M. M., Dplhain R. J. E. M., de Kuiper R. et al.: Теnidap inhibits T-cell pro-lipheration, cytokine production ant the induction of mRNA encoding TNF alpha and IFN gamma. Arthritis Rheum. 1994; 37 (suppl.): S-384. 28. Moilanen E., Alanko J., Asmawi M. Z., Vapaatalo H.: CP-66, 248, a new anti-inflammatory agent, is a potent inhibitor of leukotrien B4 and prostanoid synthesis in human polymorphonuclear leucocytes in vitro. Ecosanoids, 1988; 1: 35-39. 29. Otterness I. G., Carty T. J., Loose L. D.: Tenidap: a new drug for arthritis. In: Therapeutic approaches to inflammatory disease. Lewis A. J., Doherty N. S., Ackerman N. R. (eds), 1989; 229-241. 30. Otterness I. G., Bliven M., Downs J. T., et. al.: Inhibition of interleukin I synthesis by tenidap: a new drug for arthritis. Cytokine, 1991; 3: 277-283. 31. Otterness I. G., Pozoles P. P., Moore P. P., Pepys M. B.: C-reactive protein as an index of disease activity: comparison of tenidap, cyclophosphamide and dexamethasone in rat adjuvant arthritis. J. Rheumatol. 1991; 18: 505-511. 32. Pazoles C. J., McNiff P., Laliberte R., Gabel C. A.: Tenidap acts in vitro as an antagonist of cytokineinduced function. Arthritis Rheum., 1993; 36 (Suppl. 9); S109. 33. Pelletier J.-P., McCollum R., Di Battista J., et al.: Regulation of human normal and osteoarthritic chondrocyte interleukin-1 receptor by antirheumatic drugs. Arthritis Rheum. 1993; 36: 1-11. 34. Sipe J., Barte L., Loose L.: Modification of proinflammatory cytokine production by the antirheumatic agents tenidap and naproxen. J. Immunol., 1992; 148: 480484. 35. Smith D. M., Johnson J. A., Loeser R., Turner R. A.: Evaluation of tenidap (CP-66, 248) on human neutrophilic arachidonic acid metabolism, chemotactic potential and clinical efficacy in the treatment of rheumatoid arthritis. Agents Actions, 1990; 31: 102-109. 36. Van den Berg W., Joosten L. A. B., Uelsen M.: Suppressive effect of CP-66, 248 on murine antigeninduced arthritis. Arthritis Rheum. 1989; 32: 552. 37. Van Leeuwen M. A., Van-Rijswijk M. H., Van der Heijde D. M. F. M. et. al.: The acute-phase response in relation to radiographic progression in early rheumatoid arthritis: a prospective study during the first three years of the disease. Brit. J. Rheumatol., 1993; 32 (Suppl. 3): 9-13. 38. Wylie G. A, Tenidap European Study Group.: 24-week study of tenidap versus diclofenac in rheumatoid arthritis. Rev. Esp. Reumatol. 1993; 20 (suppl 1): 142. ГЛАВА 12 ВНУТРИВЕННЫЙ ИММУНОГЛОБУЛИН Одним из наиболее эффективных и широко апробированных методов иммунотерапии аутоиммунных заболеваний, несомненно, является лечение высокими дозами вводимого внутривенно иммуноглобулина (IVIg), который используется в клинической практике в течение уже более 10 лет (E. M. Gelfand, 1989; J. M. Dwyer, 1993; N. Ronda и соавт., 1993; S. A. Berkman и соавт., 1990). В начале 80-х годов P. Imbach и соавт. (1981) и J. Fehr и соавт. (1982) впервые использовали введение высоких доз IVIg для лечения детей с идиопатической тромбоцитопенической пурпурой. IVIg является препаратом нормального полиспецифического Ig, полученного из пула сывороток нескольких тысяч доноров. Коммерческие препараты представляют собой интактный IgG с таким же распределением по субклассам, как и в нормальной сыворотке, и периодом полужизни около 3 нед. В большинстве препаратов присутствуют только следовые количества IgM, IgA, Fc-çàâèñèìûõ агрегатов IgG и около 30% F (ab')2-F (ab')2 димеров. Поскольку IVIg получен от большого количества доноров, в препаратах содержится весь нормальный репертуар IgG, включающий антитела к экзогенным антигенам, естественные антитела и антиидиотипические антитела. Спектр заболеваний, при которых с успехом используется IVIg, чрезвычайно широк (Kaveri S.-V. и соавт., 1991): Ревматические заболевания: • СКВ • ПМ/ДМ • Ювенильный РА • РА • Синдром Фелти • АНЦА-позитивный системный васкулит • Болезнь Кавасаки • АФС Другие: • Идиопатическая тромбоцитопеническая пурпура • Аутоиммунная гемолитическая анемия • Аутоиммунная нейтропения • Аутоиммунная эритробластопения • Миастения гравис • Синдром Гийена—Барре • Хроническая воспалительная демиелинизирующая полиневропатия • Рассеянный склероз • Диффузный токсический зоб • Язвенный колит • Болезнь Крона Считается, что при рефракторных аутоимунных цитопениях, болезни Кавасаки и синдроме Гийена-Барре лечение IVIg является средством выбора. Механизмы действия (таблица 12.1) Таблица 12.1. Механизмы действия IVIg 1. обратимая блокада IgG Fc-ðåöåïòîðîâ клеток ретикулоэндотелиальной системы, предотвращающая удаление из кровяного русла циркулирующих клеток, сенсибилизированных IgG аутоантителами (R. J. Kurlander и совет., 1984); блокада Fc-peцепторов лейкоцитов, лимфоцитов (N. Ronda и соавт., 1993); 2. Fc-çàâèñèìàÿ обратная ингибиция синтеза аутоантител В-лимфоцитами (C. L. Anderson, 1989); 3. модуляция супрессорной и хелперной активности Т-лимфоцитов (E. L. Atsura и соавт., 1988); 4. подавление комплементзависимого повреждения тканей (M. Basta и соавт., 1989; 1991); 5. модификация распознавания антигена клетками-мишенями, связанная с присутствием в препарате растворимых CD4, CD8 и HLA молекул (R. Blasczyk и соавт., 1993); 6. подавление синтеза ФНО-а (А. Асhiron и соавт., 1994); увеличение синтеза антагонистов ИЛ-1 рецепторов (W. P. Arend и соавт., 1991); 7. V-çàâèñèìàÿ модуляция аутоиммунного репертуара (S.-V. Kaveri и соавт., 1991), поскольку в препарате содержатся антиидиотипические антитела, взаимодействующие с F (аb')2-фрагментами аутоантител к тиреоглобулину, нейробластоме, нейтрофильным цитоплазматическим антигенам внутреннему фактору, гликопротеину IIb/IIIa тромбоцитов, ДНК, фосфолипидам; предполагается, что быстро развивающиеся клинические эффекты IVIg связаны с пассивной нейтрализацией сывороточных аутоантител антиидиотипическими антителами, содержащимися в препарате IVIg, а долгосрочная эффективность лечения определяется V-çàâèñèìîé селекцией иммунного репертуара, опосредуемое активацией или ингибицией IVIg-ðåàêòèâíûõ клонов Т- и В-лимфоцитов (V. R. de Souza, 1993). 8. присутствие в препарате антител к суперантигенам некоторых микроорганизмов, способных индуцировать развитие аутоиммунных и токсических реакций (S. Takei и соавт., (1993); 9. изменение структуры и растворимости циркулирующих иммунных комплексов Ching-Yuang и соавт., 1989; R. Y. Lin и соавт., 1986); 10. подавление функциональной зависимости цитокинов, связанное с присутствием в препарате IVIg естественных антицитокиновых антител (J. A. Schiferli и соавт., 1991). Клиническая эффективность и схема лечения Схема применения препарата в настоящее время не стандартизована: обычно применяемая доза IVIg варьирует от 0.4 до 2 г/кг/день в течение 4-5 дней. Скорость введения раствора составляет вначале 0.5-1.0 мл/мин (10-20 капель), при отсутствии побочных эффектов в течение первых 15 мин. скорость увеличивают до 1.0-1.5 мл/мин, а затем до 2.0-2.5 мл/мин. При необходимости инфузии повторяют каждые 4 нед. РА Первое сообщение о благоприятном эффекте плацентарного гаммаглобулина при РА появилось еще в 1982 г. (J. Sany и соавт.), что стимулировало проведение небольших клинических испытаний IVIg при этом заболевании (J. Mitropoulou и соавт., 1982; H. Becker и соавт., 1987). B. Tumiata и соавт. (1992) использовали IVIg для лечения 10 больных РА, не отвечающих на противовоспалительную терапию. В начале лечения применяли стандартную схему, а поддерживающая терапия заключалась в назначении IVIg по 0.4 г/кг/день 1 раз в месяц в течение 6 мес. Улучшение отмечено у 9 больных, у большинства — после 2-й инфузии. Однако после завершения лечения в течение нескольких недель развивалось обострение. Констатирована хорошая переносимость лечения, побочные эффекты были минимальными и проявлялись тошнотой, рвотой, кожной сыпью, снижением АД во время инфузии. P. Spath и соавт. (1992) провели двойное слепое контролируемое исследование IVIg/àëüáóìèí у 32 больных с ранним активным РА (длительность 6-36 мес.). Препарат или плацебо вводили по 0.5 г/кг/день 2 дня подряд с интервалом в 4 нед. до 6 мес. Отмечено, что непосредственно после введения препарата наблюдалось умеренное улучшение, но при длительном лечении достоверных различий с плацебо не было. Эти результаты существенно отличаются от тех, которые получены при позднем РА и в открытых испытаниях. ЮХА По данным E. D. Silverman и соавт. (1990), IVIg эффективно контролировал артрит и внесуставные проявления у 10 из II больных с системным вариантом ЮХА, позволил снизить дозу ГК. Кроме того, на фоне лечения отмечена положительная динамика лабораторных показателей (СОЭ, количество тромбоцитов, концентрация IgG и др.). При иммунологическом исследовании установлено, что лечение способствует нормализации синтеза ИЛ-2 и пролиферативного ответа лимфоцитов на митоген лаконоса (K. S. Barron и соавт., 1992). Многоцентровое исследование в рамках 1/11 фаз клинических испытаний IVIg у 25 больных с полиартикулярным вариантом ЮХА было проведено E. Gianini и соавт. (1994). Больные получали препарат в дозе 1.5-2.0 г/кг (максимально 100 г) 2 раза в месяц в течение 2 мес., а затем ежемесячно в течение 6 мес. Положительный клинический эффект отмечен у 75% больных, а переносимость лечения была очень хорошей. A. M. Prieur и соавт. (1990) использовали IVIg (по 300-400 мг/кг/день 1 раз в неделю) для лечения 16 больных, у 13 из которых был системный, а у 3— полиартикулярный вариант ЮХА. По данным этих авторов, лечение в некоторых случаях приводило к очень быстрой положительной динамике клинических и особенно лабораторных проявлений болезни, позволило снизить дозу ГК. Однако у некоторых больных с непрерывнорецидивирующим течением клинический эффект был менее очевиден. СКВ В 1984 г. G. Gaedicke и соавт. впервые сообщили о купировании лихорадки, кожного васкулита и язв слизистой оболочки полости рта на фоне лечения IVIg у 2 детей с СКВ. Проведено открытое испытание эффективности IVIg (вначале 2г/кг в течение 4-5 дней, затем по 0.4 г/кг 1 раз в месяц в течение 5 мес.) у 7 больных СКВ, рефракторных к глюкокортикоидной и цитотоксической терапии (K. Akashi и соавт., 1990). У 3 больных улучшилась функция почек, уменьшилась тромбоцитопения, отмечалась положительная динамика некоторых других клинических симптомов. У 1 пациента динамики не выявлено, а у 3 больных через 3 мес. лечения возникли серьезные осложнения: у 1 пациента с волчаночным нефритом развилась острая почечная недостаточность, у 1 — отмечены нарастание протеинурии и отрицательная динамика иммунологических показателей активности болезни, у 1 больного с кортикостероидрезистентной гемолитической анемией и гипокомплементемией развился диффузный пролиферативный гломерулонефрит. C. Francioni и соавт. (1994) провели открытое испытание IVIg у II больных СКВ, рефрактeрных к лечению ГК. Препарат вводили 1 раз в месяц по 0.4 г/кг в течение 5 дней, общая продолжительность лечения составила 6-24 мес. Положительная динамика, проявляющаяся в увеличении уровня гемоглобина, комплемента, числа тромбоцитов (в 2 случаях) и снижении СОЭ, ЦИК и АНФ, отмечен у всех II больных. У пациентов с поражением почек уменьшилась протеинурия и увеличился клиренс креатинина. Побочные эффекты отсутствовали. Таким образом, по данным авторов, лечение IVIg позволяет контролировать активность болезни и снизить дозу ГК. Сходные результаты получены N. I. Abdou и соавт. (1994), которые использовали IVIg в стандартной дозе для лечения 12 больных активной СКВ. На фоне лечения отмечена достоверная положительная динамика активности процесса: SLEDAI индекс активности снизился в среднем с 30 до 7 баллов, у половины больных удалось уменьшить дозу ГК на 50%. Кроме того, констатирована отчетливая положительная динамика уровня антител к ДНК и концентрации С3а анафилотоксина. Имеются многочисленные наблюдения, свидетельствующие об эффективности IVIg в отношении купирования отдельных проявлений заболевания, включая тромбоцитопению (W. P. Maier и соавт., 1990; E. J. T. Terborg и соавт., 1992), АФС (L. O. Carreras и соавт., 1989; R. Kaaja и соавт., 1993; D. Foley-Nolan и соавт., 1994; G. S. Sturfelt и соавт., 1990), цереброваскулит, проявляющийся психозом (A. Corveta и соавт., 1989; Y. Tomer и соавт., 1992; F. C. Hall и соавт., 1992), васкулитную нейропатию (T. P. Enevoldson и соавт., 1991; F. C. Hall и соавт., 1992), рефракторное поражение кожи (D. Foley-Nolan, Н. С. Goot Н. С., 1994). ПМ/ДМ Эффективность IVIg при ПМ/ДМ продемонстрирована в нескольких неконтролируемых (C. M. Roifman и соавт., 1987; P. Cherin и соавт., 1991; B. Lang и соавт., 1991) и одном контролируемом (M. C. Dalakas и соавт., 1993) исследовании. По данным M. Dalakas и соавт. (1993), у большинства ( у 9 из 12) больных ДМ на фоне введения IVIg наблюдалось существенное клиническое улучшение, которого не зарегистрировано в контрольной группе. При этом у 5 больных, которым проводились серийные мышечные биопсии, отмечалась положительная динамика иммуногистохимических нарушений, проявляющаяся в снижении экспрессии молекул класса I и II ГКГ и межклеточных молекул адгезии (IСАМ-1) на мембране миоцитов. Кроме того, выявлена регрессия воспалительных изменений и микроциркуляторных нарушений, а также исчезновение компонентов мембраноатакующего комплекса (С5-9) в пораженных сосудах (M. Basta и M. C. Dalakas, 1994). Имеется несколько сообщений об эффективности IVIg при ювенильном ДМ C. M. Roifman и соавт., 1987; K. S. Barron и соавт., 1992). В одном исследовании IVIg в дозе 1 г/кг/день в течение 2 дней каждый месяц в течение 6 мес. назначали 5 больным, у которых сохранялась мышечная слабость, несмотря на длительную предшествующую терапию ГК и цитостатиками. На фоне лечения отмечена положительная динамика мышечной силы и снижение уровня креатинфосфокиназы, у всех больных удалось существенно снизить дозу ГК. В другом исследовании IVIg (0.4 г/кг/день в начале в течение 4 дней подряд, а затем ежемесячно) использовали для лечения 6 больных ювенильным ДМ, резистентным к высоким дозам ГК (от 0.5 до 2мг/кг/день). У 5 пациентов удалось существенно снизить дозу ГК — в среднем с 23.5 до 9.5 мг/кг/день, у всех 6 больных отмечено снижение концентрации мышечных ферментов. По мнению P. Cherin, IVIg особенно эффективен у больных миозитом, достоверно связанным с вирусной инфекцией или индуцированным лекарственными препаратами. Кроме того, его следует использовать у лиц, которым противопоказано лечение ГК в высоких дозах (например, у больных пожилого возраста). Лечение IVIg не эффективно при ДМ, связанном с злокачественными новообразованиями. Cистемные васкулиты Недавно было предпринято открытое исследование эффективности IVIg у 14 больных гранулематозом Вегенера и микроскопическим полиартериитом, 9 из которых были рефрактерны к предшествующей терапии, а 5—не получали лечения, В течение 10-месячного наблюдения после завершения лечения обострение развилось у 2 больных, остальные для поддержания ремиссии нуждались в значительно меньших дозах ГК и цитотоксиков. Интересно, что уменьшение титров АНЦА после лечения коррелировало со способностью препарата IVIG блокировать активность антител in vitro (D. R. W. Jayne и соавт., 1992). В другом исследовании, проведенном той же группой авторов (D. R. W. Jayne и соавт., 1991), IVIg использовали для лечения 7 больных системными васкулитами (без тяжелого гломерулонефрита), в сыворотках которых обнаружены АНЦА, в том числе 4 — гранулематозом Вегенера, 1 — ревматоидным васкулитом и 1 - микроскопическим полиартериитом. Препарат назначали в дозе 0.4 г/кг/день в течение 5 дней, У всех больных отмечено улучшение, причем в 6 случаях достаточно устойчивое. Через 2-3 нед. терапии наблюдалось уменьшение титров АНЦА, концентрации ОФБ. В течение 6-18 мес. наблюдения у 3 больных развилось обострение. P. Tuso и соавт. (1992) сообщили о выраженном улучшении у 2 больных АНЦА-позитивным васкулитом и тяжелым гломерулонефритом, не отвечающих на ЦФ. Положительная клиническая динамика коррелировала со снижением титров АНЦА. Улучшение на фоне лечения IVIg описано также у 3 больных кожным васкулитом и у 1 — системным васкулитом (A. Antonelli и соавт., 1992), C. Richter и соавт. (1994) применили IVIg (30 мг/день в течение 5 дней) для лечения 15 больных с различными формами АНЦА-ассоциированных васкулитов, плохо отвечающих на стандартную терапию. У 6 человек отмечено улучшение, касающееся в первую очередь кожного синдрома, но не поражения глаз, почек и перикардита. Повторные курсы лечения также были неэффективны. Интересно, что в F (ab')2-ôðàãìåíò IVIg in vitro эффективно блокировал активность АНЦА (антител к протеиназе 3), однако выраженность ингибиции не коррелировала с клинической эффективностью лечения. Особенно широко используется IVIg при болезни Кавасаки — остром лихорадочном заболевании, встречающемся преимущественно у детей и характеризующемся диффузным воспалением слизистых, индуративным отеком кистей и стоп, полиморфной сыпью и негнойной шейной лимфаденопатией. Наиболее серьезным осложнением болезни Кавасаки является поражение сердечно-сосудистой системы с развитием аневризм коронарных и других крупных артерий, коронарного тромбоза, миокардита, перикардита, нарушений ритма сердца, поражения митрального клапана. В многочисленных исследованиях было показано, что лечение IVIg приводит к достоверному снижению частоты развития аневризм коронарных артерий и быстрому исчезновению других признаков системного воспаления. При этом однократное введение 2 г/кг IVIg не уступает по эффективности стандартному курсу лечения, заключающемуся во введении препарата в дозе 400 мг/кг в течение 4 дней (K. Furusho и соавт., 1983; K. S. Barron и соавт., 1990; J. W. Newburger и соавт. 1991). Другие заболевания Недавно появилось сообщение об успешном применении IVIg в лечении 3 больных с редким аутоиммунным неврологическим заболеванием — синдромом "скованного человека" (stiff-man). Этот синдром характеризуется выраженной скованностью мышц туловища и конечностей, болезненными спастическими сокращениями и связан с гиперпродукцией антител к нейтротрансмиттеру у-аминобутириловой кислоты (E. W. Karlson и соавт., 1994). Несколько контролируемых исследований посвящено оценке эффективности IVIg при так называемом синдроме хронической усталости (chronic fatigue syndrome) — заболевании предположительно вирусной природы, проявляющемся необъяснимой инвалидизирующей усталостью, лимфаденопатией, лихорадкой, другими неспецифическими симптомами, развивающимися на фоне разнообразных иммунных нарушений. По данным A. Lloyd и соавт. (1990), введение IVIg в дозе 2 г/кг/день 3 раза в течение месяца привело к достоверному улучшению клинических и иммунологических показателей. Однако результаты, полученные P. K. Paterson и соавт. (1990), свидетельствуют о неэффективности IVIg (1 г/кг/день через каждые 30 дней в течение 6 мес.) при этом синдроме. Побочные эффекты Терапия IVIg является относительно безопасным методом терапии, однако у отдельных больных развиваются побочные реакции, ограничивающие возможность дальнейшего лечения. Наиболее часто наблюдаются побочные эффекты, зависящие от скорости инфузии препарата. К ним относятся головные боли, лихорадка, ознобы, затруднение дыхания, боли в животе и спине, умеренная гипотензия. Каждый больной имеет собственный уровень толерантности к скорости инфузии препарата, который следует тщательно оценивать при проведении терапии. Для уменьшения выраженности этих реакций показано профилактическое назначение НПВС, антигистаминных препаратов или небольших доз ГК. Истинные анафилактические реакции при введении IVIg встречаются довольно редко. Они могут быть связаны с выработкой IgE-àíтител к IgA и особенно характерны для больных с селективным IgAиммунодефицитом или общим вариабельным иммунодефицитом (A. W. Burks и соавт., 1986). У больных с гипогаммаглобулинемией описано развитие иммунокомплексного кожного васкулита, однако при аутоиммунных заболеваниях этого осложнения не наблюдалось. Описано развитие симптомов, напоминающих асептический менингит (M. Casteels-Van Daele и соавт., 1990; S. P. Rao и соавт., 1992) и гемолитической анемии, связанной с присутствием агглютининов в препарате IVIg (A. G. Brox и соавт., 1987). Особую потенциальную опасность представляет трансмиссия инфекционных агентов. Это наблюдается крайне редко, поскольку обработка плазмы и добавление различных предохраняющих веществ и полиэтиленгликоля в препарат иммуноглобулина подавляет инфекционную активность. Однако недавно появились сообщения о заражении вирусом гепатита С больных, которым проводили инфузии IVIg (R. Laser и соавт., 1994). Важным фактором, лимитирующим возможность широкого использования IVIg в клинической практике, является высокая стоимость лечения. Цена препаратов IVIg, выпускаемых западными компаниями, колеблется от 25 до 100 долларов за грамм, в то время как терапевтическая доза препарата, используемая при аутоиммунных заболеваниях, составляет 2 г/кг/мес. Таким образом, стоимость месячного курса лечения одного больного достигает 5000 долларов. Список литературы к главе "Внутривенный иммуноглобулин" 1. 2. 3. 4. 5. 6. 7. 8. 9. Abdou N. J., Yesenosky P., Chou A. et al.: Intravenous human immunoglobulin (IVIG) therapy in active SLE: a six month in vivo and in vitro study. Arthritis Rheum. 1994; 37: (suppl.): S-406. Achiron A., Margalit R., Hershkoviz R. et al.: Intravenous immunoglobulin treatment of experimental T cell-mediated autoimmune disease. Upregulation of T cell proliphera-tion and downregulation of tumor necrosis factor alpha secretion. J. Clin. Invest. 1994; 93: 600-605. Akashi К., Nagasawa К., Mayuni Т., et al.: Succesful treatment of refractery systemic lupus erythematosus with intravenous immunoglobulun. J. Rheumatol. 1990: 17: 375-379. Anderson C. L: Human IgG Fc receptors. Clin. Immunol. Immunopathol. 1989; 53: 563-0571. Anderson J. P., Anderson U. G.: Human Intravenous immunoglobulin modulates monokine production in vitro. Immunology 1990; 71: 372-376. Antonelli A., Agostini G., Agostini S.: Preliminary results of intravenous immunoglobu-lins in treating patients with vasculitis. Clin. Ter. 1992; 141: 33-36. Barron K. S., Murphy D. J., Silverman E. D. et al.: Treatment of Kawasaki syndrome: a comparison of two dosage regimens of intravenous immune globulin. J. Pediatr. 1990; 117: 638-644. Barron K. S., Sher M. R., Silverman E. D.: Intravenous immunoglobulin therapy: magic or black magic. J. Rheumatol. 1992; 19 (suppl. 33): 94-97. Basta M., Dalakas M. C.: High-dose intravenous immunoglobulin exert its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J. Clin. Invest. 1994; 94: 1729-1735. 10. Basta M., Langlois P. F., Marques M. et al.: High-dose intravenous immunoglobulin modifies complementmediated in vivo clearence. Blood 1989; 74: 326-333. 11. Basta M., Fries L., Frank M. M.: High-dose intravenous immunoglobulin (IVIG) inhibit im vitro u[take of CD4 fragments onto sensitized erythrocytes. Blood 1991; 77: 376-380. 12. Brasczyk R., Westhoff U., Grosse-Wilde H.: Soluble CD4, CD8, and HLA molecules in commercial immunoglobulin preparation. Lancet 1993; 341: 789-790. 13. Brox A. G., Cournoyer D., Sternbock M., Spull E.: Hemolitic anaemia following intravenous gammaglobulin administration. Amer. J. Med. 1987; 82: 633-635. 14. Becker H., Mitropoulou G., Hemke K. Immunomodulating therapy of rheumatoid arthritis by high dose intravenous immunoglobulin. Klin. Wochenshr. 1989: 67: 286-289. 15. Berkman S. A., Lee H. L, Gale P. P.: Clinical uses of IVIg. Ann. Int. Med. 1990, 112; 287-292. 16. Burks A. W., Sampson H. A., Buckley R. H.: Anaphylactic reactions after gamma globulin administration In patients with hypogammaglobulinemia. Detection of IgE antibodies to IgA. New Engl. J. Med. 1986; 314: 560-564. 17. Carreras L. O., Perez G. N., Vegs H. R., Casavilla F.: Lupus anticoagulant and reccurent fetal loss: succesful treatment with gammaglobulin. Lancet 1988; ii: 393-394. 18. Casteels-Van Daele M., Wijndaele L., Hunnick K.: Intravenous immune globulin and acute aseptic meningitis. New Engl. J. Med. 1990; 323: 614-615. 19. Cherin P., Herson B., Wechsler J. et al.: Effect of intravenous gammaglobulin therapy in chronic refractory polymiositis and dermatomyositis. An open study with 20 adult patients. Am. J. Med., 1991; 91: 162-168. 20. Corveta A., Delia Vitta R., Gabrielli A. et al. : Use of high-dose intravenous immunoglobulin in systemic lupus erythematosus: report of three cases. Clin. Exp. Rheumatol., 1989; 7: 295-299. 21. Dalakas M. C., Illa I., D'Ambrosi J. M. et al.: A controled trial of high dose of IVIG as treatment lor dermatomiositis. New Engl. J. Med. 1993; 329: 1993-2000. 22. De Souza V. R., Kaveri S. A., Kazatchkine M. D. Intravenous immunoglobulin in the treatment of autoimmune and inflammatory disease. Clin. Exp. Rheumatol. 1993; II (Suppl. 9): S33-S36. 23. Dwyer J. M.: Manipulating the immune system with intravenous immune globuline. New Engl. J. Med., 1992; 326: 107-116. 24. Enevoldson T. P., Wiles C. M.: Severe vasculitic neuropathy in systemic lupus erythematosus and response to cyclophosphamide. J. Neurol. Neurosurg. Psychiatry 1991; 54: 468 469 25. Fehr J., Hofmann V., Kappeler V. : Transient reversal of thrombocytopenia in idlopathic thrombocytopenic purpura by high-dose intravenous gamma-globulin. New Engl. J. Med. 1982; 306: 1254-1258. 26. Folan-Nolan D., Gooi H. C.: Current rheumatological uses of intravenous immunoglobulin. Brit. J. Rheumatol. 1994; 33: 1002-1006. 27. Francioni C., Galeazzi M., Fioravanti A. et al.: Long term I. V. Ig treatment in systemic lupus erythematosus. Clin. Exp. Rheumatol., 1994; 12: 163-168. 28. Furusho K., Nakano H., Shinomiya K.: High dose intravenous gammaglobulin for Kawasaki disease. Lancet 1984; 2: 1055-1058. 29. Gaedicke G., Teller W. M., Kohne E. et al.: Immunoglobulin therapy in systemic lupus ery-thematosus-two case report. Blut 1984; 48: 387-390. 30. Gelfand E. M.: Untervention in autoimmune disorders: creation of a Niche for intravenous gamma-globulin. Clin. Immunol. Immunopathol. 1989; 53: S1-6. 31. Gianini E., Lovell D. J., Silverman E. D., et al.: Intravenous immune globulin In polyarticular IRA. Results of a phase I/II trial. Arthritis Rheum., 1994; 37 (Suppl.): S276. 32. Hall F. C., Watts R. A., Ginsberg L.: Treatment of a case of systemic lupus erythematosus by pooled immunoglobulin as an alternative to immunosuppression. J. Neurol. Neurosurg. Psychiatry 1992; 55: 84. 33. Imbach P., Baraudun S., d'Appuzo V., Baumgartner C.: High-dose intravenous gamma-globulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981; 1: 128-131. 34. Jayne D. R. W. et. al.: ANCA anti-idiotype antibodies and the treatment of systemic vasculitis with IVIG. J. Autoimmunity 1993; 6: 207-219. 35. Jayne D. R. W., Davies M. J., Fox C. J. V. et al.: Treatment of systemic vasculitis with pooled intravenous immunoglobulins. Lancet 1991; 337: 1137-1139. 36. Kaaja R., Jutkunen H., Ammala P., Palosuo T., Kurki P.: Intravenous immunoglobulin treatment of pregnant patients with reccurent pregnancy losses associated with antiphospholipid antibodies. Acta Obstet. Gynecol. Scand. 1993: 72: 63-66. 37. Karlson E. W., Sudarsky L., Ruderman E. et al.: Treatment of stiff-man syndrome with intravenous immune globulin. Arthritis Rheum. 1994; 37: 915-918. 38. Kaveri S.-V., Dietrich G., Huber V., Kazatchkine M. D.: Intravenous immunoglobulins (IVIgG) in the treatment of autoimmune disease. Clin. Exp. Immunol., 1991; 86: 192-198. 39. Kurlander R. J., Ellison D. M., Hall J.: The blockade of Fc receptor mediated clearence of immune complexes in vivo by monoclonal antibody (2. 4G2) directed against Fc receptors on murine leukocytes. J. Immunol. 1984; 133: 855-862. 40. Lang B. A., Laxer R. M., Murphey G. et al.: Treatment of dermatomiositis with intravenous gammaglobulin. Am. J. Med. 1991; 91: 169-171. 41. Laxer R., Calabrese L, H., Klassen L. W.: Use of intravenous gammaglobulin (IVIg) in autoimmune disease, 58th. Annual. Sci. Meeet. Amer. Coll. Rheumatol. Clinical. Symposium "Use of Intravenous gammaglobulin in autoimmune disease", 1994, Minneapolis. 42. Lloyd A., Hickie I., Wakefield D. et al.: A double-blind placebo-controlled trial of intravenous immunoglobulin therapy in patients with chronic fatigue syndrom. Am. J. Med. 1990,89,561-568. 43. Maier W. P., Gordon D. S., Howard R. F. et al.: Intravenous immunoglobulin therapy in systemic lupus erythematosus-associated thrombocytopenia. Arthritis Rheum., 1990; 33: 1233-1234. 44. Newburger J. W., Takahashi M., Beiser A. S. et al.: A single intravenous infusion of gamma globulin as compared with four intravenous infusion of gamma globulin in the treatment of acute Kawasaki syndrome. New Engl. J. Med. 1991; 324: 1633-1639. 45. Peterson A. M., Adieff A., Deire M. et al.: A controlled trial of intravenous Igg in chronic fatigue syndrome. Am. J. Med. 1990, 89, 554-560. 46. Prieur A. M., Adieff A., Debre M. et al.: High dose immunoglobulin therapy in severe juvenile chronic arthritis: long-term follow-up in 16 patients. Clin. Exp. Rheumatol. 1990; 8: 603-609. 47. Rao S. P., Teitelbaum J., Miller S. T.: Intravenous immune globulin and aseptic meningitis. Amer. J. Dis. Childhood 1992; 146: 539-540. 48. Richter С., Schnabel A., Dsernok E. et al.: Treatment of ANCA-associated vasculitis with high-dose intravenous immunoglobulin. Arthritis Rheum. 1994; 37 (suppl.): S-353. 49. Ronda N., Hurez U., Kazatchkine M. D.: Intravenous immunoglobulin therapy (IVIg) of autoimmune and inflammany disease. Vox Sang., 1993, 64, 65-72. 50. Rossi F., Jayne D. R. W., Lockwood C. M. et al.: Anti-idiotypes against antineutrophil cytoplasmatic antigen autoantibodies in normal human polyspecific IgG for therapeutic rise and in the remission sera of patients with systemic vasculitis. Clin. Exp. Immunol. 1991; 83: 1-6. 51. Roifman C. M., Schaffer P. M., Wachsmuth S. E. et al.: Reversal of chronic polymiositis following intravenous immune serum globulin therapy. JAMA 1987: ; 258: 513-515. 52. Schifferli J. A., Sanrot J. U., Didierjean L.: Immunomodulatory effects of intravenous immunoglobulin. J. Rheumatol. 1991, 18, 937-939. 53. Silverman E. D., Laxer R. M., Greenwald M. et al.: Intravenous gamma globulin therapy in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1990; 33: 1015-1022. 54. Spath P., Affentranger P., Schwartz H. A. et al. High-dose intravenous immunoglobulin therapy in patients with early rheumatoid arthritis (RA): preliminary results from a placebo-controled double-blind study. XVIII ILAR Congr. Rheumatol., Barcelona, 1993: 148 (321 abst.). 55. Sturfelt G., Mousa F., Jonsson H. et al.: Reccurent cerebral infarction and the antiphos-pholipid syndrome: effect of intravenous gammaglobulin in patient with systemic lupus erythematosus. Ann. Rheuum. Dis., 1990; 49: 939-941. 56. Takei S., Arora Y. K., Walker S. M. Intravenous immunoglobulin containes specific antibodies inhibotory to activation of T cells by staphylococcal toxin superantigen. J. Clin. In vest., 1993; 91: 602-607. 57. Ter Borg E. J. T., Kallenberg C. G. M. : Treatment of severe thrombocytopenia in SLE with intravenous gammaglobulin. Ann. Rheum. Dis., 1992; 51: 1149. 58. Tomer Y., Shoenfeld Y.: Succesful treatment of psychosis secondary to SLE with high dose intravenous immunoglobulin. Clin. Exp. Rheumatol., 1992; 10: 391-393. 59. Tumiati В., Casoli P., Veneziani M., Rinaldi G.: High-dose immunoglobulin therapy as an immunomodulatory treatment of rheumatoid arthritis. Arthritis Rheum., 1992;-35: 1126-1133. 