ОСНОВЫ ДИЭЛЕКТРИЧЕСКОЙ СПЕКТРОСКОПИИ Гусев Ю.А.
advertisement

Физический факультет
Казанского государственного университета
Гусев Ю.А.
ОСНОВЫ ДИЭЛЕКТРИЧЕСКОЙ
СПЕКТРОСКОПИИ
Учебное пособие
Казань 2008
Предисловие
Метод диэлектрических измерений впервые в Казанском государственном
университете был применен Непримеровым Н.Н. для определения диэлектрической
постоянной парамагнитных солей (1954). Далее сотрудник кафедры радиоэлектроники
Седых Н.В. применял данный метод для исследования растительных белков (1960). В
дальнейшем методика измерения диэлектрических параметров в широком частотном
диапазоне от 50Гц до 37ГГц развивались научным сотрудником кафедры
радиоэлектроники Гусевым Ю.А., в результате чего впервые им была обнаружена область
релаксации аминокислот в водных растворах (1975).
Последующие работы выпускников кафедры радиоэлектроники лаборатории
диэлектрической спектроскопии по изучению диэлектрической релаксации связанной
воды в дисперсных системах (Гусев А.А., 1979), водно-спиртовых смесей (Фельдман
Ю.Д., 1982), систем целлюлоза-вода (Зуев Ю.Ф., 1985), молекулярных кристаллов
(Гончаров В.А., 1987), нефтяных дисперсных систем (Сараев Д.В., 2006), водных
растворов изопропилового спирта (Лунев И.В., 2007) были проведены на временных
диэлектрических спектрометрах, работающих на принципиально иных основах по
сравнению с предыдущей экспериментальной техникой.
Поскольку все большее количество ученых различных областей физики, химии,
биологии и др. применяют для исследования своих объектов методы диэлектрической
спектроскопии, возникла необходимость подготовить данное учебное пособие. Для его
создания были использованы работы Ахадова Я.Ю. («Диэлектрические параметры чистых
жидкостей», 1999), работы, опубликованные в сборнике F. Kremer, A. Schonhals,
“Broadband Dielectric Spectroscopy”, Springer-Verlag Berlin Heidelberg, 2003, оригинальные
работы автора и его учеников.
Данное учебное пособие может быть полезно студентам старших курсов,
занимающимся изучением структуры и свойств вещества, аспирантам, научным
сотрудникам, работающим в областях физики, химии, биологии, медицины, геологии, и
другим.
Автор благодарен младшему научному сотруднику кафедры радиоэлектроники
Васильевой М.А. за помощь и критику при подготовке данной работы.
2
Оглавление
Предисловие..............................................................................………………
Введение....................................................................................………………
Принятые обозначения ..........................................................………………..
Глава 1. Теория диэлектрической поляризации в статическом
электрическом поле
1.1.
Неполярные и полярные диэлектрики.................………………
1.2.
Поляризация диэлектриков ..................................……………….
1.3.
Феноменологические (макроскопические) характеристики
диэлектриков ........................................................ ………………
1.3.1.
Среднее макроскопическое поле в диэлектрике………………..
1.3.2.
Диэлектрическая проницаемость и диэлектрическая
восприимчивость……………………………………………..……
1.4.
Поляризуемость ......................................................……………….
1.4.1.
Электронная поляризация..................................………………
1.4.2.
Атомная поляризация.........................................……………….
1.4.3.
Ориентационная поляризация...........................……………….
1.5.
Локальное поле Лоренца......................................………………
1.6.
Формула Клаузиуса-Мосотти для неполярных жидкостей и газов
1.7.
Уравнение Дебая................................................... ………………
1.8.
Опытное определение поляризуемости и дипольного момента
молекулы………………………………………………………….
1.9.
Локальное поле Онзагера .................................... ………………
1.10.
Теория Кирквуда-Фрёлиха.................................……………….
1.11.
Дальнейшее развитие теории диэлектрической поляризации
Глава 2. Релаксационные свойства жидких диэлектриков
2.1.
Дипольная релаксация ......................................... ………………
2.2.
Дисперсионные уравнения ..................................……………….
2.3.
Линейные соотношения по Дебаю ..................... ………………
2.4.
Макроскопическое и микроскопическое времена релаксации...
2.5.
Распределение времен релаксации……………………………..
2.6.
Функция распределения Фрёлиха…………..…………………..
2.7.
Распределение Коула-Коула……………………………………..
2.8.
Распределение Девидсона-Коула……………………………….
2.9.
Уравнение Фаусса-Кирквуда …………………...........................
2.10.
Определение времени релаксации по данным одночастотных
измерений .......................................................... ………………..
2.11.
Диэлектрическая релаксация с двумя временами
релаксации……………………………………………………….
2.12.
Связь между диэлектрической релаксацией, вязкостью и
структурой жидкости ........................................………………..
2.13.
Диэлектрическая релаксация и термодинамические функции
Глава 3. Методы измерения диэлектрической проницаемости и
диэлектрических потерь
3.1.
Резонансные методы.............................................. ………………..
3.1.1.
Метод биений ....................................................………………..
3.1.2.
Метод расстройки контуров.............................………………..
3.1.3.
Метод куметра...........................................……………………..
3.1.4.
Методы измерения в объемном резонаторе... ………………..
2
5
6
9
9
10
11
11
12
14
15
15
16
19
20
21
22
24
26
30
31
31
33
39
42
44
45
47
49
51
52
55
59
60
63
63
64
65
67
67
3
3.2.
3.3.
3.3.1.
3.3.2.
3.3.3.
3.3.4.
Коаксиальные методы ........................................ ………………..
Волноводные методы ......................................... ………………..
Метод короткого замыкания ........................... ………………..
Балансные методы ........................................... ………………..
Диапазон частот 250 ÷ 3000 МГц .................. ……………….
Измерения диэлектрической проницаемости на частотах:
9584,6 МГц, 163,93 МГц, 37037,0 МГц ……………………..
Методы, использующие волны в свободном пространстве………
Широкодиапазонные и высокоточные методы измерения
диэлектрической проницаемости ..........................………………
Временная спектроскопия жидкостей ....................……………....
Метод одного отражения (толстого образца) ……………......
Метод тонкого образца…………….............……………..........
Метод сосредоточенной емкости.............……………..............
93
108
115
115
116
Заключение..............................................................................……………….
Литература……………………………………………………………………
119
120
3.4.
3.5.
3.6.
3.6.1.
3.6.2.
3.6.3.
69
71
72
74
76
85
91
4
Введение
Исследование диэлектрических свойств жидких и твердых диэлектриков - одна
из наиболее фундаментальных и сложных проблем науки, имеющая большое
теоретическое и практическое значение. В настоящее время центральное место в
работах, посвященных изучению межмолекулярных взаимодействий в жидкостях и
твердых телах, занимают исследования молекулярной структуры жидкости и динамики
ее перестройки в ходе теплового движения. Для решения этих задач используются
современные физические и физико-химические методы исследования: дифракционные
и термодинамические методы, диэлектрическая, акустическая и релеевская
спектроскопия, инфракрасная спектроскопия, спектроскопия ядерного резонанса и т. д.