60. Tuso P., Moudgil A., Hay J. et al.: Treatment of antineutrophilcytoplasmatic antibody positive systemic vasculitis and glomerulonephritis with pooled intravenous gamma-globulin. Am. J. Kidney Dis. 1992; 20: 504-508. ГЛАВА 13 МЕТОДЫ АФЕРЕЗА, ЭКСТРАКОРПОРАЛЬНАЯ ФОТОХИМИОТЕРАПИЯ, ДРЕНАЖ ГРУДНОГО ЛИМФАТИЧЕСКОГО ПРОТОКА, ОБЩЕЕ ОБЛУЧЕНИЕ ЛИМФАТИЧЕСКИХ УЗЛОВ 13.1. Методы афереза Под термином "аферез" понимают разделение крови на составные части с последующим удалением одной из них или более. Извлечение плазмы с помощью афереза получило название "плазмаферез" (или плазмозамещение). Основными вариантами афереза, которые наряду с плазмаферезом используются в ревматологии, являются лимфоцитаферез (извлечение лимфоцитов), каскадная фильтрация плазмы (использование 2 фильтров или более для последовательного или дифференциального удаления плазмы), иммуносорбция (перфузия плазмы с антителами через твердую фазу, содержащую носитель, связывающий соответствующие антитела). В таблице 13.1. представлены сведения о возможности использования плазмафереза для лечения ревматических и неревматических заболеваний человека. Таблица 13.1. Использование плазмафереза при заболеваниях человека Заболевания Ревматические заболевания: • РА • феномен Рейно • СКВ • ССД • синдром Шегрена • ПМ/ДМ • системные васкулиты Неврологические заболевания: • миастения гравис • полинейропатия • синдром Гийена—Барре Заболевания печени: • фулминантный гепатит • печеночная недостаточность • первичный билиарный цирроз • хронический активный гепатит Гематологические болезни: • парапротеинемия • макроглобулинемия (гипервязкость) • криоглобулинемия • идиопатическая тромбоцитопеническая пурпура • тромботическая тромбоцитопеническая пурпура • резус-несовместимость • аутоиммунная гемолитическая анемия • пернициозная анемия • пересадка костного мозга Болезни почек: • нефриты • отторжение трансплантата Злокачественные новообразования Другие заболевания: • инсулинзависимый диабет • инсулиновый аутоиммунный синдром • бронхиальная астма Потенциально патогенные субстанции, удаляемые из плазмы Криоглобулины, аутоантитела, иммунные комплексы Аутоантитела, криоглобулины, иммунные комплексы Токсины, связанные с белком Аутоантитела Парапротеины, криоглобулины Аутоантитела, иммунные комплексы Аутоантитела, иммунные комплексы Антитела, иммунные комплексы Аутоантитела • • • • • • язвенный колит аутоиммунный тиреоидит болезнь Аддисона пузырчатка гиперлипидемия отравления Липопротеиды Токсины, яды Среди перечисленных заболеваний основными показаниями к плазмаферезу являются следующие: тромботическая тромбоцитопеническая пурпура, острая криоглобулинемия, особенно в сочетании с тяжелым поражением почек, до того, пока начнут действовать цитостатики, синдром Гудпасчера, синдром Гийена— Барре, синдром повышенной вязкости, миастения гравис, наследственная гиперхолестеринемия. Механизмы действия плазмафереза связывают с улучшением функциональной активности ретикулоэндотелиальной системы, удалением аутоантител, ЦИК и воспалительных медиаторов из кровяного русла. В ревматологии аферез начали использовать начиная с 70-х годов. (J. V. Jones и соавт., 1976; J. H. Klippel, 1984). Однако, несмотря на большое число исследований, результаты, касающиеся эффективности плазмафереза при системных ревматических заболеваниях, достаточно противоречивы. СКВ Результаты контролируемого исследования, проведенного у 20 больных СКВ с умеренной активностью, свидетельствуют об отсутствии различий между больными, получившими плазмаферез, и больными, получившими ложную процедуру (N. Wei и соавт., 1985). По данным многоцентрового слепого контролируемого исследования (86 больных, продолжительность наблюдения 136 нед.), включение плазмафереза в стандартную схему лечения больных волчаночным нефритом с использованием ГК и ЦФ не улучшает непосредственные результаты лечения и не влияет на выживаемость больных. (E. G. Lewis и соавт., 1992). Однако у больных, резистентных к цитотоксическим препаратам, использование плазмафереза в некоторых случаях дает очевидный клинический эффект (D. J. Wallace, 1992). Полагают, что проведение сеансов плазмафереза при СКВ наиболее оправданно у больных с криоглобулинемией, повышением вязкости крови, тромботической тромбоцитопенической пурпурой, тяжелым васкулитом (Y. Tatner и соавт., 1987), с резистентными к ГК и цитостатикам формами пролиферативного нефрита, а также аутоиммунной гемолитической анемией, АФС (S. Kobayashi и соавт., 1992) и геморрагическим волчаночным пневмонитом (R. W. Erikson и W. Emlen, 1994). РА По данным контролируемых исследований при PA (I. L. Dwosh и соавт., 1983), различия между результатами, полученными при интенсивном плазмаферезе и ложных процедурах, отсутствуют. В серии исследований было показано, что лимфоцитаферез обладает существенно более высокой эффективностью, чем плазмаферез (J. Karsh и соавт., 1981; I. L. Dwosh и соавт., 1983). Системные васкулиты Частота обострений и 7-летняя выживаемость у больных узелковым периартериитом и с синдромом Черджа—Строс, получавших ГК или ГК в сочетании с процедурами пламазфереза, была одинаковой (L. Guillevin и соавт., 1992). По мнению авторов, показанием к применению плазмафереза при системных васкулитах является острое, прогрессирующее течение заболевания, проявляющееся быстропрогрессирующим нефритом и тяжелым васкулитом. АФС По данным R. H. W. Derksen и соавт. (1987), использование плазмафереза в объеме 2.5 л позволяет снизить уровень антифосфолипидных антител (аФЛ), который однако возвращается к исходному в течение 1-7 дней. В других исследованиях было показано, что падение уровня аФЛ различных изотипов после окончания курса лечения, сохраняется в течение нескольких недель (S. В. Ingram и соавт., 1987; G. Frampton и соавт., 1987). Имеются единичные наблюдения, свидетельствующие об эффективности комбинированного лечения плазмаферезом и иммунодепрессантами у женщин с акушерской патологией. D. Fulcher и соавт. (1989) на фоне 4 процедур плазмафереза достигли стабилизации кровотока у плода. G. Frampton и соавт. (1987), использовав по 2 сеанса плазмафереза в неделю (в сроки между 16-й и 34-й неделями беременности) в сочетании с ГК и препаратами, подавляющими агрегацию тромбоцитов, достигли нормального родоразрешения у женщин с АФС. Плазмаферез в сочетании с иммунодепрессивной и антикоагулянтной терапией применяли для лечения больных с множественными тромботическими осложнениями, включая церебральную окклюзию. Так, по данным S. B. Ingham и соавт. (1987), на фоне этой терапии наблюдалось снижение титров аФЛ и некоторое уменьшение неврологических проявлений. Сходные результаты получены D. P. Briley и соавт. (1989) у 3 больных с неврологическими нарушениями. Однако имеются наблюдения развития повторных венозных тромбозов, инсультов после такого лечения, несмотря на снижение уровня аФЛ. Создается впечатление, что комбинация плазмафереза с цитотоксической терапией позволяет поддерживать низкий уровень аФЛ достаточно длительное время, однако пока неясно, приводит ли это к реальному уменьшению риска развития повторных тромботических осложнений. ПМ/ДМ Данные об эффективности экстракорпоральных процедур у больных ПМ/ДМ противоречивы. В открытых испытаниях и отдельных клинических наблюдениях сообщается о весьма положительном влиянии плазмафереза на течение заболеваний, в то время как на основании результатов контролируемого исследования сделан вывод о неэффективности этой процедуры. По данным P. C. Dau и соавт. (1981), на фоне интенсивного плазмафереза положительная динамика клинических и лабораторных показателей, а в отдельных случаях и клиническая ремиссия, достигнуты у 32 из 35 больных ПМ/ДМ, резистентных к ГК и цитотоксическим препаратам. Методика плазмафереза заключалась в еженедельном удалении объема плазмы, эквивалентного 56% массы тела (проводилось до 10 процедур) с последующим удлинением интервалов между сеансами (до 4 нед.). К особенностям протокола этого исследования следует отнести введение 10 г в/м иммуноглобулина после каждого сеанса плазмафереза. Кроме того, все больные получали довольно высокие дозы ЦФ (2.5 мг/кг), а в случае развития побочных эффектов переходили на прием хлорбутина (0.12 мг/кг). Основным осложнением, наблюдавшимся в процессе лечения, был опоясывающий герпес, который развился у 20% больных. Противоположные результаты получены в недавно проведенном слепом контролируемом исследовании (F. M. Miller и соавт., 1992), в котором сравнивали эффективность плазмафереза (замена 1 объема плазмы — 40-50 мл/кг с замещением 5% альбумином, 4 раза в неделю), лимфоцитафереза (5-10õ10 9 клеток) и ложных процедур у 39 больных ПМ/ДМ. Больные были рандомизированы на 3 группы по 13 человек. В каждой из сравниваемых групп только у 3 человек отмечалась положительная динамика мышечного синдрома. У 3 больных, леченных лимфоцитаферезом, и у 1 больного, которому проводили плазмаферез, развилось обострение, а у остальных 26 пациентов динамика отсутствовала. Таким образом, полученные результаты свидетельствуют о том, что у кортикостероидрезистентных больных ПМ/ДМ эффективность экстракорпоральных процедур не выше, чем ложного афереза. Обращает на себя внимание важное отличие работы P. C. Dau от протокола контролируемого исследования, которое исключало прием цитотоксических препаратов. Можно предположить, что проведение плазмафереза следует зарезервировать для больных с тяжелым, резистентным к другим методам лечения ПМ/ДМ, и его необходимо сочетать с применением цитотоксических препаратов. Рекомендации по применению: 1. Следует удалять не менее 40 мл/кг плазмы 3 раза в неделю в течение 3 нед. или 60 мл/кг/день в течение 6 дней. 2. Во всех случаях, за исключением тромботической тромбоцитопенической пурпуры, для замещения потери жидкости следует использовать раствор альбумина. 3. Необходимо иметь в виду, что некоторые белки присутствуют в более высокой концентрации во внесосудистом пространстве, чем во внутрисосудистом. Например, во внутрисосудистом пространстве находится 75% пула IgM и только 25% пула IgG. 4. Следует сочетать процедуры плазмафереза с интенсивной глюкокортикоидной и цитотоксической терапией. В день проведения плазмафереза больные должны принимать ГК и цитотоксические препараты не до, а после процедур. Побочные эффекты Суммарная частота осложнений при проведении плазмафереза колеблется от 4.5 до 25% ( в среднем 0.7% на процедуру). Наиболее частыми осложнениями являются кардиогенный шок, анафилактические и цитратные реакции. При возмещении удаленной плазмы раствором альбумина возможно развитие гипогаммаглобулинемии. В то же время при корректном проведении процедур плазмафереза частота инфекционных осложнений не увеличивается, по крайней мере у больных волчаночным нефритом. (M. A. Pohl и соавт. 1991). Необходимо иметь в виду возможность развития синдрома "рикошета" (удаление плазменных белков стимулирует их синтез), который особенно часто наблюдается у больных с активным воспалительным процессом (J-C. Bystryn и coавт. 1970; R. H. W. M. Derksen и соавт. 1984). Например, в отсутствие цитотоксической терапии после проведения плазмафереза наблюдается двукратное увеличение концентрации антител. Пульс-синхронизация Разработана схема, получившая название "пульс-синхронизация" (или "стимуляция-удаление"), которая, как полагают, способствует увеличению эффективности цитотоксической терапии (H. H. Euler и соавт., 1992, 1994). Суть этого метода заключается в отмене на 4 нед. поддерживающей терапии ГК и цитотоксическими препаратами, что вызывает стимуляцию пролиферации лимфоидных клеток и развитие синдрома "рикошета". Последний купируется 3 циклами интенсивного плазмафереза и высокими дозами ЦФ. Предполагается, что эта схема позволяет добиться более эффективной элиминации патологических клонов клеток, синтезирующих аутоантитела. H. H. Euler и соавт. (1994) сообщили о быстром клиническом улучшении у 14 больных СКВ, леченных плазмаферезом (3õ60 мл/кг) и ЦФ (36 мг/кг). Эффективность метода пульс-синхронизации требует дальнейшего подтверждения. Селективный аферез Селективный аферез предполагает использование абсорбционных колонок, несущих лиганды, способные селективно удалять крупные молекулы (аутоантитела, иммунные комплексы, другие белки), принимающие участие в развитии тканевого повреждения. Имеются данные о применении при ревматических заболеваниях различных типов иммуносорбентов, способных удалять перекрестно реагирующие органонеспецифические аутоантитела (антитела к ДНК, аФЛ) и иммунные комплексы или и те, и другие субстанции. К ним относятся иммуносорбенты, приготовленные на основе активированного угля, нагруженного ДНК для удаления антител к ДНК (D. S. Ter-man и соавт., 1979), декстрансульфата, способного удалять антитела к ДНК и антитела к кардиолипину (S. Aotsuka и соавт., 1990), фенилаланина (антитела к ДНК и иммунные комплексы) (M. Schneider и соавт., 1990), белка А и антител к иммуноглобулинам человека (иммуноглобулины и иммунные комплексы). По данным H. Tsuda и соавт. (1989), пропускание плазмы больных через колонки с декстрансульфатом позволяет на 40-50% снизить сывороточный уровень антител к ДНК и антител к кардиолипину. К. Suzuki и соавт. (1991) использовали для лечения 6 больных СКВ декстрансульфатцеллюлозные колонки с автоматической регенерацией (SL-01). После 2-4 процедур у всех больных отмечено существенное снижение уровня антител к ДНК, у 3 — уменьшение протеинурии, а у 4 — увеличение количества лимфоцитов. T. Ishizuka и соавт. (1993) продемонстрировали благоприятный эффект этой процедуры у больных с диффузным пролиферативным волчаночным нефритом. Недавно для лечения РА и СКВ начали применять экстракорпоральную иммуноабсорбцию на колонках с белком A (IMRE Corporation) или антителами к IgG (Ig-Therasorb, Baxter) C. W. Wiesenhutter и ñîàâò. (1994) провели процедуры с использованием колонок с белком А (по схеме вначале 1 раз в месяц, а затем 2 раза в месяц) у 6 больных с системным РА, резистентных к базисной терапии (включая ЦФ и Цс А). УЗ больных достигнут очевидный клинический эффект, коррелирующий со снижением титров IgGPÔ. M. Gaubitz и соавт. (1994) лечили 6 больных острой СКВ с помощью IgG-èììóíîñîðáåíòà. На фоне лечения отмечено снижение концентрации сывороточного IgG, антител к ДНК, аФЛ и антител к SS-A/Ro, а также снижение клинической активности заболевания в среднем с 21 до 12 баллов по SLAM. Побочных эффектов не наблюдалось. 13.2. 13.2. Экстракорпоральная фотохимиотерапия (ЭФХТ) ЭФХТ (фотаферез) — новый метод иммунотерапии, при котором лейкоциты периферической крови, сенсибилизированные 8-метокси-прозаленом, подвергаются длинноволновому ультрафиолетовому облучению, а затем возвращаются больному. ЭФХТ была разработана в 1981 г. для лечения диссеминированной кожной Тклеточной лимфомы (R. L. Edelson и соавт., 1989). Механизм действия Прозалены — трициклические ароматические вещества (фурокумарины), обладающие способностью сильно абсорбировать ультрафиолетовое излучение с длиной волны 250-300 нм (P. Song и K. Tapley, 1979). Наиболее часто используемым аналогом прозаленов является метоксален (8-метоксипрозален; 8-МОП). Оптимальной для активации прозалена является область ультрафиолетового спектра с длиной волны 320-400 нм. Под влиянием ультрафиолета 8-МОП фотоактивируется и приобретает способность ковалентно связываться с клеточными молекулами и образовывать фотоаддукты с ДНК, которые ингибируют синтез ДНК. Кроме того, 8-МОП способен модифицировать структуру и других макромолекул, вызывая образование фотоаддуктов с мембранными фосфолипидами и белками сыворотки. Предполагается, что окисление липидов лежит в основе фототоксичности ЭФХТ, проявляющейся эритемой, отеком и образованием кожных пузырьков. Антипролиферативный и иммуномодулирующий эффекты ЭФХТ продемонстрированы в серии экспериментальных и клинических исследований. По данным M. I. Perez и соавт. (1989), отторжение кожного трансплантата у мышей замедляется при реинфузии специфичных к коже Т-клеток, обработанных 8МОП/УФО. C. L. Berger и соавт, (1989) обнаружили, что реинфузия 8-МОП/УФЩ спленоцитов от старых MRL-мышей молодым сингенным мышам приводила к замедлению у последних синтеза антител к ДНК, развития гепатоспленомегалии, увеличению продолжительности жизни животных. На модели экспериментального аллергического энцефаломиелита было показано, что ЭФХТ подавляет прогрессирование патологического процесса за счет индукции клон-специфических Т-супрессоров. У больных пузырчаткой клиническое улучшение на фоне ЭФХТ ассоциируется с увеличением количества Т-клеток с "супрессорным" фенотипом в периферической крови (A. H. Rook и соавт., 1989). По данным B. Vowels и соавт. (1991) моноциты, подвергнутые ЭФХТ, синтезируют в избыточном количестве ФНО-а. Предполагается, что все эти эффекты ЭФХТ могут объяснить положительное действие процедуры при ССД. Известно, что при ССД наблюдается активация Т-лимфоцитов, которые синтезируют ТРФ-b. Последний играет важную роль в прогрессировании склеротического процесса. Предполагается, что усиление синтеза ФНО-а моноцитами в процессе ЭФХТ может приводить к подавлению образования ТРФ-b. Схема лечения Больной перорально принимает метоксален (0.6 мг/кг), через 2 ч проводится лейкоцитаферез, полученные лимфоциты облучаются УВ-А и возвращаются больному (R. Edelson и соавт., 1987). Поскольку биодоступность 8-МОП после перорального приема варьирует в широких пределах, рекомендуется мониторировать концентрацию препарата в сыворотке до и в процессе процедуры ЭФХТ. Оптимальная длина волны УФО для активации 8-МОП составляет 320-400 нм. Для проведения процедур ЭФХТ разработана специальная система (UVAR, Therakos. USA). Клинические испытания Псориатический артрит Фотаферез (2 процедуры в месяц в течение 5-15 мес. был использован для лечения 5 больных с длительно текущим псориатическим артритом (H. Wilfert и соавт. 1990). У 4 больных наблюдалось умеренное уменьшение клинических симптомов, которое, по мнению авторов, может быть связано не только с истинной эффективностью лечения, но и с плацебо-эффектами. РА S. E. Malawista и соавт. (1991) провели предварительное испытание эффективности ЭФХТ у 7 больных. Процедуры проводили сначала 1 раз в неделю, затем 2 раза в неделю. Клиническое улучшение отмечено у 3 больных. C. J. Menkes и соавт. (1992) использовали ЭФХТ для лечения 7 больных РА и отметили клиническое улучшение у всех пациентов в конце первой недели лечения. J. Laing и соавт. (1993) недавно разработали для лечения синовита коленного сустава модификацию метода фотохимиотерапии, основанную на использовании внутрисуставного облучения аргоновым лазером. СКВ M. Knobler и соавт. (1992) изучали эффективность ЭФХТ у 10 больных СКВ, 8 из которых закончили исследование. У 7 — отмечен существенный эффект, проявляющийся в снижении общей активности болезни (индекс клинической активности в целом по группе уменьшился с 7 до 1) и особенно уменьшении кожных проявлений заболевания и артрита. У большинства больных удалось снизить дозу ГК и цитостатиков. Побочные эффекты практически отсутствовали. У 5 больных клиническая ремиссия поддерживалась в течение 30 мес. ССД и ювенильный ДМ Сравнение эффективности ЭФХТ и D-ïåíèöèëëàìèíà (750 мг/день) проведено у 80 больных ССД. 6месячный курс лечения закончили 29 больных, получавших ЭФХТ, и 22 больных, леченных D-ïåíèöèëëàìèном. Положительная динамика кожного счета отмечена только на фоне лечения ЭФХЦ (A. H. Rook и соавт., 1992; B. Freunlich и соавт., 1990). F. X. Di Spalto и соавт. (1993) использовали ЭФХТ для лечения 9 больных активной прогрессирующей ССД в течение 6-21 мес. Отмечены существенное уменьшение выраженности кожных и мышечно-скелетных лроявлений, феномена Рейно, одышки, дисфагии, артралгий, стабилизация функции легких. По данным A. DeWilde и соавт. (1992), на фоне лечения ЭФХТ наблюдается клиническое улучшение у больных ювенильным ДМ. 13.3. Дренаж грудного лимфатического протока (ДГЛП) ДГЛП — метод лечения, заключающийся в удалении большого количества циркулирующих лимфоцитов (M. B. Yunis, 1988; Н. В. Бунчук, 1985). В настоящее время ДГЛП практически не используется в клинической практике, однако его эффективность при РА рассматривается как важное подтверждение патогенетического значения клеточных иммунных реакций в патогенезе этого заболевания. Клиническое применение E. Paulus и соавт. (1977) использовали ДГЛП у 9 больных с тяжелым РА в течение 19-105 дней с удалением во время процедур в среднем 46õ10 12 лимфоцитов. В результате лечения отмечено статистически достоверное улучшение, включающее снижение суставного счета, длительности утренней скованности, увеличение силы сжатия кисти, однако после окончания терапии в сроки между 2-й и 12-й неделями активность заболевания вернулась на прежний уровень. У 3 больных развились побочные эффекты, включающие септицемию. Позднее J. H. Vaughan и соавт. (1984) использовали ДГЛП для лечения 5 больных РА в течение 1-2 мес. У 2 больных было достигнуто выраженное клиническое улучшение, длившееся в течение 9 мес., а у 2 больных — частичный клинический эффект. Таким образом, ДГЛП представляется потенциально эффективным методом лечения РА. Однако его широкое использование нецелесообразно из-за технической сложности процедуры, невозможности достижения стойкой ремиссии и существования более простых и эффективных методов, индуцирующих развитие лимфопении. 13.4. Общее рентгеновское облучение Метод, широко применяющийся при лечении лимфогранулематоза и неходжкинских лимфом. Механизм действия Общее рентгеновское облучение вызывает длительную иммуносупрессию. В экспериментальных исследованиях было показано, что она эффективно подавляет развитие адъювантного (D. J. Schumacher и соавт., 1981) и коллагенового (W. J. McCune и соавт., 1981) артритов, уменьшает выраженность протеинурии, приводит к снижению уровня антител к ДНК и увеличению продолжительности жизни у NZB/NZW мышей, а у MRL/lpr мышей предотвращает развитие гломерулонефрита (D. J. Schurman и соавт., 1981). Это послужило основанием для использования общего рентгеновского облучения для лечения больных тяжелым РА и другими ревматическими заболеваниями. Клиническое применение Впервые этот метод был использован для лечения РА D. E. Trentham и соавт., (1981) и B. L. Kotzin и соавт., (1981), Облучению подвергали шейные, подмышечные, медиастинальные лимфатические узлы, средостение и субдиафрагмальные лимфатические узлы. Доза облучения колебалась от 2000 до 3000 рад, а длительность лечения — от 5 до 13 нед. В целом к 6-му месяцу у 80% больных отмечено достоверное клиническое улучшение — уменьшение числа отекших и болезненных суставов и длительности утренней скованности, которое длилось до 1 года. Сходные клинические результаты были получены позднее другими авторами, в том числе в процессе двойных слепых контролируемых исследований (S. Strober и соавт., 1985; J. G. Hanly и соавт., 1986). Так, по данным S. Strober и соавт. (1985), облучение в высоких дозах (2000 рад) приводит к более значимому уменьшению утренней скованности, боли и других клинических показателей, чем облучение в дозе 200 рад. J. G. Hanley и соавт. (1986) сравнивали эффективность дозы 750 и 2000 рад у 20 больных РА. Оба режима вызывали уменьшение продолжительности утренней скованности, болезненности суставов, окружности проксимальных межфаланговых суставов по сравнению с базальным уровнем. Интересно, что сила сжатия кисти достоверно возросла у больных, получавших 750 рад, а выраженность боли достоверно уменьшилась при использовании дозы 2000 рад. Оценка отдаленных результатов показала, что у большинства больных клиническое улучшение сохраняется в течение 12 мес. и более, однако выраженность его не достигает 50%. По данным лабораторных исследований, у подавляющего большинства больных РА отмечалось развитие лимфопении, преимущественно за счет СD4+Т-лимфоцитов, снижение пролиферативного ответа лимфоцитов на митогены в отсутствие достоверной динамики уровня РФ, других показателей гуморального иммунитета и СОЭ. При исследовании синовиальной жидкости выявлено существенное снижение числа лейкоцитов, концентрации иммуноглобулинов, в то время как титры РФ оставались без изменений. Имеются отдельные сообщения о потенциальной эффективности рентгеновского облучения при СКВ (Е. Field и соавт., 1984). Интересно, что при СКВ рентгеновское облучение, как правило, вызывает снижение титров антител к ДНК и АНФ (A. Tanay и соавт., 1986) Рентгеновское облучение было применено у 3 больных ПМ/ДМ с очень тяжелым обострением, не поддающимся терапии высокими дозами ГК и цитостатиков (S. H. Morgan и соавт., 1985). Во всех случаях отмечалось временное улучшение или даже ремиссия, развитие которых коррелировало с лимфопенией. После окончания процедур эффект сохранялся в течение 3-6 мес., после чего развивалось обострение. У 2 из 3 больных в процессе лечения имели место инфекционные осложнения, в 1 случае приведшие к смерти пациента. Побочные эффекты (таблица 13.2.) Таблица 13.2. Побочные эффекты обшего рентгеновского облучения у 57 больных РА (M. B. Yunis, 1988} Побочный эффект Частота, % Недомогание, слабость, желудочно-кишечные симптомы 100 Ксеростомия 77 (22) Алопеция 57 (21) Похудание Лейкопения, костномозговая супрессия Смертельный исход Бактериальные инфекции Герпетическая инфекция Сердечно-легочная недостаточность/инфаркт миокарда Другие (выпадение зубов, гипотиреоз, амилоидоз, саркома Капоши, кандидоз пищевода) 30 (33) 25 18 18 18 9 33 Применение общего рентгеновского облучения часто сопровождается побочными реакциями. Практически у всех больных наблюдаются общее недомогание, потеря аппетита, сухость во рту, нередко развиваются инфекционные осложнения. Отмечена высокая частота смертельных исходов, достигающая почти 20%. Наиболее часто причиной смерти больных являются инфекционные осложнения, реже — поражение сердечно-сосудистой системы. Полагают, что летальность больных после общего рентгеновского облучения выше при РА, чем при лимфопролиферативных заболеваниях. Это связывают с более пожилым возрастом больных, предшествующей терапией иммунодепрессивными препаратами и воздействием самого заболевания. Список литературы К главе "Методы афереза, экстракорпоральная фотохимиотерапия, дренаж грудного лимфатического протока, общее рентгеновское облучение" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. Aotsuka S., Funahashi Т., Tani N. et al.: Absorbtion of anti-DNA antibodies by Immobilized polyanionic compounds. Clin. Exp. lmunol., 1990; 79: 215-220. Berger C. L, Perez M., Laroche L., Edelson R.: Inhibition of autoimmune disease in murine model of systemic lupus erythematosus induced by exposure to syngenic photoinac-tivated lymphocytes. J. Invest. Derm., 1990; 94: 52-57. dark W. F., Dau P. C., Euler H. H. et al.: Plasmapheresis and subsequent pulse cyclophos-phamide alone in severe lupus: design of the lupus plasmapheresis study group trial. J. Clin. Apheresis., 1991; 6: 40-47. Dau P. O.: Plasmapheresis in idiopathic inflammatory myopathy. Arch. Neurol. 1981; 38: 544-552. Derksen R. H. W, Hasselaar P., Blokzijl L., De Groot P. G.: Lack of efficiency of plasma exchange In removing antiphospholipid antibodies. Lancet 1987; 2: 222. DeWilde A., DiSpalto F. X., Geller A. et al.: Exstracorporeal photochemotharapy as adjunc-tive treatment in juvenile dermatomiositis. Arch Dermatol. 1992; 128: 1656-1657. Di Spalto F. X., Cottril C., Cahil C. et al.: Extracorporeal photochemotherapy in progre-sive systemic sclerosis. Int. J. Derm. 1993; 32: 417-420. Dwosh I. L., Giles A. R., Ford P. M. et al.: Plasmapheresis therapy in rheumatoid arthritis. A controlled, double-blind, crossover trial. N. Engl. J. Med. 1983; 308: 1124-1129. Edelson J. A., Berger C., Gasparro F. et al.: Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. New Engl. J. Med. 1989; 316: 297-303. Erickson R. W., Emlen W.: Treatment of hemorrhagic lupus pnevmonotis with plasm-spheresis. Report of three cases. Arthritis Rheum. 1994; 37 (suppl.): S-323. Euler H. H., Schroeder J. O.: Antibody depletion and cytotoxic drug therapy in severe systemic lupus erythematosus. Transfus Sci. 1992; 13: 167-184. Euler H. H., Schroeder J. O., Harten P. et al.: Treatment-free remission in systemic lupus erythematosus following plasmapheresis with subsequent pulse cyclophosphamide. Arthritis Rheum. 1994; 37: 17841794. Field E., Strober S., Shemesh 0. et al.: Total lymphoid irradiation for the treatment of membranoproliferative lupus glomerulonephritis. Clin. Res. 1984; 32: 436A. Freunlich В., Rook A. H., Edelson R. L. et al.: Extracorporeal photochemotherapy in the treatment of systemic sclerosis. Arthritis Rheum. 1990; 33: S35 (Suppl. 9). Gaubitz M., Schmitz-Linneweber B., Reh H. et al.: Treatment of systemic lupus erythematosus by IGimmunoadsorbtion. Arthritis Rheum. 1994; 37 (suppl.): S-407. Guillevin L., Fain 0., Lhote F. et al.: Lack of superiority of steroids plus plasma exchange to steroids alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome. A prospective, randomozed trial in 76 patients. Arthritis Rheum. 1992; 35-208-215. Hanly J. S., Hassan J., Moriarty M. et al.: Lymphoid irradiation in intractable rheumatoid arthritis: a double-blind, randomized study comparing 750-rad treatment with 2, 000-rad treatment. Arthritis Rheum., 1986; 29: 16-25. Jones J. V., Gumming R. H., Bucknall R. C., Asplin C. M.: Plasmapheresis in the management of acute systemic lupus eryhematosus? Lancet 1976; 1: 709-711. Karsh J., Klippel J. H., Plotz P. H. et al.: Lymphapheresis in rheumatoid arthritis. A randomized trial. Arthritis Rheum. 1981; 24: 867-873. 20. Kinoshita M., Aotsuka S., Funahashi T. et al.: Selective removal of anti-double-stranded DNA antibodies by immunoabsorbtion with dextran sulphate in a patient with systemic lupus erythematosus. Ann. Rheum. Dis. 1989; 48: 856-860. 21. Klippel J. H.: Apheresis-biotechnology and the rheumatic disease. Artritis Rheum., 1984; 27: 1081-1085. 22. Кnobler R., Graninger W., Graninger W. et al.: Extracorporeal photochemoterapy for the treatment of systemic lupus erythematosus. Arthritis Rheum. 1992; 35: 319-324. 23. Kobayasahl S., Tamura N., Tsuda H. et al.: Imnnunoabsorbent plasmapheresis for a patient with antiphospholipid syndrome during pregnancy. Ann. Rheum. Dis. 1992; 51: 399-401. 24. Kotzin B. L, Strober S.: Recersal of NZB/NZW disease with total lymphoid irradiation. J. Exp. Med. 1979; 50: 371-378. 25. Kotzin B. L., Strober S., Engelman E. G. et al.: Treatment of intractable rheumatoid arthritis with total lymphoid irradiation. N. Engl. J. Med. 1981; 305: 969-976. 26. Lewis E. J., Hunsicker L. G, Lan S. P. et al.: A controlled trial of plasmapheresis therapy in severe lupus nephritis. N. Engl. J. Med., 1992; 326: 1373-1379. 27. Liang T. J., Ike R. W., Griffits C. E. et al.: A pilot study of the effect of oral 8-methoxyprsoralen and intraarticular ultraviolet light on rheumatoid synovitis. Arthritis Rheum. 1993; 36 (suppl.): S-57. 28. Malawista S., Trock D. H., Edelson R. L.: Treatment of rheumatoid arthritis by extracor-poleal photochemotherapy. Arthritis Rheum., 1991; 34: 646-654. 29. McCune W. J., Buckley J. A., Belli J. A. et al.: Partial supression of type II collagen-induced arthritis by fractioneted total lymphoid irradiation. Clin. Res. 1981; 29: 160A. 30. Miller F. W., Leitman S. F., Cronin M. E. et al.: Controlled trial of plasma exchange and leukapheresis in polymiositis and dermatomiositis. N. Engl. J. Med., 1992; 326: 1380-1384. 31. Menkes C. J., Andreu G., Heshmati F., Hilliquin P.: Extracorporeal photochemotherapy, Brit. J. Rheumatol. 1992; 31: 789-790. 32. Morgan S. H., Bernstein R. M., Coppen J. et al.: Total body irradiation and the course of polymiositis. Arthritis Rheum. 1985; 28: 831-835. 33. Paulus H. E., Machleder H. I., Levine S. et al.: Lymphocyte involvment in rheumatoid arthritis-studies during thoracic duct drainage. Arthritis Rheum. 1977; 20: 1249-1262. 34. Perez M. I., Edelson R., Laroche L., Berger C.: Inhibition of antiskin allograft immunity by infussions with syngenic photoinactivated effector lymphocytes. J. Invest. Derm. 1989; 92: 669-676. 35. Pohl M. A., Lan S.-P., Berl S.-P.: Plasmapheresis does not increase the risk for infection in immunosuppressed patients with severe lupus nephritis. Ann. Int. Med., 1991; 114:924-929. 36. Rook A. H., Freundlich B., Jegasothy B. V. et al.: Treatment of systemic sclerosis with extracorporeal phototherapy: results of multicentric trial. Arch. Derm., 1992; 128:1517-1520. 37. Schroeder J. O., Euler H., Loffler H.: Synchronization of plasmapheresis and pulse cyclophosphamide in severe systemic lupus erythematosus. Ann. Int. Med., 1987; 107:344-346. 38. Schneider M., Berning Т., Waldendorf M. et al.: Immunoabsorbent plasma perfusion in patients with systemic lupus erythematosus. J. Rheumatol. 1990; 17: 900-907. 39. Schurman D. J., Hirshman H. R., Strober S.: Total lymphoid and local join irradiation in the treatment of adjuvant arthritis. Arthritis Rheum. 1981; 24: 38-46. 40. Schumaher D. J., Hirshman H. P., Strober S.: Total lymphoid and local joint irradiation in the treatment of adjuvant arthritis. Arthritis Rheum., 1981; 24: 38-44. 41. Song P., Tapley K.: Photochemistry and photobiology of prosalens. Photochem. Photobiol, 1979: 29: 11771197. 42. Stafford F. J., Klippel J. H.: Apheresis and the rheumatic disease. Bull. Rheum. Dis., 1993; 42; 3. 43. Strober S., Tanay A., Field E. et al.: Efficacy of total lymphoid irradiation in intractable rheumatoid arthritis. Ann. Intern. Med„ 1985; 102: 441-458. 44. Suzuki К., Нага M., Harigai M. et al.: Continouos removal of anti-DNA antibody, using a new extracorporeal Immunoadsorbtion system, in patients with systemic lupus erythematosus. Arthritis Rheum. 1991; 34: 1546-1552. 45. Tanay A., Schiffman G., Strober S.: Effect of total lymphoid irradiation on levels of serun autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 1986; 29: 26-31. 46. Tanter Y., Rifle G., Chalopin J. M. et al.: Plasma exchange In central nervous system involvment of systemic lupus erythematosus. Plasma Ther. Transfus Technol., 1987; 8: 161-168. 47. Taylor A., Gasparro F. P.: Extracorporeal photochemotherapy for cutaneous T-cell lymphoma and other disease. Semin. Hematol. 1992; 29: 132-141. 48. Terman D. S., Buffaloe G., Mattioli C. et al.: Extracorporeal immunoabsorbtion: initial experience in human systemic lupus erythematosus. Lancet 1979; II: 824-826. 49. Trentham D. E., Belli J. A., Anderson R. J., et al.: Clinical and Immunologic effects of fractionated total lymphoid irradiation in refractory rheumatoid arthritis. N. Engl. J. Med., 1981; 305: 976-982. 50. Tsuda H., Taniguchi 0., Mokumo C., et. al.: Utilisation of dextran sulfate column plasmapheresis to remove anticardiolipin antibodies. Jap. J. Artif. Organs. 1989; 18: 7-10. 51. Wallace D. J.: Plasmapheresis in lupus. Lupus. 1993; 2: 141-143. 52. Wallace D. J. : Plasmapheresis for lupus nephritis. N. Engl. J. Med., 1992: 327: 1029. 53. Wiesenhutter C. W.: Treatment of severe refractory rheumatoid arthritis patients with offline extracorporeal protein A immunoadsorbtion columns. Arthritis Rheum., 1994; 37: 4P (R5). 54. Wei N., Klippel J. H., Huston D. P. et al.: Randomised trial of plasma exchange in mild systemic lupus erythematosus. Lancet 1985: 1: 17-22. 55. Wilfert H., Honigsmann H., Steiner G. et al.: Treatment of psoriatic arthritis by extracorporeal photochemotherapy. Br. J. Dermatol., 1990; 122: 225-232. 56. Vaughan J. H., Fox R. I., Abresch R. J. et al.: Thoracic duct drainage in rheumatoid arthritis. Clin. Exp. Immunol. 1984; 58: 645-653 57. Vovels В., Cassin M., Boufal M. et al.: Extracorporeal photochemotherapy induced production of tumor necrosis factor and IL-6 adherent peripheral blood mononuclera cells. J. In vest. Dermatol. 1991; 86: 14891495. 58. Yunus M. B.: Investigational therapy in rheumatoid arthritis: a critical review. Semin. Arthritis Rheum., 1988; 17: 163-184. ГЛАВА 14 МЕТОДЫ ИММУНОТЕРАПИИ Несмотря на существование многообразных подходов к лечению РА, это заболевание по-прежнему остается неблагоприятным в отношении как ранней инвалидизации, так и отдаленного прогноза (T. Pincus, 1994). По данным проспективных исследований выживаемость у некоторых больных РА приближается к таковой при инсулинзависимом сахарном диабете, трехсосудистом поражении коронарных артерий и лимфогранулематозе (J. P. Vandenbroucke и соавт., 1984; T. Pincus и L. F. Calahan, 1990). Средняя продолжительность жизни больных РА уменьшается на 7 лет у мужчин и на 3 года у женщин. Поэтому неудивительно, что многочисленные методы иммунотерапии, разрабатывающиеся в первую очередь для нужд трансплантологии и онкологии, проходят клиническую апробацию именно при РА. Современные представления о патогенезе РА основываются на нескольких взаимодополняющих концепциях, учитывающих участие CD4+ Т-лимфоцитов (G. S. Panayi и соавт., 1992), моноцитов/макрофагов, синтезирующих цитокины, обладающие провоспалительной активностью (F. M. Brennan и соавт., 1992; W. P. Arend и J.-M. Dayer, 1994), роль автономных неиммунных механизмов, определяющих опухолеподобный рост синовиальной ткани, приводящий к деструкции хряща (N. Zvaifler и G. S. Firestein, 1994; W. J. Koopmam и S. Gay, 1993). О важной роли CD4+T лимфоцитов свидетельствуют следующие факты: 1) необычно высокая аккумуляция активированных (экспрессирующих антигены класса II ГКГ) СD4+Т-клеток в синовиальном инфильтрате на самых ранних стадиях РА; 2) увеличение чувствительности к РА у носителей некоторых аллелей Bl-ëîêóñà класса II ГКГ (HLA-DRB1*0401, *0404, *0405, *0408, *0101, *0402, *1001), которые экспрессируют общую аминокислотную последовательность, локализованную в области молекулы ГКГ, связывающей антигенный пептид; 3) клиническое улучшение у больных РА, инфицированных ВИЧ или леченных с помощью методов, вызывающих элиминацию Т-клеток (дренаж грудного лимфатического протока, общее рентгеновское облучение, ЦсА и др.); 4) участие Т-клеток в развитии экспериментальных артритов, напоминающих РА: коллагенового, адъювантного, артрита у MRL/lpr мышей. Другой аспект патогенеза РА связан с расшифровкой роли моноцитов/макрофагов. Установлено, что в синовиальной жидкости и тканях суставов при РА содержится избыточное количество цитокинов, главным образом макрофагального происхождения (ИЛ-1, ФНО-a, ГМ-КСФ, ИЛ-6), при минимальном содержании Тклеточных цитокинов, таких, как ИЛ-2, ИЛ-3, ИЛ-4 и ИФ-у. Первым обнаруженным в синовиальной мембране цитокином, индуцирующим развитие воспаления и разрушение хряща, был ИЛ-1, который благодаря именно этому эффекту сначала был назван "катаболин". Вскоре было показано, что наряду с ИЛ-1 ФНО-а также обладает способностью стимулировать хондроциты и вызывать деградацию хрящевой ткани. В серии экспериментальных исследований было установлено, что ИЛ-1 и ФНО при введении в сустав экспериментальным животным проявляют синергическую активность, индуцируют развитие транзиторного синовита с лейкоцитарной инфильтрацией суставной ткани. При этом одной из причин лейкоцитарной инфильтрации синовиума является способность этих цитокинов усиливать экспрессию молекул адгезии (IСАМ-1, ELAM-1 и V-CAM) на мембранах эндотелиальных клеток сосудов синовиальной оболочки и их лейкоцитарных лигандов (глава 1), индуцировать синтез хемотаксических факторов (ИЛ-8 и моноцитарный активирующий фактор). Кроме того, эти цитокины стимулируют продукцию фактора роста фибробластов и медиаторов воспаления ПГЕ2. С помощью разнообразных методических подходов, включающих использование соответствующих ДНК-зондов, позволяющих оценивать экспрессию иРНК цитокинов, биологических и иммунохимических методов, было показано, что ИЛ-1-b и ÔÍÎ-a активно синтезируются клетками синовии. При этом основным источником цитокинов в синовиальной мембране являются клетки моноцитарно-макрофагального ряда. Примечательно, что ФНО вырабатывается "палисадными" клетками, обнаруживаемыми в избыточном количестве на стыке между паннусом и суставным хрящом, то есть в той зоне, с которой начинается деструкция сустава. В экспериментальных исследованиях было показано, что у трансгенных мышей, которым был инокулирован ген ФНО-а человека, в возрасте 4 нед. развивается артрит, напоминающий ревматоидный, который может быть предотвращен введением моноклональных антител к ФНО-а (J. Keffler и соавт., 1991). Кроме того, моноклональные антитела к ФНО подавляют развитие экспериментального коллагенового артрита (R. Williams и соавт., 1991). ÈË-1-b и ФНО-а являются мощными индукторами синтеза ИЛ-6, концентрация которого тесно коррелирует с клиническими и лабораторными параметрами активности воспалительного процесса при РА. Кроме того, ИЛ-6 наряду с ÈË-1-b принимает участие в развитии остеопороза, характерного для РА. Способность ИЛ-6 регулировать дифференцировку Вклеток в плазматические клетки также может иметь патогенетическое значение, так как может вызывать синтез РФ и гипергаммаглобулинемию. Обнаружена тесная корреляция между увеличением концентрации ОФБ в плазме и скоростью прогрессирования суставной деструкции. Другой цитокин, ГМ-КСФ, наряду с ИФ-у и ФНО-а принимает участие в процессах клеточной активации и вызывает гиперэкспрессию молекул класса II ГКГ на мембранах различных клеток. Эти, а также некоторые другие эффекты ГМ-КСФ (дегрануляция нейтрофилов, стимуляция ангиогенеза) свидетельствуют о его важной роли в патогенезе PA (J. A. Hamilton, 1993). Сведения, касающиеся возможной роли провоспалительных цитокинов в развитии РА, суммированы в таблице 14.1. Таблица 14.1. Патогенные эффекты провоспалительных цитокинов при РА Эффекты Цитокины ИЛ-1 ФНО-а ИЛ-6 ГМ-КСФ Активация МН/МФ + + + Мононуклеарно-клеточная инфильтрация + + + + Фиброз/гиперплазия синовиума + + +/Разрушение хряща + + +/+/ Резорбция кости + + ? +/Наконец, в последние годы получены многочисленные экспериментальные факты, свидетельствующие о важной роли неиммунных механизмов в прогрессировании патологического процесса при РА. Так, например, при изучении артрита у MRL/lpr мышей отмечено, что гиперплазия синовиальных выстилающих клеток, ассоциирующаяся с деструкцией сустава, наблюдается в отсутствие инфильтрации Т-клетками, В-клетками или плазматическими клетками. Синовиоциты у больных РА обладают свойствами трансформированных клеток, а в синовиальной жидкости и тканях обнаружено увеличение уровня тромбоцитарного фактора роста и фактора роста фибробластов. Примечательно, что некоторые цитокины обладают способностью вызывать длительно сохраняющиеся фенотипические изменения клеток. Например, индуцированный ФНО-а синтез ПГЕ2 фибробластами устойчиво персистирует в течение более чем 20 нед. в отсутствие повторной стимуляции ФНОа, а другой цитокин — ИФ-а вызывает очень длительное подавление синтеза коллагена фибробластами. Совсем недавно W. J. Koopman (1994) сформулировал концепцию, согласно которой Т-зависимый иммунный ответ, в котором участвуют активированные неизвестным этиологическим агентом СD4+Т-лимфоциты и моноциты/макрофаги, играет ведущую роль на ранних стадиях РА, в то время как на поздней стадии болезни начинают преобладать автономные, независимые от Т-клеток патологические процессы, вероятно, связанные с нарушением взаимодействия синовиальных клеток и компонентов внеклеточного матрикса. Таким образом, при РА и, вероятно, других аутоиммунных ревматических заболеваниях иммунотерапевтические методы лечения должны быть в первую очередь направлены на раннее селективное подавление активации Т-лимфоцитов и макрофагов, ЭК и снижение синтеза или инактивацию провоспалительных цитокинов (см. также главу 1). Частично эти задачи решаются с помощью НПВС, ГК. и базисных противоревматических препаратов, механизмы действия которых рассмотрены в предыдущих главах монографии. Однако ни одно из существующих в настоящее время лекарственных средств или их комбинаций не позволяет эффективно подавить прогрессирование иммунопатологического процесса при воспалительных ревматических заболеваниях. Все это вместе взятое и послужило основанием для разработки новых подходов к иммунотерапии этих болезней. Конкретные подходы к иммунотерапии связаны с использованием моноклональных антител к широкому спектру мембранных антигенов мононуклеарных клеток и эндотелия, антител к цитокинам, естественных лигандов цитокиновых рецепторов и растворимых антагонистов цитокинов или химических веществ, обладающих иммуномодулирующей активностью. Предполагается, что введение антител может не только вызывать элиминацию соответствующих клеток-мишеней, но и вести к изменению их функциональной активности (H. Waldman, 1989). Практически все рассмотренные ниже методы прошли экспериментальную проверку на лабораторных моделях аутоиммунной патологии (коллагеновый типа II артрит, волчаночноподобное заболевание у NZB/NZW и MRL мышей и др.) (C. Auffray и соавт. 1991; Steinmann, 1990; D. Wofsy и N. L. Cameron, 1990), а также 1 или II фазу клинических испытаний. 14.1. Моноклональные антитела 14.1.1. Антитела к CD4 Наиболее обширные исследования, включающие уже более 300 больных РА, касаются использования моноклональных антител к CD4 (F. C. Breedvald и R. R. P. De Vireis, 1992). CD4 — молекула мембранного гликопротеина с молекулярной массой 55 kD, который экспрессируется преимущественно на Т-лимфоцитах, обладающих хелперной/индукторной активностью, и в меньшей степени на моноцитах и эозинофилах. Естественным лигандом для CD4 являются молекулы класса II ГКГ, экспрессирующиеся на мембранах АПК, которые также контактируют с Т-клеточными рецепторами (ТКР). Тесный физический контакт между CD4, ТКР и CD3, опосредуемый молекулами класса II ГКС, инициирует активацию CD4+ Т-лимфоцитов, в реализации которой участвует протеинкиназа (р56), ассоциирующаяся с цитоплазматической частью молекулы CD4. Предполагается, что связывание антител с CD4 позволяет, во-первых, предотвратить ассоциацию CD4 с TKP/CD3, что необходимо для активации Т-лимфоцитов; во-вторых, вызывает реактивацию протеинкиназы (ð56), что в свою очередь приводит к развитию рефрактерности к последующей активации клеток через TKP/CD3 комплекс; в-третьих, индуцирует апоптоз этих клеток, что может иметь значение для формирования состояния толерантности к мишеневым антигенам. (M. K. Newell и соавт., 1990; E. Chou и соавт., 1992). В настоящее время в клинической практике используются 3 типа моноклональных антител к CD4: 1) мышиные моноклональные антитела (M-T151) (C. Reiter и соавт., 1991; G. Horneff и соавт., 1991); их основным недостатком является антигенность, потенциально приводящая к снижению эффективности и увеличению риска анафилактических реакций при повторных циклах лечения; 2) химерные антитела (сМ-Т412; Centocor, США), состоящие из антигенсвязывающего фрагмента мышиных антител к CD4, соединенного с константным участком IgG1 каппа человека; химерные антитела менее антигены, чем мышиные, и имеют большую продолжительность жизни; 3) приматизированные антитела, состоящие из вариабельного участка иммуноглобулина макаки и константного участка молекулы иммуноглобулина человека (PRIMATIZED IDECCE9.1; IDEC Pharm. Comp. и SmithKline Beecham, США). Введение мышиных анти-CD4-антител вызывает быстрый клиренс CD4+ Т-лимфоцитов. Через 30 мин. после инфузии 90% клеток исчезают из циркуляции, но через 24 ч число клеток постепенно увеличивается, достигая 50% от исходного. После 7-дневного курса лечения низкий уровень CD4+T-ëèìôoöèòoâ сохраняется примерно в течение 2 мес. Введение химерных антител вызывает более длительную элиминацию CD4+Tëèìôoöèòîâ, чем введение мышиных антител, что, как уже отмечалось, связано с большей (в 5-6 раз) продолжительностью жизни химерных антител. Уменьшение количества CD4+T-клеток наблюдается в течение 6-18 мес. и всегда коррелирует с вводимой дозой препарата (P. Van der Lubbe и соавт., 1993). Кроме того, на фоне лечения развивается своеобразный феномен, заключающийся в исчезновении CD4-маркера с мембраны Тлимфоцитов. Клиническая эффективность В большинство исследований были включены больные с тяжелым активным РА, рефракторным к базисным препаратам. Схема лечения заключалась в назначении препарата антител в дозе от 10 до 100 мг в день (общая доза 70-700 мг, курс лечения составлял 5-7 дней). Суммарный анализ клинических результатов лечения РА свидетельствует о несомненной эффективности, не зависящей от типа применяемых антител. В целом клиническое улучшение, длящееся в среднем до 30 мес. после одного курса терапии, наблюдалось у 60-75% больных (G. Horneff и соавт., 1991; C. Reiter и соавт., 1991; D. Wendling и соавт., 1991). Наиболее существенными клиническими эффектами были уменьшение числа воспаленных суставов, снижение выраженности синовита (G. R. Burmester и F. Emmrich, 1991), уменьшение индекса Ричи, увеличение силы сжатия кисти. Описана положительная динамика некоторых внесуставных проявлений РА (язвы голени). Van der Lubbe и соавт. (1993) использовали антитела сМ-Т412 для лечения 32 больных РА. Препарат назначали в дозе 10, 50 или 100 мг в течение 7 дней. Отмечена хорошая переносимость лечения; снижение концентрации CD4+T-ëèìôoöèòoâ не приводило к развитию признаков иммунологической недостаточности и не коррелировало с клинической эффективностью. При анализе клинических данных отмечено, что снижение более чем на 20% индекса Ричи и числа воспаленных суставов наблюдалось у 19 из 29 больных, более чем на 30% — у 7 пациентов. У 9 больных снижение активности сохранялось более 4 мес. E. H. S. Choy и соавт. (1994) применили ñÌ-Ò412 у 20 больных с рефрактерным РА. Вначале пациенты в течение 5 дней получали 50 мг антител, а затем в одной группе больных препарат в этой дозе вводили еженедельно (5 нед.), а в другой — ежедневно. Примерно одинаковый клинический эффект был çàðåãèñòðèðîâàí в обеих группах. В другом исследовании этих же авторов было установлено, что эффективная доза антител составляет не менее 50 мг в день в течение 5 дней. Снижение уровня CD4+ Т-клеток в синовиальной жидкости наблюдается только при длительном (не менее 5 дней) введении антител (E. H. S. Choy и соавт., 1992). W. Moreland и соавт. (1993) изучали эффективность ñÌ-Ò412 (10-700мг) в рамках 1 фазы испытаний у 25 больных с рефракторным РА. У 43% пациентов через 5 нед. лечения наблюдалось 50% улучшение суставного счета, сохранявшееся у трети больных через через 6 мес. после инфузии антител. Однако динамики СОЭ, силы сжатия кисти, продолжительности утренней скованности и интенсивности боли не отмечено. Корреляции между числом СD4+Т-лимфоцитов в циркуляции и клиническим эффектом не обнаружено; титры РФ и СОЭ, уровень иммуноглобулинов, компонентов комплемента и концентрации цитокинов (за исключением ИЛ-6) существенно не менялись. Предварительные результаты, полученные E. Hiepe и соавт. (1993), свидетельствуют о принципиальной возможности лечения антителами к CD4 больных СКВ. Введение антител в дозе 0.3 мг/кг/день в течение 7 дней 3 больным активным волчаночным нефритом, рефракторным к ГК и цитотоксическим препаратам, привело к снижению активности по индексу SLAM, уменьшению выраженности протеинурии и снижению титров антител к ДНК. Однако через 4 нед. у всех больных наступило обострение. Клинические результаты совпадают с данными экспериментальных исследований (Y. Tomer и соавт., 1994) о способности анти-СD4 предотвращать развитие экспериментальной СКВ и АФС, индуцированных моноклональными анти-ДНК антителами. H. S. Choy и соавт. (1991) представили результаты лечения одного больного с рецидивирующим полихондритом, резистентными к высоким дозам ГК и МТ. На фоне 3 введений антител в дозе по 50 мг/кг отмечена положительная динамика клинических симптомов. Побочные эффекты Побочные эффекты наблюдаются у большинства больных, но они обычно мягкие и не приводят к прерыванию лечения. Их можно разделить на 2 основные группы. Эффекты, непосредственно связанные с введением препарата (в первый день инфузии), могут проявляться преходящей лихорадкой, сочетающейся с тошнотой, рвотой, миалгией, транзиторной гипотензией, аллергическими кожными реакциями. Они ассоциируются с умеренной гиперпродукцией цитокинов, в первую очередь ИЛ-6 (L. W. Moreland и соавт., 1993; G. Horneff и соавт., 1991). Описано развитие интерстициального нефрита, приведшего к острой почечной недостаточности через 4 нед. после окончания лечения. Отдаленные побочные эффекты чаще развиваются при использовании моноклональных мышиных антител и определяются синтезом антител к мышиному иммуноглобулину. Однако, уровень этих антител, как правило, невысок, не более 2-3 мг/л. Поэтому препарат может назначаться повторно без снижения эффективности (C. Reiter и соавт., 1991; G. Hofner и соавт., 1991). Описан один больной, который погиб через 18 мес. после однократного введения антител от стафилококковой пневмонии и геморрагического инсульта. Однако этот больной страдал очень тяжелым РА и в течение длительного времени получал различные базисные противоревматические препараты. 14.1.2. Антитела к CDw52 (САМРАТН-1Н; Burroughs Wellcome. Великобритания) ÑÀÌÐÀÒÍ-1H-IgG1, представляющие собой гуманизированные моноклональные антитела к специфическому гликопротеину CDw52, присутствующему на мембранах лимфоцитов и моноцитов (G. Hale и соавт., 1990). В рамках 1 фазы клинических испытаний J. D. Issacs и соавт. (1992) оценили различные режимы лечения у 7 больных рефракторным РА. Препарат вводили внутривенно в течение 5 дней по 4мг/день, а затем 5 дней по 8мг/день. Второй курс длился 5 дней, доза антител составила 40 мг/день. У 7 больных был получен определенный клинический эффект, сохраняющийся в течение 18 мес. Е. Soriano и соавт. (1993) оценили действие однократного введения GAMPATH (3-100 мг) у 7 больных с рефракторным РА. В 5 из 7 случаев зарегистрирован кратковременный клинический эффект (по критериям Паулюса) и снижение числа лимфоцитов. У всех больных введение препарата сопровождалось побочными явлениями (у 2 пациентов лечение было приостановлено из-за бронхоспазма). М. Е. Weinblat и соавт. (1993) провели испытание однократного введения GAMPATH (1-100 мг) у 13 больных РА и отметили временное клинический эффект в 7 случаях. При лабораторном исследовании выявлено развитие лимфопении (СDЗ+Т-клетки, СD4+Т-клетки, CD8+T-клетки), сохраняющейся в течение 1-8 мес., без изменения числа моноцитов (CD14+), В-лимфоцитов (CD19+) и ЕК-клеток (CD16+). При этом динамика концентрации СD4+Т-лимфоцитов не коррелировала с активностью процесса и клинической эффективностью. Недавно J. D. Issacs и соавт. (1994) представили предварительные результаты II фазы клинического испытания GAMPATH, проведенного у 41 больного РА в течение 4 мес. Установлена примерно одинаковая клиническая эффективность различных доз препарата (100, 250 и 400 мг). У большинства больных в первый день введения антител наблюдались побочные эффекты, проявляющиеся гриппоподобными симптомами в сочетании с умеренным снижением АД ( у 16 больных). У 1 больного развился гемолитико-уремический синдром. Несмотря на персистирующее снижение концентрации CD4+ Т-лимфоцитов, инфекционные осложнения наблюдались не часто. У 11 больных в течение первых 10 дней лечения появились признаки легкой герпетической инфекции. Лишь у 4 пациентов в более поздний период развились более серьезные инфекционные осложнения (кокцидомикоз — у 1, вирусная пневмония у 1, цитомегаловирусная инфекция — у 1, кандидоз слизистых — у 1). По данным Mathieson и соавт. (1990), использование GAMPATH-1Н позволяет достигнуть длительной ремиссии у больных с тяжелым ревматоидным васкулитом. 14.1.3. Антитела к СD5-иммуноконъюгат (СD5 Плюс) (ХОМА Corporation, США) Антитела СD5-иммуноконъюгат представляют собой мышиные моноклональные антитела (IgG1), конъюгированные с рицином А иммунотоксином, являющимся мощным ингибитором белкового синтеза, что позволяет добиться более выраженной элиминации мишеневых клеток (V. H. Byers и соавт., 1990). Они специфичны в отношении молекулы CD5, которая экспрессируется на Т- лимфоцитах и определенной популяции В-лимфоцитов, принимающих участие в синтезе некоторых типов аутоантител (РФ и антитела к ДНК). Strand и соавт. (1993) применили антитела к CD5 у 79 больных активным РА по следующей схеме: 0.20 мг/кг/день и 0.33 мг/кг/день (максимальная дневная доза 25 мг/кг/день). При этом 76 из 79 больных получили по крайней мере 4 дозы. При анализе результатов установлено, что клинический эффект развивается весьма быстро (к 8-му дню лечения) и более очевиден у больных с ранним (до 3.5 года), чем с поздним РА. Побочные эффекты проявлялись в основном конституциональными симптомами, небольшими отеками, сыпью, миалгиями и развивались сразу после введения антител. Их частота была выше при использовании высокой дозы препарата (0,33 мг/кг/день). В 6 случаях они явились причиной прекращения лечения. По мнению авторов, достоинством лечения антителами к CD5 является быстрое достижение и достаточно длительное сохранение клинического эффекта терапии. Недавно N. J. Olsen и соавт, (1994) обобщили результаты многоцентрового исследования антител к CD5, в которое вошли 109 больных с ранним (менее 8 лет) РА. Установлено, что клинический эффект коррелировал со степенью снижения количества CD3+ Т-лимфоцитов, однако в целом он был не очень выражен. Существенным ограничивающим фактором явился иммунный ответ на вводимые антитела. По данным M. Wacholtz и соавт. (1992), введение антител к CD5 в дозе 0,1 мг/кг/день 4 больным волчаночным нефритом позволило достигнуть стойкой ремиссии у 1 больного и временного улучшения у 2. 14.1.4. Антитела к ФНО-а Разработано 2 типа антител к ФНО-a: 1) химерные (мышь/человек) моноклональные антитела (сА2), состоящие из константной области IgGk человека и Fv-ôðàãìåíòà высокоаффинных мышиных антител к ÔÍÎ-a человека; 2) рекомбинантные гуманизированные моноклональные антитела к ÔÍÎ-a. Механизм действия антител связывают с их способностью нейтрализовывать ФНО-а во внутрисосудистом пространстве и синовиуме и тем самым подавлять развитие воспалительных реакций. Химерные моноклональные антитела (сА2; Centocor, США) Для первичной оценки переносимости и эффективности у 20 больных РА, резистентных к базисной терапии, препарат вводили по 10 мг/кг 2 раза или по 5 мг/кг 4 раза (общая доза 20 мг/кг) в течение 8 нед. (M. Elliot и соавт., 1993). Отмечена хорошая переносимость лечения, практическое отсутствие побочных эффектов и существенная положительная динамика клинических (индекс Ричи, утренняя скованность, счет боли, суставной счет, данные опросника состояния здоровья) и лабораторных (снижение уровня СРБ, САБ и ИЛ-6) показателей. Анализ предварительных результатов повторных курсов лечения (из 7 больных 4 — закончили 4 повторных цикла терапии) свидетельствует о стойком клиническом эффекте, сохраняющемся в течение 12 мес. после введения 20мг/кг антител (M. Elliot и соавт., 1994). Недавно были подведены итоги многоцентрового рандомизированного двойного слепого контролируемого исследования эффективности лечения (доза антител варьировала от 1 до 100 мг/кг/день), в которое вошли 73 больных РА. Полученные результаты подтвердили мнение о том, что инактивация ФНО-а является высокоэффективным и нетоксичным методом лечения PA (M. J. Elliot и соавт., 1994). Создалось впечатление, что темпы клинического улучшения и его выраженность при лечении антителами были существенно выше, чем при лечении базисными противоревматическими препаратами. В другом исследовании была продемонстрирована возможность повторных введений антител, которые эффективно подавляют обострения PA (M. J. Elliot и соавт., 1994). Установлено, что лечение антителами ñÀ2 сопровождается выраженным снижением концентрации СРБ и САБ, коррелирующим со снижением уровня ИЛ-6 (M. J. Elliott и соавт., 1993). На фоне лечения отмечено развитие своеобразной побочной реакции, заключающейся в появлении в сыворотках больных, которым проводились повторные курсы, антител к ДНК и аФЛ, но в отсутствие клинических проявлений СКВ. Гуманизированные моноклональные антитела (CDP571 lgG4; Celltech, Великобритания) Проведено двойное слепое контролируемое испытание антител CDP571 у 36 больных PA (E. C. C. Rankin и соавт., 1994). Больные были рандомизированы на несколько групп в зависимости от доз антител (0.1, 1.0 и 10 мг/кг антител) или плацебо. Положительный клинический эффект проявлялся в течение первой недели после введения препарата, нарастал при увеличении дозы и длился 8 нед. O. Williams и соавт. (1994) показали, что сочетанное введение антител к ФНО-а и антител к CD4 вызывает существенно более выраженное подавление экспериментального коллагенового артрита, чем введение только антител к ФНО-а. Эти результаты могут иметь очень важное значение для разработки подходов к комбинированной терапии РА. 14.1.5. Антитела к CD54 (IСАМ-1 (BIRRR1. Boehringer Ingelheim, Германия) Проведена I/II фаза испытаний моноклональных мышиных антител к ICAM-1 (A. Kavanaugh и соавт., 1994) у 32 больных РА. Препарат ввводили внутривенно в течение 5 или 1-2 дней. В группе 5-дневного режима к 8-му дню улучшение наблюдалось у 13 из 24 больных. У 3 пациентов эффект сохранялся до 90-го дня. Побочные явления наблюдались у большинства больных, но были умеренными и включали лихорадку, головные боли, тошноту. Недавно появились сообщения об определенной эффективности терапии у 7 из 10 больных ранним РА (давность заболевания меньше 1.5 года). Интересно, что у 3 больных клиническое улучшение сохранялось длительное время (5-8 мес.) после завершения лечения, а у 1 пациента удалось добиться развития длительной ремиссии (более 5 мес.) (A. Kavanaugh и соавт., 1994). В целом в группе больных с ранним РА степень клинического улучшения была выше, чем у больных с поздним, рефракторным к предшествующей терапии PA (A. Kavanaugh и соавт., 1994). 14.2. Цитокиновые рецепторы и антагонисты цитокиновых рецепторов 14.2.1. Рецептор ФНО (ФНО: Fс) (Immunex, США) Препарат представляет собой рекомбинантный ФНО- рецептор человека, состоящий из 2 идентичных цепей мономера р80 ФНО, соединенный Fc-ôðàãìåíòîì IgG1 человека. Благодаря этому препарат имеет очень высокую авидность к ФНО-а. W. Moreland и соавт. (1994) недавно представили результаты 1 фазы клинического испытания ФНО: Fс у 16 больных с активным РА, которые ранее не отвечали ни на один базисный противоревматический препарат. ФНО: Fс назначали вначале по 4, 8, 16 или 32 мг/м2 два раза в неделю в течение 4 нед. В целом по группе зарегистрировано достоверное улучшение большинства клинических показателей, а побочные реакции практически отсутствовали, за исключением небольшой сыпи в месте инъекции препарата. 14.2.2. DAB486 ИЛ-2 (Seragen, США) DAB486 ИЛ-2 — молекула, полученная генноинженерным путем, состоящая из полипептидного фрагмента ( молекулярная масса 68 kD) ИЛ-2 и АДФ-рибозилтрансферазы дифтерийного токсина (D. P. Williams и соавт., 1987). Последний, связываясь с фактором элонгации 2, подавляет синтез белка. Эта гибридная молекула интернализуется в клетки, экспрессирующие высокоаффинные ИЛ-2Р и не реагирует с клетками, экспрессирующими низкоаффинные ÈË-2Ð (р55) и ÈË-2Ð с промежуточной аффинностью (р75). Основным свойством DАВ486ИЛ-2 является цитотоксическая активность по отношению к клеткам, экспрессирующим высокоаффинные ÈË-2Ð. Поскольку последние экспрессируются на Т-и В-лимфоцитах, моноцитах и некоторых опухолевых клетках только после их активации, DАВ486ИЛ-2 селективно разрушает только активированные клетки иммунной системы. Предполагается, что элиминация только ИЛ-2Р-позитивных активированных клеток приводит к селективной иммуносупрессии, поскольку не затрагивает "покоящиеся" лимфоциты, клетки "памяти" и неактивированные лимфоциты. L. Sewel и соавт. (1993) провели I/II фазы клинического испытания DAB486 ИЛ-2 (препарат вводили внутривенно в дозах 0.1, 0.07 или 0.04 мг/кг в день в течение 7 дней 19 больным РА, рефракторным к МТ). 13 больным проведен дополнительный курс лечения. Клинический эффект был зарегистрирован у 9 из 19 пациентов, получавших высокие/средние дозы DAB486HA-2. Побочные эффекты включали повышение уровня печеночных ферментов (55%), тошноту и рвоту (30%), аллергические реакции и тромбоцитопению (5%). У всех больных была обнаружена продукция антител к препарату, которые, однако, не ослабляли его цитотоксической активности. Недавно разработана улучшенная версия препарата — DAB389 ИЛ-2, который представляет собой более короткий пептидный фрагмент ИЛ-2, состоящий из 97 аминокислот. Установлено, что этот препарат обладает в 10 раз более выраженной цитотоксической активностью и аффинностью к ÈË-2Ð, имеет более широкий терапевтический индекс, чем DAB486 ИЛ-2. В экспериментальных исследованиях продемонстрирована более высокая активность DAB389-ИЛ-2 на модели коллагенового артрита. Данные клинических исследований в рамках 1/11 фаз клинических испытаний также свидетельствуют об эффективности DAB398 ИЛ-2 при PA (K. L. Sewel и соавт., 1994) и о возможности комбинированной терапии DAB398 ИЛ-2 в сочетании с МТ (J. M. Kremer и соавт., 1994). 14.2.3. Растворимые ИЛ-2Р (ANAKIRA; Synergen, США) Проведено 7-недельное двойное слепое контролируемое исследование рекомбинантного препарата ANAKIRA (20, 70 или 200 мг 7, 3 или 1 раз в неделю, а затем по 1 разу в неделю), в которое вошло 175 больных PA (G. Gampion и соавт., 1994). В целом отмечены эффективность препарата и удовлетворительная переносимость, причем ежедневное применение препарата было более эффективно, чем еженедельное. 25 больных выбыли из исследования: 13 — из-за побочных явлений, 12 — из-за отсутствия эффекта. Наиболее частой побочной реакцией было поражение кожи, которое послужило причиной отмены препарата у 8 больных. 