Среди этих методов важное место принадлежит диэлектрической
спектроскопии, получившей в последнее десятилетие значительное теоретическое и
экспериментальное развитие. Исследования диэлектрических свойств жидких систем
позволяют получать обширную информацию об их молекулярной структуре,
межмолекулярных взаимодействиях, кинетике и механизмах молекулярных
процессов. Решение этих задач необходимо для понимания и, следовательно,
управления химическими и технологическими процессами, подавляющее большинство
которых протекает в жидких фазах. Знание диэлектрических свойств жидкостей
необходимо для разработки ряда радиотехнических систем, развития методов
анализа и контроля химического состава и решения многих практических проблем
промышленности и технологии.
Диэлектрические параметры жидкостей представляют интерес для широкого
круга специалистов, работающих в различных областях физики, химии, биологии,
медицины. Этим объясняется большое число опубликованных работ, в которых
излагаются сведения о диэлектрической проницаемости, диэлектрических потерях и
релаксационных явлениях в чистых жидкостях и растворах в широком диапазоне частот
и температур.
Возможности
современной
компьютерной
техники
позволили
автоматизировать проведение эксперимента, использовать новые методы измерения
диэлектрических параметров, повысить точность измерения и разработать систему
оценки достоверности данных о диэлектрических свойствах веществ.
Развитие экспериментальных методов измерения диэлектрических свойств дали
возможность расширить перечень объектов исследования, диапазон частот и
интервал температур.
Учебное пособие включает в себя теоретическую часть, методы измерения
диэлектрических параметров и оценки погрешностей измерения. Оно предназначено
для студентов, аспирантов, научных работников, физиков, химиков, биологов и
инженерно-технических работников научно-исследовательских институтов и
лабораторий.
Учебное пособие состоит из трех глав. В первых двух главах, носящих вводный
характер, приведены основные понятия теории диэлектриков. Рассмотрены основные
теоретические соотношения, применяемые при описании и анализе равновесных и
динамических свойств индивидуальных жидкостей. Описаны макроскопические
величины, характеризующие диэлектрические свойства жидкостей и их взаимосвязь с
молекулярными параметрами. Рассмотрены релаксационные процессы и их связь с
термодинамическими свойствами.
В
третьей
главе
описываются
основные
методы
измерения
диэлектрической проницаемости и потерь, приводятся погрешности эксперимента,
даются практические советы. Уделено особое внимание обработке
экспериментальных данных и оценке ошибки эксперимента.
5
Принятые обозначения
С - электроемкость
С1, С2 - вклад первого и второго релаксационных процессов в диэлектрической
релаксации
с - скорость света
D - электростатическая индукция (смещение)
E - напряженность среднемакроскопического электрического поля в диэлектрике
E0 - напряженность внешнего электрического поля
e - элементарный электрический заряд
F - напряженность локального электрического поля
∆F - изменение свободной энергии активации диэлектрической релаксации
∆F1, ∆F2, ∆F3 - изменение свободной энергии активации диэлектрической релаксации
для первой, второй и третьей областей поглощения
fm - предельная частота, соответствующая максимуму поглощения
fm1 , fm2 , fm3 - предельная частота, соответствующая первой, второй и третьей областям
поглощения
G -проводимость
CAS - Chemical Abstracts Servis (регистрационный номер реферативного журнала
Американского химического общества)
g - структурный фактор Кирквуда
H - напряженность магнитного поля
∆H - теплота активации диэлектрической релаксации
∆H1, ∆H2, ∆H3 - теплота активации диэлектрической релаксации для первой, второй и
третьей областей поглощения
h - постоянная Планка
I - ток
k - постоянная Больцмана
к - коэффициент поглощения (α/β)
L - индуктивность
M - молярная масса
m - магнитный момент
N - число частиц в единице объема
NA - число Авогадро
n - показатель преломления
n*=n(1-jk) - комплексный показатель преломления
P -поляризация (электрический дипольный момент единицы объема)
Pe - электронная поляризация
Pa - атомная поляризация
Po - ориентационная поляризация
PM - молекулярная поляризация
PR - молекулярная рефракция
Q - добротность
Q - электрический заряд
R - газовая постоянная
r - коэффициент отражения
∆S - энтропия активации диэлектрической релаксации
6
∆S1, ∆S2 , ∆S3 - энтропия активации диэлектрической релаксации для первой, второй, и
третьей областей поглощения
Тпл - температура плавления (С°)
Тот - температура замерзания
Ткип - температура кипения
V - объем
z - полное внутреннее сопротивление
zc - волновое сопротивление
z0 - волновое сопротивление вакуума
a -поляризуемость
ae - электронная поляризуемость
aa - атомная поляризуемость
ad - деформационная поляризуемость
a* - комплексная поляризуемость
a' - действительная часть комплексной поляризуемости
a" - мнимая часть поляризуемости
d - коэффициент поглощения
α - коэффициент распределения времен релаксации по Коулу-Коулу
β - параметр распределения времен релаксации по Коулу-Коулу
β - фазовая постоянная
β0 - коэффициент объемного расширения
γ=α+jβ - комплексная постоянная
δ - угол потерь
tgδ=ε"/ε' - тангенс угла потерь
ε0 - диэлектрическая проницаемость вакуума
ε - предельная низкочастотная диэлектрическая проницаемость
ε1, ε2, ε3 - предельная низкочастотная диэлектрическая проницаемость первой, второй и
третьей областей поглощения
ε∞ - предельная низкочастотная диэлектрическая проницаемость
ε∞1 , ε∞2 , ε∞3 – предельная высокочастотная диэлектрическая проницаемость для
первой, второй и третьей областей поглощения
ε*=ε'-jε" - комплексная диэлектрическая проницаемость
ε' - действительная часть диэлектрической проницаемости
ε" - мнимая часть диэлектрической проницаемости (диэлектрические потери)
ε'1, ε'2, ε'3 - действительная часть диэлектрической проницаемости в первой, второй и
третьей областях поглощения
ε"1, ε"2, ε"3 - диэлектрические потери в первой, второй и третьей областях поглощения
η - вязкость
λc - критическая длина волны
λo - длина волны в волноводе
λd - длина волны в диэлектрике
λ - длина волны в свободном пространстве
λm - предельная длина волны, соответствующая максимуму диэлектрических потерь
μ0 - дипольный момент свободной молекулы
μж - дипольный момент молекулы в жидкости
τ - макроскопическое время релаксации
τ1 , τ2 , τ3 - время релаксации в первой, второй и третьей областях поглощения
7
τμ - молекулярное время релаксации
ρ - плотность
χ - диэлектрическая восприимчивость
ω - круговая частота
ωm - предельная круговая частота, соответствующая максимуму диэлектрических
потерь
σ' - поверхностная плотность связанных зарядов
σ - поверхностная плотность свободных зарядов
8
ГЛАВА 1
ТЕОРИЯ ДИЭЛЕКТРИЧЕСКОЙ ПОЛЯРИЗАЦИИ В
СТАТИЧЕСКОМ ЭЛЕКТРИЧЕСКОМ ПОЛЕ
Результаты теоретических и экспериментальных работ, посвященных
диэлектрическим свойствам диэлектриков, обобщены в нескольких книгах и
монографиях [5—21]. С целью раскрытия физического смысла приведенных в книге
экспериментальных и расчетных параметров в этой главе кратко рассмотрены основные
уравнения, описывающие диэлектрические свойства жидкостей. В ограниченном
объеме теоретической части невозможно подробно описать все физические и
математические допущения, лежащие в основе каждого уравнения. Эти вопросы
подробно рассмотрены в литературе.