14.2.4. Антагонист ИЛ-1P (Synergen, США: Immunex Res., США) Проведено двойное слепое контролируемое исследование рекомбинантной формы естественного антагониста ИЛ-1 рецептора у 175 больных PA (M. E. Lebsack и соавт., 1993). Лечение было прервано из-за неэффективности лишь в 6% случаев. Побочные реакции также отмечены лишь у 6% больных: у 3 пациентов — инфекция мягких тканей, у 1 — боли в грудной клетке, у 1 — простатит, у 1 — мочевая инфекция. Установлена оптимальная доза препарата: 20-200 мг 7 раз в неделю). Drevlon и соавт. (1993) провели двойное слепое контролируемое испытание внутрисуставного введения препарата у 16 больных РА в дозе 25, 100, 250- или 500 мг. Зарегистрировано достоверное уменьшение окружности коленного сустава при введении дозы 250 мг, признанной оптимальной. Побочные эффекты в виде контактного дерматита развились только у 2 больных. 14.3. Вакцинация пептидом ТКР Vb 17 (Immune Response Corporation, США) L. W. Moreland и ñîàâò. (1994) провели изучение переносимости, эффективной дозы и биологического действия пептида, имеющего 17 общих аминокислот с Т-клеточным рецептором Vbl7, которым иммунизировали 15 больных РА. Схема лечения: 4 дозы пептида (10, 30, 100 или 300 мкг) в неполном адъюванте Фрейнда, затем через 4 нед. бустерное введение пептида. Определенный клинический эффект достигнут у всех больных через 48 нед. при отсутствии побочных реакций. Через 6 нед. у половины больных отмечено появление Т-клеточной пролиферации при стимуляции пептидом. Кроме того, у пациентов, получивших высокую дозу препарата, снизилось количество ИЛ-2Р+Vb17 Т-лимфоцитов. 14.4. Препараты ИФ Механизм действия: 1. подавление синтеза эйкозаноидов макрофагами (J. Browning, 1987); 2. подавление синтез ИЛ-1 активированными моноцитами/макрофагами (F. M. Rowe и соавт., 1987; S. Ruschen и соавт., 1989); 3. подавление ИЛ-4-зависимой пролиферации В-лимфоцитов; 4. индукция синтеза ингибитора С 1; 5. активация и дифференцировка Т-супрессоров и естественных супрессорных клеток (T. Noma и M. E. Dorf, 1985); 6. ингибиция синтеза коллагена, поздней пролиферации фибробластов; 7. ингибиция миграции макрофагов/моноцитов; 8. десентизация С5а рецепторов. У больных РА наблюдается очень низкая концентрация ИФ-у в синовиальной жидкости (G. S. Firestein и соавт., 1990). Предполагают, что дефект синтеза ИФ-y является одним из факторов неконтролируемой гиперпродукции ИЛ-1 при РА. По данным S. M. Wahl и соавт. (1991), ИФ-у подавляет развитие артрита, индуцированного мембраной стрептококка. РА Эффективность ИФ-у при РА вначале была продемонстрирована в краткосрочных открытых испытаниях в рамках фазы II, в которое вошло 922 больных (E. M. Lemmel и соавт., 1987), а затем и в нескольких двойных слепых контролируемых испытаниях (III фаза) (Е. М. Lemmel и соавт., 1988; G. W. Cannon и соавт., 1989; Е. М. Veys и соавт., 1988; K. P. Machold и соавт., 1992), а также длительных проспективных (12-24 мес.) исследованиях (R. Sprekeler и соавт., 1990; G. W. Cannon и соавт., 1990). M. Lemmel и соавт. (1988) провели 28-дневное испытание ИФ-у в дозе 50 мг (20 дней), затем по 50 мг через день у 40 больных РА в сравнении с 39 больными, получавшими плацебо; отмечен статистически достоверный клинический эффект у больных, леченных ИФ-у. G. M. Cannon и соавт., (1989) представили результаты лечения 105 больных, среди которых 54 — получали ИФ-у (по 100 мг 5 дней в неделю); группу плацебо составил 51 пациент. Различия в эффективности лечения между основной и контрольной группами в пользу основной были достоверными только по результатам Стенфордского опросника состояния здоровья; к концу испытания СОЭ существенно увеличилась в обеих группах. Е. М. Veys и соавт. (1988) сравнили эффективность ИФ-y и плацебо у 26 больных РА, разделенных на группы по 13 человек. Продолжительность лечения составила 24 нед., а доза ИФ-у — 100 мг в день в течение 5 дней, а затем 2 раза в неделю. Отмечено достоверное уменьшение суставного счета в группе, получавшей ИФ-у. E. M. Lemmel и соавт. (1992) обобщили результаты многоцентрового двойного слепого контролируемого исследования, в которое вошли 107 больных РА, леченных ИФ-у, и 116 больных, получавших плацебо. Продолжительность лечения составила 3 мес., а схема лечения заключалась во введении 50 мг рекомбинантного ИФ-у (2õ10 7 IU/ìã) подкожно 7 раз в неделю (3 нед.), 3 раза в неделю (4 нед.) и 2 раза в неделю (5 нед.). Отмечено статистически значимое улучшение всех клинических параметров и возможность снижения дозы ГК у больных РА, получавших ИФ-у, по сравнению с пациентами, получавшими плацебо. За исключением умеренного повышения температуры тела и гриппоподобных симптомов, различий между основной и контрольной группами по частоте побочных эффектов не отмечено. Однако в длительном проспективном наблюдении за больными (2- 5 лет), закончившими 12-недельное контролируемое испытание (G. M. Cannon и соавт., 1990, 1991), отмечено прогрессирующее снижение эффективности лечения ИФ-у. Эти результаты указывают на необходимость продолжения исследований, направленных в первую очередь на отработку оптимальных терапевтических схем лечения ИФ-у больных PA (E. M. Veys и соавт., 1992) и возможностей комбинации ИФ-у с другими базисными противоревматическими препаратами. Имеются данные об определенном терапевтическом эффекте ИФ-y У БОльных ЮХА (Pernice и соавт., 1989). Положительные результаты были зарегистрированы у 7 из 9 детей, получавших препарат в дозе 0.5 мкг/кг ежедневно в течение 3 нед., а затем 3 раза в неделю в течение 3 нед. ССД В открытом неконтролируемом исследовании ИФ-у у 10 больных ССД в течение 6 мес. (A. Kahan и соавт., 1989) препарат вводили 1 раз в день внутримышечно вначале в дозе 10 мкг с постепенным увеличением до 100 мкг/день. Через 6 мес. зарегистрированы достоверная положительная динамика кожных и мышечноскелетных проявлений, некоторое улучшение состояния почек при отсутствии уменьшения выраженности феномена Рейно и улучшения функционального состояния легких. Проведена 1/11 фазы испытания ИФН-у при ССД, длившегося 18 нед. В исследование было включено 19 больных с быстропрогрессирующей ССД. 14 больных получали препарат по крайней мере 16 нед. (B. Freundlich и соавт., 1992). ИФ-у вводили внутримышечно 3 раза в неделю: 11 больных получили 0.1 мг/м2, 8 — 0.5мг/м2 На фоне лечения отмечена достоверная положительная динамика кожного синдрома. Однако у 3 — развился почечный криз, у 1 — инфаркт миокарда. Частыми побочными эффектами были гриппоподобные симптомы (лихорадка, озноб, головные боли, слабость, миалгии), которые развивались в течение 1-2 ч после введения препарата и длились 13 ч. Лечение было прервано у 5 больных, у 1 — из-за очень тяжелых гриппоподобных симптомов, у 1 — из-за инфаркта миокарда, у 3 — из-за почечного криза. Таким образом, вопрос об эффективности ИФ-у при ССД остается открытым. Shiozawa и соавт. (1993) провели 12-недельное двойное слепое контролируемое исследование ИФ-а в низких дозах (5х10 5 IU 2 раза в неделю) при РА и отметили статистически достоверную положительную динамику суставного синдрома, снижение уровня СРБ и количества тромбоцитов. Однако в исследованиях других авторов было показано, что лечение высокими дозами ИФ-а, напротив, может приводить к обострению PA (Verrazzi и соавт., 1994). С. Ferri и соавт. (1993) провели контролируемое перекрестное исследование эффективности ИФ-а у 26 больных смешанной криоглобулинемией, большинство из которых были позитивны по вирусу гепатита С. Схема лечения заключалась в назначении ИФ-а в дозе 2õ10 6 ME ежедневно в течение месяца, затем через день в течение 5 мес. На фоне лечения у 20 больных зарегистрировано достоверное уменьшение выраженности пурпуры, снижение концентрации трансаминаз и криоглобулинов и исчезновение вируса гепатита С у 2. После прекращения лечения наблюдалось развитие обострения. По данным P. Cohen и соавт. (1994), обобщивших результаты многоцентрового исследования, положительный клинический эффект достигнут у 57% больных. Однако после прекращения лечения у подавляющего большинства пациентов очень быстро развивалось обострение. Отмечена высокая частота и тяжесть побочных эффектов. По мнению авторов, эти данные свидетельствуют о необходимости разработки альтернативных методов лечения смешанной криоглобулинемии. В то же время R. Misiani и соавт. (1994) обнаружили благоприятной действие ÈÔ-à 2а у 26 больных смешанной криоглобулинемией, которое проявлялось в исчезновении признаков кожного васкулита, маркеров гепатита С, уменьшении концентрации криоглобулинов и титров РФ. Проведено открытое исследование влияния ИФ-а (1õ10 6 ME) на ксеростомию у 10 больных с синдромом Шегрена (S. Shiozawa и соавт., 1993). У 6 больных на фоне лечения отмечен определенный клинический эффект. В отдельных клинических наблюдениях описаны положительные результаты лечения ИФ-а язвенного поражения слизистой полости рта при болезни Бехчета (V. Hamuryudan и соавт., 1990) и глазных проявлений заболевания (J. M. Durand и соавт., 1993). При проведении лечения препаратами ИФ (особенно ИФ-а) необходимо помнить об их потенциальной способности индуцировать развитие волчаночноподобных синдромов (L. E. Ronnblom и соавт., 1990; P. J. Schilling и соавт., 1991; L. E. Ronnblom и соавт., 1991), артрита (Y. Ueno и соавт., 1992; P. D. W. Kielly и F. E. Bruckner, 1994) или серологических нарушений, характерных для СКВ (M. R. Ehrenstein и соавт., 1993). Таким образом, в настоящее время в ревматологии используется широкий спектр методов иммунотерапии, однако для окончательного ответа на вопрос о месте того или иного подхода в лечении определенных аутоиммунных ревматических заболеваний необходимы специальные контролируемые исследования больших групп больных и в течение длительного времени. Последнее особенно важно для оценки влияния новых методов лечения на отдаленный прогноз заболеваний и развитие отсроченных побочных эффектов, связанных с длительной иммуносупрессией, в первую очередь инфекционных осложнений и онкологической патологии. Список литературы к главе "Иммунотерапия в ревматологии" 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. Arend W. P., Smith M. F., Janson R. W., Joslin F. G.: IL-I receptor antagonist and IL-I beta production in human monocytes are regulated differently. J. Immunol. 1991; 147: 1530-1535. Auffray С., Pitatier-Tonneau D., Kroemer G.: CD4-target immune intervention: a strategy for the therapy of AIDS and autoimmune disease. Trends Biotech. 1991; 9: 124-131. Breedveld F. C., De Vries R. R. P.: Anti-CD4 antibodies in rheumatoid arthritis. Clin. Exp. Rheumatol. 1992; 10: 325-326. Brennan F. M., Maini R. N., Feidman M.: TNF-alpha — a pivotal role in rheumatoid arthritis. Brit. J. Rheumatol., 1992; 31: 293-298. Browning J.: Interferons and rheumatoid arthritis: Insight into interferon biology?. Immunol. Today 1987; 8: 372-374. Burmester G. R., Emmrich F.: Anti-CD4 therapy in rheumatoid arthritis. Clin. Exp. Rheumatol., 1993; II (Suppl. 8): S139-S145 Byers V. S., Henslee P. J., Blazar B. D. et al.: Use of anti-pan-T lymphocyte ricin A immune toxin in steroid resistant acute graft-versus-host disease. Blood. 1990; 75: 1462-1432. Campion G.,Witt K., Musikis P. et al.: The effect of dose and dose frequency on disease activity in patients with rheumatoid arthritis (RA) with the subcutaneous administration of recombinant human Interleukin receptor antagonist (anakira). Brit. J. Rheumatol., 1994. (Suppl. 1); 33: 182. Cannon G. W., Schindler J. D., Emkey R. D. et al.: Double-blind trial of recombinant inter-feron- gamma versus placebo in rheumatoid arthritis. Arthritis Rheum. 1987; 30 (suppl.): S-18. Cannon G. W., Emkey R. D., Denes A. et al.: Prospective two-year following of recombinant interferongamma in rheumatoid arthritis. J. Rheumatol. 1990; 17 (suppl. 30): 304-310. Choy E., Adjaye J., Kingsiey G., Panayi G.: Chimeric anti-CD4 antibody acts by causing apoptosis (programmed cell death) of human lymphocytes. Arthritis Rheum., 1992; 35: S44 (abst.). Choy E. H. S., Chicanza I. C., Kingsley G. H. et al. Treatment of rheumatoid arthritis with single dose or weekly pulses of chimeric anti-CD4 monoclonal antibody. Scand. J. Immunol., 1992; 36: 291-298. Choy E. H. S., Pitxalis C., Bijl J. A. et al. The importance of dose and dosing regimen of anti-CD4 monoclonal antibody (mAb) in the treatment of rheumatoid arthritis. Brit. J. Med., 1994. (Suppl. 1); 33: 294. Choy E. H. S., Chikanza I. C., Kingsley G. H., Panayi G. S.: Chimeric anti-CD4 monoclonal antibody for relapsing polychondritis. Lancet, 1991; 338: 450. Cohen P., Nguyen Q. T., Roulot D. et al.: Treatment od essential mixed cryoglobulinemia by interferon alpha. An open multicenter prospective trial. Arthritis Rheum. 1994; 37 (Suppl.): S270. Dreviow B., Capezio J., Lovis R. et al. Phase I study of recombinant human interleukin-I receptor (rhu IIIR) administered intra-articulary in active rheumatoid arthritis. , Arthritis Rheum. 1993; 36 (suppl.): S-39. Durand J. M., Kaplanski G., Soubeyrand J.: Beneficial effects of interferon-alfa2b in Behcet's disease. Arthritis Rheum. 1993; 36: 1025-1026. Ehrenstein M. R., McSween E., Swana M. et al.: Appearance of anti-DNA antibodies in patients treated with interferon-alpha. Artritis Rheum. 1993; 36: 279-280. Elliott M., Maini R. N., Feldman M. et al.: Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alfa. Arthritis Rheum. 1993. 36: 1681-1690. Elliot M. J., Maini R. N., Feldman M. et al.: Randomised double-blind comparison of chimeric monoclonal 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. antibody to tumar necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet 1994; 344: 1105-1110. Elliott M. J., Maini R. N., Feldmann M. et al.: Repeated therapy with monoclonal antibody to tumor necrosis factor alpha (cA2) in patients with rtheumatoid arthritis. Lancet 1994; 344: 1125-1127. Ferrazi V., Jorgensen С., Sany J.: Alpha-interferon therapy for leukemia or active viral hepatitis in patients with rheumatoid arthritis. Arthritis Rheum. 1994; 27 (suppl.): S-246. Ferri C., La Civita L., Longombardo G. et al.: Treatment of mixed cryoglobulinemia by alpha-interferon: a controled crossover trial. XVIII ILAR Cogr. Rheumatol., 1993; 454 (abst.): 208. Firestein G. S., Alvaro-Garcia J. M., Maki R.: Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J. Immunol. 1990; 144: 3347-3353. Freundlich В., Jimenez S. A., Steen V. et al.: Treatment of systemic sclerosis with recom-binant interferongamma: a phase I/II clinical trial. Arthritis Rheum., 1992; 35: 1134-1142. Hale G., Xia M.-Q., Tighe H. P. et al.: The CAMPATH-1H antigen (CDw52). Tissue antigens 1990; 35: 118-127. Hamuryudan V., Yurdakul S., Serdaroglu S. et al.: Topical alpha interferon in the treatment of oral ulcers in Behcet's syndrome: a preliminary report. Clin. Exp. Rheumatol. 1990; 8: 51-54. Hamuryudan V., Morab F., Yurdakui S. et al.: Systemic interferon alpha2b treatment in Bechcet's syndrome. J. Rheumatol. 1994; 21: 1098-1100. Hiepe E., Thiele B., Brink U. et al.: Succesful treatment of severe systemic lupus erythe-matosus with the anti-CD4 monoclonal antibody max. 16H5. XVIII ILAR Congr. Rheumatol., 1993; 464 (abst.): 212. Horneff G., Krause A., Kalden J. R. et al. Elevated levels of circulating tumor necrosis factor-alpha, interferon-gamma and interleukin 2 in systemic reactions induced by anti-CD4 therapy in patients with rheumatoid arthritis. Cytokine. 1991; 3: 1-2. Horneff G., Burmester G. R., Emmrich F., Kalden J. R.: Treatment of rheumatoid arthritis with anti-CD4 monoclonal antibody. Arthritis Rheum 1991; 36: 291-298. Issacs J. D., Watts R. A., Hazleman B. L., Hale G. et. al.: Humanised monoclonal antybody therapy for rheumatoid arthritis. Lancet 1992; 340: 748-752. Isaacs J. D., Manna V. K., Hazleman B. L. et al.: CAMPATH-1H IN RA A Study of multiple IV dosing. Brit. J. Med. 1994. (Suppl. 1): 33: 154 (abst.). Kahan A., Amor B., Menkes C. J., Strauch G.: Recombinant interferon-gamma In the treatment of systemic sclerosis. Amer. J. Med., 1989; 87: 273-277. Kavanaugh A., Davis L., Nichols L. et al.: Treatment of refractory rheumatoid arthritis with monoclonal antibody to intercellular adhesion molecule 1. Arthritis Rheum. 1994; 37: 992-999. Kavanaugh A., Jain R., McFarlin J. et al.: Anti-CD54 (Intercellular adhesion molecule-1; ICAM-1) monoclonal antibody therapy in early rheumatoid arthritis. Arthritis Rheum., 1994; 37 (Suppl.): S220. Keffer J., Probert L., Cazlaris H. et al.: Transgenic mice expressing human tumor necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991: 10: 4025-4031. Kiely P. D. W., Bruckner F. E.: Acute arthritis following interferon-alpha therapy. Arthritis Rheum. 1994; 33: 502-503. Koopman W. J., Gay S.: Do nonimmunologically mediated pathways play a role in the pathogenesis of rheumatoid arthritis. Rheum. Dis. Clin. North. Amer. 1993; 19: 107-122. Kremer J. M., Petrillo G. F., Rigby W. F. C. et al.: Phase I/II, open trial of DAB398IL-2 administered to patients with active rheumatoid arthritis receiving treatment with methotrexate. Arthritis Rheum. 1994; 37 (suppl.): S-341. Lebsack M. E., Paul C. C., Martindale J. J., Catalano M. A.: A dose and regimen-ranging of IL-1 receptor antagonist in patients with rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S-39. Lemmel E. M., Franke M., Gaus W. et al.: Results of a phase-II clinical trial on the treatment of rheumatoid arthritis with recombinant interferon-gamma. Rheumatol. Int. 1987; 7: 127-132. Lemmel E. M. and German Lymphokine Study Group.: Double blind controlled phase III multicenter study trial with interferon gamma in rheumatoid arthritis. Rheumatol. Int. 1992; 12: 175-185. Machold K. P., Neumann K., Smolen J. S.: Recombinat human interferon gamma in the treatment of rheumatoid arthritis: double blind placebo control study. Ann. Rheum. Dis., 1992; 51: 1039-1043. Margolies L. W., Heck G. R. et al.: Soluble tumor necrosis factor receptor (sTNFR): results of a phase I dose-escalation study in patients with rheumatoid arthritis. Arthritis Rheum.. 1994, (Suppl.): 37: R27. Mathieson P. W., Cobbol S. P., Hale G., Waidman H.: Monoclonal antibody therapy in systemic vasculitis. New Engl. J. Med. 1990; 323: 250-254. Misiani R., Bellavita P., Fenili D. et al.: Interferon alfa-2a therapy in cryoglobulinemia associated with hepatitis С virus. New Engl. J. Med. 1994; 330: 751-756. Moreland L. W., Pratt P. W., Sanders M. E., Koopman W. J.: Experience with a chimeric monoclonal antiCD4 antibody in the treatment of refractory rheumatoid arthritis. Clin. Exp. Rheumatol. 1993; II (Suppl. 8): S153-S159. Moreland L. W., Heck I. W., Koopman W. J. et al.: Vbl7 T-cell rexeptor vaccine: results of a phase I dose- finding study in patients with rheumatoid arthritis. Arthritis Rheum. 1994 (Suppl.); 37: S-28. 50. Newell M. K., Haughn L. J., Maroun C. R., Julius M. H.: Death of mature T cells separate ligation of CD4 and the T cell receptor for antigen. Nature 1990; 347: 286-289 51. Noma T, Dorf M. E.: Modulation of supressor T cell induction with gamma-interferon. J. Immunol. 1985; 135: 3655-3660. 52. Olsen N. J., Cush J. J., Lipsky P. E. et al.: Multicenter trial of an anti-CD5 immunoconjugate in rheuimatoid arthritis. Arthritis Rheum., 1994; 37 (Suppl.): S295. 53. Panayi G. S., Lanchbury J. S., Kingsley G. H.: The importance of the T cell in initiating and maintaning the chronic synovitis of rheumatoid arthritis. Arthritis Rheum. 1992; 35: 729-735. 54. Pincus Т., Callahan L. F.: Remodeling the pyramid or remodeling the paradigm concerning rheumatoid arthritis-lessons from Hodgkin's disease and coronory artery disease. J. Rheumatol. 1990; 17: 1582-1585. 55. Rankin E. C. C., Choy E. H. S., Kassimos D. et al.: A double blind, placebo controlled ascending dose trial of the recombinant humanised anti-TNF alpha antibody CDP571 in patients with rheumatoid arthritis: a preliminary report. Arthritis Rheum., 1994; 37 (Suppl.): S295. 56. Reiter C., Kakavand B., Rieber E. P. et al. Treatment of rheumatoid arthritis with monoclonal CD4 antibody M-T151. Clinical results and immunopharmacological effects in an open study, including repeated administration. Arthritis Rheum., 1991; 34: 525-536. 57. Ronnblom L. E., Alm G. V., Oberg K. E.: Possible induction of systemic lupus erythematosus by interferon alpha treatment in patient with a malignant carcinoid tumour. Int. J. Med. 1990; 227: 207-210. 58. Ronnblom L. E., Aim G. V., Oberg K. E.: Autoimmunity after alpha interferon therapy for malignant carcinoid tumors. Ann. Intern. Med. 1991; 115: 178-183. 59. Rowe F. M., Edwards J., Cozens P. J.: Increased interleukin-I levels from monocyte of rheumatoid arthritis patients during early stages of the rheumatoid arthritis are down regulated in vitro by the addition of interferon-gamma. J. Leukocyte Biol. 1987; 42: 600-601. 60. Ruschen S., Lemm G., Warnatz H.: Spontaneous and LPS-stimulated production of intra-cellular IL-lbeta by synovial macrophages in rheumatoid arthritis is Inhibited by IFN-gamma. Clin. Exp. Immunol. 1989; 76: 246-251. 61. Sewell K. L., Parker K. C., Woodworth T. G. et. al.: DAB486 IL-2 fusion toxin in refractory rheumatoid arthritis. Arthritis Rheum. 1993; 36: 1223-1233. 62. Sewel K. L., Moreland L. W., Furst D. E. et al.: Phase II, open trial of DAB398IL-2 administered up to four times a year to patients with active rheumatoid arthritis. Arthritis Rheum. 1994; 37 (suppl.): S-341. 63. Shiozawa S., Morimoto I., Tanaka Y., Shiozawa K. A.: Preliminary study on the interferon-alpha treatment for xerostomia of Sjogren's syndrome, Brit. J. Rheumatol. 1993; 32: 52-54. 64. Shiozawa S., Shiozawa К., Kita M. et al. A preliminary study of the effect of alpha-interferon treatment on the joint inflammation and serum calcium in rheumatoid arthritis. Brit. J. Rheumatol. 1992; 31: 405-408. 65. Soriano E., Davies J., Maddison P. J., Hall N. D. Preliminary experience with single infusion Campath-1H in the treatment of refractory rheumatoid arthritis. rit. J. Rheumatol. 1993 (Suppll); 32: 54 (abst.). 66. Steinman L.: The use of monoclonal antibodies for the treatment of autoimmune disease J. Clin. Immunol. 1990; 10 (suppl. 6): S30-S38. 67. Strand V., Lipsky P. E., Cannon G. W. et al.: Effects of administration of an anti-CD5 plus immunoconjugate in rheumatoid arthritis. Resuls of two phase II studies. Arthritis Rheum. 1993; 36: 620630. 68. Sturfelt G., Mousa F., Jonsson H. et al.: Reccurent cerebral infarction and the antiphos-pholipid syndrome: effect of intravenous gammaglobulin in patient with systemic lupus erythematosus. Ann. Rheum. Dis. 1990; 49: 939-941. 69. Tomer Y., Blank M., Scoenfeld Y.: Supression of experimental antiphospholipid syndrome and systemic lupus erythematosus in mice by anti-CD4 monoclonal antibodies. Athritis Rheum. 1994; 37: 1236-1244. 70. Ueno Y., Sohma Т.: Alpha interferon induced nodular rheumatoid arthritis in renal cell carcinoma. Ann, Intern. Med. 1992; 117: 266-267. 71. Van der Lubbe P., Reiter C., Breedveld F. C. et al. Chimeric CD4 monoclonal antibody cM-T412 as a therapeutic approach to rheumatoid arthritis. Arthritis Rheum. 1993; 36: 1375-1379. 72. Vandenbroucke J. P., Hazevoet H. M., Cats A.: Survival and death in rheumatoid arthritis: a 25 year prospective follow up. J. Rheumatol. 1984; II: 158-161. 73. Veys E. M., Mielants H., Verbuggen G., Keyser F.: Intervention with immunomodulatory agents: new pharmacological development. Bailliere's Clin. Rheumatol. 1992; 6: 455-484. 74. Veys E. M., Millants U., Jerbrugger G. et al.: Recombinant IFN-gamma in rheumatoid arthritis. A doubleblind study. Clin. Exp. Rheumatol. 1987; 5 (suppl. 2): 53. 75. Wacholtz M., Lipsky P. E.: Treatment of lupus nephritis with CD5 plus, an immunoconjugate of an antiCD5 monoclonal antibody and ricin A chain. Arthritis Rheum., 1992; 35: 837-839. 76. Wahl S. M., Allen J. B., Ohura D. E. et al.: IFN-gamma inhibits inflammatory cell recruitment and evolution of bacterial cell wall-induced arthritis. J. Immunol. 1991; 146: 95-100. 77. Waidmann H.: Manipulation of T-cell responses with monoclonal antibodies. Annual Rev. Immunol. 1989; 7: 407-444. 78. Weinblat M. E., Coblyn J., Maer A. et al.: Sustained lymphocyte supression after single dose CAMPATH1H infusion: an 8 month follow-up. Arthritis Rheum. 1993; 36 (suppl.): S-40. 79. Wendling D., Wijdenes J., Racadot E. et al.: Therapeutic use of monoclonal anti-CD4 antibody in rheumatoid arthritis. J. Rheumatol., 1991; 18: 325-327. 80. Williams D. P., Parker K., Bacha P. et al. Diphteria toxin receptor binding domain substitution with interleukin-2: genetic construction and properties of a diphteria toxin-related interleukin-2 fusion. Protein Engin., 1987; 1: 493-498. 81. Williams P. O., Maini R. N., Feldman R. N. Anti-tumor necrosis factor (TNF) treatment inhibits the progression of established collagen-induced arthritis. Arthritis Rheum. 1991 (suppl); 34: S67. 82. Williams R. O., Mason L. J., Feldman M., Maini R. N.: Synergy bet ween anti-CD4 and antitumor necrosis factor in the amelioration of established collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1994; 91: 2762-2766. 83. Wotsy D., Carteron N. L.: CD4 antibody in systemic lupus erythematosus. Semin Immunol. 1990; 2: 419425. 84. Zvaifler N. J., Firestein G. S.: Pannus and pannocytes. Alternative models of Joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994; 37: 783-789. ГЛАВА 15 ДРУГИЕ ПРЕПАРАТЫ И МЕТОДЫ ЛЕЧЕНИЯ 15.1. Левамизол и другие производные имидазола Гидрохлорид левамизола — стабильное кристаллическое вещество с молекулярной массой 240 D, хорошо растворимое в воде, обладающее антигельминтной, а также противовоспалительной (в супратерапевтических дозах) и мягкой иммуномодулирующей активностью. Фармакологические свойства Левамизол быстро абсорбируется в ЖКТ, после перорального приема 150 мг максимальная концентрация препарата в сыворотке достигается в течение 1-2 ч. Время полужизни препарата в плазме составляет 4 ч, вещество быстро метаболизируется в печени и полностью выводится из организма в течение 48 ч. В процессе гидролиза вещества образуется активный метаболит (ОМР1), обладающий свободными сульфгидрильными группами. Механизм действия Механизмы действия левамизола неизвестны. По данным экспериментальных исследований левамизол обладает способностью стимулировать дифференцировку предшественников Т-лимфоцитов в зрелые Тлимфоциты и восстанавливать подавленные эффекторные функции Т-лимфоцитов и фагоцитирующих клеток. Отмечено благоприятное влияние левамизола на течение иммунопатологаческого процесса у NZB/NZW мышей: увеличение выживаемости, снижение протеинурии, подавление активности нефрита, снижение титров АНФ, увеличение пролиферации Т-лимфоцитов и клеток селезенки, увеличение числа антителообразующих клеток. Клиническое применение РА Symoens и соавт. (1983) обобщили данные 13 контролируемых исследований эффективности левамизола, в которые вошли 1356 больных РА, многие из которых имели тяжелый РА и нуждались в базисной терапии. Препарат назначали в дозе 150 мг/день 1, 3 или 7 раз в неделю. В целом отмечен положительный клинический эффект, который проявлялся через 6 нед.-3 мес. терапии и достигал максимума в сроки между 6-м и 12-м месяцем. Иногда клиническому улучшению предшествовало умеренное обострение. В целом препарат оказался эффективным у 50-70% больных. Клиническое улучшение ассоциировалось со снижением СОЭ, уменьшением концентрации острофазовых белков, титров РФ. Кроме того, на фоне лечения отмечена нормализация некоторых иммунных показателей, отражающих фагоцитоз и функцию Т- и В-лимфоцитов, однако корреляции между динамикой иммунологических и клинических показателей не прослеживалось. Клиническая эффективность левамизола не зависела от пола, возраста, массы тела, длительности и активности заболевания. Однако ответ на левамизол и переносимость лечения были выше у больных с ранним РА, не получавших базисные препараты. Другие заболевания В открытых неконтролируемых исследованиях продемонстрирована определенная эффективность левамизола при ювенильном РА, СКВ, анкилозирующем спондилоартрите, синдроме Рейтера, синдроме Шегрена, ССД, ПМ/ДМ (J. Symoens и соавт., 1983; Н. В. Бунчук, 1985). Имеются сообщения об успешном применении левамизола при кортикостероидзависимом нефротическом синдроме (T. J. Beattie и соавт., 1991). Побочные эффекты Левамизол вызывает многочисленные побочные эффекты, что ограничивает возможности его клинического применения (J. Symoens и соавт., 1979). Основными побочными эффектами являются следующие: 1) сенсорно-неврологические: изменение вкусовых ощущений, бессонница, головные боли, головокружение возбуждение; 2) желудочно-кишечные: тошнота, потеря аппетита, расстройство стула; 3) идиосинкразические: агранулоцитоз, кожная сыпь, лихорадка, стоматит. Большинство побочных эффектов левамизола сравнительно нетяжелые, обычно развиваются в течение первого месяца лечения. Левамизол не обладает почечной и печеночной токсичностью. Сенсорно-неврологические нарушения Эти побочные эффекты, вероятно, связаны с фармакологическим действием препарата на нервную систему. Они развиваются не часто и редко выражены в такой степени, что это приводит к прерыванию лечения. Поражение ЖКТ Частота желудочно-кишечных нарушений очень низкая и, вероятно, в большей степени связана с сопутствующей терапией. Развития пептических язв и геморрагий ЖКТ не описано. Идиосинкразические реакции Эти реакции имеют самое важное клиническое значение и наиболее часто развиваются именно у больных ревматическими заболеваниями. Самым тяжелым осложнением лечения является агранулоцитоз, который наблюдается у 1-2% больных РА. Наиболее высок риск агранулоцитоза у женщин, страдающих серопозитивным РА, являющихся носителями HLA-B27. Агранулоцитоз развивается в сроки от нескольких недель до нескольких месяцев от начала лечения, его особенностью является внезапное исчезновение нейтрофилов из кровяного русла. Однако регенераторная способность костного мозга сохраняется, полностью кроветворение нормализуется в течение 1-2 нед. после отмены препарата. Агранулоцитоз следует отличать от лейкопении, при которой не наблюдается деструкции лейкоцитов. В последнем случае число лейкоцитов нормализуется, несмотря на продолжение лечения. При лейкопении в отличие от агранулоцитоза не происходит столь выраженного снижения числа лейкоцитов и нейтрофилов, отсутствуют изменения в костном мозге и не наблюдается увеличения носительства HLA-B27. Самой частой идиосинкразической реакцией является кожная сыпь. Эта реакция описана почти исключительно у больных РА, иногда она выражена в такой степени, что приходится прервать лечение. Стоматит развивается реже, иногда бывает изолированным, но чаще сочетается с кожной сыпью и агранулоцитозом. Обычно он быстро исчезает после прекращения лечения и редко бывает причиной отмены препарата. Гриппоподобные (flu-like) симптомы являются одной из наиболее частых причин прерывания лечения. Они напоминают проявления вирусной инфекции и характеризуются лихорадкой, миалгией, ознобом, плевритом, артралией, болями в горле, головными болями и др. Нередко гриппоподобные симптомы предшествуют развитию агранулоцитоза. Описано несколько очень редких побочных реакций, включающих "сухой" синдром, волчаночноподобный синдром и лимфаденопатию. Тактика лечения Начинать лечение следует с дозы 150 мг/нед. При отсутствии эффекта в течение 6 мес. недельную дозу препарата увеличивают по 150 мг через день 3 раза в неделю. Частота приема левамизола не должна превышать 3 раза в неделю, а дневная доза — 150 мг. Доза 50 мг/нед. менее эффективна, чем 150 мг/нед., но позволяет почти полностью избежать тяжелых побочных эффектов. В процессе лечения необходим очень тщательный гематологический контроль. Общий анализ крови рекомендуется делать на следующий день после приема препарата. Снижение количества нейтрофилов на 25% является основанием для прерывания лечения. Как уже отмечалось, при агранулоцитозе отмена препарата приводит к нормализации крови в течение 7-14 дней. Для профилактики инфекционных осложнений целесообразно назначать антибактериальные препараты. Использование высоких доз ГК и переливание крови не рекомендуется. Хотя абсолютных противопоказаний к назначению левамизола не существует, препарат не следует применять у больных с лекарственными цитопениями в анамнезе и у пациентов, получающих высокие (более 10 мг/сут) дозы ГК и другие базисные противоревматические препараты. Несмотря на доказанный базисный эффект левамизола при РА, в настоящее время этот препарат редко используется в клинической практике, в первую очередь из-за побочных эффектов. Появились сообщения о препарате тиломизол (3-(p-chlorophenol)-thiazole [3,2-a]benzimidazole-2-acetic), который сходен с левамизолом по химической структуре и механизмам действия, но менее токсичен. В опытах in vitro было показано, что тиломизол усиливает пролиферацию лимфоцитов, синтез ИЛ-2 и ИФ-у лимфоцитами (C. M. Rogers и соавт., 1985; S. C. Gilman и соавт., 1987). Кроме того, тиломизол подавляет развитие артрита, индуцированного коллагеном, уменьшает отек суставов и тормозит образование эрозий при адъювантном артрите (S. G. Gilman и соавт., 1987). Предварительные данные, полученные в рамках фазы I и II клинических испытаний, свидетельствуют об эффективности препарата при РА. Другими препаратами, обладающими близкой к левамизолу биологической активностью, является имутиол (diethyidithiocarbamate, DTC), а также антабус (Disulfiram), являющийся предшественником DTC (C. S. Pickett, C. D. Johnston, 1977). Имутиол индуцирует созревание Т-лимфоцитов из "нулевых" клеток, усиливает иммунный ответ, стимулированный антигенами и митогенами, увеличивает синтез ИЛ-2, активность ЕК-клеток и др. (G. Renox и M. Renox, 1977; 1980; V. Churg и ñîàâò., 1986; G. Renoux и ñîàâò., 1980). Кроме того, имутиол является хелатором тяжелых металлов, проявляет противораковую, антигрибковую и антибактериальную активности. C. F. Corke и соавт. (1986) провели открытое 6-месячное исследование эффективности имутиола (250 мг/день) при лечении 12 больных РА. На фоне лечения отмечено уменьшение суставного индекса и выраженности боли, в том числе и у 1 больного, рефрактерного к пеницилламину и хлорамбуцилу. В процессе лечения наблюдалось увеличение числа СDЗ+Т-лимфоцитов и нормализация сниженного соотношения СD4+/СD8+Т-лимфоцитов. Иммуномодулирующая активность, проявляющаяся в способности зависимым от дозы образом ингибировать хемотаксис нейтрофилов и подавлять пролиферативный ответ лимфоцитов при стимуляции растительными митогенами, обнаружена и у других производных имидазола. Некоторые из этих препаратов, такие, как циметидин (H. Permin и соавт., 1981), фенитоин (K. A. Grindulis и соавт., 1986), метронидазол (J. A. Harkness и соавт., 1982; D. A. S. Marshal и соавт., 1992), фенфлюмизол (K. Christensen, 1986), имидазол-2гидроксибензоат (M. Fumagalli и соавт., 1986), тифламизол (N. R. Ackerman и соавт., 1985), клотримазол (R. Wyburn-Mason, 1976; J. A. Wotjulewski и соавт., 1980; W. B. Dennison и соавт., 1990) использовались 'для лечения больных РА. Однако эффективность всех этих препаратов продемонстрирована только в открытых исследованиях или в отдельных клинических наблюдениях и не была подтверждена в контролируемых испытаниях. Важным фактором, лимитирующим использование этих препаратов при РА, является высокая частота побочных эффектов при длительном приеме. 15.2. Колхицин Колхицин — алкалоид, выделенный из семян и корнеплодов Colchicum autamnale, принадлежащего к семейству Liliaceae. Этот алкалоид рассматривается как наиболее активное и специфическое противовоспалительное средство для лечения подагры, в настоящее время используется для лечения и других ревматических заболеваний. Фармакологические свойства При пероральном приеме колхицин быстро всасывается, достигая максимальной концентрации в плазме через 0.5-2 ч. Колхицин накапливается в почках, печени, селезенке и пищеварительном тракте, но не обнаруживается в сердце, мозге и скелетной мускулатуре. Основными клетками-мишенями для колхицина являются лейкоциты, в которых концентрация препарата в 10 раз превышает сывороточную. Метаболизм колхицина происходит в печени и заканчивается образованием демитилированных неактивных метаболитов, 10-20% в неизмененном виде выделяется с мочой. После однократного приема концентрация колхицина в плазме достигает стабильного уровня в течение 0.5 -24 ч, в клетках препарат сохраняется 10 дней. Механизм действия Колхицин, как и другие алкалоиды винка (например винбластин), оказывает токсическое действие на микротрубочки. Препарат образует димеры с тубулином, что вызывает предотвращение сборки субъединиц тубулина в микротрубочки. Это в свою очередь приводит к подавлению митоза клеток в метафазе (Р. А. В. Кеа1егs и G. B. Mason, 1981; L. Wilson и соавт., 1974). Нарушение сборки микротрубочек в лейкоцитах ведет к ингибиции подвижности, хемотаксиса, прилипания лейкоцитов, предотвращает их диапедез в зону воспаления (C. A. Dinarello и соавт., 1976). К другим противовоспалительным эффектам колхицина относятся подавление высвобождения гистамина из гранул тучных клеток, синтеза хемотаксических факторов лейкоцитами, в том числе ЛТВ4 (Reibman и соавт., 1986; Y. Ouyang и соавт., 1989). Колхицин подавляет секрецию иммуноглобулинов, что коррелирует с обнаружением ультраструктурных изменений в аппарате Гольджи и эндоплазматическом ретикулуме (J. C. Antoine и соавт., 1986). Кроме того, колхицин ингибирует клеточные иммунные реакции, что, как полагают, связано с блокированием ИФ-у-зависимой экспрессии HLA-Dr (Y. A. Mekory и соавт., 1989). По сравнению с НПВС колхицин обладает низкой общей противовоспалительной активностью. В экспериментальных исследованиях было показано, что колхицин в отличие от НПВС не подавляет развитие реакции Артюса и существенно не влияет на развитие отека, индуцированного каррагенином. Один из основных механизмов действия колхицина связан с влиянием на взаимодействие лейкоцитов и ЭК. Колхицин замедляет перекатывание (rolling) лейкоцитов вдоль эндотелиальной выстилки микрососудов (H. Asako и соавт., 1992), что связывают со способностью препарата ингибировать экспрессию L-ñåëåêòèíà на мембране лейкоцитов и межклеточных молекул адгезии-1 (IСАМ-1) на поверхности эндотелиальных клеток и Е-селектинзависимое прилипание нейтрофилов к ЭК (Molad и соавт., 1992). Клиническое применение (таблица 15.1.) Таблица 15.1. Заболевания, при которых отмечена эффективность колхицина (по I. P. Famaey, 1988) Заболевание Комментарий Острый подагрический артрит, профилактика обострений Средство выбора хронического подагрического артрита Пирофосфатная артропатия и гидроксиаппатитная Эффект менее выражен, чем при подагре артропатия Саркоидозный артрит Эффект непредсказуем РА Положительный эффект в отдельных клинических наблюдениях Псориатический артрит Результаты противоречивы Периодическая болезнь Доказана эффективность (профилактика обострений и развития амилоидоза) Болезнь Бехчета Положительный эффект в отдельных клинических наблюдениях Амилоидоз Результаты противоречивы ССД То же Кожные васкулиты, синдром Свита, рецидивирующий Положительный эффект в отдельных полихондрит клинических наблюдениях Подагра При остром приступе подагры колхицин назначают по 0.5 мг каждый час до купирования приступа (максимально до 5 мг) или развития побочных эффектов. По другой схеме колхицин назначают по 1мг 4 раза в день. В последующем лечение препаратом продолжают в меньшей дозе (она зависит от выраженности клинических симптомов), снижая ее по 1-2 мг в день и увеличивая промежутки между приемами. При достаточно раннем назначении колхицина эффективность лечения достигает 90%. Боль и покраснение сустава купируются в течение 12 ч, а другие симптомы — в течение 48-72 ч. Поддерживающая терапия колхицином (0,5-1 мг/день иногда 3 дня в неделю) может проводиться в течение нескольких недель или даже месяцев без существенного риска токсичности, связанной с аккумуляцией препарата; она эффективно предотвращает обострение подагры по крайней мере у 90% больных. У пациентов с почечной и печеночной недостаточностью дозу колхицина следует уменьшить. Колхицин не влияет на метаболизм мочевой кислоты. Известно, что любые гипоурикемические препараты в начале лечения могут вызывать острый подагрический приступ за счет мобилизации депозитов уратов. Для профилактики этих обострений используют небольшие дозы колхицина (0.5-1 мг) в течение нескольких недель или месяцев, особенно в случае тофусной подагры (до исчезновения тофусов). Профилактическое назначение колхицина показано больным хронической подагрой перед оперативным лечением. Псориатический артрит (ПА) Проведено два двойных слепых контролируемых исследования, в одном из которых (P. Seidman и соавтюю.,1987) лечение колхицином 1.5 мг/сут.) привело к достоверному увеличению силы сжатия кисти, уменьшению индекса Ричи, суставного счета, уменьшению болей в суставах при отсутствии влияния на поражение кожи. Однако, по данным других авторов (R. J. R. McKendry и соавт., 1993) колхицин при ПА неэффективен. Пирофосфатная артропатия При пирофосфатной артропатии препарат назначают по той же схеме, что и при подагре, однако эффективность лечения ниже. Предполагается, что при пирофосфатной артропатии предпочтительнее внутривенное введение препарата. Однако в большинстве случаев более эффективными средствами для лечения этой патологии являются НПВС (J. Spilberg и соавт., 1972; M. R. Tabatabai и N. A. Cummings, 1980). Саркоидоз Колхицин в дозе 1.5-3 мг/день положительно влияет на течение саркоидозного артрита (H. Kaplan, 1963; Enrenfeld и соавт., 1984). Семейная средиземноморская лихорадка (периодическая болезнь) Основные симптомы периодической болезни — остро развивающаяся высокая лихорадка, сочетающаяся с артритом, перитонитом, плевритом или перикардитом, кожной сыпью, а характерным осложнением является амилоидоз почек, приводящий к почечной недостаточности. Колхицин в дозе 1-3 мг/день позволяет уменьшить частоту и тяжесть обострений периодической болезни (R. C. Goldstein и соавт., 1974). Кроме того, лечение колхицином замедляет прогрессирование амилоидоза. D. Zemer и соавт. (1986) установили на основании наблюдений за 1070 больными, что колхицин предотвращает развитие амилоидоза и дальнейшее нарушение функции почек у больных с протеинурией, но не с нефротическим синдромом. В другом исследовании D. Zemer и соавт. (1991), в которое вошло 350 детей с периодической болезнью, было показано, что профилактический прием колхицина (1-2 мг/день в течение 6-13 лет) позволяет добиться полной ремиссии заболевания у 64% больных, частичной ремиссии —у 31% и полностью предотвратить развитие амилоидоза у 5%. Ни у кого из детей не отмечалось побочных реакций, которые заставили бы прервать лечение, а также задержки роста, развития или подавления детородной функции. Болезнь Бехчета При болезни Бехчета лечение колхицином (0.5-1.5 мг/день) позволяет уменьшить частоту и тяжесть обострений и замедлить прогрессирование болезни (E. Aktulga и соавт., 1980). Амилоидоз Имеются данные о том, что колхицин подавляет образование сывороточного амилоидного белка (SAA) (E. Tatsura и соавт., 1984). По данным S. A. Cohen и соавт. (1987), у больных первичным амилоидозом (53 человека), леченных колхицином (по 0.5-1-мг/день), выживаемость составила 17 мес., в то время как у нелеченных больных (29 человек) — всего 6 мес. Однако в процессе проспективного рандомизированного исследования (R. A. Kyle и соавт., 1985) оказалось, что комбинация мелфалана и преднизолона предотвращает прогрессирование амилоидоза более эффективно, чем колхицин. A. Escalante и соавт. (1991) описали больного анкилозирующим спондилоартритом, у которого лечение колхицином вызвало регрессию почечного амилоидоза. Имеются данные об эффективности колхицина при амилоидозе, связанном с воспалительными заболеваниями кишечника (S. Meyers и соавт.,1988). ССД Теоретическим основанием для применения колхицина при ССД является способность препарата подавлять накопление коллагена, блокируя конверсию проколлагена в коллаген. Этот эффект связывают с нарушением транспорта коллагена, в котором принимают участие микротрубочки цитоскелета. Кроме того, колхицин усиливает синтез коллагеназы. Имеются данные о положительном влиянии колхицина на кожные проявления ССД. Эффективность колхицина была выше у больных с ранней ССД (до 5 лет от начала болезни), получивших общую дозу колхицина 1400 мг. (D. Alarcon-Segovia и соавт., 1979). Однако, по данным M. Guttadauria и соавт. (1977), прием колхицина в дозе 1 мг/день в течение длительного времени не оказывает существенного влияния на прогрессирование кожных и висцеральных проявлений ССД, в том числе на функцию легких. Другие заболевания Имеются отдельные сообщения о благоприятном эффекте колхицина у больных ДМ с кальцинозом (J. Taborn и соавт., 1978), при постоперационных келоидных рубцах, при поражении позвоночных дисков, а также при лейкоцитокластическом васкулите (J. P. Callen, 1987; S. Plotnick и соавт., 1989), уртикарном васкулите, смешанной криоглобулинемии (F.Invernizzi и G. Monti, 1993), синдроме Свита (S. Suehisa и H. Tagami, 1981), рецидивирующем полихондрите (A. D. Askari, 1984). Побочные эффекты Побочные эффекты колхицина могут быть суммированы следующим образом: 1. Частые, обычно нетяжелые: • желудочно-кишечные: тошнота, рвота диарея боли в животе • локальное • повреждение вены (при в/в) 2. Редкие, более тяжелые: • геморрагический гастроэнтерит • апластическая анемия • ДВС-синдром • неврологические нарушения • миопатия • печеночная недостаточность • острая почечная недостаточность • некроз тканей 3. Редкие, нетяжелые: • алопеция • стоматит • азоспермия • аменорея У больных подагрой введение полной терапевтической дозы колхицина (1 мг 4-6 раз в день) в 80% случаев вызывает желудочно-кишечные нарушения, включая боли и спазмы в животе, диарею, реже тошноту и рвоту. Частота этих проявлений ниже при внутривенном введении препарата. При длительном профилактическом применении низких доз колхицина (1 мг/день) побочные эффекты, как правило, развиваются через несколько месяцев или лет. Наиболее частыми являются лейкопения, апластическая анемия, миопатия, выпадение волос. Миопатия проявляется проксимальной мышечной слабостью, часто сочетается с увеличением уровня КФК. После отмены препарата наблюдается быстрое исчезновение мышечных симптомов и нормализация лабораторных показателей. 15.3. Гормоны тимуса В настоящее время получено в чистом виде и охарактеризовано несколько гормонов тимуса или их синтетических фрагментов, пригодных для клинического применения (E. M. Veys и соавт., 1992). Основные иммунологические свойства препаратов тимуса представлены в обзоре T. L. K. Low и A. L. Goldstein (1984) и заключаются в первую очередь в иммуностимулирующей активности, продемонстрированной как in vivo, так и in vitro. Однако точки приложения препаратов тимуса, определяющие их клиническую эффективность при ревматических заболеваниях, не установлены. Для лечения заболеваний человека за рубежом использовано 2 гормона тимуса — тимопоетин или его синтетический фрагмент (тимопентин) и тимулин. Тимопоетин — полипептид, состоящий из 49 аминокислот. Пентамерный пептид — тимопентин (ArgLys-Asp-Val-Tyr) (ТР-5), соответствующий остаткам тимопоетина в положении между 32-36 аминокислотами, сохраняет биологическую активность нативной молекулы (Goldstein и соавт., 1979). В серии открытых и контролируемых испытаний ТР-5 при РА было показано, что внутривенное введение (в течение 10 мин.) этого препарата в дозе 50 мг 3 раза в неделю приводит к достоверному, по крайней мере кратковременному (3 нед.), снижению активности заболевания. Однако, после окончания лечения в течение ближайших 4 нед. наступает обострение (E. M. Veys и соавт.). Тимулин — вещество, изолированное в начале из сыворотки свиньи, а затем — человека, имеет следующую аминокислотную последовательность: Glu-Ala-Lys-Ser-Glu-Gly-Ser-Asn. Синтезированный на этой основе пептид имеет биологическую активность, аналогичную таковой нативного препарата. Проведено 2 двойных слепых контролируемых исследования тимулина при РА. В одном из них тимулин в дозе 1 или 5 мг либо плацебо (2-3 раза в неделю) вводили 49 больным РА. Тимулин в дозе 5 мг был достоверно эффективнее плацебо в отношении таких параметров активности РА, как индекс Ричи, сила сжатия кисти, индекс Ли и возможность снижения дозы ГК. Во втором исследовании, в котором тимулин (10 мг подкожно) или плацебо назначали ежедневно 30 больным РА в течение 6 мес. какого-либо действия этого препарата по сравнению с плацебо не отмечено. Таким образом, эффективность препаратов тимуса в лечении РА не доказана. Положительным свойством этих препаратов является практическое отсутствие побочных эффектов. Имеются данные о том, что сочетанное применение МТ и препарата тимуса (тактивин) позволяет несколько повысить эффективность лечения больных РА (Р. М. Балабанова, 1994). 15.4. Антибактериальные препараты 15.4.1. Тетрациклиновые препараты Механизм действия Механизмы, лежащие в основе базисного эффекта препаратов тетрациклина при РА, связывают с их противовоспалительным и иммунодепрессивным действием. Так, например, имеются данные о способности тетрациклиновых производных инактивировать металлопротеиназы в синовиальной ткани (R. A. Greenwald и соавт., 1987; M. P. Vincenti и соавт., 1994), подавлять активность фосфолипазы А2 (W. Pruzanski и соавт., 1992) и окислительную активацию латентной коллагеназы. Кроме того, тетрациклины ингибируют функциональную активность нейтрофилов (в том числе нейтрофильный хемотаксис, способность вызывать необратимое повреждение клеток, фагоцитарную активность, образование реактивных форм кислорода) (R. R. Martin и соавт., 1974; A. Forgsen и соавт., 1974; W. L. Gabler и H. R. Creamer, 1991) и Т-лимфоцитов (E. Ingham и соавт., 1991; M. Kloppenburg и соавт., 1992). По данным М. Kloppenburg и соавт. (1992), миноциклин зависимым от дозы образом ингибирует пролиферацию клонов CD4+ Т-лимфоцитов и СD8+Т-лимфоцитов, выделенных из синовиальной ткани больных РА, стимулированных анти-СDЗ и синтез ИФ-у Т-лимфоцитами. Антиартритическая активность миноциклина была продемонстрирована при изучении 2 моделей хронического артрита: коллагенового и адъювантного артрита у крыс (F. C. Breedveld и D. E. Trentham, 1988). Химически модифицированная форма тетрациклина, лишенная антибактериальной активности, в комбинации с флурбипрофеном предотвращала потерю костной ткани при адъювантном артрите (R. A. Greenwald и соавт. 1992), а оксациллин уменьшал тяжесть экспериментального остеоартрита (L. P. Yu и соавт., 1992). Клинические испытания Клиническая эффективность миноциклина при РА была продемонстрирована в серии открытых (F. C. Breedveld и соавт., 1990; P. Langevitz и соавт., 1992) и контролируемом исследованиях (M. Kloppenburg и соавт., 1994). M. Kloppenburg и соавт. (1994) провели двойное слепое 26-недельное контролируемое испытание миноциклина (200 мг/сут) у 80 больных активным РА, которые ранее применяли хотя бы один базисный препарат. У пациентов, леченных миноциклином, отмечалось более значительное улучшение 9 из 14 параметров клинической активности, чем у больных, получавших плацебо (индекс Ричи, число воспаленных суставов, СОЭ, уровень гемоглобина, СРБ, IgM РФ). При этом нормализация лабораторных показателей была выражена в большей степени, чем клинический эффект. Частота побочных реакций была выше у больных, леченных миноциклином, чем у пациентов, получавших плацебо. Основными побочными эффектами были желудочно-кишечные расстройства и головокружение, которые быстро исчезали после отмены препарата. Отмечена корреляция между клинической эффективностью миноциклина и снижением уровня ИЛ-6 (M. Kloppenburg и соавт., 1995). 15.4.2. Рифадин Получены данные о возможности использования рифадина для лечения PA (S. E. Gabriel и соавт. 1990), однако в исследовании N. L. Cox и соавт. (1992) было показано, что хотя лечение рифадином (по 300 мг) и изониазидом (по 150 мг 2 раза в день) и вызывает снижение уровня СРБ и СОЭ, это не ассоциируется с какойлибо положительной динамикой клинических симптомов. 15.4.3. Сульфаметоксазол/триметоприм Сульфаметоксазол/триметоприм — комбинированный препарат, содержащий 2 действующих вещества — сульфаниламид (сульфаметоксазол) и производное диаминопиримилина — триметоприм. Сочетание 2 препаратов, оказывающих бактериостатическое действие, обеспечивает бактерицидную активность в отношении грамположительных и грамотрицательных микроорганизмов. Сульфаметоксазол нарушает биосинтез дигидрофолиевой кислоты, а триметоприм блокирует восстановление дигидрофолиевой кислоты. Имеются данные об иммуносупрессивной активности препарата, подавлении функциональной активности нейтрофилов (R. Anderson и соавт., 1988; D. E. Roberts и J. G. Curd, 1990). Появились сообщения об использовании сульфаметоксазола/триметоприма для лечения гранулематоза Вегенера (R. A. DeRemee, 1988). 15.5. Лобензарит Лобензарит представляет собой двунатриевую соль 4-хлор-2,2' иминодибензоата, обладает иммуномодулирующей активностью. Механизм действия связывают с подавлением ДНК полимеразы а в Влимфоцитах. Механизм действия 1. ингибиция синтеза антител, включая антитела к ДНК (S. Hirohata, 1992) и замедление прогрессирования аутоиммунного поражения почек у NZBxNZW F1 мышей (Y. Takeda и соавт., 1988); 2. подавление поликлональной В-клеточной активации и прогрессирование аутоиммунного процесса у MRL мышей (M. Michara и соавт., 1987; J. Abe и соавт., 1989); 3. подавление экспрессии HLA-DR на мембране ЭК, стимулированных ИФ-у, ингибиция прилипание Тклеток к ЭК, индуцированная ИФ-у и ИЛ-1 (A. Kawakai и соавт., 1991); подавление экспрессии HLADR на мембране макрофагов; 4. подавление спонтанного синтеза IgM РФ и IgM и синтеза IgM РФ при стимуляции белком A (S. Hirohata и соавт., 1992); 5. подавление синтеза ИЛ-2 CD4+T-клетками, стимулированными анти-CD3. Эффективность лобензарита была продемонстрирована при РА в двойном слепом контролируемом исследовании (Y. Shiokawa и соавт., 1984) и в открытом испытании при СКВ (S. Hirohata и соавт., 1994). 15 больных СКВ получали лобензарит в дозе 40 мг 2 раза в день в течение 2 нед., а затем по 80 мг 2-3 раза в день в течение 12 нед. На фоне лечения отмечено увеличение числа лимфоцитов в периферической крови, нормализация соотношения CD4+T-ëèìôoöèòû/CD8+T-ëèìôoциты, снижение уровня антител к ДНК. У отдельных больных развились побочные реакции, включая умеренное нарушение функции печени, желудочнокишечные расстройства, головокружение. 15.6. Бромокриптин Бромокриптин (2-бром-а-эргокриптин) — полусинтетическое производное алкалоидов спорыньи, является специфическим агонистом дофаминовых рецепторов, подавляет синтез гормонов передней доли гипофиза, особенно пролактина. Обоснование к применению Пролактин рассматривается как один из наиболее важных иммуностимулирующих гормонов, преодолевающих иммуносупрессивные эффекты кортизола (I. Berczi, 1992). Пролактиновые рецепторы присутствуют на клетках нервной и эндокринной систем, а также на Т-и В-лимфоцитах (L. J. Jara и соавт., 1991). Показано, что для транслокации пролактина в ядро требуется ИЛ-2-зависимая активация Т-лимфоцитов. При этом пролактин выступает в роли кофактора для ИЛ-2 зависимой пролиферации Т-хелперов. (C. V. Clevenger и соавт., 1991), а антитела к пролактину ингибируют ИЛ-2- зависимую клеточную пролиферацию (D. P. Hartmann и соавт., 1989). Более того, пролактин стимулирует В-клетки (R. J. Cross и соавт., 1989) и индуцирует синтез иммуноглобулинов и антиядерных антител у здоровых и больных СКВ (M. A. Gutierez и соавт., 1993). Пролактин обладает митогенной активностью в отношении Т-лимфоцитов. Специфическое связывание пролактина с рецепторами может усиливаться или подавляться ЦсА (D. H. Russell и соавт., 1983). Активированные лимфоциты продуцируют пролактиновые пептиды, которые оказывают аутокринное действие, усиливая иммунный ответ (D. Montgomery и соавт., 1987). Примечательно, что структура пролактина напоминает структуру ИЛ-6, а пролактиновые рецепторы принадлежат к семейству рецепторов гемопоэтических факторов роста, включающих ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6 и гранулоцитарномакрофагальный колониестимулирующий фактор (R. Hooghe и соавт., 1993). Все это вместе взятое свидетельствует о том, что пролактин относится к семейству цитокинов. Синтез пролактина гипофизом модулируется различными гормонами и нейропептидами гипоталамуса. В свою очередь некоторые нейрогормоны (серотонин, тиролиберин, вазоактивный интестинальный пептид), а также ИЛ-1 и ИЛ-6 увеличивают синтез пролактина (M. Yamaguchi и соавт., 1990). В экспериментальных исследованиях было показано, что развитие адъювантного артрита у крыс может быть подавлено бромокриптином, ингибирующим синтез пролактина гипофизом (I. Berczi и соавт., 1984). При этом артрит может быть вновь индуцирован у животных, леченных бромокриптином, после введения пролактина. Показано также, что пролактин ускоряет развитие иммунопатологического процесса у линий мышей аутоиммунных линий NZBxNZW F1 со спонтанно развивающимся волчаночноподобным заболеванием (R. McMurray и соавт., 1991). Трансплантация гипофиза NZBxNZW мышам приводит к более раннему развитию гипергаммаглобулинемии и увеличению концентрации антител к ДНК. При этом в селезеночных клетках наблюдается гиперэкспрессия иРНК ИЛ-4 и ИЛ-6. Известно, что ИЛ-6 играет важную роль в созревании В-клеток, а у мышей со спонтанно развивающимся волчаночноподобным аутоиммунным синдромом и у больных СКВ отмечается увеличение концентрации ИЛ-6 в сыворотке и гиперэкспрессия гена ИЛ-6 в лимфоцитах. По данным M. Blank и соавт. (1994), бромокриптин подавляет развитие экспериментально индуцированного АФС у мышей за счет усиления активности неспецифических Т-супрессоров. У больных с гиперпролактинемией развиваются разнообразные аутоиммунные реакции. (C. Ferraci и соавт., 1983), а у пациентов с различными аутоиммунными заболеваниями (СКВ, синдром Шегрена и др.) обнаруживается увеличение концентрации пролактина. (L. J. Jara и соавт., 1991; J. M. Anaya и соавт., 1994). У больных РА базальный уровень пролактина в пределах нормы, однако при изучении суточных колебаний уровня пролактина были выявлены его изменения, проявляющиеся в развитии гиперпролактинемии в период максимальной активности болезни (I. C. Chikanza и G. S. Panaui, 1991). Кроме того, при РА обнаружен необычно высокий синтез пролактина в ответ на введение тиреотропина (C. Jorgensen и соавт., 1993). Бромокриптин использовали для лечения рефракторной болезни Рейтера (G. Bravo и соавт., 1992) и болезни Рейтера, ассоциирующейся со СПИДом (F. Medina-Rodriguez и соавт., 1993), псориатического артрита с положительным клиническим эффектом. Описана больная СКВ с тяжелым поражением ЦНС, не отвечающая на высокие дозы ГК, ЦФ и плазмаферез, у которой назначение бромокриптина в сочетании с IVIg индуцировало развитие ремиссии (C. E. Rabinovich и соавт., 1990). Недавно R. W. McMurray и соавт. (1994) провели открытое исследование эффективности бромокриптина (5-7.5мг/день) при СКВ. На фоне лечения отмечено снижение индексов активности СКВ (SLAM, SLEDAI), уменьшение концентрации IgG и увеличение концентрации С3-компонента комплемента. Обострение, наступившее у больных после прекращения лечения, ассоциировалось с увеличением уровня пролактина. По данным G. Bravo и соавт. (1994), у больных ювенильным анкилозирующим спондилитом отмечается корреляция между увеличением концентрации ИЛ-6 и гиперпролактинемией. На фоне лечения бромокриптином наблюдалось снижение уровня пролактина, коррелирующее с нормализацией концентрации ИЛ-6 и острофазовых белков и развитием клинической ремиссии, В то же время комбинация бромокриптина с ЦсА при РА не сопровождалась увеличением эффективности лечения (M. Dougados и соавт., 1988). 15.7. Микофенолат мофетил (Mycophenolat mofetil, RS-61443-000) Препарат представляет собой морфолиноэтиловый эфир микофеноловой кислоты (МФК) (Syntex Research, США). Механизм действия Препарат вызывает обратимую ингибицию активности инозинмонофосфат дегидрогеназы, что приводит к подавлению биосинтеза пуринов в Т-и В- лимфоцитах. (H. J. Lee и соавт., 1985). Предполагается, что, поскольку функциональная активность лимфоцитов в большей степени, чем других быстро делящихся клеток, зависит от синтеза пуринов, препарат дает более выраженный антипролиферативный эффект в отношении лимфоцитов (A. C. Allison и соавт., 1991) и проявляет цитостатическую, а не цитотоксическую активность. При изучении иммунологической активности микофенолата выявлены следующие основные эффекты: 1. ингибиция Т- и В- клеточной пролиферации в ответ на антигенную и митогенную стимуляцию, подавление первичного и вторичного антительного ответа (E. M. Eugui и соавт., 1991), образования цитотоксических Т-лимфоцитов; ингибиция пролиферации и дифференцировки промоноцитов; 2. ослабление миграции лимфоцитов в зону воспаления за счет ингибиции гликозилирования молекул адгезии; 3. зависимое от дозы подавление адъювантного и стрептококкового артрита у крыс, а также других экспериментальных аутоиммунных заболеваний (диабет, уретроретинит). Клинические испытания В настоящее время имеются данные об использовании препарата более чем у 600 больных РА, некоторые из которых продолжают лечение в течение 3,5 года и более. Оптимальная доза составляет 2 г/день (C. M. Franklin и соавт., 1993), максимально допустимая — 4 г/день. Клинический анализ свидетельствует об улучшении у многих больных, резистентных к базисной терапии. Зарегистрировано статистически достоверное уменьшение числа воспаленных и отекших суставов, улучшение состояния по общей оценке врача и больного (M. H. Schiff и соавт., 1991), снижение титров РФ (R. Golblum и соавт., 1991), количества СD2-клеток (Т-лимфоциты) без заметного снижения количества Т-хелперов, Т-супрессоров и Влимфоцитов. Сыворотки больных приобретали способность ингибировать пролиферативый ответ лимфоцитов доноров на различные митогены. У 62% больных отмечено развитие кожной анергии. Однако, несмотря на выраженную иммуносупрессию, не наблюдалось увеличения чувствительности к инфекционным осложнениям. Результаты недавно проведенного многоцентрового 36-недельного контролируемого исследования, в которое вошли 217 больных с рефракторным РА, подтверждают эффективность препарата при лечении РА (C. M. Franklin и соавт., 1993). По данным M. H. Schiff и соавт. (1993), многие больные РА, рефракторные к не менее чем 3 базисным противоревматическим препаратам, отвечают на микофенолат и могут принимать его длительное время без развития тяжелых побочных эффектов и резистентности. Побочные эффекты: поражение ЖКТ (анорексия, тошнота, рвота, понос, боли в животе, диспепсия), головная боль, головокружение, лихорадка, слабость, бессонница, частое мочеиспускание, язвы во рту, кожная сыпь. Очень редко наблюдалась небольшая лейкопения, герпетическая инфекция. 15.8. Гидрохлорид амиприлозы (Amiprilose hydrochloride) Вещество представляет собой 1,2-0-изопропилидин-3-0-3'[N'N'-диметиламиноnпропил] ,dãëþêîôóðàíèë гидрохлорид. Рассматривается как новый класс синтетических моносахаридов, обладающих иммуномодулирующей и противовоспалительной активностью в сочетании с низкой токсичностью. Под названием терафектин (Therafectin) выпускается компанией Greenwich Pharmaceuticals, США). Противовоспалительная активность амиприлозы обнаружена на модели адъювантного артрита, экспериментального моноартрита, каррагенинового отека и коллагенового (тип II) артрита. Клиническая эффективность амиприлозы продемонстрирована в 12недельном многоцентровом двойном слепом контролируемом исследовании, включавшем 221 больного PA (W. G. Riskin и соавт., 1989). Лечение амиприлозой (6 г/день) приводило к достоверному снижению числа воспаленных истекших суставов, увеличению силы сжатия кистей, снижению потребности в анальгетиках. В целом эффект зарегистрирован у 41% больных. По другим параметрам (утренняя скованность, СОЭ, СРБ и РФ) различий с плацебо не отмечено. Переносимость лечения была удовлетворительной, не закончили лечение изза развития побочных эффектов 7 из 98 больных. Недавно эффективность и низкая токсичность амиприлозы были подтверждены в длительном (13 года) двойном слепом контролируемом исследовании (J. R. Caldwell и соавт., 1993) с участием 286 больных РА. Наиболее частые побочные эффекты: головные боли, небольшие желудочнокишечные расстройства и зрительные нарушения. 