1.1. Неполярные и полярные диэлектрики
Электрический диполь — это пара зарядов, равных по абсолютной величине, но
противоположных по знаку и расположенных близко друг к другу.
Дипольный момент — произведение величины одного заряда на расстояние между
ними:
r
r
(1.1)
μ = q⋅d
Дипольный
момент
представляется
вектором,
направленным
от
отрицательного заряда к положительному. Дипольные моменты возникают при
асимметрии положительных и отрицательных зарядов в системе. Положительные
заряды связаны с ядрами атомов, т.е. они локализованы. Изменения плотности
положительных зарядов вызываются структурными трансформациями молекул
(локальные движения, вращение групп и т.д.). Изменения плотности отрицательных
зарядов в системе определяются электронами, которые делокализованы. Степень
делокализации зависит от химической структуры.
Система из положительных и отрицательных зарядов, образуемая молекулой,
может рассматриваться как единый диполь или как система диполей, ориентированных
вдоль молекулярных осей или групп. Векторная сумма дипольных моментов этой
системы образует молекулярный дипольный момент
N
r
r
M = ∑ μi
. (1.2)
i
Значения дипольных моментов принято выражать в следующих единицах: 10-18
электростатических единиц заряда на сантиметр. Эта единица называется "дебай" и
обозначается "Д".
Все диэлектрики в зависимости от строения молекул можно разделить на две
большие группы - полярные и неполярные. В неполярных диэлектриках центры
положительных и отрицательных зарядов молекулы совпадают и ее дипольный
момент равен нулю. К неполярным молекулам относятся, например, СН4, ССl4, С6Н6.
В полярных диэлектриках молекулы представляют собой диполи, обладающие
электрическим моментом, возникающим за счет смещения электрических зарядов из
положения равновесия в свободных атомах в результате химической связи (Н2О, НС1,
NН3 и т.д.). Молекулы-диполи полярного диэлектрика участвуют в тепловом движении.
9
В результате теплового движения электрические моменты диполей полярного
диэлектрика хаотически распределены по направлениям, и векторная сумма
дипольных моментов равна нулю.
1.2. Поляризация диэлектриков
При внесении диэлектрика в электрическое поле происходит процесс поляризации
диэлектрика. Поляризация диэлектрика вызывается смещением электрических
зарядов под действием сил внешнего и внутреннего электрических полей. Поэтому в
любом элементарном объеме диэлектрика возникает отличный от нуля суммарный
дипольный момент. Диэлектрик, находящийся в таком состоянии, называется
поляризованным. При этом на поверхности диэлектрика или в его объеме
образуются связанные электрические заряды. Поверхностные заряды образуют как бы
заряженный конденсатор, который создает электрическое поле в вакууме,
направленное противоположно направлению внешнего поля. Это поле называется
деполяризующим полем. Под действием электрического поля связанные заряды могут
лишь немного смещаться из положения равновесия, т. к. не могут покинуть пределы
молекул, в состав которых они входят.
Состояние поляризованного диэлектрика характеризуется векторной величиной,
называемой поляризованностью P:
r
1
P=
ΔV
N
∑μ
i =1
i
,
(1.3)
где P — поляризованность, ∆V — объем, N — число диполей в объеме ∆V, μi —дипольный
момент i-го диполя.
Рассмотрим конденсатор из плоскопараллельных пластин с площадью S и
расстоянием между пластинами d. Предположим, что распределение плотности
зарядов однородное по всему объему диэлектрика. В электрическом поле общий
r
дипольный момент M электрических диполей диэлектрика, заполняющего объем V
между пластинами, можно представить в виде
r
r
M = P ⋅ S ⋅ d . (1.4)
Так как V=S d, то
r
r M
P=
.
(1.5)
V
Вектор поляризации может быть определен как момент электрических диполей в
единице объема диэлектрика.
r
Установим связь между поляризованностью P и поверхностной плотностью
σ' связанных зарядов, расположенных на поверхности диэлектрика. Вырежем в плоской
пластине диэлектрика элементарный объем в виде цилиндра вдоль силовых линий поля,
перпендикулярных граням диэлектрика. Пусть площадь торцевых поверхностей
цилиндра ΔS, поверхностная плотность связанных зарядов σ'. На торцевых поверхностях
выделенного объема будут сосредоточены связанные заряды (рис. 1)
Δq = σ ′ΔS (1.6)
разного знака. Эти заряды создают в цилиндре электрический дипольный момент,
10
модуль которого
μ = Δq ⋅ d = σ ′ΔS ⋅ d = σ ′ΔV . (1.7)
r
Тогда поляризованность P выделенного участка диэлектрика находим, разделив
электрический момент цилиндра на его объем:
P=
μ
ΔV
=
σ ′ΔV
ΔV
= σ ′ . (1.8)
Таким образом, поверхностная плотность связанных зарядов численно равна
поляризованности:
σ ′ = P . (1.9)
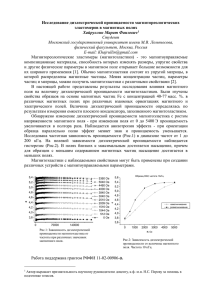
Рис.1. Связь между вектором поляризованности и поверхностной плотностью связанных
зарядов
Для описания диэлектрических свойств вещества существует два подхода:
феноменологический, который включает в себя описание макроскопических свойств
и характеристик диэлектрика, и микроскопический, включающий описание свойств и
явлений, протекающих в диэлектрике на атомно-молекулярном уровне.
1.3. Феноменологические (макроскопические) характеристики диэлектриков
1.3.1. Среднее макроскопическое поле в диэлектрике
В электрическом поле напряженность внутри диэлектрика Е можно
рассматривать как наложение двух полей в вакууме — внешнего E0 и деполяризующего
(поля связанных зарядов) Е1:
r r
r
E = E0 + E1 , (1.10)
или в скалярной форме
E = E0 − E1 . (1.11)
11
Результирующее поле Е в диэлектрике всегда меньше внешнего поля. Это поле
называется средним макроскопическим (или просто средним) полем диэлектрика.
Среднее поле в диэлектрике представляет собой усредненный по объему результат
суперпозиции внешнего поля и электрических полей молекулярных диполей
диэлектрика.
1.3.2. Диэлектрическая проницаемость и диэлектрическая восприимчивость
Диэлектрическая проницаемость является важнейшей характеристикой
диэлектрика. Величину диэлектрической проницаемости можно определить
несколькими путями. Величина, определяемая отношением емкости конденсатора с
диэлектриком С к емкости того же конденсатора без диэлектрика С0, называется
диэлектрической проницаемостью:
ε=
С
. (1.12)
С0
Так как емкость конденсатора с диэлектриком
С=
q
, (1.13)
U
где q — заряд на одной пластине, U — разность потенциалов между пластинами, то
емкость конденсатора в вакууме
С0 =
q0
. (1.14)
U
Из (1.12, 1.13) и (1.14) получаем
ε=
q
, (1.15)
q0
т.е. диэлектрическая проницаемость определяется отношением заряда конденсатора с
диэлектриком q к заряду того же конденсатора в вакууме q0.
Диэлектрическая проницаемость равна отношению напряженности внешнего
электрического поля Е0 к напряженности Е среднего поля в диэлектрике
ε=
E0
.
E
(1.16)
Известно, что при введении диэлектрика в конденсатор его емкость возрастает от
величины С0 в вакууме до величины С с диэлектриком. Отношение изменения
емкости ΔС = С - С0 к емкости С0 называется диэлектрической восприимчивостью:
χ=
C − C0 ΔC
=
.
C0
C0
(1.17)
С учетом (1.12) имеем
12
χ=
C
−1 = ε −1 ;
C0
(1.18)
χ = ε −1 ;
(1.19)
ε = χ +1 .
(1.20)
Диэлектрическая восприимчивость χ характеризует способность всего объема
диэлектрика поляризоваться в электрическом поле единичной напряженности.
Установим связь между напряженностью среднего макроскопического поля Е ,
внешним электрическим полем Е 0 , поляризацией Р , диэлектрической
проницаемостью ε и диэлектрической восприимчивостью χ.
На основании уравнений (1.11) и (1.16) имеем
E1 = (ε − 1) E .
(1.21)
Так как напряженность поля связанных зарядов
E1 =
σ′ P
,
=
ε0 ε0
(1.22)
где ε0 — электрическая постоянная, то с учетом (1.19), (1.21) и (1.22) имеем
P = (ε − 1)ε 0 E = χε 0 E ,
или P = ε 0εE − ε 0 E .
(1.23)
(1.24)
Для характеристики электрического поля в диэлектрике вводят вектор
r
электрической индукции (вектор электрического смещения) D :
r
r
D = ε 0εE ;
(1.25)
из (1.23), (1.24) и (1.25) получаем в векторной форме
r r
r
r
r
P = D − ε 0 E = (ε − 1)ε 0 E = χε 0 E ; (1.26)
r
r r
D = ε0E + P ,
(1.27)
Найдем связь между поверхностной плотностью σ зарядов на пластинах
конденсатора с поверхностной плотностью σ' связанных зарядов на плоских гранях
диэлектрика.
Напряженность поля, создаваемого свободными зарядами на пластинах
конденсатора,
13
E0 =
σ
. (1.28)
ε0
Так как связанные заряды распределены по поверхности диэлектрика равномерно,
то их можно охарактеризовать поверхностной плотностью σ' и тогда
напряженность Е1 деполяризующего поля можно вычислять так же, как
напряженность поля плоского конденсатора, то есть
E1 =
σ′
.
ε0
Подставляя полученные выражения для Е0 и Е1 в (1.21) и сокращая на ε0, получаем
σ′=
Так как ε > 1, то
ε −1
σ .
ε
σ fσ′ .
(1.29)
(1.30)
Таким образом, поверхностная плотность зарядов на пластинах конденсатора
больше поверхностной плотности связанных зарядов на плоских гранях
диэлектрика.
1.4. Поляризуемость
В электрическом поле каждая молекула диэлектрика становится диполем с
определенной ориентацией. Поэтому электрическое поле в диэлектрике является
суперпозицией внешнего поля и полей всех молекулярных диполей. Это поле, вообще
говоря, неоднородно, однако можно рассматривать его среднее значение.
Напряженность поля, реально действующего на молекулу диэлектрика, не равна
среднему полю и представляет собой некоторое эффективное поле, которое называется
локальным полем в диэлектрике.
Под действием локального поля в молекуле индуцируется дипольный момент,
пропорциональный напряженности локального поля,
r
μ = αε 0 F .
(1.31)
где α называется поляризуемостью молекулы.
Поляризуемость определяется значением индуцированного дипольного момента,
возникающего при действии напряженности электрического поля, равной единице.
Электрический момент единицы объема
P = NαF ,
(1.32)
где N — число молекул в единице объема.
Существует три механизма возникновения у молекул эффективных
электрических дипольных моментов при внесении диэлектрика во внешнее
электрическое поле. Эти механизмы непосредственно связаны со строением
молекул.
Рассмотрим каждый из этих механизмов в отдельности.
14
1.4.1. Электронная поляризация
Под действием электрического поля электронные облака атомов и молекул
смещаются относительно положения ядер на расстояния, меньшие размеров атомов или
молекул. Атомы и молекулы при этом поляризуются в электрическом поле, приобретая
индуцированные дипольные моменты, пропорциональные напряженности
локального поля, в котором они находятся. Такая поляризация называется электронной.
Электронная поляризация диэлектрика устанавливается за время порядка 10-15 с, т.е.
характерной особенностью электронной поляризации является ее малая
инерционность. Такая поляризация происходит практически мгновенно при включении
электрического поля и отслеживает быстрые колебания его величины вплоть до частот
порядка 1015 Гц. Электронная поляризация имеет место во всех без исключения
диэлектриках. Электронная поляризуемость сферического объема, заполненного
диэлектриком может быть определена равенством
α e = 4π r 3 .
(1.33)
где r — радиус сферы.
По порядку величины электронная поляризуемость равна 10-29 м3.
1.4.2. Атомная поляризация
Атомная поляризация наблюдается у гетероатомных молекул, в которых в
результате
различной
электроотрицательности
атомов
произошло
перераспределение электронной плотности. В молекулах, состоящих из атомов
различных элементов, электроны внешних оболочек перераспределяются между
атомами, смещаясь в направлении атомов с более сильными связями. В результате
такого перераспределения электронов их суммарный заряд на одних атомах
становится избыточным по сравнению с зарядом ядра, а на других недостаточным.
Этот заряд называют эффективным зарядом. Суммарное значение эффективных зарядов
всех атомов молекулы равно нулю.
Итак, под действием электрического поля происходит смещение атомов с
отличными от нуля эффективными зарядами на них относительно друг друга.
Изменяются межъядерные расстояния, и возрастает плечо диполя. Появляется тем
самым индуцированный дипольный момент, который и обусловливает атомную
поляризацию. Атомная поляризуемость равна 10-30 м3, что на порядок меньше, нежели
электронная поляризуемость. Относительно меньше и скорость установления
атомной (иногда ее называют еще ионной) поляризации, т. е. сравнительно велико
время ее установления. На высоких частотах, соответствующих видимой и
ультрафиолетовой частям оптического диапазона, эту составляющую уже можно не
принимать во внимание.
Для не слишком высоких температур, когда электронную плотность и,
соответственно, межатомные силы можно считать постоянными, электронная и
атомная поляризуемости не зависят от температуры. Оба эффекта обусловлены
деформациями электронно-ядерной системы под действием электрического поля, и их
сумма называется деформационной поляризуемостью (случай "квазиупругих" диполей):
αd = αe + αa
.