15.9. Субриум (SubreumR) (ОМ8980) Этот препарат является лиофилизированным экстрактом, полученным при фракционировании щелочных гидролизатов нескольких линий E. Colli. Присутствующий в экстракте эндотоксин полностью разрушен в процессе гидролиза, гликопротеиновый компонент содержит главным образом кислые пептиды с молекулярной массой от 10 до 300 kD. Специальные исследования позволили выявить крайне низкую токсичность препарата. Механизм действия Механизмы действия препарата суммированы T. L. Vischer (1993). В серии экспериментальных исследований обнаружены существенные иммуномодулирующие эффекты препарата при его пероральном введении животным, в том числе увеличение резистентности к инфекции, клиренса коллоидных частиц количества колониеобразующих клеток с эритроцитами барана, снижение активности адьювантного артрита, увеличение синтеза IgA в кишечнике. Кроме того, препарат обладает способностью in vitro усиливать прилипание макрофагов и моноцитов, цитотоксичность моноцитов макрофагов против опухолевых клеток, индуцировать синтез цитокинов. Полагают, что иммуномодулирующие эффекты препарата могут быть связаны с индукцией пероральной толерантности к белковым антигенам. Данные как открытых, так и контролируемых исследований свидетельствуют об определенной эффективности субриума при РА по сравнению с плацебо (M. Rosenthal и S. Plattner, 1981; D. Brackertz и Vischer, 1989; J. P. Hauzer и Th. Appelboom, 1989). При этом показано, что оптимальные результаты достигаются при использовании препарата перорально в суточной дозе 24 мг/день. Оказалось также, что препарат по клинической эффективности не уступает ауранофину (T. L. Vischer, 1988) и D-ïåíèöèëëàìèíó (A. Verstraeten и соавт., 1990), но дает меньше побочных эффектов. При длительном лечении отмечается нарастание клинического эффекта к 12му месяцу, что сочетается с хорошей переносимостью (T. L. Vischer, 1990; M. Rosenthal и соавт., 1991). 15.10. SKF 105685 SKF 105685 представляет собой N-N-диметил-8,8-дипропил-2-азаспирол [4,5 дикан-2-пропанамин дигидрохлорид], относится к семейству азаспиранов. Являясь катионным амфильным веществом, обладает лизосоматотропной активностью. Препарат разработан компанией Smith Kline Beecham Pharm, США. Противовоспалительная и иммуномодулирующая активность продемонстрирована на нескольких экспериментальных моделях, включая адъювантный артрит (А. М Badger и B. A. Swift, 1993) и волчаночноподобное заболевание у MRL мышей (C. R. AlbrightsonWinslow и соавт., 1990). Предполагается, что терапевтическая эффективность может быть связана с активацией супрессорных клеток. 15.11. Талидомид Талидомид представляет собой aNôèòàëèäîãëþòàðèìèä. Препарат синтезирован в Германии в 1956 г., вначале использовался как седативный и снотворный препарат. В 1961 г. исключен из клинической практики в связи с тератогенным эффектом. Начиная с 1965 г. талидомид вновь стали применять для лечения лепроматозной формы проказы, а затем и некоторых ревматических и аутоиммунных заболеваний, таких, как дискоидная и системная красная волчанка, PA (O. GutierreresRodriguez и соавт., 1984), болезнь Крисчена—Вебера (Eravelly и соавт., 1977), болезнь Бехчета (T. Saylan и соавт., 1982), язвенный колит (M. R. F. Waters и соавт., 1979). Иммунные эффекты 1. селективная ингибиция синтеза ФНО моноцитами человека, индуцированная ЛПС, но не ИЛ1b или ГМ-КСФ; 2. ингибиция синтеза антител в ответ на Тзависимые антигены и отсутствие влияние на антителообразование при стимуляции Тнезависимыми антигенами; 3. снижение соотношения Тхелпер/Тсупрессор у здоровых индивидуумов; 4. подавление хронической реакции трансплантат против хозяина у экспериментальных животных и человека; 5. стабилизация лизосомальных ферментов, снижение хемотаксиса нейтрофилов in vitro, фагоцитарной активности, образования супероксидных радикалов; 6. ингибиция ангиогенеза (R. J. D'Amato и соавт., 1994). Клиническое применение По данным O. GutierrezRodriguez (1984), талидомид (6.915 мг/кг/день) индуцировал развитие длительной ремиссии 4 из 7 больных РА, а у остальных 3 — вызвал клиническое улучшение. Клинический эффект ассоциировался с нормализацией СОЭ и снижением титров РФ. E. Ato и E. I. Sato (1993) использовали талидомид для лечения 23 женшин, страдающих СКВ с тяжелым поражением кожи, резистентным к антималярийным препаратам и ГК. На фоне лечения талидомидом (300 мг/день с постепенным снижением) у 90% больных отмечена полная ремиссия кожного процесса. Побочные эффекты Сонливость и боли в животе, которые исчезали после уменьшения дозы или отмены препарата. Основным ограничением при использовании талидомида является его тератогенность. Кроме того, описано развитие необратимой периферической невропатии, зависящей от дозы и длительности лечения. 15.12. Тепоксалин (Tepoxalin) Тепоксалин представляет собой новый иммуносупрессивный препарат, имеющий следующую структуру: (5-4(-chlorophenyl)-N-hydroxy-(4-methoxyphenyl)pN-methyl-1 H-pyrazole-3-propanamide). Создан в R. W. Johnson Pharmaceutical Res. Inst. Механизм действия Препарат разрабатывался как двойной ингибитор циклооксигеназного и липоксигеназного путей арахидонового каскада (D. W. Anderson и соавт., 1990; D. C. Argentieru и соавт., 1990). В настоящее время описаны разнообразные иммунные эффекты препарата, напоминающие выявленные у ЦсА. Однако по молекулярным механизмам ингибирующего действия на Тклеточный иммунитет тепоксалин отличается от ЦсА (L. Zhou и соавт., 1994). Показано, что тепоксалин подавляет Са2+независимые пути Тклеточной активации, которые резистентны к действию ЦсА, в частности связанные с влиянием на фактор транскрипции NFAT. Имеются данные о том, что тепоксалин обладает синергизмом с субоптимальными концентрациями ЦсА в отношении подавления отторжения трансплантатов. В настоящее время проводится многоцентровое контролируемое исследование тепоксалина при РА. Предварительные результаты свидетельствуют о несомненной эффективности тепоксалина при РА. 15.13. Линомид (Linomide) Линомид является новым иммуномодулирующим препаратом, имеющим следующую структуру: (Nmethyl-N-phenyl-1, 2-dihydro-4-hydroxy-1methyl-2-oxoquinoline-3-carxamide) (Kabi Pharmacia Therapeutics). Линомид обладает способностью усиливать активность ЕКклеток, моноцитов/макрофагов и Тлимфоцитов (T. Kaland и соавт., 1985; E. L. Larsson и соавт., 1987; T. Kalland, 1990), подавляет активность аутоиммунного процесса и увеличивает продолжительность жизни MRL/lpr и NZB/NZW мышей (A. Tarkowski и соавт., 1986; A. Tarkowski и соавт., 1986). Предполагается, что механизм действия линомида связан с подавлением дистальных механизмов процесса регулируемой гибели клетки (апоптоза) (G. Kroemer и C. Martinez, 1994). Предварительные результаты свидетельствуют о возможности использования линомида (от 0.0125 до 0.8 мг/кг) при СКВ и PA (O. Nived и соавт., 1994). 15.14. Таксоиды Таксоид — природный продукт, полученный из коры тисового дерева, Taxus brevifolia, произрастающего на островах, расположенных в северозападной части Тихого океана. Существуют 2 препарата таксоида — таксол (паклитаксел; BristolMyers Squibb, США) и доцитаксил (таксотир; RhonePoulenc, США—Франция). Оба препарата обладают выраженной противоопухолевой активностью при экспериментальных опухолях. Таксол проходит III фазы клинических испытаний при раке яичников, молочной железы, легких, меланоме и лейкозах (K. Gelmon, 1994). В настоящее время молекула таксола полностью синтезирована химическим путем (K. G. Nicolaou и соавт., 1994). Механизм действия Препарат связывается с bсубъединицами тубулина, что приводит к усилению полимеризации микротрубочек и смещению динамического равновесия в сторону образования стабильных тубулиновых полимеров (S. B. Horwitz, 1992). Это вызывает подавление клеточного деления в позднюю G2митотическую фазу. Кроме того, нарушение сборки микротрубочек может приводить к нарушению различных клеточных функций, включая миграцию, фагоцитоз, хемотаксис, адгезию, внутриклеточный транспорт (R. L. Roberts и соавт., 1982; A. Iannone и соавт.. 1987; E. Rowinsky и соавт., 1990). Недавно было обнаружено, что таксол влияет на синтез ФНОa и ИЛ-1 (C. Bogdan и A. Ding, 1992; C. BottexGauthier и соавт., 1992; C. L. Manthey и соавт., 1992). Получены данные о способности таксола подавлять развитие экспериментального коллагенового артрита (C. Tang и соавт., 1993; Е. Branch и соавт., 1994; S. J. Oliver и соавт., 1994), что указывает на потенциальную возможность использования этого нового класса лекарственных препаратов для лечения воспалительных ревматических заболеваний. 15.15. Ультрафиолетовая фототерапия Фотосенсибилизация — хорошо известное осложнение СКВ. Прямое повреждающее воздействие солнечного света на кожу, особенно очевидное при подострой кожной красной волчанке, может быть причиной обострения кожного процесса при дискоидной волчанке или усиливать поражение кожи при СКВ, Кроме того, ультрафиолетовое облучение (УФО) потенциально способно вызывать обострение не только кожного синдрома, но и системного иммунопатологического процесса при СКВ. Однако недавно появились сообщения о благоприятном эффекте УФО с определенной длиной волны при СКВ. Механизм действия В спектре электромагнитной радиации ультрафиолетовое излучение по длине волны занимает промежуточное место между рентгеновским и видимым спектром. Ультрафиолетовое излучение подразделяется на УВ-С (200280 нм), УВ-В (280320 нм), и УВ-А (320400 нм). УВ-А в свою очередь подразделяется на УВ-А2 (320340 нм) и УВ-А1 (340400 нм). ÓÂ-À2 по физическим свойствам приближается к УВ-В, в то время как ÓÂ-À1 находится в пределах видимого спектра. Важно подчеркнуть, что именно облучение УВ-В индуцирует развитие поражения кожи при СКВ. В зависимости от длины волны ультрафиолетовое излучение обладает неодинаковой способностью проникать в кожу. При этом УВ-А проникает более глубоко, чем УВ-В, а ÓÂ-À1 — более глубоко, чем ÓÂ-À2. УВ-В абсорбируется главным образом хромофорами эпидермиса, а ÓÂ-À1 абсорбируется в дерме. Поскольку кровь и лимфа циркулируют в дерме, ÓÂ-À1 оказывает влияние на циркулирующие клетки, что и определяет системную фотобиологическую активность этого типа ультрафиолетового излучения. Токсическое действие УВ-В на кожу в наибольшей степени определяется повреждением ДНК (основной хромофор УВ-В протонов), приводящим к образованию иммуногенной ДНК, которая в свою очередь может индуцировать продукцию патогенных антиДНК антител. Кроме того, УВВ активирует транслокацию ядерного или цитоплазматического SSA/Ro антигена на поверхности кератиноцитов, вызывая синтез àíòèRo или связывание циркулирующих àíòèRo с антигеном и развитие воспалительного процесса, индуцирует появление в эпидермисе CDDR+клеток Лангерганса и cisóðîêàíîâîé кислоты, что, как полагают, определяет развитие УВ-Виндуцированной иммуносупрессии. Это эффект УВ-В может приводить как позитивным, так и к негативным последствиям, особенно при СКВ, при которой наблюдается тенденция к депрессии клеточного иммунитета. Фотобиологический эффект ÓÂ-À1 существенно отличается от такового УВ-В и заключается в первую очередь в воздействии не на ДНК, а на эндогенные хромофоры клеточных мембран и цитоплазмы. Это приводит к появлению в коже DR+ Тклеток, обладающих способностью модулировать клеточные иммунные реакции. В опытах in vitro было показано, что ÓÂ-À1 фотоны подавляют гуморальные иммунные реакции, активируют механизмы фоторепарации ДНК, предохраняют от окислительного повреждения. В экспериментальных исследованиях было показано, что хроническое облучение УФА1 увеличивает выживаемость NZW/B мышей и вызывает снижение уровня антител к ДНК (H. McGrath и соавт., 1987). Кроме того, ÓÂ-À1 оказывает влияние на образование простаноидов, модулирует противовирусную активность, активирует некоторые ферменты. Клинические испытания Методика проведения ультрафиолетовой фототерапии подробно изложена H. MacGrath (1994). В клинических исследованиях, охватывающих в общей сложности 55 больных СКВ (в том числе одном перекрестном контролируемом) было показано, что облучение УФА1 (5 дней в неделю в течение 3 нед.) приводит к достоверному снижению некоторых параметров активности СКВ, включая слабость, боли в суставах, скованность, лихорадку, но не влияет на головную боль и серозит. При этом клинический эффект был более заметен у больных, в сыворотках которых были обнаружены антитела к SSA/ Ro. У нескольких больных, продолживших лечение более длительное время, наблюдалось нарастание эффекта со снижением активности с 30% через 3 нед. до 70% через 8 мес. Обращает на себя внимание эффективность УФА1 в отношении кожных проявлений, в том числе подострой кожной красной волчанки, ассоциирующейся с антителами к SSA/Ro (W. L. Morison, 1994; H. McGrath, 1994) 15.16. Мерказолил (Methimazole) Синтетический антитиреоидный препарат, использующийся для лечения диффузного токсического зоба (аутоиммунной болезни Грейвса). Механизм действия при диффузном токсическом зобе связывают с подавлением пероксидазной активности тиреоцитов, приводящей к ингибиции образования тиреоидных гормонов (D. S. Cooper, 1984). Недавно появились сообщения об иммуномодулирующей активности препарата, проявляющейся в подавлении транскрипции генов класса 1 ГКГ тиреоцитов (M. Saji и соавт., 1992). По данным D. S. Singer и соавт. (1994), лечение метимазолом уменьшает экспрессию молекул класса 1 ГКГ и предотвращает развитие заболевания у мышей с волчаночноподобным заболеванием, индуцированным иммунизацией моноклональными антителами к ДНК, экспрессирующими патогенный 16/6 идиотип. Предварительные результаты свидетельствуют о том, что лечение метамизолом замедляет развитие волчаночноподобных проявлений у NZB/NZW мышей. 15.17. Витамин E Витамин E (атокоферол) — представитель семейства веществ, называющихся токоферолами, состоящих из полностью насыщенной 16-углеродной фитоновой цепи, соединенной 2-метил, 6-хроманоловым ароматическим кольцом. В зависимости от степени и места метилирования хроманолового кольца могут образовываться различные типы токоферолов, из которых наиболее мощной биологической активностью обладает атокоферол. Витамин Е обладает антиоксидантной активностью (C. W. Burton и K. U. Ingold, 1989): предотвращает неконтролируемое перекисное окисление мембранных липидов и влияет на образование промежуточных продуктов циклооксигеназного и липоксигеназного каскадов. Продемонстрированы разнообразные иммунные эффекты (a-токоферола, проявляющиеся в усилении клеточных и гуморальных иммунных реакций (R. P. Tergendy, 1989), и цитопротективное действие, связанное со способностью витамина Е действовать как скавенджер реактивных кислородных радикалов, стабилизировать мембраны, препятствуя их перекисному окислению (C. K. Chow, 1991). Кроме того, витамин Е ингибирует индуцированное различными агонистами прилипание моноцитов к ЭК, что связывают с подавлением экспрессии Е-селектина (R. Faruqi и соавт., 1994). Проведено двойное слепое контролируемое исследование эффективности высоких доз витамина Е (541 ME 3 раза в день) в сравнении с 150 мг диклофенака у 41 больного РА, в котором показана сходная эффективность и частота побочных эффектов обоих препаратов в течение 3 нед. терапии. Обсуждается роль витамина Е в реализации противовоспалительной активности рыбьего жира. Витамин Е иногда применяется для лечения поражения кожи при дискоидной или системной красной волчанке. Препарат более эффективен при недавно развившемся поверхностном поражении кожи и при использовании его в больших дозах (8002000МЕ/день). Витамин Е дает положительный инотропный эффект, с особой осторожностью его следует применять у больных с артериальной гипертензией и сахарным диабетом. 15.18. Витамин D При хронических воспалительных заболеваниях (туберкулез, саркоидоз, ревматоидный артрит) наблюдается локальная гиперпродукция 1,25-дигидроксивитамина Д (кальцитриол) (M. E. Hayes и соавт., 1989). Кальцитриол проявляет следующие иммунные эффекты: 1. потенцирует ингибирующее действие ЦсА в отношении подавления Т-хелперов у больных PA (P. Gepner и соавт., 1989); 2. подавляет митогенез лимфоцитов (W. F. C. Rigby и соавт., 1984); 3. регулирует продукцию цитокинов, в том числе ИЛ-2 Т-лимфоцитами (A. K. Bhalla и соавт., 1986; Rigby W. F. C. и соавт., 1987); 4. подавляет экспрессию HLADR и CD4 антигена моноцитами, и представление (презентирование) антигенов АПК (W. F. C. Rigby и соавт., 1990); 5. подавляет акцессорную функцию моноцитов (W. F. C. Rigby и M. G. Waugh, 1992); 6. подавляет развитие экспериментального адъювантного артрита (M. C. Boissier и соавт., 1989); 7. ингибирует синтез иммуноглобулинов Влимфоцитами больных СКВ (M. LinkerIsraeli и соавт., 1995). Проведено 6месячное открытое исследование эффективности 1,25-дигидроксивитамина Д (Rocatrol, Hoffman-LaRoche) у 10 больных псориатическим артритом (D. Huckins и соавт., 1990). Доза препарата вначале составляла 0.5 мг/день, затем она была увеличена (по 0.25 мг/день) до 2 г в день. На фоне лечения у 4 больных отмечено выраженное, а у 3— умеренное клиническое улучшение. У 2 больных развилась гиперкальциурия. 15.19. Гранулоцитарномакрофагальный колониестимулирующий фактор (ГМ-КСФ) и гранулоцитарный колониестимулирующий фактор (ГКСФ) ГМ-КСФ — гликопротеин, обладающий способностью in vitro стимулировать пролиферацию и дифференцировку клетокпредшественников нейтрофилов, эозинофилов и макрофагов, а Г-КСФ — только гранулоцитов. Оба препарата получены в рекомбинантной форме и используют в клинической практике для лечения заболеваний, ассоциирующихся с нейтропенией. Имеется несколько сообщений об успешном использовании ГМ-КСФ или Г-КСФ для лечения нейтропении у больных с синдромом Фелти (G. Joseph и соавт., 1991; J. S. Pixley и соавт., 1993; H. Markusse и соавт., 1990; T.Ito и соавт., 1992; H. Nielsen и J. Mejer, 1992; K. Bhalla и соавт., 1993), лейкопении у больных СКВ, связанной с использованием высоких доз ЦФ (I. Maldonato и соавт., 1994), и индуцированной препаратами золота нейтропении у больных PA (D. A. Collins и соавт., 1993). Однако при РА на фоне лечения нормализация числа лейкоцитов иногда ассоциируется с обострением артрита (E. G. E. De Vries и соавт., 1991; M. Yasuda и соавт., 1994), коррелирующим с увеличением концентрации острофазовых белков и ИЛ-6. Имеются данные о благоприятном эффекте комбинированной терапии МТ и ГМ-КСФ при синдроме Фелти с СD4+Т-клеточной лимфопенией (Y. Hoshina и соавт., 1994). 15.20. Пентоксифиллин Пентоксифиллин (3,7-Диметил-1-(5-оксогексил)-ксантин) обладает способностью подавлять активность фосфодиэстеразы, способствует накоплению цАМФ. Терапевтическая активность связана с сосудорасширяющим действием, уменьшением агрегации тромбоцитов, повышением деформируемости эритроцитов и снижением вязкости крови. Появились сообщения о следующих иммуномодулирующих эффектах пентоксифиллина: 1. подавление высвобождения ФНО макрофагами (L. Morrone и соавт., 1994); 2. ингибирование in vitro пролиферативного ответа лимфоцитов здоровых и больных РА при стимуляции фитогемагглютинином и синтез ФНО-а (JM. Anaya и соавт., 1994); 3. подавление активации нейтрофилов за счет ингибиции синтеза ФНО-а при экспериментальной эндотоксемии (D. Van Leenan и соавт., 1993). 15.21. Кетотифен Кетотифен — препарат, ингибирующий высвобождение медиаторов из тучных клеток и являющийся антагонистом гистамина. Тучным клеткам придают большое значение в развитии ССД. Медиаторы тучных клеток (гистамин, гепарин и др.) дают профибротический эффект (H. M. Glaman, 1989). В ранней, воспалительной фазе ССД в коже обнаруживаются активированные фибробласты и увеличение содержания тучных клеток. Медиаторы тучных клеток способствуют пролиферации фибробластов и синтезу коллагена in vitro. Показано, что кетотифен подавляет развитие фиброза у линии мышей с плотной кожей, рассматривающихся как модель склеродермии (M. Walker и соавт., 1990). Имеются данные об эффективности кетотифена у больных прогрессирующей ССД, резистентной к D-ïåíèöèëëàìèíó (B. L. Gruber и соавт., 1990). Проведено двойное слепое контролируемое исследование кетотифена у 24 больных ССД в течение 6 мес. (B. L. Gruber и L. D. Kaufmann, 1991). Больные получали 3 мг кетотифена 2 раза в день. Не отмечено существенных различий с плацебо по клиническим параметрам, функции легких и общей оценке состояния больных, кроме кожного зуда. 15.22. Лефлуномид (Leflunomid) Лефлуномид (Hoechst AG Werk Kalle-Albert) — производное изоксазола, является пролекарством, метаболизирует в кишечнике и печени с образованием активного вещества А77 1726, обладающего противовоспалительной и иммуномодулирующей активностью (R. R. Bartell и соавт., 1993). Препарат сильно связывается с белком и имеет продолжительность полужизни около 15 дней. Стабильная концентрация в сыворотке на фоне постоянного приема достигается через 16 недель. Механизм действия 1. подавление высвобождения гистамина тучными клетками и реактивных кислородных радикалов лейкоцитами 2. подавление прилипания лейкоцитов к ЭК 3. подавление миграции лейкоцитов в ткани 4. подавление ранних этапов активации Т-лимфоцитов за счет ингибиции экспрессии ИЛ2Р и рецепторов для трансферина на Т-лимфоцитах 5. подавление синтеза ИЛ-2, ИЛ-3, ИЛ-4, ГМ-КСФ и ФНО-а Молекулярный механизм действия лефлуномида связывают с его способностью подавлять биосинтез пуринов и активность тирозинкиназы. В экспериментальных исследованиях было показано, что лефлуномид подавляет развитие адъювантного и протеогликанового артритов (R. R. Barlett и соавт., 1985), оказывает благоприятный эффект на течение болезни у MRL/lpr мышей и у крыс с экспериментальным аутоиммунным тубулоинтерстициальным нефритом (G. H. Thomas и соавт., 1991), предотвращает синтез антител к ацетилхолиновым рецепторам и развития паралича у животных при экспериментальных аутоиммунных заболеваниях нервной системы. Клинические испытания Эффективность лефлуномида была продемонстрирована Z. Dorniian и соавт. (1993) в 6-месячном двойном слепом контролируемом исследовании у 402 больных активным РА. Установлено, что лечение препаратов в дозе 10 мг в день и 25 мг в день вызывает достоверное улучшение следующих параметров по сравнению с плацебо: суставной счет, количество отекших и болезненных суставов, общая эффективность по оценке врача и больного. Побочные эффекты зависели от дозы и проявлялись болями в животе, расстройством стула, кожной сыпью, обратимой алопецией, похуданием. Безопасность и потенциальная эффективность лефлуномида (доза 5, 10 или 25 мг в день) по сравнению с плацебо была оценена у 350 больных РА в рамках фазы II клинических испытаний (B. Rozman и соавт., 1994). Длительность испытания составила 18 месяцев. Продемонстрирована определенная эффективность и низкая токсичность препарата в дозе 20 мг в день. По данным В. Rozman и соавт. (1994) однократный еженедельный прием лефлуномида в дозе 100 или 200 мг по эффективности не уступает ежедневному приему препарата в дозе 10 и 25 мг в день. Проводится фаза II клинических испытаний препарата HR 325, который химически сходен с активным метаболитом лефлуномида А77 1726. Список литературы к главе "Другие препараты и методы лечения" 1. 2. 3. Балабанова Р. М: Препараты тимуса в комплексной терапии ревматоидного артрита. Клиническая фармакология и терапия 1994, No. 4, стр. 29-31. Бунчук Н. В.: Средства и методы преимущественно иммунотропного действия. В кн: Патогенетическая терапия ревматических заболеваний. В. А. Насонова, Я. А. Сигидин. М. Медицина. 1985; стр. 107-140. Abe J., Nakamura К., Nakano Т. et al.: A novel remissioninducing drug having preventive effects on the autoimmune disorders developed in MRL/L mice (abstract). Arthritis Rheum. 1989; 32 (Suppl. 4); S55. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. Aktulga E., Altac M., Muftuoglu A. et al.: A doubleblind study of colchicine in Behcet's disease. Haematologia. 1980; 65: 399. AlarconSegovia D., Ibanez G., Kershenobisch D. et al: Treatment of scleroderma by modification of collagen metabolism. A double blind trial of colchicine versus placebo. J. Rheumatol. Suppl. 1, 1974; 97. AlarconSegovia D., RamosNiembro F., Ibanez G. et al.: Longterm evaluation of colchicine in the treatment of scleroderma. J. Rheumatol., 1979; 6: 705. AlbrightsonWinslow C. R., Brickson B. B., King A. et al.: Beneficial effects of longterm treatment with SKF 105685 in murine lupus nephritis. J. Pharm. Exp. Ther., 1990; 255: 382-387. Allison A. C., Muller C. D., Almquist S. J. et al.: In vitro immunosupressive effects of mycophenoloc acid and an ester prodrug. RS61443. Transp. Proc., 1991: 23 (Suppl. 2): 10-14. Anaya J.M., Gutierrez M., Silvera L. H., Espinoza L. R.: Effects of pentoxifylline (PTX) on TNFalpha and IL10 production in stimulatedmononuclear cells from rheumatoid arthritis. Arthritis Rheum. 1994; 37 (suppl.): S339. Anaya J. M., Gutierrez M. A., Scopelitis E. et al.: Hyperprolactinemia in primary Sjogren's syndrome. Clin. Res. 1994. Anderson R., Grabow G., Dosthuizen R. et al.: Effects of sulfametoxozole and trimethoprim on human neutrophil and lymphocyte function in vitro: in vivo effects of cotrimazole. Antimocrob. Agents Chemoter, 1980; 17: 322-326. Anderson D. W., Argentieri D. C., Ritchie D. M. et al.: Gastrointestinal profile of tepoxalin, orally active dual cyclooxygenase/lipoxygenase inhibitor with potent antiinflammatory activity. FASEB J. 1990; 4 (suppl. 3): A1122. Antoine J. C., Maurice M., Feldman G. et al.: In vitro and in vivo effects of colchicine and vinblastine on the secretory process of antibodyproducing cells. J. Immunol. 1980; 78: 230-232. Argentieri D. C., Anderson D. W., Ritchie D. M. et al.: Tepoxalin inhibits prostaglandin and leukotriene production in adjuvant arthritis and dog's knee joints challenged with sodium urate and immune complexes. FASEB J. 1990; 4 (suppl. 3): 11-22. Asako H., Kubes P., Baethge B. A. et. al.: Colchicine and methotrexate reduce leukocyte adherence and emigration in rat mesenteric venules. Inflammation 1992; 16: 45-56. Atka E., Sato E. I.: Treatment of the cutaneous lession of systemic lupus erythematosus with thalidomide. Clin. Exp. Rheumatol. 1993; II: 487-493. Badger A. M., Swift B. A.: Therapeutic activity of SKF 105685, a novel azaspirane with suppressor cell inducing activity. Clin. Exp. Rheumatol., 1993; II (Suppl. 8): S107-109. Bartlett R. R., Anagnosotpulos H., Zielinski Т., et al.: Effects of leflulomide on immune responses and models of inflammation. Springer Sem. Immunopath. 1993; 14: 381-394. Beattie T. J. and British association for paediatric nephrology: Levamisole for corticosteroiddependent nephrotic syndrome in childhood. Lancet 1991; 337: 1555-1557. Berczi I., Nagy E., Asa S. L., Kovacs K.: The influence of pituitrary hormones on adjuvant arthritis. Arthritis Rheum. 1984; 27: 682-688. Berсzi I.: The immunology of prolactine. Reprod. Endocrinol. 1992; 10: 196-219. Bhalla A. K., Amento E. P., Krane S. M.: Differential effects of I, 25dihidroxyvitamin D3 on human lymphocytes and monocyte/macrophages: inhibition of interleukin2 and augmentation of interleukinI production. Cell Immunol. 1986; 98: 311-322. Bhalla К., Ross R., Jeter E. et al.: GCSF improves granulocytopenia in Felty's syndrome without flareup of arthritis. Am. J. Hematol. 1993; 42: 230-231. Blank M., Krause I., Teitenbaum D. et al.: Bromocriptine immunomodulation of experimental SLE and primary antiphospholipid syndrome via induction of nonspecific T supressot cells. Lupus 1995; 4 (suppl. 2): 16. Bogdan C., Ding A.: Taxol, a microtubulestabilizing antineoplastic agent, induces expression of tumor necrosis factor alpha and interleukinI in macrophages. J. Leukoc. Bi ol. 1992; 52: 119-121. Boissier M. C., Chipcchia G., Fournier C.: Association of cyclosporin A and calcitriol in the treatment of adjuvant arthritis, (abstract). Arthritis Rheum. 1989; 32 (suppl. 4): S55. Bottex-Gauthier С., Condemine F., Picot F., Vidal D.: Effects of taxol on the macrophage function: Interactions with some immunological parameters. Immunopharmacol. Immunotoxicol. 1992; 14: 3961. Brackertz D., Vischer T. L: OM8980 in rheumatoid arthritis: a sixmonth doubleblind placebocontrol study. J. Rheumatol., 1989; 16: 19-23. Branh E., Tang C., Banquerigo M. L: Regretion of collageninduced arthritis with taxol, a microtubule stabilizer. Arthritis Rheum. 1994; 37: 839-845. Bravo G., Zazueta B., Lavalle C.: An acute remission of Reiters's syndrome in male patients treated with bromocriptine. J. Rheumatol. 1992; 19: 747-750. Bravo G., Zazueta B., Felix A. et al.: Juvenile ankylosing spondililitis (JAS): direct relationship between hyperprolactinemia and interleukin6 (IL6). Arthritis Rheum. 1994; 37 (suppl.): S428. Breedvald F. C., Trentham D. E.: Supression of collagen and adjuvant arthritis by a tetracycline. Arthritis Rheum. 1988; 31 (suppl.): R9. 33. Breedvald F. C., Dijkmans B. A. C., Mattie H.: Minocycline treatment for rheumatoid arthritis: an open dose finding study. J. Rheumatol. 1990; 17: 43-46. 34. Burton G. W., Ingold K. U.: Vitamine E as an in vitro and in vivo antioxidant. Ann. NY Acad. Sci. 1989; 570: 7-22. 35. Caldwell J. R., Bonebrake R., Gruhn W., Mcllwain H. H.: Longterm efficacy and safety of amiprilose hydrochloride (AM) in a doubleblind, randomized withdrawal study in patients with rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S215. 36. Callen J. P.: Oral colchicine therapy is effective for chronic cutaneous leukocytoclastic vasculitis (abstract). Arthritis Rheum. 1987; 30 (suppl. 4): S106. 37. Chikanza I. C., Panayi G. S.: Hypothalamicpitutary mediated modulation of immune function: prolactine as a neuroimmune peptide. Br. J. Rheumatol. 1991; 30: 203-207. 38. Christensen К.: A doubleblind placebo controlled evaluation of fenflumizole in rheumatoid arthritis. Scand. J. Rheumatol., 1986; 15: 80-84. 39. Chow C. K.: Vitamine E and oxidative stress. Free Radical Biol. Med. 1991; II: 215-232. 40. Churg V., Florentin L., Renoux G.: Effects of imuthiol administration to normal and immunodeficient mice on ILI and IL2 production and immune responses regulated by these medication. Int. J. Immunopharmacol., 1986; 2: 163-169. 41. Claman H. N.: On scleroderma: must cells, endothelial cells, and fibroblasts. JAMA, 1089; 162: 12061209. 42. Clevenger C. V., Russel D. H., Appasamy P. M., Prystowsky M. B.: Regulation of interleukin2 driven Tlymphocytes prolipheration by prolactin. Proc Natl. Acad. Sci. USA 1990; 87: 6460-6464. 43. Cohen S. A., Rubinow A., Anderson J. J. et al: Survival of patients with primary (AL) amyloidosis. Colchicinetreated cases from 1976 to 1983 compared with cases seen in previous years (1961 to 1973) . Am. J. Med., 1987; 82: 1182. 44. Collins D. A., Tobias J. H., Hill R. P., Bourke B. E.: Reversal of goldinduced neutropenia with granulocyte colonystimulating factor. Brit. J. Rheumatol. 1993; 32: 518-520. 45. Cooper D. S.: Antithyroid drugs.: New Engl. J. Med., 1984; 311: 1353. 46. Corke C. F., Gul V., Huskisson E. C. et al.: Imuthiol (sodium diethyidithiocarbamate) and rheumatoid arthritis. Int. J. Immunother., 1986; 2: 163-169. 47. Сох N. L., Prowse M. N., Maddison M. C., Maddison R. J.: Treatment of early theumatoid arthritis with rifampicin. Ann. Rheum. Dis., 1992; 51: 32-34. 48. Cross R. J., Campbell J. L., Rozman T. L.: Potentiation of antibody responsiveness after transplantation of syngenic pituitary gland. J. Neuroimmunol. 1989; 25: 29-35. 49. D'Amato R. J., Loughnan M. S., Flynn E., Folkman J.: Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA., 1994; 91: 4082-4085. 50. Dennison W. B., Loeser R., Turner R. A. et. al.: A double blind placebo controlled trial of low dose clotrimasole in rheumatoid arthritis. J. Rheumatol. 1990; 17: 1003-1007. 51. DeRemee R. A.: The treatment of Wegener's granulomatosis with trimetoprim/sulfamethoxazol: illusion or vision. Arthritis Rheum. 1988; 31: 1068-1072. 52. De Vries E. G. E., Willemse P. H. B., Biesma B. et al.: Flareup of rheumatoid arthritis .during GMCSF treatment after chemotherapy. Lancet 1991; 338: 517-518. 53. Dinarello C. A., Chusid M. J., Fauci A. S. et. al.: Effect of prophylactic colchicine therapy on leukocyte function in patients with familial Mediterrian fever. Arthritis Rheum., 1976;19: 618. 54. Domiian Z, Popovic M, Miadenovic V., et al.: Efficacy and safety of leflunomide in the treatment of patients with rheumatoid arthritis. Arthritis Rheum. 1993; 36: S 56. 55. Dougados M., Duchesne L., Amor B.: Bromocriptine and cyclosporine combination therapy for rheumatoid arthritis. Arthritis Rheum. 1988; 31: 1333-1339. 56. Eravelly J., Waters M. F. R.: Thalidomide in WeberChristian disease, (letter). Lancet, 1977; 1: 251. 57. Escalante A., Ehresmann G. R., Quismorio F. P.: Regretion of reactive systemic amyloidosis due to ankylosing spondilitis following the administration of colchicine. Arthritis Rheum. 1991; 34: 920-922. 58. Eugui E. M., Mircovoch A., Allison A. C.: Lymphocyteselective antiprolipherative and immunosupressive activity of mycophenolic acid and its morpholinoethyl ester (RS61443) in rodents. Transpl. Proc., 1991; 23 (Suppl. 2): 15-18. 59. Famaey J. P.: Colchicine in therapy. State of the art and new perspectives for an old grug. Clin. Exp. Rheumatol., 1988; 6: 305-317. 60. Faruqui R., de la Motte C., DiCorleto P. E.: Alphatocopherol inhibits agonistinduced monocytic cell adhesion to cultured human endothelial cells. J. Clin. Invest. 