(1.34)
15
1.4.3. Ориентационная поляризация
Рассмотрим полярный диэлектрик, каждая молекула которого имеет постоянный
электрический дипольный момент (т.н. "твердые" диполи). В отсутствие внешнего поля,
в результате теплового движения, диполи ориентированы хаотично и суммарный
вектор поляризации диэлектрика равен нулю.
Во внешнем поле за счет сил электрического взаимодействия диполя с внешним
полем каждый диполь будет стремиться ориентироваться по полю, однако тепловое
движение препятствует ориентации диполей. В результате совместного действия этих
двух факторов устанавливается некоторое равновесное распределение с
r
преимущественной ориентацией по полю (векторная сумма ∑ μ i становится
отличной от нуля). Как и всегда в таких случаях, равновесное распределение
описывается формулой Больцмана [7, 13, 15, 16].
В области слабых электрических полей ( μF«kT ) избыточная концентрация
молекулярных диполей, ориентированных вдоль электрического поля, обусловливает
результирующий момент всего диэлектрика Р.
В поле F потенциальная энергия диполя
rr
U = − μF = − μF cosθ ,
(1.35)
r
r
где θ — угол между векторами μ и F . Очевидно, что при распределении числа
диполей ориентированных под углом θ к вектору напряженности внешнего
электрического поля по закону Больцмана, средняя величина индуцированного за счет
ориентации дипольного момента на одну молекулу соответствует среднему значению
интегрального выражения [7, 15]:
π
μ∫e
− μ F cos θ
kT
sin θ d θ ≅
0
μ 2F
3 kT
.
(1.36)
Приведем более наглядный, хотя и упрощенный, вывод для величины
ориентационной поляризуемости, исходящий из учета всего лишь шести
равновероятных при отсутствии электрического поля ориентаций. Если принять, что
r
направление вектора E соответствует оси Z, то ориентации будут такими: +Z; -Z; +Y;
-Y; +Х; -X. Заметим, что кроме первых двух ориентаций все другие имеют cosθ=0 и
соответственно U=0, а также не дают вклада в суммарный индуцированный
момент объема. Поэтому при подсчете последней величины учтем лишь ориентации по
полю и против поля.
Для молекул, дипольные моменты которых ориентированы по полю (θ = 0),
U = − μF .
(1.37)
Для молекул, ориентированных против поля (θ =π),
U = μF .
(1.38)
В электрическом поле в соответствии с распределением Больцмана изменяется
концентрация молекул, дипольные моменты которых ориентированы по полю (n+) и
против поля (n-). Они определяются выражениями:
16
μF
μF
N kT
n+ =
e
6
N −
n− = e kT
6
(1.39)
где N - общее число молекул, k - постоянная Больцмана, Т - абсолютная
температура.
В случае μFloc « kТ (потенциальная энергия взаимодействия диполя с полем
много меньше энергии теплового движения молекул) экспоненциальные функции
можно разложить в ряды Мак-Лорена, ограничиваясь только двумя первыми членами
разложения:
n+ =
μF
N
(1 +
)
kT
6
n− =
и
μF
N
(1 −
) .
kT
6
(1.40)
Внешнее поле изменяет число диполей, ориентированных в различных
направлениях. Числа молекул, диполи которых ориентированы вдоль направления
внешнего поля и в противоположном направлении, различны.
Избыточная концентрация молекул, ориентированных вдоль электрического поля,
Δn = n+ − n− =
NμF
.
3kT
(1.41)
Тогда поляризованность диэлектрика
P = (n+ − n− ) μ = Δnμ .
(1.42)
С учетом (1.41) и (1.42) имеем
P=
Nμ 2 F
3kT
.
(1.43)
Эффективный (средний по ориентациям) дипольный момент, приходящийся на
одну молекулу диэлектрика,
⟨μ⟩ =
P μ 2F
=
.
N 3kT
(1.44)
Молекулу полярного диэлектрика можно характеризовать ориентационной
поляризуемостью α0, которая определяется следующим образом:
⟨ μ ⟩ = α 0ε 0 F
,
(1.45)
поскольку
P = Nα 0 F
.
(1.46)
Из равенств (1.43) и (1.46) имеем
17
μ2
α0 =
3kT
,
(1.47)
где εО — электрическая постоянная.
Следовательно, при μF«kТ величина ориентационной поляризуемости обратно
пропорциональна температуре и пропорциональна квадрату дипольного момента
молекулы. По порядку величины при комнатной температуре α 0 ≈ 10 −28 м 3 .
Ориентационная поляризация инерционна и, хотя она преобладает для полярных
диэлектриков в постоянных и медленно изменяющихся электромагнитных полях,
ее практически можно не учитывать уже для самой длинноволновой части оптического
диапазона.
Когда μF»kТ, все дипольные моменты выстраиваются вдоль внешнего поля и
дальнейшее его увеличение уже не изменяет поляризованности диэлектрика.
Поляризованность достигает насыщения. Значение поляризованности в этом
состоянии зависит только от значения дипольного момента молекулы и от
концентрации молекул N:
PH = N μ .
(1.48)
График зависимости поляризованности диэлектрика от внешнего поля приведен
на рис. 2. В области μF«kТ зависимость близка к линейной.
В другом предельном случае (μF»kТ ) Р = const (насыщение). Во всем диапазоне
напряженностей поля зависимость индуцированного по ориентационному
механизму момента единицы объема (или молекулярной поляризуемости) задается
известной функцией Ланжевена и приведена на рис.2.
Рис.2. Зависимость поляризованности диэлектрика P от напряженности локального поля F
Естественно, в полярных диэлектриках все три механизма поляризации (два
механизма деформационных и один ориентационный) проявляются одновременно:
P = Pe + Pa + P0
;
(1.49)
α = αe + αa + α0 ;
P = (α d +
Pd = Pe + Pa ;
μ2
) NF
9ε 0 kT
Pe =
N
αe ;
3ε 0
(1.50)
;
Pa =
(1.51)
N
αa ;
3ε 0
(1.52)
18
P0 =
Nμ 2
9ε 0 kT
P=
;
(1.53)
μ2
N
(α l + α a +
).
3ε 0
3kT
Строгий вывод выражений (1.49 — 1.54) приведен в работах [7, 14, 16].
1.5. Локальное поле Лоренца
Локальным полем называют результирующую напряженность поля F ,
действующего на молекулу вещества. Локальное поле по Лоренцу описывается
следующей моделью (рис. 3).
Рис.3. Модель локального поля Лоренца
Предполагается, что выделенная молекула А окружена воображаемой сферой с
радиусом порядка 100 диаметров молекулы. За пределами сферы диэлектрик считается
непрерывным с диэлектрической проницаемостью ε. Поле, действующее на молекулу в
сфере, выражается через величину поляризации Р. Если допустить, что молекулы из
данной сферы удалены (кроме А), при сохранении поляризации вне ее неизменной, то
поле, действующее на молекулу А, обусловлено средним макроскопическим полем Е,
полем, создаваемым связанными зарядами на поверхности сферы Е1, и полем,
создаваемым всеми молекулами внутри сферы за исключением рассматриваемой
молекулы Е2:
F = E + E1 + E2 .
(1.55)
Расчет по электростатической теории показывает, что с одной стороны поле E1,
создаваемое связанными зарядами на поверхности выделенной сферы, определяется как
[13, 16]
E1 =
P
.
3ε 0
(1.56)
С другой стороны, можно полагать, что поле Е2, которое производят молекулы,
находящиеся внутри сферы, в изотропной среде благодаря хаотическому
распределению молекул равно нулю:
Тогда
E2 = 0 .
(1.57)
P
.
3ε 0
(1.58)
F =E+
19
r
r
Между поляризацией и средним полем существует соотношение P = (ε − 1)ε 0 E ,
подставив из которого значение Р в (1.58), получим
F =(
ε +2
3
)E .
(1.59)
1.6. Формула Клаузиуса—Мосотти для неполярных жидкостей и газов
Электрический момент единицы объема диэлектрика (1.32)
P = NαF ,
где N — число молекул в единице объема.
Напомним, что
F =(
ε +2
3
)E
и
P = (ε − 1)ε 0 E ,
Таким образом, локальное поле связано со средним полем, приведенным выше
соотношением (1.58) или (1.59).
Пользуясь тремя приведенными выше соотношениями, получим выражение для
поляризации единицы объема (удельной поляризации) Р:
NαF = (ε − 1)ε 0 E ;
P = Nα (
P=
ε +2
3
(1.60)
) ;
(1.61)
ε − 1 Nα
=
.
ε + 2 3ε 0
(1.62)
Легко также получить выражение для молекулярной поляризации Рм. Для этого
умножим обе части (1.62) на М/ρ — величину, соответствующую числу молей (M—
молярная масса, ρ — плотность вещества) в единице объема. Получим
PM =
где
NA =
N
ρ
⋅ M - число Авогадро
ε − 1 M N Aα
⋅
=
ε +2 ρ
3ε 0
,
(1.63)
(1.64)
Молекулярная поляризация не зависит от числа молекул в единице объема.
Формула (1.63) называется формулой Клаузиуса—Мосотти. Эта формула
применима только к изотропным однородным средам, что очевидно из
предположений, которые были приняты при ее выводе. Из нее вытекает одно из
важных соотношений между электронным строением и оптическими свойствами
вещества.
20
Согласно электромагнитной теории Максвелла, показатель преломления среды n
связан с ее диэлектрической проницаемостью на предельно высоких частотах ε∞
соотношением
ε = ε ∞ = n 2 . (1.65)
При оптических частотах f= 10 15 -10 1 6 Гц все поляризуемости, кроме
электронной, равны нулю и следует учитывать лишь электронную поляризуемость αе, в
результате чего получим соотношение для молекулярной рефракции R:
R=
n2 −1 M NA
⋅ = αe
n2 + 2 ρ 3ε
,
(1.66)
именуемое формулой Лорентц—Лоренца.
Формула
Лорентц—Лоренца
оправдывается
для
диэлектрических
проницаемостей неполярных или слабополярных жидкостей. Такие жидкости обычно
имеют диэлектрическую проницаемость, значения которой лежат в пределах 2-2,5 и,
соответственно, показатели преломления, значения которых лежат от 1,4 до 1,6.
Из (1.63) вытекает механизм, обусловливающий зависимость диэлектрической
проницаемости слабополярных жидкостей от температуры. В уравнение Клаузиуса—
Мосотти входят в этом случае деформационная поляризуемость молекул (не
зависящая от температуры) и число молекул в единице объема, пропорциональное
плотности, которая уменьшается при нагревании (эффект теплового расширения).
Последний эффект приводит к уменьшению диэлектрической проницаемости при
возрастании температуры. Дифференцируя уравнение (1.63) по температуре, получаем
температурный коэффициент изменения диэлектрической проницаемости:
1 dε (ε − 1)(ε + 1) 1 dρ
⋅
=
⋅ ⋅
3ε
ε dT
ρ dT
или
. (1.67)
1 dε
(ε − 1)(ε + 1)
⋅
=−
β0 ,
ε dT
3ε
(1.68)
где β0 — коэффициент объемного расширения.
1.7. Уравнение Дебая
В полярных жидкостях поле, действующее на молекулу, отличается от поля,
вычисленного Лоренцем для неполярных молекул, дополнительным вкладом
ориентационной поляризуемости. Любая молекула жидкости находится также и в
поле соседних молекул, направление которого изменяется вместе с изменением их
ориентации.
Использовав формулу внутреннего поля Лоренца и учтя ориентационную
поляризуемость молекул, Дебай [7] предложил уравнение для поляризации
полярной жидкости:
P = (α d + α 0 ) NF
.
(1.69)
Подставляя для ориентационной поляризации единицы объема α0 выражение (1.47)
21
α0 =
μ2
,
3kT
получаем уже приведенное ранее (1.51):
P = (α d +
μ2
3kT
) NF ,
и, учитывая (1.59)
F =(
Кроме того, по (1.26)
ε +2
3
)E .
P = (ε − 1)ε 0 E ,
и, исключая из (1.51), (1.26) Р и Е, получаем
ε −1 N
μ2
=
(α +
)
ε + 2 3ε 0 d 3kT
(1.70)
для единицы объема или, умножая обе части равенства на М/ρ,
ε −1 M NA
μ2
⋅
=
(α d +
)
3kT
ε + 2 ρ 3ε 0
(1.71)
для грамм-моля.
Деформационная поляризуемость для слабополярных молекул может быть
приближенно получена из выражения
α d ≅ 1,05
n 2 − 1 M 3ε 0
⋅
⋅
n2 + 2 ρ N A
, (1.72)
в котором атомная составляющая деформационной поляризации принята равной 5%
от электронной.
Уравнение (1.71) называется уравнением Дебая—Ланжевена. Оно хорошо
выполняется для газов, а также применимо к сильно разбавленным растворам
полярных жидкостей в неполярных растворителях. Формула (1.71) неприменима к
полярным жидкостям, так как при ее выводе используется выражение для
внутреннего поля Лоренца, в котором не учитывается влияние ближайших соседних
молекул [13—15].
1.8. Экспериментальное определение поляризуемости и дипольного момента
молекулы
Определение дипольного момента молекулы может производиться путем
измерения зависимости диэлектрической проницаемости слабополярной жидкости или
раствора от температуры.
Уравнение Дебая—Ланжевена имеет вид (1.71)
22
P=
ε −1 M N A
μ2
⋅
=
(α d +
) ,
3kT
ε + 2 ρ 3ε 0
что можно представить как
P = A+ B T ,
(1.73)
где А — деформационная поляризация, не зависящая от температуры:
A=
NA
αd ;
3ε 0
В — ориентационная поляризация, зависящая от температуры:
B=
NA μ2
.
9ε 0 kT
(1.74)
Зависимость Р от 1/Т представляет собой прямую линию с углом наклона к оси
1/Т, равным β = агсtgB (рис.4).
Рис.4. Зависимость молекулярной поляризации слабополярной жидкости от температуры Т
Экстраполируя прямую линию к точке 1/T=0, можно получить величину А , т. е.