1994; 94:592-600. 61. Ferraci C., Boghen M., Cesano A.: Thyroid autoimmunity in hyperprolactinemia disorders?: Acta Endocrinol. 983; 104: 35-411. 62. Forgren A., Schmeling D., Quie P. G.: Effect of tetracycline on the phagocytic function of human leukocytes. J. Infect. Dis., 1974; 130: 412-415. 63. Franklin C. M., Goldblum R., Robinson C. et al.: Doseranging, placebocontrolleed trial of mycophenolat moffelit (MYC) in refractory rheumatoid arthritis. Arthritis Rheum. 1993; 36 (suppl.): S57. 64. Fumagalli M., Cunietti E., Vainai G. et al.: Controlled clinical trial of imidazole2hydroxybenzoate (ITF182) versus sulindac in patients with rheumatoid arthritis. Clin Ther., 1986; 8: 292-300. 65. Gabriel S. E., Conn D. L., Luthra H.: Rifadin therapy in rheumatoid arthritis. J. Rheumatol., 1990; 17: 163166. 66. Gelrnon К.: The taxoids: paclitaxel and docetaxel. Lancet 1994; 344: 1267-1271. 67. Gepner P., Amor B., Fournier C.: 1,25dihydroxyvitamin D3 potentiates the in vitro inhibitory effects of cyclosporin A on T cells from rheumatoid arthritis patients. Arthritis Rheum. 1989; 32: 31-36. 68. Geusens P., Wouters C., Nijs J., Jiang Y., Dequeker J.: Londterm efffect of omega3 fatty acid supplementation in active rheumatoid arthritis. Arthritis Rheum., 1994; 37: 824-829. 69. Gilman S. C., Carson R. P., Lewis A. J.: Immunomodulatory activity of WY18, 251 (3 (pchlorophenol)thiazole[3, 2a]benzimidazole2acetic). J. Immunopharmacol., 1985; 7: 479-488. 70. Gilman S. C., Carson R. P., Daniels J. F. et al.: Immunological abnormalities in rats with adjuvantinduced arthritis. II. Effect of antiarthritis therapy on immune function in relation to disease involvment. Int. J. Immunopharmacol. 1987; 9: 916. 71. Goldblum R., Rees M. M. C., Eugui E., Schiff M. H.: Immunologic changes in patients with rheumatoid arthritis (RA) treated for one year with new DMARD, mycophenolate mofelit (MycoM). Arthritis Rheum., 1991; 34: S157 (abst.). 72. Goldstein R. C., Schwabe A. D.: Prophylactic colchicine therapy in familial Mediterranean fever: a controlled doubleblind study. Ann. Intern. Med., 1974; 81: 792. 73. Greenwald R. A., Golub L. M., Lavietes B. et. al.: Tetraciclines inhibit human synovial collagenase in vitro and in vivo. J. Rheumatol. 1987; 14: 28-32. 74. Greenwald R., Moak S. A., Ramamurthy N. S., Golub L. M.: Tetraciclines suppress matrix metalloproteinase activity in adjuvant arthritis and in combonation with flurbiprofen, ameliorate bone damage. J. Rheumatol. 1992; 19: 927-938. 75. Grindulis K. A., Nichol F. E., Oldham R.: Phenytoin in rheumatoid arthritis. J. Rheumatol. 1986; 13: 10351039. 76. Grosshans E., Geneviere I.: Thalidomide therapy for inflammatory dermatoses. (Review). Int. J. Dermatol., 1984; 23: 598-602. 77. Gruber B. L., Kaufman L. D.: A doubleblind randomized controlled trial of ketotifen versus placebo in early diffuse scleroderma. Arthritis Rheum., 1991; 34: 362-366. 78. Grube B. L., Kaufman L. D.: Remission in progressive early diffuse scleroderma induced by ketotifen: evidence for the role of mast cells in disease pathogenesis. Am. J. Med., 1990; 89: 392-395. 79. Gutierrez-Rodriguez O.: Talidomide. A promising new treatment for rheumatoid arthritis. Arthritis Rheum., 1984; 27: 1118-1121. 80. Gutierrez M. A., Cabrera O. E., Cuellar M. L. et al.: Prolactin induced autoantibody formation by PBL of normal individuals and SLE patients. Arthritis Rheum. 1993; 36: S197. 81. Guttadauria M., Diamond H., Kaplan D.: Colchicine in the treatment of scleroderma. J. Rheumatol. 1977; 4: 272. 82. Harkness J. A., Griffen A. J., Heinrich I. et al.: A doubleblind comparative study of metronidazole and placebo in rheumatoid arthritis. Rheumatol. Rheabil., 1982; 21: 231-234. 83. Hartman D. P., Holaday J. W., Benton E. W.: Inhibition of lymphocyte prolipheration by antibodies to prolactine. FASEB J 1989: 3: 2194-2202. 84. Hauzer J. P., Appelboom Th.: Doubleblind placebocontrolled study of OM8980 in rheumatoid arthritis. Rheumatol. Int. 1989; 9: 71-76. 85. Hayes M. E., Denton J., Freemont A. J. Mawer E. B.: Synthesis of the acive metabolite of vitamin D, 1, 25 (OH)2D3 by synovial fluid macrophages in arthritis disease. Ann. Rheum. Dis. 1989; 48: 723-729. 86. Hirohata S., Shinohara S., Inoue T. et al.: Regulation of B cell function by lobenzarit, a novel diseasemodifying antirheumatic drug. Arthritis Rheum., 1992; 35: 168-175. 87. Hirohata S.: Regulation of in vitro antiDNA antibody production by a novel disease modifying antirheumatoc drug, Lobenzarit. Clin. Exp. Rheumatol. 1992; 10: 357-363. 88. Hirohata S., Ohnishi К., Sagawa A.: Treatment of systemic lupus erythematosus with lobenzarite: an open clinical trial. Clin. Exp. Rheumatol. 1994; 12: 261-265. 89. Horwitz S. B.: Mechanism of action of taxol. Trends Pharmacol. Sci. 1992; 13: 134-136. 90. Hoshina Y., Moriuchi J., Nakamura Y. et al.: CD4+T cellmediated leukopenia of Felty's syndrome succesfully treated with granulocytecolonystimulating factor and methotrexate. Arthritis Rheum l994; 37: 289-299. 91. Huckins D., Felson D. T., Holick M.: Treatment of psoriatic arthritis with oral 1, 25dihydroxyvitamin D3: a pilot study. Arthritis Rheum. 1990; 33: 1723-1727. 92. Iannone A., Wolberg G., ReynoldsVaughn R., Zimmerman T. P.: Taxol inhibits Nformylmethionylleucylphenylalanine)FMLP)induced neutrophil polarisation and H202 production while decreasing [3H]FMLP binding. Agents Action 1987; 21: 278-280. 93. Ingham E., Turnbull L., Kearney J. N.: The effect of minocycline and tetracycline on the mitotic response of the human peripheral blood lymphocytes. J. Antimicrob. Chemotherapy. 1991; 27: 607-617. 94. Invernizzi F., Monti G.: Colchicine and mixed cryoglobuline mia. Arthritis Rheum. 1993; 36: 722-723. 95. Ito Т., Mlyari Y., Kuwabara T. et al.: Granulocytecolony stimulating factor corrects granulopenia In Felty's syndrome. Am. J. Hematol. 1992; 40: 318-319. 96. Jara L. J., Lavalle C., Fraga A. et al.: Prolactine, immunoregulation and autoimmune disease. Semin. Arthritis Rheum. 1991; 20: 273-284. 97. Jorgensen C., Bressot N., Lefeber P. et al.: Dysregulation of pituitary adrenal axis and pituitary prolactin synthesis in rheumatoid arthritis. Arthritis Rheum. 1993; 36: S175. 98. Joseph G., Neustadt D. H., Hamm J. et al.: GMCSF in the treatment of Felty syndrome. Am. J. Med. 1991; 37: 55-56. 99. Kaland Т.: Regulation of NKprogenitor. Studies with a novel immunomodulator with distinct effect at precursor level. J. Immunol. 1990; 144: 4472-4478. 100. Kawakami A., Eguchi K., Ueki Y. et al.: Effect of lobenzarit disodium on human endothelial cells. Arthritis Rheum., 1991; 34: 296-303. 101. Keater R. A. B., Mason G. B.: Inhibition of microtubule polimerisation by tubulincolchicine complex.: inhibition of spontaneous assembly. Can. J. Biochem. 1981; 59: 361-370. 102. Kroemer G., Martinez C.: Pharmacological inhibition of programmed lymphocyte death. Immunol. Today 1994; 15: 235-242. 103. Kloppenburg M., Breedveld F, C., Terwiel J. P. et al.: Minocycline in active rheumatoid arthritis (RA): a doubleblind placebocontrolled trial. Arthritis Rheum., 1994; 37: 629-636. 104. Kloppenberg M., Miltenburg A. M. M., Verdonk M. J. A. et al.: Minocycline inhibits T cell prolipheration and interferon gamma (IFNgamma) production after stimulation with antiCD3 monoclonal antibodies. Br. J. Rheumatol., 1992; 31 (Suppl. 2): 41. 105. Kloppenburg M., Verwejj C. L., Breedvald F. C., Dijkmans B. A. C: Effect of minocycline on acute phase proteins and serum interleukin6 levels in rheumatoid artritis patients. Brit. J. Rheumatol. 1995; 34 (suppl. 1): 96. 106. Kyle R. A., Greipp P. R., Carton J. P., Gertz A.: Primary systemic amyloidosis. Copparison of melphalan/prednisone versus colchicine. Am. J. Med., 1985; 79: 708. 107. Langevitz P., Zemer D., Book M., Pras M.: Treatment of resistant rheumatoid arthritis with minocycline: an open trial. J. Rheumatol. 1992; 19: 1502-1504. 108. Lee H.J., Pawlak K., Nguyen R. K. et al.: Biochemical differences among four inosinate dehydrogenase inhibitors, mycophenolic acid, ribavirin, tiazofurin, and selenazourin, studied in mouse lymphoma cell culture. Cancer, 1985; 45: 5512. 109. Maldonato I., Lara N., Benitez R. et al.: GMCSF twice monthly cyclophostamide (Ctx) pulses im severe lupus nephritis. Preliminary report. Arthritis Rheum. 1994; 37 (suppl.): S180. 110. Manthey C. L., Brandes M. E., Perera P. Y., Bogel S. N.: Taxol increases steadystate levels of lipopolysaccharideinducible genes and protein tyrosine phosphorilation in murine macrophages. J. Immunol. 1992; 149: 2459-2465. 111. Marshal D. A. S., Hunter J. A., Cappel H. A.: Double blind, placebo controlled study of metronidazole as a disease modifying agent in the treatment of rheumatoid arthritis. Ann. Rheum. Dis., 1992; 51: 758-760. 112. Martin R. R., Warr G. A., Couch R. B. et al.: Effects of tetraciclines on leucotaxis. J. Infect. Dis., 1979; 129: 110-116. 113. McMurray R. W., Weidensaul D., Allen S. H., Walker S. E.: Efficacy of bromocriptine (BCR) in an open label therapeutic trial for systemic lupus erythematosus. Arthritis Rheum. 1994; 37: S369. 114. Mekory Y. A., Chowers Y., Drucker I., Klajman A.: Inhibition of delyed hypersensitivity reaction by colchicine: colchicine inhibits interferongamma induced expression of HLADR on gutepoithelial cell line. Clin. Exp. Immunol. 1989; 78: 230-232. 115. Meyers S., Janowitz H. D., Dumaste V. V. et al.: Colchicine therapy of the renal amyloidosis of ulcerative colitis. Gastroenterology 1988; 94: 1503-1507. 116. Mcgrath H.: UltravioletA1 irradiation decreases clinical disease activity and autoantibodies in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol., 1994; 12: 129-135. 117. McGrath H., Bak E., Michalski J. P.: UltravioletA light prolongs survival and improves immune function in (New Zeland black x New Zeland white) F1 hybrid mice. Arthritis Rheum., 1987; 30: 557-561. 118. McKendry R. J. R., Kraag G., Siegel S., A1Awadhi A.: Therapeutic value of colchicine in the treatment of patients with psoriatic arthritis. Ann. Rheum. Dis. 1993; 52: 826-828. 119. McMurray R. W., Hoffman R. W., Walker S. E.: In vivo prolactine manipulation alters in vitro IL2, IL4, and IFNgamma mRNA levels in female B/W mice. Clin. Res. 1991; 39: 734A (abst.). 120. McMurray R. W., Keiser D., Izui S., Walker S. E.: Hyperprolactinemia accelerates disease activity in the male NZB/NZW mouse model of SLE. Arthritis Rheum. 1991; 34 (suppl.): S163. 121. McMurray R. W., Keiser D., Kanuckel K. et al.: The influence of prolactine on disease activity in the female B/W mouse. J. Immunol. 1991; 147: 3780-3787. 122. Michara M., Nakano Т., Ohsugi Y.: An immunomodulating antirheumatic drug, lobenzarit disodium (CCA): inhibition of policlonal Bcell activation and prevention of autoimmune disease in MRL/Mplpr/lpr mice. Clin. Immunol. Immunopathol. 1987; 45: 366-374. 123. Molard Y., Reinman J., Levin R. I., Cronstein B. N.: A new mode of action tor an old drug: colchicine decreases surface expression of adhesion molecules on both neutrophils (PMNs) and endothelium (abstract). Arthritis Rheum. 1992 (suppl. 9): S35. 124. Montgomery D., Zukovski C., Shan G.: Con A stimulated murine splenocyte produce a factor with prolactine like bioactivity and immunoreactivity? Biochem. Biophys Res. Commun 1987: 145: 692-698. 125. Morrone L., Fernandes L., Schumacher R. et al.: Pentoxifilline inhibits the particleinduced production of tumor necrosis factor by raw 264&7 macrophages in culture. Arthritis Rheum. 1994; 77 (suppl.): S168. 126. Muller-Fassbender H. R.: A 6month randomized dose range study of OM8980 in rheumatoid arthritis. Br. J. Rheumatol., 1993; 32: 746-750. 127. Nicolaou K. C., Yang Z., Liu J. J. et al.: Total synthesis of taxol. Nature 1994; 367: 630-634. 128. Nielsen H., Mejer J.: Remission of neutropenia in case of Felty's syndrome by rhGCSF. Scand. J. Rheumatol. 1992; 21: 305-307. 129. Nived O., Sturfelt G., Nilsson B. et al.: Clinical pilot study of roquimex (linomide) in patients with autoimmune disease. Int. J. Immunother. 1994; 2: 49-59. 130. Ouyang Y., Wang W., Bhuta S., Chang Y.H.: Mechanism of action of colchicine VI: effect of colchicine on generation of leukotriene B4 by human polymorphonuclear leukocytes. Clin. Exp. Rheumatol. 1989; 7: 397-402. 131. Permin H., Stahl Skov P., Norm S, et al.: Possible role of histamine in rheumatoid arthritis. Allergy 1981; 36: 435-436. 132. Pixley J. S., Yoneda K. Y., Manalo P. B.: Sequential administration of cyclophosphamide and granulocytecolony stimulating factor relieves impaired myeloid maturation in Felty's syndrome. Am. J. Hematol. 1993; 43: 304-306. 133. Plotnick S., Huppert A. S., Kantor G.: Colchicine and leukocytoclastic vasculitis (letter). Arthritis Rheum. 1989; 32: 1489-1490. 134. Prickett C. S., Johnston C. D.: The in vivo production of carbon disulfide from tetraethylthiuramdisulfide (Antabuse). Biochem. Biophys. Acta, 1977; 12: 542-546. 135. Pruzanski W., Greenwald R. A., Steet I. P. et al.: Inhibition of enzymatic activity of phospholipase A2 by minocycline and doxycicline. Biochem. Pharmacol., 1992; 44: 1165-1170. 136. Rabinovich C. E., Schanberg L., Kredich D.: Intravenous immunoglobulin and bromocriptine in the treatment of refractory neuropsychiatric SLE. Arthritis Rheum. 1990; 33 (Suppl): R22. 137. Reibman J., Haines K. A., Rich A. M. et al.: Colchicine inhibits ionophoreinduced formation of leukotriene B4 by human neutrophils: the role of microtubules. J. Immunol. 186; 136: 1027-1031. 138. Renoux G., Renoux M.: Thymuslike activities of sulfur derivatives on Tcell differentiation. J. Exp. Med., 1977; 145: 466-471. 139. Renoux G., Renoux M.: The effects of sodium diethyldithiocarbamate, azathyoprine, cyclophosphamide or hydrocortisone acetate administered alone or in association for weeks on the immune responses of Balb/c mice. Clin Immunol. Immunopathol., 1980; 2: 49. 140. Renoux G., Touraine J. L., Renoux M.: Induction of human null cells into T lymphocytes by the serum of mice treated with sodium diethyldithiocarbamate. J. Immunopharmacol., 1980; 2: 49. 141. Rigby W. F. C., Dinome S., Fanger M. V.: Regulation of lymphokine production and human T lymphocyte activation of 1, 25dihydroxyvitamin D. J. Clin. Invest. 1987; 79: 1659-1664. 142. Rigby W. F. C., Waugh M. G., Graziano R. F.: Regulation of human monocyte HLADR and CD4 antigen expression and antigen presentation by 1, 25dihydroxyvitamin D. Blood 1990; 76: 189-197. 143. Rigby W. F. C., Waugh M. G.: Decreased accessory cell function and costimulatory activity by 1, 25dihydroxyvitamin D3treated monocyte. Arthritis Rheum. 1992; 35: 110-119. 144. Riskin W. G., Gillings D. B., Scarlett J. A.: Amiprilose Hydrochloride for rheumatoid arthritis. Ann. Int. Med., 1989; 111: 455-465. 145. Roberts R. L., Nath J., Friedman M. M., Gallin J. I.: Effects of taxol on human neutrophils. J. Immunol. 1982; 129: 2134-2141. 146. Roberts D. E., Curd J. G.: Sulfonamides as antiinflammatory agents in the treatment of Wegener's granulomatosis. Arthritis Rheum. 1990; 33: 1590-1593. 147. Rogers C. M., Rogers T. J., Gilman S. C.: Effects of WY188251 (3 (pchlorophenil)thiazole[3, 2a]benzimidazole2acetic acid), levamisole and indometacin in the generation of murine T suppressor cell in vitro. J. Immunopharmacol., 1985; 7: 479-488. 148. Rosental M. S. Plattner S.: The treatment of rheumatoid arthritis with OM8980, a bacterial immunostimulating agent. Z. Rheumatol. 1981; 49: 228-231. 149. Rosental M., Bahous I., Ambrosini G.: Longterm treatment of rheumatoid arthritis with OM8980: a retrospective study. J. Rheumatol., 1991; 18: 1790-1793. 150. Rozman B., Domian Z., Popovic M., et al.: Long term administration of leflulomide to patients with rheumatoid arthritis. Arthritis Rheum 1994; 37: S 339. 151. Rozman B., Domian Z., Popovic M., et al.: Weekly administration of leflulomide to patients with active rheumatoid arthritis. Arthritis Rheum. 1994; 37: S 339. 152. Rowinsky E., Cazenave L., Donehower R.: Taxol, a novel investigatlonal antimicrotubule agent. J. Natl. Cancer Inst. 1990; 82: 1247-1257. 153. Russell D. H., Kibler R., Matrisian L. et al.: Prolactin receptors on human T and В lymphocytes: antagonism ot prolactine binding by cyclosporine. J. Immunol. 1985; 134: 3027-3031. 154. Saji M., Moriarty J., Ban T. et al.: MHC class I expression in rat thyroid cells Is regulated by hormones, methimazole, and iodide, as well as interferon. J. Clin. Endocrinol., 1992: 75: 871. 155. Sampio E. P., Sarno E. N., Galilly R. et al.: Thalidomide selectively inhibits tumor necrosis factoralpha production by stimulated human monocytes. J. Exp. Med. 1991; 173: 699-703. 156. Saylan Т., Saltic I.: Thalidomide in the treatment of Behcet's syndrome (letter). Arch. Derm., 1982; 118; 536. 157. Seidman P., Fjellner B., Johannesson A.: Psoriativ arthritis treated with oral colchicine, J. Rheumatol. 1987: 14: 771-779 158. Schiff M. H., Goldblum R., Rees M. M. C.: New DMARD, mycophenolate mofetil (MycoM), effectively treats refractery rheumatoid arthritis (RA) patients for one year. Arthritis Rheum., 1991; 34: S89 (abst.). 159. Schiff M. H., Goldblum R., Fine G. D.: Refractory rheumatoid arthritis treated with new second line agent mycophenolate mofetil for two years. Arthritis Rheum. 1993; 36 (suppl.): S216. 160. Singer D. S., Kohn L. D., Zinger H., Mozes E.: Methimazole prevents induction of experimental systemic lupus erythematosus in mice. J. Immunol., 1994; 153: 873-880. 161. Symoens J., Schuermans Y.: Levamisole. In: AntiRheumatic drugs. Ed. E. C. Huskisson. Fraeger. 1983,569-596. 162. Takeda Y., Abe J., Nakamura K. et al.: A novel remissioninducing drug having preventing effects on the autoimmune disorders in NZB/WF1 mice. Arthritis Rheum. 1988; 31 (Suppl. 4): S40. 163. Tang С., Banquerigo M. L., Brahn E.: Regression of collageninduced arthritis with taxol, a microtubule stabilizer. Arthritis. Rheum. 1993 (suppl.); 36: S45. 164. Tarkowski A., Gunnarsson K., Nilsson L. A. et al.: Succesful treatment of autoimmunity in MRL/1 mice with LS 2616, a new immunomodulator. Arthritis Rheum. 1986; 29: 1405-1408. 165. Tarkowski A., Gunnarson K., Stalhandske Т.: Effects of LS 2616 administration upon autoimmune disease of (NZBxNZB) Fl hybrid mice. Immunol. 1986; 59: 589-594. 166. Tergendy P. P.: Vitamin E, immune response, and disease resistance. Ann. NY Acad Sci, 1989; 570: 335344. 167. Van Leenan D., van der Poll Т., Levi M. et al.: Pentoxifylline attenuates neutrophil activation In experimental endotoxemia in chimpanzees. J. Immunol. 1993; 151: 2318-2325. 168. Verstraeten A., Sileghem A., Dequeker J.: OM8980 and dpenicillamine in the treatment of rheumatoid arthritis: a 12month doubleblind randomized study. Scand. J. Rheumatol., 1990; 19: 422431. 169. Vischer T. L. Subreum (OM8980): an antiarthritic E. Coli extract. Clin. Exp. Rheumatol., 1993; II (Suppl. 8): S121-123. 170. Vischer T. L.: A doubleblind multicenter study of OM8980 and auranofin in rheumatoid arthritis. Ann. Rheum. Dis., 1988; 47: 582-587. 171. Waters M. R. F., Laing A. B. D., Ambikarpathy A., LennardJones J. E.: Treatment of ulcerative colitis with thalidomide. Br. Med. J. 1979; 1: 792. 172. Wilson L., Bamburg J. R., Mizel S. B. et al.: Intraction of drugs with microtubule proteind. Fed. Proc. 1974; 33: 158-166. 173. Wotiulewski J. A., Gow P. J., Walter J. et al.: Clotrimazol in rheumatoid arthritis. Ann. Rheum. Dis., 1980; 39: 469-472. 174. WyburnMason R.: Clotrimazole in rheumatoid arthritis (letter). Lance, 1976: 1: 489. 175. Yamaguchi M., Matsuzaki N., Hirita K.: lnterleukin6 possibly induced IL1 in the pituitary gland stimulates the release of gonadotropins and prolactin. Acta endocrinol. 1990; 122: 201-205. 176. Yasuda M., Kihara Т., Wada T. et al.: Granulocyte colonystimulating factor induction of improved leukocytopenia with inflammatory flare in a Felty's syndrome. Arthritis Rheum. 1994: 37: 145-146. 177. Yu L. P., Smith G. N., Brandt K. D. et al.: Reduction of severe of canine osteoarthritis by prophylactic treatment with oral doxycycline. Arthritis Rheum. 1992; 35: 1150-1159. 178. Zemer D., Pras M., Sohar E. et al.: Colchicine in the prevention and treatment of the amiloidosis of familial Mediterranean fever. New Engl. J. Med., 1986; 314: 1001. 179. Zemer D., Livneh A., Danon Y. L. et al.: Longterm colchicine treatment in children with familial mediterraneal fever. Arthritis Rheum. 1991; 34: 973-977. 180. Zhou L., Ritchie D., Wang E. Y. et al: Tepoxalin, a novel immunosuppressive agent with a different mechanism of action from cyclosporin A. J. Immunol. 1994; 153: 5026-5037. ЗАКЛЮЧЕНИЕ. ПЕРСПЕКТИВЫ ФАРМАКОТЕРАПИИ РЕВМАТИЧЕСКИХ ЗАБОЛЕВАНИЙ Несмотря на существенные успехи, достигнутые в лечении ревматических болезней, фармакотерапия этих заболеваний продолжает оставаться одним из наиболее сложных разделов клинической медицины. Это явилось мощным стимулом для разработки новых лекарственных средств с противовоспалительной и иммуномодулирующей активностью и более оптимальных схем лечения этих заболеваний (W. P. Arend, J.M. Dayer, 1995; J. R. Kalden и B. Manger, 1995; C. Richardson и P. Emery, 1995). Наиболее важной группой противоревматических препаратов несомненно остаются НПВС, универсальным механизмом действия которых является блокада синтеза ЦОГ — ключевого бифункционального фермента, участвующего в регуляции синтеза ПГ (глава 2). Этот фермент состоит из трех независимых субъединиц: домена, напоминающего эпидермальный фактор роста, связанного с мембраной основного участка и ферментного домена. Особый интерес представляет открытие нескольких изоформ ЦОГ, играющих различную роль в регуляции синтеза ПГ. ЦОГ-1 является структурным ферментом, участвующим в синтезе ПГ, регулирующих нормальные физиологические процессы, в то время как экспрессия ЦОГ-2, усиливаясь под влиянием провоспалительных стимулов, определяет синтез ПГ, инициирующих развитие воспалительных реакций. В недавних исследованиях было показано, что существующие в настоящее время НПВС в большей степени подавляют активность ЦОГ-1, чем ЦОГ-2, что позволяет объяснить природу основных побочных эффектов этих препаратов (язвенное поражение ЖКТ, нарушение функции почек и др.). Все это вместе взятое создало теоретические предпосылки для разработки нового класса фармакологически активных веществ, обладающих способностью селективно ингибировать ЦОГ-2, которые продемонстрировали свою эффективность на различных моделях воспаления и низкую ульцерогенную активность. Интересно, что в отличие от ряда НПВС, являющихся производными карбоксиловых кислот, большинство селективных ингибиторов ЦОГ-2 содержат в составе молекулы сульфонамидную группу. Один из этих препаратов мелоксикам (Boehringer Ingelheim, Австрия) уже заканчивает фазу III клинических испытания и зарекомендовал себя как весьма эффективный и малотоксичным противовоспалительный препарат. Наконец, результаты исследования фармакологической активности ацетоминофена, обладающего выраженной жаропонижающей и анальгетической, но минимальной противовоспалительной активностью, позволили предположить существование еще одной изоформы циклооксигеназы, ЦОГ-3, регулирующей синтез ПГ в головном мозге. Разработка селективных ингибиторов ЦОГ-3 создаст предпосылки для создания новых эффективных жаропонижающих и анальгетических препаратов. Большим достижением в изучение молекулярных механизмов действия НПВС является расшифровка трехмерной пространственной структуры ЦОГ-1. (D. Picot и соавт., 1994). Оказалось, что участок молекулы с циклооксигеназной активностью представляет собой длинный узкий гидрофобный канал, в верхней части которого присутствуют молекулы тирозина в положении 385 и серина в положении 530. Было также установлено, что некоторые НПВС (например флурбипрофен) ингибирует ЦОГ-1 за счет вытеснения арахидоната из верхней части канала. При этом Sèçîìåð флурбипрофена за счет карбоксильной группы, взаимодействует с аргинином в положении 120, тем самым помещая второе фенильное кольцо в пределы Ван дер Ваальсовых контактов тирозина в положении 358. Необратимая ингибиция ЦОГ-1 аспирином определяется ацетилированием серина в положении 530. Высказано предположение о том, что в пределах этого узкого канала существуют и другие участки связывания, с которыми могут взаимодействовать различные НПВС. Очевидно, что расшифровка молекулярных механизмов взаимодействия НПВС и ферментов арахидонового каскада имеет исключительно важное значение для создания новых химических веществ с антипростагландиновой активностью. Важный дополнительный механизм, определяющий противовоспалительную активность НПВС связан с действием на фактор транскрипции NFkB, которые регулирует экспрессию генов провоспалительных цитокинов. Определенные преимущества по сравнению с другими НПВС имеет набуметон (SmithKline Beecham, Великобритания), который реже вызывает развитие побочных эффектов. Набуметон не является кислотой, он адсорбируется в тонком кишечнике и в нативной форме не влияет на активность ЦОГ. При поступлении в кровяное русло препарат метаболизирует в печени с образованием активного вещества 6метокси2нафтилацетиловой кислоты. Положительным свойством набуметона является отсутствие билиарной экскреции и печеночной рециркуляции, что исключает возможность билиарного рефлюкса активного вещества в желудок. Кроме того, в кислом содержимом желудка набуметон остается в ионизированной форме, что предотвращает его диффузию и накопление в эпителиальных желудочных клетках. Предполагается также, что набуметон в большей степени ингибирует синтез ЦОГ-2, чем ЦОГ-1, он не влияет на аггрегационную способность тромбоцитов и не нарушает функцию почек. Другой подход к уменьшению выраженности побочных эффектов НПВС, а возможно и к увеличению их противовоспалительной активности связан с разработкой комбинированных препаратов, содержащих простагландин Е1 (мизопростол), который обладает цитопротектиной активностью. В настоящее время клиническую апробацию проходят препараты, в состав которых входят мизопростол в сочетании с диклофенаком (Атротек, Searle, США) (В. А. Насонова и соавт., 1995) и мизопростол в сочетании с напросином. Эффективным вспомогательным методом воздействия на воспалительный процесс является локальная терапия с использованием мазей, кремов и гелей, содержащих НПВС. Особенно хорошо зарекомендовал себя крем "Долгит" (PRO. MED. CS, Чешская республика), приготовленный на основе ибупрофена (В. А. Насонова и соавт., 1995). В последние годы в ревматологии все шире используются новые иммуномодулирующие препараты, в основе действия которых лежит ингибиция синтеза провоспалительных и иммунорегуляторных цитокинов, различные олигопептиды вещества, полученные с помощью методов генной инженерии и моноклональные антитела, направленные против мембранных антигенов иммунокомпетентных клеток и провоспалительных цитокинов, изучаются возможности генной терапии (глава 14). Среди лекарственных средств наиболее активно изучается циклоспорин А (Сандиммун, Sandoz Pharma, Швейцария), который нашел применение для лечения РА, СКВ, ССД, болезни Бехчета, ПМ/ДМ и даже гранулематоза Вегенера (глава 10). Предполагается также, что циклоспорин А может быть важным компонентом комбинированной терапии РА и других ревматических заболеваний (P. Tugwell и соавт., 1994). Выраженной иммунной активностью обладает лефлуномид (Behringer Ingelheim, Австрия), действие, которого связывают с подавлением синтеза цитокинов. Проведенные исследования в рамках фазы III клинических испытаний свидетельствуют о несомненной эффективности и низкой токсичности этого препарата. Перспективным препаратом является тенидап (Pfizer, США) (глава II) — представитель нового класса химических веществ — оксиндолов (Е. Л. Насонов и соавт., 1994). Наряду с антициклооксигеназной активностью, тенидап ингибирует синтез ИЛ-1, ИЛ-6 и ФНО-а, снижает уровень острофазовых белков, подавляет продукцию металлопротеиназ хондроцитами. Имеются данные о том, что в некоторых случаях тенидап можно использовать для монотерапии РА вместо стандартного лечения, предполагающего применение комбинации НПВС и какоголибо "базисного" противоревматического препарата. Учитывая уникальный механизм действия тенидапа высказано предположение о том, что лечение этим препаратом может быть прообразом "базисной" терапии остеоартрита. Установлено также, что подавлением синтеза провоспалительных цитокинов лежит в основе фармакологической активности солей золота, МТ и, возможно, сульфасалазина. Ниже суммированы некоторые новые подходы к экспериментальной терапии ревматических заболеваний, находящиеся в стадии экспериментальных или преклинических исследований (G. W. Duff, 1994; J. R. Kalden и B. Manger, 1995; W. P. Arend и J.M. Dayer, 1995; M. P. Vincenti и соавт., 1994; C. Evans и P. D. Robbins, 1994). 1. "Антивоспалительные" цитокины • ИФ-y • ТФР-b • ИЛ-4 • ИЛ-10 • ИЛ-13 2. Растворимые цитокиновые рецепторы • ИЛ-1 • ФНО • ИЛ-8 3. Антагонисты цитокиновых рецепторов • природный антагонист ИЛ-1 • рекомбинантные мутантные цитокины, блокируюшие соответствуюшие интокиновые рецепторы 4. Моноклональные антитела к цитокинам 5. Аутоантигенные пептиды, индуцирующие толерантность (коллаген типа II) 6. Другие ингибиторные механизмы • ингибитор ИЛ-1 конвертирующего фермента • ингибитор высвобождения цитокинов • ингибитор транскрипции генов цитокинов • пост-рецепторные ингибиторы • антисмысловые олигонуклеотиды, комплементарные иРНК провоспалительных цитокинов • индукторы апоптоза • ингибиторы ангиогенеза • селективные ингибиторы металлопротеиназ • ингибиторы синтетазы окиси азота • другие низкомолекулярные ингибиторы В настоящее время активно разрабатываются специфичные методы подавлением воспалительных реакций, связанные с использованием препаратов, селективно ингибирующих синтез цитокинов или их связывание с клеточными рецепторами. Например, недавно было показано, что ИЛ-1 b синтезируется в цитоплазме в виде биологически неактивного крупного предшественника, который превращается в активную внеклеточную форму под влиянием специфического фермента, получившего название ИЛ-1 b конвертазы. Создано химическое соединение (ацилоксиметилкетон), ингибирующее активность этого фермента и продемонстрирована его эффективность на модели экспериментального артрита. Разработаны методы генной терапии артрита (C. Evans и P. D. Robbins, 1994), в которых комплементарная ДНК антагониста ИЛ-1 рецептора вводилась в коленный сустав кролика с помощью аденовирусного вектора. Многообещающие результаты получены в процессе лечения больных РА моноклональными антителами к ФНО-a. Новый аспект лечения РА связан с формированием оральноиндуцированной толерантности к потенциально аутоантигенным компонентам суставного матрикса. Установлено, что экспериментальный аутоиммунный артрит может быть предотвращен при введении животным пептидного фрагмента коллагена типа II (G. Ku и соавт., 1993) и этот эффект опосредован активной периферической иммуносупрессией (H. S. G. Thompson и соавт., 1993). Результаты предварительных клинических испытаний свидетельствуют о том, что пероральный прием коллагена типа II у части больных РА вызывает развитие полной ремиссии в отсутствии существенных побочных эффектов (D. E. Trentham и соавт., 1993). Наряду с разработкой новых лекарственных препаратов, не менее важным направлением является создание рациональных схем лечения с использованием уже известных противоревматических средств и особенно проведение агрессивной комбинированной терапии "базисными" препаратами уже на ранних этапах развития болезней (P. Emery, 1995). Таким образом, в конце 20 века в лечении ревматических заболеваний достигнут очевидный прогресс, и возможности их фармакотерапии неуклонно расширяются. Это дает основания надеяться, что начало 21 века будет ознаменовано решающими успехами в лечении этих широко распространенных тяжелых хронических воспалительных заболеваний человека. Список литературы к заключению "Перспективы фармакотерапии ревматических заболеваний" 1. Насонова В. А., Муравьев Ю. В., Насонов Е. Л. и др: Многоцентровая оценка эффективности и переносимости локальной терапии кремом долгит у больных остеоартрозом. Терапевт, архив 1995; 6: 48-50. 2. Насонов Е. Л.: Новые подходы к лечению аутоиммунных болезней. Клин. фармакол. терапия 1994; 4: 16-21. 3. Насонов Е. Л.