деформационную поляризацию. Наименее инерционная часть деформационной
поляризации - электронная - может быть определена с помощью уравнения (1.66)
Лорентц—Лоренца из измерений коэффициента преломления n в видимой части
оптического диапазона, деформационная поляризация для слабополярных молекул
приблизительно равна 1,05 электронной поляризации.
В свою очередь, так как
N Aμ2
= tgβ = B ,
9ε 0 k
μ =3
то
где
3
ε0k
NA
ε 0k
NA
B,
= 4,27 ⋅10 −9 ,
(1.76)
(1.77)
23
и
μ =3
ε 0k ⋅ B
NA
= 4,27 ⋅10 −9 tgβ Кл ⋅ м . (1.78)
При корректной постановке эксперимента погрешность метода составляет 0,5-5 %
и определяется точностью измерения приращений диэлектрической проницаемости и
систематическими погрешностями.
1.9. Локальное поле Онзагера
Было отмечено, что выражение для поля Лоренца (1.59) не учитывает вклад
источников поля, находящихся внутри выделенного сферического объема, в центре
которого находится диполь.
Онзагер считает, что поле, действующее на молекулу со стороны ближайших
соседей, зависит от направления дипольного момента этой молекулы. Под действием
поля соседних молекул рассматриваемая молекула получает дополнительный
индуцированный момент, и этот момент воздействует на соседние молекулы. Такое
дополнительное поле изменяет состояние поляризации соседних молекул, т.е. сама
молекула активно участвует в формировании внутреннего поля, действующего на нее
[24].
Онзагер с учетом этого предложил для полярных диэлектриков модель локального
поля, существенно отличающуюся от модели Лоренца (рис. 5).
Допустим, что сферическая полость имеет радиус а и объем ее равен объему,
приходящемуся на одну молекулу. В центр сферы помещена молекула с точечным
диполем μ, имеющая изотропную поляризуемость α. Сфера погружена в непрерывный и
однородный диэлектрик с диэлектрической проницаемостью ε.
Рис.5. Модель Онзагера:
а) поле в полости G; б) реактивное поле внутри полости ( μ — дипольный момент в полости)
Мысленно удалим молекулу L; в диэлектрике останется сферическая полость.
r
Если в диэлектрике действует однородное электрическое поле E , то в этой полости
r
также будет действовать однородное поле G , которое назовем полем полости. Поле
r
r
r
полости G отличается от поля E по величине и оно больше E по причине
r
r
образования связанных зарядов на поверхности полости. Поле G направлено вдоль E
r
и ориентирует дипольную молекулу по направлению E .
Если мысленно удаленную полярную молекулу вернуть в центр полости, то ее
дипольный момент окажет воздействие на соседние молекулы в виде некоторого поля,
поляризующего эти молекулы. В свою очередь это дополнительное изменение их
поляризации приводит к появлению добавочного поля, действующего со стороны
окружения на рассматриваемую молекулу.
Разность между измененным полем и приложенным к образцу макроскопическим
24
r
r
r
полем E называется реактивным полем R . Направление поля R совпадает с
направлением дипольного момента молекулы L; это поле поляризует молекулу, но не
ориентирует ее. Таким образом, полное поле внутри сферы (локальное поле) слагается
r
r
из поля полости G и реактивного поля R .
Молекула, находящаяся в центре сферы, имеет дипольный момент μ:
μ = μ0 + α e F ,
(1.79)
где μ0 - собственный дипольный момент полярной молекулы, αe - поляризуемость
смещения, αeF - индуцированный момент молекулы.
Пользуясь законами электростатики, можно определить зависимости G и R от
полного дипольного момента молекулы μ, поляризуемости α и диэлектрической
проницаемости: [24, 13, 16]
F =G+R ;
(1.80)
F=
G=
3ε
E ;
2ε + 1
(1.81)
R=
2(ε − 1) μ
;
2ε + 1 α 3
(1.82)
3ε
2(ε − 1) μ
E+
.
2ε + 1
(2ε + 1) α 3
(1.83)
Полный дипольный момент молекулы
μ = μ *+
ε (ε ∞ − 1) 3
α E ,
2ε + ε ∞
(1.84)
где μ* - действительный дипольный момент молекулы, равный сумме собственного
дипольного момента и момента индуцированного реактивным полем:
μ* =
(ε ∞ + 2)(2ε + 1)
μ0 .
3(2ε + ε ∞ )
(1.85)
Уравнения Онзагера, связывающие μ и α с ε и N, имеют вид
(ε − ε ∞ )(2ε + ε ∞ )
N μ 20
=
⋅
;
3ε 0 3kT
ε (ε ∞ + 2) 2
ε ∞ − 1 Nα
.
=
ε ∞ + 2 3ε 0
(1.86)
(1.87)
Здесь квадрат показателя преломления заменен на значение предельной
высокочастотной диэлектрической проницаемости ε∞.
Формула (1.86) упрощается в следующих случаях:
25
1. При больших значениях ε, т.е. в случае сильно полярных жидкостей, когда ε»ε∞:
ε≅
Nμ 02 (ε ∞ + 2) 2
. (1.88)
2ε 0
9kT
2. Для слабо полярных жидкостей, т.е. при (ε - ε∞ ) « ε∞ :
μ2
ε (ε ∞ + 2)
ε −1 ε ∞ −1
−
≅N
⋅ 0
.
ε∞ + 2 ε∞ + 2
(2ε + ε ∞ )(ε + 2) 3ε 0 kT
(1.89)
Теория Онзагера удовлетворительно объясняет экспериментальные данные по
измерению диэлектрической проницаемости полярных жидкостей в широком интервале
температур, но для сильно полярных жидкостей дает заниженные значения е.
Например, для воды предсказывается ε = 31, тогда как на опыте ε = 78,2.
Модель Онзагера обладает следующими недостатками:
а) молекула упрощенно представляется сферой радиуса а;
б) окружение выделенной молекулы принимается непрерывным и изотропным;
в) диполь представляется точечным и помещенным в центр сферы, что явно не
справедливо для любой полярной молекулы, в которой распределение зарядов носит
сложный характер.
1.10. Теория Кирквуда — Фрёлиха
В этой теории рассматривается не одна молекула, а шаровой объем В с радиусом r,
достаточно большим по сравнению с размерами молекулы. Этот объем является частью
полярного диэлектрика А, имеющего форму шара с настолько большим радиусом,
чтобы за пределами сферы В среду можно было считать непрерывной и обладающей
макроскопической диэлектрической проницаемостью ε (рис. 5а).
Рис. 5а. Модель Кирквуда
По Кирквуду в центре области В находится i-я молекула, и она создает в ней
r
r
момент M i * . Эта же молекула с моментом μ i , индуцирует в области А дипольный
r
r
r
r
момент M i * . Если нам известно M i * , то момент M i можно выразить через M i * ,
решая электростатическую задачу. Допустив, что радиусы областей А и В стремятся к
бесконечности, Кирквуд получил
r
Mi =
3
3ε r ∗
M .