Белоусов Бю Ю., Насонова В. А.: Тенидап, новый препарат для лечения ревматоидного артрита. Клин. фармакол. терапия 1994; 4: 52-56. 4. Arend W. P., Dayer JM.: Inhibition of the production and effects of interleukinI and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995; 2: 151-160. 5. Duff G. W.: Cytokines and acute phase proteins in rheumatoid arthritis. Scand. J. Rheumatol. 1994; 23 (suppl. 100): 919. 6. Emery P.: The disease course, prognosis and outcome of rheumatoid arthritis. Rheumatology in Europe. 1995; 24 (suppl. 2): 169-171. 7. Evans С, Robbins P. D.: Prospects for treating arthritis by gene therapy. J. Rheumatol. 1994; 21: 779-782. 8. Kalden J. R., Manger B.: New therapeutic approaches in autoimmune rheumatic disease, with special emphasis on rheumatoid arthritis. Brit. J. Rheumatol. 1995; 34: 193-196. 9. Ku G, Kronenberg M, Peacock D. J. et al.: Prevention of experimental autoimmune arthritis with a peptide fragment of type II collagen. Eur. J.lmmunol. 1993; 23: 591-599. 10. Richardson C., Emery P.: New therapies for rheumatoid arthritis. Brit. J. CIin. Pharmacol. 1995; 49: 14. 11. Thompson H. S. G., Harper N, Bevan D. J., Staines N. A.: Supression of collagen induced arthritis by oral administration of type II collagen: changes in immune and arthritic responses mrdiated by active peripheral supression. Autoimmunity 1993; 16: 189-199. 12. Trentham D. E., DynesiusTrentham R. A., Orav E. J., et al.: Effects of oral administration of type II collagen on rheumatoid arthritis. Science 1993; 261: 1727-1730. 13. Vincenti M. P., Clark I. M., Brinckerhoff C. E.: Using inhibitors of metalloprotelnasrs to treat arthritis: easier said than done? Arthritis Rheum 1994; 37: 1115-1126. ПРИЛОЖЕНИЕ. КРАТКАЯ ХАРАКТЕРИСТИКА ОСНОВНЫХ ПРОТИВОРЕВМАТИЧЕСКИХ ПРЕПАРАТОВ АРТРОТЕК (ARTHROTEC) Международное название — arthrotec. Состав и форма выпуска. Активное вещество — 50 мг диклофенака натрия и 200 мг мизопроcтола. Таблетки с кишечным покрытием. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. SEARLE, США. АЗАТИОПРИН (AZATHIOPRIN) Международное название — azathioprin. Состав и форма выпуска. Таблетки 0.05 г по 50 или 100 шт. в упаковке. Фармакологическое действие и клиническое применение. Цитотоксический препарат (стр. 120). Производитель. Имуран (Imuran) WELLCOME, Великобритания. АНСАИД (ANSAID) см. Флурбипрофен. АПРАНАКС (APRANAX) Международное название — naproxen natrium. Состав и форма выпуска. Активное вещество — напроксена натриевая соль. Таблетки 0.275 и 0.55 г по 10 и 30 шт. в упаковке. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. SYNTEX США АСПИРИН (ASPIRIN) см. Ацетилсалициловая кислота. АУРАНОФИН (AURANOFIN) Международное название — auranofin Состав и формы выпуска. Активное вещество — ауранофин. Таблетки 0.003 г по 10 или 30 шт. в упаковке. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 118). Производитель. Ауропан (Auropan) KRKA, Словения (в сотрудничестве с Smith Kline, США); Гольдар (Goldar), CADILA, Индия. АУРОТИОМАЛЯТ НАТРИЕВАЯ СОЛЬ Международное название — natrium aurothiomalat. Состав и форма выпуска. Активное вещество — ауратиомалят натрия. Раствор для инъекций (1 ампула содержит 0.01, 0.02 г или 0.05 г активного вещества), ампулы 0.5 мл по 10 шт. в упаковке. Фармакологические свойства и клиническое применение. Базисный противоревматический препарат (стр. 118). Производитель. Тауредон (Tauredon) BYK GULDEN, Германия; Миокризин (Myochrisine) MERK, США. АЦЕМЕТАЦИН (ACEMETACIN)* Международное название — acemetacin. Состав и форма выпуска. Активное вещество — ацеметацин. Капсулы 60 мг. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Эмфлекс (Emflex) E. MERCK PHARM., США. АЦЕТИЛСАЛИЦИЛОВАЯ КИСЛОТА (ACIDUM ACETILSALICYLICUM) Международное название — acidum acetilsalicylicum. Состав и форма выпуска. Активное вещество — ацетилсалициловая кислота. Таблетки 0.3, 0.325 и 0.5 г. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. АСПИРИН (ASPIRIN) BAYER, Германия и др. БРОМОКРИПТИН (BROMOCRIPTIN)** Международное название — bromocriptin. Состав и форма выпуска. Активное вещество — бромокриптина метансульфонат. Фармакологическое действие и клиническое применение. Специфический ингибитор секреции пролактина (стр. 291). Производитель. Ибупрофен (lbuprofen) FARMOS, Финляндия, HAFSLUND NYCOMED, Австрия, POLFA, Польша; Мотрин (Motrin) UPJOHN, США ИМУРАН (IMURAN) см. Азатиоприн, ИНДОМЕТАЦИН (INDOMETACIN) Международное название — indometacin. Состав и форма выпуска. Активное вещество — индометацин. Таблетки, драже, капсулы. 0.025 г и 0.05 г. Таблетки ретард 0.075 г. Раствор (1 мл — 0.03 г.) в ампулах. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Индометацин (Indometacine) CHINOIN, Венгерская республика; PHARMACHIM, Болгария; Метиндол (Metindol) POLFA Пабьяницкий, Жешувский, Варшавский фармацевтические заводы (Польша). ИНТЕРФЕРОН а2а (INTERFERON а2а)** Международное название — interferon alpha2a. Состав и форма выпуска. Активное вещество — интерферон а2а. Порошок для инъекций (1 флакон 18 млн. ME активного вещества, 0.009 г хлорида натрия, 0.005 мг альбумина) по 5 флаконов в упаковке. Фармакологическое действие и клиническое применение. Цитокин, полученный генноинженерным путем, обладающий противоопухолевой, противовирусной и иммуномодулирующей активностью. Применение в ревматологии (стр. 271). Побочное действие. Гипертермия, вялость, лихорадка, озноб, потеря аппетита, мышечная боль, головная боль, артральгии, потливость; редкогепатит, парестезии, нейропатия, протеинурия, лейкопения, тромбоцитопения, аутоиммунные реакции. Производитель. РоферонА (RoferonA) ROCHE, Швейцария. КАПТОПРИЛ (CAPTOPRIL)** Международное название — captopril. Состав и форма выпуска. Активное вещество — каптоприл. Таблетки 0.025 г. и 0.05 г. Фармакологическое действие и клиническое применение. Блокирует ангиотензинпревращающий фермент, что ведет к подавлению продукции антиотензина II, который оказывает суживающие действием на артериальные и венозные сосуды. Снижает общее периферическое сосудистое сопротивление, уменьшает постнагрузку, снижает АД. Основными показаниями к назначению являются артериальная гипертензия; хроническая сердечная недостаточность. Применение в ревматологии (стр. 139). Побочное действие. Препарат, как правило, хорошо переносится. Побочные эффекты легкие и преходящие: головокружения, головная боль, усталость, тошнота. Иногда ортостатическая гипертония, атения, кожная сыпь, и др. Очень редко волчаночноподобный синдром, агранулоцитоз, Производитель. Капотен (Capoten) BRISTOLMYERS SQUIBB, США. КЕНАЛОГ (KENALOG) см. Метилпреднизолон. КЕТОПРОФЕН (KETOPROFEN) Международное название — ketoprofen. Состав и форма выпуска. Активное вещество — кетопрофен. Капсулы по 0.95 г по 24 шт. в упаковке. Таблетки ретард по 0.2 г по 14 шт. в упаковке. Свечи ректальные 0.1 г по 12 шт. в упаковке. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Профенид (Profenid) RHONEPOULENC RORER, СШАФранция. КЕТОРОЛАКА ТРОМЕТАМИН (KETOROLACA TROMETAMIN) Международное название — ketorolaca trometamin. Состав и форма выпуска. Активное вещество — кеторолака трометамин. Таблетки 10 мг. Раствор 1мл (15 и 30 мг.) в ампулах. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Торадол (Toradol) SYNTEX, Швейцария, КЕТОТИФЕН (KETOTIFEN) Международное название — ketotifen. Состав и форма выпуска. Активное вещество — кетотифен. Таблетки и капсулы 1 мг. Сироп 02 мг кетотифена в 1 мл в виде гидрогенфумарата. Фармакологическое действие. Тормозит высвобождение гистамина и других медиаторов воспаления из тучных клеток, что связано с его способностью блокировать H1 гистаминовые рецепторы и ингибировать фосфодиэстеразу, в результате чего повышается уровень цАМФ. Применение â ревматологии (стр. 304). Побочное действие. Сонливость, сухость во рту, легкое головокружение. Производитель. Галитифен (Galitifen) ICN GALENICA, Югославия, Задитен (Zaditen) KRKA, Югославия в сотрудничестве с Sandoz, Швейцария; Позитан (Pozitan) POLFA Познанский фармацевтический завод (Польша). КИСЛОТА ФОЛИЕВАЯ (ACIDIC FOLICUM) Международное название — folic acid. Состав и форма выпуска. Активное вещество — фолиевая кислота. Таблетки 0.0008 г. Фармакологическое действие и клиническое применение. Применяется для снижения частоты побочных эффектов на фоне лечения метотрексатом (стр. 151). КЛИНОРИЛ (CLINORIL) см. Сулиндак. КОЛХИЦИН (COLCHICIN) Международное название — colchicin. Состав и форма выпуска. Активное вещество — колхицин. Таблетки 0.001 г. Фармакологическое действие и клиническое применение. Противовоспалительный препарат, подавляет развитие амилоидоза (стр. 282). КОТРИМОКСАЗОЛ (COTRIMOXAZOL) Международное название — cotrimoxazol. Состав и форма выпуска. Активные веществасульфаметоксазол и триметоприм. Таблетки 0.4 и 0.8 г. Фармакологическое действие и клиническое применение. Оказывает бактерицидное действие в отношении ряда грамположительных и грамотрицательных микроорганизмов. Сульфаметоксазол нарушает синтез дигидрофолиевой кислоты в бактериальных клетках, а триметоприм препятствует превращению дигидрофолиевой кислоты в тетрагидрофолиевую. Производитель. Бактрим (Bactrim) ICN Galenica, Югославия; ROCHE, Швейцария; Бисептол (Biseptol) POLFA, Польша; Септрин WELLCOME, Великобритания. КРИЗАНОЛ (CRYSANOLUM) Международное название — crysanol. Состав и форма выпуска. Препарат содержит 70% ауротиопропансульфоната кальция и 30% глюконата кальция. Выпускается в виде 5% масляного раствора по 2 мл в упаковке по 25 ампул. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 118). Производитель. Россия КУПРЕНИЛ (CUPRENIL) см. Пеницилламин. ЛЕВАМИЗОЛ (LEVAMISOL) Международное название — левамизол. Состав и формы выпуска. Активное вещество — левамизола гидрохлорид. Таблетки 0.05 г по 2 шт. в упаковке. Таблетки 0.15 г по 1 штуке в упаковке. Фармакологическое действие и клиническое применение. Оказывает базисное противоревматическое действие (стр. 278). Производитель. Декарис (Decaris) Gedeon Richter, Венгерская республика (по лицензии Jensen, Бельгия). ЛЕЙКЕРАН (LEUKERAN) см. Хлорамбуцил. ЛЕЙКОВОРИН (LEUCOVORIN) Международное название — leucovorin. Состав и форма выпуска. Активное вещество — лейковорин. Порошок для инъекций 0.01 г, 0.025 г во флаконах по 10 шт. в упаковке. Фармакологические свойства и клиническое применение. Препарат позволяет миновать заблокированную метотрексатом реакцию биосинтеза фолиевой кислоты, применяется для уменьшения токсических реакций на фоне лечения метотрексатом (стр. 151). Производитель. LACHEMA, Чехия. ЛЕЙКОМАКС (LEUCOMAX) Состав и форма выпуска. Активное вещество — рекомбинантный человеческий гранулоцитраномакрофагальный колониестимулирующий фактор в виде стерильного лиофилизированного порошка для растворения. Порошок для инъекций 50 мкг (0.55õ10*6 ед); 150 мкг (1.67õ10*6 ед); 400 мкг (4.44õ10*6 ед); 500 мкг (5.55õ10*6 ед); 700 мкг (7.77õ10*6 ед), 1500 мкг (16.7õ10*6 ед) во флаконах. Фармакологическое действие и клиническое применение. Препарат оказывает выраженное влияние на кроветворение и функциональную активность лейкоцитов, стимулирует пролиферацию и дифференцировку предшественников кроветворных клеток. Стимулирует рост гранулоцитов, моноцитов, Т-лимфоцитов, не влияя на созревание В-лимфоцитов. В ревматологии используется для лечения нейтропении при синдроме Фелти и для коррекции цитопенических осложнений, возникающих на фоне лечения базисными препаратами (стр. 303). Побочное действие. Подъем температуры, сыпь. Редко — гипотензия, тошнота, рвота, отеки, боли в груди и костях, диарея. Крайне редко — аллергические реакции, сердечная недостаточность, суправентрикулярная аритмия, спутанность сознания, судороги, одышка, отек легких, анафилаксия, ангионевротический отек, бронхоспазм, полисерозит. Производитель. Совместное производство SANDOZ, Швейцария, SCHERING PLOUGH, США МЕТИЛПРЕДНИЗОЛОН (METHYLPREDNISOLON) Метилпреднизолон депо Международное название — methylprednisolon acetat. Состав и форма выпуска. Активное вещество — метилпреднизолон в виде ацетата. Флаконы, ампулы, шприцы; для внутримышечного и внутрисуставного применения. Метилпреднизолон таблетки Состав и форма выпуска. Активное вещество — метилпреднизолон. Таблетки 0.004, 0.016, 0.032 и 0.1 г. Метилпреднизолон инъекции Международное название — methylprednisolon sodium succinate. Состав и форма выпуска. Сухое вещество в ампулах. Фармакологические свойства и клиническое применение. Глюкокортикоидный препарат (стр. 84). Производитель. ДепоМедрол (Depomedrol)/Ìåäðîä (Medrol)/Coлюмедрол UPJOHN, США; Метипред (Methypred) ORION, Финляндия; Урбазон суспензия/Урбазон (URBASON suspensia) HOECHST, Германия. МЕТОКЛОПРАМИД (METHOCLOPRAMID) Международное название — methoclopramid Состав и формы выпуска. Активное вещество — метоклопрамид. Таблетки 0.01 г по 50 шт. в упаковке. Раствор для приема внутрь (1 мл препарата содержит 0.005 г активного вещества), ампулы 2 мл по 5 шт. в упаковке. Фармакологическое действие и клиническое применение. Оказывает противорвотное действие, успокаивает икоту, устраняет тошноту. Оказывает регулирующее влияние на функцию ЖКТ, понижает двигательную активность пищевода, ускоряет опорожнение желудка, а также ускоряет продвижение пищи по тонкому кишечнику, не усиливая перистальтику и не вызывая диареи. Является антагонистом дофаминовых рецепторов. Дозировка. 510 мг 34 раза в день. При парентеральном введении суточная доза 0.51 мг/кг тела. Побочное действие. В начале лечения усталость, сонливость, головокружение, головная боль, депрессия, акатизия. Редко при длительном приеме в высоких дозах — галакторея, гинекомастия, нарушение менструального цикла. Производитель. Клометол (Klometol) ICN GALENICA, Югославия; Метоклопрамид (Metoclopramid) POLFA, Польша; Реглан (Reglan) ALKALOID, Македония; Церукал (Cerucal) AWD, Германия. МЕТОТРЕКСАТ (METHOTREXATE) Международное название — methotrexate. Состав и форма выпуска. Активное вещество — метотрексат. Таблетки 0.025 г, 0.01 г по 100 шт. в упаковке. Лиофилизированный порошок 0.005, 0.01, 0.05, 0.5, 1.0 г во флаконах по 10 шт. в упаковке. Раствор для инъекций (1 ампула содержит 0.005 г или 1 г активного вещества) ампулы по 10 шт. в упаковке. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 143). Производитель. Метотрексат LACHEMA, Чехия; МетотрексатЭбеве (MethotrexateEbewe) EBEWE Австрия; Трексан (Trexan) FARMOS Финляндия; Ревматрекс (Rheumatrex) LEDERLE, США. МИЗОПРОСТОЛ (MISOPROSTOL)** Международное название — misoprostol. Состав и форма выпуска. Активное вещество — мизопростол. Таблетки 0.2 г по 60, 100, 120 шт в упаковке. Фармакологическое действие и клиническое применение. Синтетический аналог простагландина Е1, используется для профилактики и лечения гастропатии, индуцированной НПВС (стр. 74). Дозировка. 0.2 г 34 раза в день во время еды и на ночь. Побочное действие. Боль в животе, метеоризм, тошнота, рвота, аллергические реакции (кожная сыпь, зуд, отек Квинке), меноррагии, метроррагии. Производитель. Сайтотек (Cytotec) SEARLE, США. МИНОЦИКЛИН (MINOCYCLIN) Международное название — minocyclin hydrochloride. Состав и форма выпуска. Активное вещество — миноциклина гидрохлорид. Капсулы 0.05 г и 0.1 г. Фармакологическое действие и клиническое применение. Полусинтетическое производное тетрациклина. Испытывается в качестве базисного противоревматического средства (стр. 289). Производитель. Миноцин (Minocin) LEDERLE, США. МОТРИН (MOTRIN) см. Ибупрофен. НАБУМЕТОН (NABUMETON)* Международное название — nabumeton. Состав и форма выпуска. Активное вещество — набуметон. Таблетки 0.5 г и 0.75 г. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Релафен (Relafen) SMITHKLINE ВЕЕСНАМ, США. НАКЛОФЕН (NAKLOFEN) см. Диклофенак натрия. НАПРОКСЕН (NAPROXEN) Международное название — naproxen. Состав и форма выпуска. Активное вещество — напроксен. Таблетки 0.125, 0.25, 0.375, 0.5 и 1.0 г. Суспензия для парентерального применения, свечи ректальные. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат, производное пропионовой кислоты (стр. 60). Производитель. Напроксен (Naprpxen) POLFA Пабьяницкий фармацевтический завод (Польша); Напросин (Naprosyn) SYNTEX, Швейцария, KRKA, Словения (в сотрудничестве с Syntex). ОКСАПРОЗИН (OXAPROZIN)* Международное название — oxaprozin. Состав и форма выпуска. Активное вещество — оксапрозин. Капсулы 0.6 г. по 100 шт. в упаковке. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Дайпро (Daypro) SEARLE, США. ОМЕПРАЗОЛ (OMEPRASOL) Международное название — omeprasol. Состав и форма выпуска. Активное вещество — омепразол. Капсулы по 10 шт. в упаковке. Фармакологическое действие и клиническое применение. Препарат, подавляющий желудочную секрецию, ингибитор "протонного" насоса, тормозит активность Н+К+АТФазы в париетальных клетках желудка и блокирует тем самым секрецию соляной кислоты. Язвенная болезнь желудка и двенадцатиперстной кишки, в том числе гастропатия, индуцированная НПВС (стр. 74). Препарат принимают по 20 мг 1 раз в сутки (перед завтраком). Побочное действие. Изредка тошнота, диарея, запоры, боли в животе, метеоризм, головная боль, головокружение, слабость, кожная сыпь. Производитель. Лосек (Losec), ASTRA, Швеция; Омепрол (Omeprol) ZDRAVLE, Югославия. ОРТОФЕН (ORTOPHENUM) см. Диклофенак натрия. ПАНАДОЛ (PANADOL) см. Парацетамол. ПАРАЦЕТАМОЛ (PARACETAMOL) Международное название — paracetamol. Состав и форма выпуска. Активное вещество — парацетамол. Таблетки 0.325 и 0.5 г. Капсулы 0.5 г. Фармакологическое действие и клиническое применение. Противовоспалительный, жаропонижающий и анальгетический препарат. Дозировка. Препарат принимают по 2 шт. (максимальная суточная доза 8 таблеток). Побочное действие. Тошнота, боли в эпигастрии, аллергические реакции, очень редко анемия, тромбоцитопения, гепатотоксичность. Производитель. Ацетаминофен (Acetaminofen) WATSON LABORATORIES, США; Панадол (Panadol) STERLING HEALTH, Великобритания. ПЕНИЦИЛЛАМИН (PENICILLAMIN) Международное название — penicillamin. Состав и форма выпуска. В 1 таблетке с оболочкой содержится 0.25 г. 3,3диметилцистеина по 100 таблеток в упаковке. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 130). Производитель. Купренил (Cuprenil) POLFA Кутновский фармацевтический завод, Польша. ПЕНТОКСИФИЛЛИН (PENTOXIFYLLIN) Состав и форма выпуска. Активное вещество — пентоксифиллин. Драже, пролонгированные драже, раствор для инъекций в ампулах. Фармакологическое действие и клиническое применение. Препарат оказывает сосудорасширяющее действие, улучшает микроциркуляцию, повышает гибкость эритроцитов, уменьшает их адгезию, уменьшает вязкость крови, обладает иммуномодулирующей активностью (стр. 304). Пентоксифиллин используется при широком круге ревматических заболеваний, для которых характерно нарушение микроциркуляции. Дозировка. Внутривенно 100400 мг (в тяжелых случаях) в 250500 мл физ. рра или 5% глюкозы в течение 90180 мин. Перорально 3001600 мг в день. Побочное действие. При парентеральном введении возможно снижение АД. При приеме внутрь — диспепсические явления, тошнота, рвота, сердцебиение, тахикардия, гиперемия кожных покровов, головокружение, головная боль и др. Производитель. Агапурин (Agapurin) SLOVAKOFARMA, Словакия; Оксилин (Oxylin) ORION, Финляндия; Пентилин (Pentilin) KRKA, Словения: Трентал (Trental) HOECHST, Германия. ПЕРКЛЮЗОН (PERCLUZON) Международное название — clofeson. Состав и форма выпуска. Активные вещества — клофезон, эквимолярные соотношения клофезамида и фенилбутазона, которые оказывают противовоспалительное, анальгетическое и жаропонижающее действие, их эффект взаимно усиливается. 1 капсула содержит 0.2 г клофезона в упаковке 30 капсул, 1 свеча содержит 0.4 г клофезона, в упаковке 10 свечей. 1 г мази содержит 0.05 г клофезона, тубы по 30 г. Фармакологическое действие и клиническое применение. Противовоспалительное, болеутоляющее средство. Дозировка. 12 капсулы 23 раза в день в течение 57 дней до 6 недель. Побочное действие. Диспепсические явления и горький вкус во рту. Производитель. POLFA Познанский фармацевтический завод (Польша), PLIVA, Хорватия. ПИРОКСИКАМ (PIROXICAM) Международное название — piroxicam. Состав и форма выпуска. Активное вещество — пироксикам. Таблетки (0.01 è 0.02 ã. Ðàñòâîð 0.02 и 0.4 г в ампулах. Свечи ректальные 0.02 г. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60) Производитель. Фельден (Felden) PFIZER, США; Пироксикам (Piroxicam) POLFA Познанский фармацевтический завод, Елена Гура АО (Польша); HARMACHIM, Болгария; Роксикам (Roxicam) ZDRAVLE, Югославия; Эразон (Erazon) KRKA, Словения. ПЛАКВЕНИЛ (PLAQUENIL) Международное название — hydroxychloroquin. Состав и форма выпуска. 1 таблетка содержит 0.2 г гидроксихлорохина. Таблетки по 30 и 100 шт. в упаковке. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 169). Производитель. SANOFI, Франция ПОЛЬКОРТОЛОН (POLCORTOLON) см. Триамсинолон. ПРЕДНИЗОЛОН (PREDNISOLON) Международное название — prednisolon. Состав и форма выпуска. 1 таблетка содержит 0.005 г преднизолона. Фармакологическое действие и клиническое применение. Глюкокортикоидный препарат (стр. 84). Производитель. GEDEON RICHTER, Венгерская республика, преднизолон, POLFA Елена Гура АО, Польша. ПРОФЕНИД (PROFENID) см. Кетопрофен. РАНИТИДИН (RANITIDIN) Международное название — ranitidin. Состав и форма выпуска. Активное вещество — ранитидина гидрохлорид. Таблетки. Раствор в ампулах. Фармакологическое действие и клиническое применение. Блокатор Н2гистаминовых рецепторов второго поколения, подавляющий базальную и стимулированную продукцию соляной кислоты. Применяется при лечении язвенной болезни желудка и двенадцатиперстной кишки, в том числе индуцированных НПВС гастропатий (стр. 74). Дозировка. 0.15 г 2 раза в день (утром и вечером) или 0.3 г перед сном. Побочное действие. Редко — головная боль, головокружение, усталость, кожная сыпь, тромбоцитопения, алопеция. Производитель. Зантак (Zantac) GLAXO, Великобритания; Ранигаст (Ranigast) POLFA, Польша; Ранисан (Ranisan) ZDRAVLE, Югославия; Ранитидин (Ranitidin) PHARMACHIM, Болгария. РЕВОДИНА 25 (REVODINA 25) см. Диклофенак натрия. РЕВОДИНА РЕТАРД (REVODINA RETARD) см. Диклофенак натрия. РЕЛАФЕН (RELAFEN)* см. Набуметон. РЕОПИРИН (RHEOPIRIN) Состав и форма выпуска. Раствор для инъекций в ампулах, содержащий 0.75 г натрия фенилбутазона, 0.75 г амидазофена. Драже (0.125 г фенилбутазона, 0.125 г амидазофена). Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60) Производитель. GEDEON RICHTER, Венгерская республика. РОКСИКАМ (ROXICAM) см. Пироксикам. РОФЕРОНА (ROFERONA)** см. Интерферон а2а. САЙТОТЕК (CYTOTEC) см. Мизопростол. САНДИММУН (SANDIMMUN) см. Циклоспорин А. САНДОГЛОБУЛИН (SANDOGLOBULIN) Международное название — immunoglobulin. Состав и форма выпуска. 1 флакон лиофилизированного препарата содержит 1, 3 или 6 г иммуноглобулина человека. 5 флаконов лиофилизированного вещества и 5 флаконов растворителя в упаковке. Фармакологические свойства и клиническое применение (стр. 230). Производитель. SANDOZ, Швейцария. СОЛЮМЕДРОЛ (SOLUMEDROL) см. Метилпреднизолон. СУЛИНДАК (SULINDAC) Международное название — sulindac. Состав и форма выпуска. Активное вещество — сулиндак. Таблетки 0.1, 0.15, 0.2, 0.3 и 0.4 г по 60 и 100 шт. в упаковке. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат, относящийся к производным (стр. 60). Производитель. Клинорил (Clinoril) MERCK SHARP DOHME, США СУЛЬФАСАЛАЗИН (SULPHASALASIN) Международное название — salazosulfapyridin. Состав и форма выпуска. Таблетки по 0.5 г салазосульфапиридина. Таблетки по 50 шт. в упаковке. Фармакологическое действие и клиническое применение. Базисный противоревматический препарат (стр. 196). Производитель. KRKA, Словения. СУРГАМ (SURGAM) Международное название — acidum tiaprofenicum Состав и форма выпуска. В 1 таблетке содержится 0.3 г тиапрофеновой кислоты. Упаковка — 30 таблеток. Ректальные суппозитории 0.3 г по 6 и 12 штук. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Дозировка. 1 таблетка 2 раза в день, при необходимости 4 таблетки. Производитель. ROLJSSEL UCLAF, Франция. ТАУРЕДОН (TAUREDON) см. Ауротиомалят натриевая соль. ТЕНИДАП (TENIDAP)* Международное название — tenidap. Состав и форма выпуска. Активное вещество — тенидап. Капсулы 0.01, 0.02, 0.04, 0.06, 0.08, 0.120 г. Фармакологические свойства и клиническое применение. Противовоспалительный препарат (стр. 222). Производитель. Енаблекс (Enablex) PFIZER, США. ТЕНОКСИКАМ (TENOXICAM) Международное название — tenoxicam. Состав и форма выпуска. Активное вещество — теноксикам. Таблетки 0.02 г. Капсулы 0.02 г. Свечи ректальные 0.02г. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Тилкотил (Tilcotil) ROCHE, Швейцария. ТИЛКОТИЛ (TILCOTIL) см. Теноксикам. ТОКОФЕРОЛ АЦЕТАТ (TOCOPHEROL АСЕТАТ)** Международное название — tocopherol acetat. Состав и форма выпуска. Активное вещество — токоферола ацетат, являющийся синтетическим препаратом витамина Е. Драже. Фармакологические свойства и клиническое применение (стр. 302). Производитель. СантЕГал (SantEGal) GALENICA, Югославия; Эвитол (EVITOL) KRKA, Словения. ТОЛЕКТИН (TOLECTIN) см. Толметин. ТОЛМЕТИН (TOLMETIN SODIUM) Международное название — tolmetin. Состав и форма выпуска. Активное вещество — толметин в форме дигидрата натрия. Таблетки 0.2 г по 20 и 50 шт. в упаковке. Таблетки 0.4 г по 20 шт. в упаковке. Свечи 0.4 г по 10 шт. в упаковке. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Толектин (Tolectin) MACNEIL LAB., США; CILAG, Швейцария. ТОРАДОЛ (TORADOL) см. Кеторолака трометамин. ТРИАМСИНОЛОН (TRIAMCINOLON) Международное название — triamcinolon. Состав и форма выпуска. Активное вещество — триамсинолон. Таблетки 0.004 г по 20, 50 и 250 шт. в упаковке. Фармакологическое действие и клиническое применение. Противовоспалительный препарат (стр. 84). Производитель. Кеналог (Kenalog) BRISTOLMAYERS SQUIBB, США; KRKA, Словения; Полькортолон (Polcortolon) POLFA, Польша. УРБАЗОН (URBASON) см. Метилпреднизолон. ФЕНИЛБУТАЗОН (PHENYLBUTAZON) Международное название — phenylbutazon. Состав и форма выпуска. Активное вещество — фенилбутазон. Драже 0.2 г. Фармакологические свойства и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. WEMER PHARMA, Германия. ФЛУГАЛИН (FLUGALIN) см. Флурбипрофен. ФЛУРБИПРОФЕН (FLURBIPROFEN) Международное название — flurbiprofen. Состав и форма выпуска. Активное вещество — флурбипрофен. Таблетки 0.05, 0.1 г. Капсулы 0.2 г. Ректальные суппозитории 0.1 г. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Флугалин (Flugalin); BOOTS, Великобритания; АНсайд (Ansaid) UPJOHN, США ХЛОРАМБУЦИЛ (CHLORAMBUCIL) Международное название — chlorambucil. Состав и форма выпуска. Активное вещество — хлорамбуцил. Таблетки 0.002 и 0.005 г по 25 шт. в упаковке. Фармакологическое действие и клиническое применение. Алкилирующий препарат, обладающий базисной противоревматической активно стью (стр. 185). Производитель. Хлорбутин; Лейкеран (Leukeran) WELLCOME, Великобритания. ЦИКЛОСПОРИН А Международное название — ciclosporin. Состав и форма выпуска. 1 мл перорального раствора содержит 0.1 г циклоспорина. Флаконы для перорального приема по 120 мл. 1 капсула содержит 0.025, 0.05 и 0.1 г циклоспорина по 50 и 100 шт. в упаковке. Фармакологические свойства и клиническое применение (стр. 204). Производитель. Сандиммун (Sandimmun) SANDOZ, Швейцария ЦИКЛОФОСФАМИД (CYCLOPHOSPHAMID) Международное название — cyclophosphamid. Состав и форма выпуска. Активное вещество — циклофосфамид. Таблетки 0.05 г. Порошок 0.1 г, 0.2 г, 0.5 г в ампулах. (Фармакологические свойства и клиническое применение. Цитотоксический препарат (стр. 183). Производитель. Эндоксан (Endoxan) BOSNALIJER, Босния и Герцеговина; Цитоксан (Cetoxan) BristolMyers Squib, США. ЦИТАРАБИН (CYTARABIN)** Международное название — cytarabin. Состав и форма выпуска. Активное вещество — цитарабин. Фармакологическое действие и клиническое применение. Цитотоксический использующийся для лечения системных ревматических заболеваний (стр. 193). Производитель. Цитозар (Cytosar) UPJOHN, США. препарат, иногда ЭТОДОЛАК (ETODOLAC)* Международное название — etodolac. Состав и форма выпуска. Активное вещество — этодолак. Капсулы 0.200 г и 0.300 г. Фармакологическое действие и клиническое применение. Нестероидный противовоспалительный препарат (стр. 60). Производитель. Лодин (Lodine) Ayerst Lab., США. * Препарат не зарегистрирован в России ** Экспериментальный метод лечения ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ Адгезии молекулы, 25 Аденозин, 50 Азатиоприн, 190193 механизм действия, 190 побочные эффекты, 192 клиническое применение при СКВ, 191 при РА, 191 при ПМ/ДМ, 192 при других заболеваниях, 192 Азота окись, 51 Алкилирующие агенты, 182 Амиприлоза, 295 Анакира, 268 Анафилотоксины, 41 Антималярийные препараты, 169 фармакологические свойства, 169 механизм действия, 169 побочные эффекты, 177 клиническое применение при СКВ, 173 при РА, 174 при других заболеваниях, 176 Антитела, моноклональные, к СD4+Тклеткам, 259 к CDw52, 263 к CD5, 264 к ФНО-а, 264 к CD54, 266 Ауранофин, 121 Аутоиммунитет, 5355 Аферез, 241247 Аферез, селективный, 246 Буцилламин, 137 Бромокриптин, 291 Витамин Е, 302 Витамин D, 302 Воспаление, 1718 Гистамин, 50 Главный комплекс гистосовместимости, 12 Глюкокортикоиды, 84117 механизм действия, 90 резистентность, 94 побочные эффекты, 97 клиническое применение, 95 при РА, 103 при СКВ, 104 при ПМ/ДМ, 105 альтернирующий режим, 106 пульстерапия, 108 Гранулоцитарно-макрофагальный колониестимулирующий фактор, 303 ДАВ 486 ИЛ, 296 Дефлазакорт, 112 D-ïåíèöèëëàìèí, 130142 механизм действия, 130 клиническое применение при РА, 132 при ССД, 133 Дренаж грудного лимфатического протока, 250 Золота препараты, 118 механизм действия, 119 клиническое применение при РА, 121 при других заболеваниях, 119 побочные эффекты, 123 Иммунные комплексы, 55 Иммунотерапия, 256 Интегрины, 26 Интерлейкины, 28 ИЛ-1, 37 ИЛ-2, 33 ИЛ-3, 29 ИЛ-4, 34 ИЛ-5, 34 ИЛ-6, 34 ИЛ-7, 34 ИЛ-8, 37 ИЛ-9, 34 ИЛ-10, 34 ИЛ-11, 35 Интерфероны, 35 клиническое применение, 269 Иммуноглобулин внутривенный, 230 механизм действия, 231 побочные эффекты, 236 клиническое применение при васкулитах, 235 при ПМ/ДМ, 234 при РА, 232 при СКВ, 233 Кальцитриол, 302 Каптоприл, 139 Кетотифен, 304 Кинины, 52 Кислота арахидоновая, 42 Кислород, 46 Колониестимулирующие факторы, 32 Колхицин, 289 механизм действия, 283 клиническое применение, 283 побочные эффекты, 287 клиническое применение, 189 побочные эффекты, 190 клиническое применение, побочные эффекты, 183 при РА, 186 при СКВ, 185 при васкулитах, 186 фармакологические свойства, 183 Цитозинарабинозид, 193 Циклоспорин А, 204221 механизм действия, 205 побочные эффекты, 214 при РА, 208 при СКВ, 214 при синдроме Шегрена, 213 при болезни Бехчета, 210 при ПМ/ДМ, 210 при ССД, 211 при других заболеваниях, 213 Левамизол, 278 Лейкотриены, 44 Лефлуномид, 305 Лимфоциты, II Т-лимфоциты, II В-лимфоциты, 14 Линомид, 298 Лобензарит, 290 Макрофаги, 19 Метотрексат, 143 фармакологические свойства, 143 механизм действия, 144 клиническое применение, при РА, 151 при ювенильном артрите, 156 при СКВ, 156 при ССД, 157 при ПМ/ДМ, 157 при васкулитах, 158 побочные эффекты, 159 Месна, 187 Миноциклин, 289 Микофенолат мофетил, 293 Мерказолил, 301 механизм действия, 222 клиническое применение, 225 Тепоксалин, 298 Тетрациклиновые препараты, 289 Тимуса гормоны, 287 Тиопронин, 138 Тромбоциты, 21 Нейтрофилы, 18 Нестероидные противовоспалительные препараты, 60 классификация, 60 механизм действия, 61 клиническое применение, 67 побочные эффекты, 72 НПВПгастропатия, 73 Пентоксифиллин, 304 Пиритинол, 138 Плазмаферез, 241 Пролактин, 291 Простагландины, 42 Протеиназы, 47 Пульссинхронизация, 246 при серонегативных спондилоартропатиях, 199 побочные эффекты, 200 Субриум, 295 Сульфаметоксазол/триметоприм, 290 SKF105685,296 Рапамицин, 217 Рентгеновское облучение, 250 клиническое применение, 251 побочные эффекты, 252 Рецепторы клеточные, Тклетки, 13 ИЛ-2, 33 ИЛ-1, 37,39 ФНО-a, 40 Рифадин, 290 Роста факторы, 28 Сакролимус, 217 Сандиммун, 204 Селектины, 26 Серотонин, 50 Сульфасалазин, 196 фармакологические свойства, 196 механизм действия, 196 клиническое применение при РА, 199 Таксол, 299 Талидомид, 297 Такролимус, 215 Тенидап, 222 Фактор активации тромбоцитов, 46 Фактор некроза опухоли, 36 Фолиевая кислота, 151 Фототерапия ультрафиолетовая, 299301 механизм действия, 300 клиническое применение, 301 Фотохимиотерапия, экстракорпоральная, 247 механизм действия, 247 клиническое применение, 249 FK506,215 Хлорамбуцил, 189 Цитокины, 2840 Циклофосфамид, 183 механизм действия, 183 Эндотелиальная клетка, 23 Эндотелины, 52