⋅
ε + 2 2ε + 1 i
(1.90)
Замена области с одной молекулой, имеющей момент μ i , на сферическую область В
26
с моментом M i * устраняет противоречие, связанное с предположением Онзагера о
сферической молекуле и непрерывности диэлектрика вблизи молекулы.
В области А, помещенной в электрическое поле Е0, создается среднее
макроскопическое поле
r
3 r
E=
E0 . (1.91)
ε +2
Кирквуд принимает, что локальное поле, действующее на молекулу, равно полю
полости Онзагера:
3ε
F=
E.
(1.92)
2ε + 1
Это локальное поле ориентирует молекулы, и средний дипольный момент,
r
обусловленный ориентацией молекул в поле E , определяется как
⟨μ⟩ E =
ε
μM *
⋅
⋅ E , (1.93)
2ε + 1 3kT
где М* - средняя величина электрического момента области В в направлении
дипольного момента молекул, находящихся в центре этой области.
При
определении
усредненного
момента
μ
E
статистическим
методом
вычисляется потенциальная энергия шаровой области, помещенной в электрическое
поле, и энергия межмолекулярного взаимодействия.
Полный средний момент молекулы μ
μ
E
и индуцированного момента μ
ин
f
в направлении поля Е складывается из
. С учетом (1.90—1.93) имеем
⟨ μ ⟩ f = ⟨ μ ⟩ E + ⟨ μ ⟩ ин =
ε μM *
E + αF ,
2ε + 1 3kT
(1.94)
где α— поляризуемость молекулы.
Для поляризации диэлектрика, используя (1.92), (1.94), получаем
P = N ⟨μ⟩ =
3ε μM *
(
+ α ) NE .
2ε + 1 3kT
(1.95)
С учетом соотношения электростатики (ε − 1)ε 0 E = P имеем
ε −1 =
3ε
N
μM *
⋅
(α +
).
3kT
2ε + 1 3ε 0
(1.96)
Отсюда для диэлектрической проницаемости полярной жидкости
(ε − 1)(2ε + 1) N
μM *
=
(α +
).
9ε 0
3ε 0
3kT
(1.97)
Считая, что короткодействующее взаимодействие имеет место лишь между
рассматриваемой молекулой и Z ее ближайшими соседями, получаем
27
μM = μ 2 (1 + ⟨cos γ ⟩ ) = μ 2 g
,
(1.98)
где g - структурный фактор, являющийся постоянной величиной для данного сорта
молекул, cos γ — среднее значение косинуса угла между направлениями диполей
двух соседних молекул.
Подстановка (1.98) в уравнение (1.97) с учетом (1.64) приводит к выражению
(ε − 1)( 2ε + 1) M
N
gμ 2 .
⋅
=
(α +
)
ρ 3ε 0
9ε 0
3kT
Структурный фактор g является мерой локального упорядочения в жидкости.
Положительные отклонения g от единицы (g>1) возникают, когда короткодействующие
силы, препятствующие вращениям, способствуют взаимно параллельной ориентации
диполей соседних молекул, а отрицательные отклонения (g<1) обусловлены
антипараллельной ориентацией диполей. Если g=1, это означает, что фиксированное
положение одного диполя не оказывает влияния на положение других диполей при
отсутствии дальнодействующих электростатических сил [27,28]. Большие
отклонения g от единицы обычно указывают на существование молекулярных
ассоциаций.
Структурный фактор g связан с эффективными моментами молекулы в жидкости
μж и в газообразном состоянии μ0 следующим образом:
μж
= g;
μ0
μж =
(1.100)
(2ε + 1)(ε ∞ + 2)
μ 0 . (1.101)
3(2ε + ε ∞ )
Исходя из приближенно тетраэдрической структуры для жидкой воды, Кирквуд
вычислил значение g, подстановка которого в уравнение (1.57) дает для воды при 25°С
величину диэлектрической проницаемости, всего на 0,4% отличающуюся от
наблюдаемого значения. Однако при 83°С вычисленная из μ и g диэлектрическая
проницаемость воды оказалась уже на 12% выше наблюдаемой. Аналогичные расчеты
для пяти различных спиртов привели к расхождениям с экспериментальными
значениями на 10—29%. Теория Онзагера, как указывалось ранее, дает еще более
существенное расхождение с опытом (в несколько раз).
Уравнение Кирквуда [29] для бинарного раствора, содержащего
соответственно мольные доли x1 и x2 соединений 1 (неполярного) и 2 (полярного),
записывается в форме
(ε − 1)(2ε + 1) M
⋅
= P1 x1 + P2 x2 .
9ε
ρ
(1.102)
где М/ρ — молекулярный объем растворителя, а молярные поляризации P1 и P2
определяются следующим образом:
P1 =
N
3εε 0
α1
;
P2
N
gμ 2
(α 2 +
) . (1.103)
3ε 0
3kT
28
Уравнение Кирквуда для поляризации полярной составляющей раствора имеет вид
(ε − 1)(2ε + 1) (ε ∞ − 1)(2ε ∞ + 1) P2 x2
=
−
9ε
9ε ∞
V
. (1.104)
Уравнения Онзагера—Фрелиха [9, 24] для растворов полярной жидкости В в
неполярном растворителе А записывается в виде
ϕA
ε − ε ∞A
ε − ε ∞B
N μ 2B
,
+ϕB
= B
2ε + ε ∞ A
2ε + ε ∞ B
3εε 0
(1.105)
где φA и φB - объемные доли компонентов, εA, εB - их высокочастотные диэлектрические
проницаемости, ε — диэлектрическая проницаемость раствора, μB — дипольный момент
молекулы полярного компонента.
Фрелих [9] и Коул [30] вывели уравнения, довольно близкие к уравнениям
Онзагера и Кирквуда. Эти и другие уравнения, а также предположения, на которых они
основаны, и вывод самих уравнений подробно описаны и обсуждаются в работах [13,
14, 16, 18].
Уравнения Сыркина [31] для полярных жидкостей и их растворов в неполярных
растворителях записываются следующим образом:
ε − ε∞ ε + 2
N 2
⋅
μ ;
=
ε ∞ + 2 2ε + 1 9kT
(1.106)
ε − 1 M 1 x1 + M 2 x2
⋅
− x1 R1 − x2 R2
N
ε +2
ρ
=
( x1μ1 + x2 μ 2 ) .
ε −1 2
9
kT
ε
0
1− (
)
ε +2
(1.107)
Уравнение для поляризации чистой полярной жидкости:
⎡ (ε − 1)(ε + 2) (n 2 − 1)(n 2 + 2) ⎤ M
Nμ 2
−
⎢
⎥ ρ = 9ε kT = P .
8ε
8n 2
⎣
⎦
0
(1.108)
Для бинарных растворов полярных жидкостей оно имеет вид [47, 48]
P0 =
⎡ x M + x 2 M 2 ⎤ ⎡ (ε − 1)(ε + 2) ( n 2 − 1)( n 2 + 2) ⎤
Nμ 2
2
2
−
⋅ ( x1 μ1 + x 2 μ 2 ) = ⎢ 1 1
⎥ . (1.109)
⎥⋅⎢
ρ
8ε
8n 2
9ε 0 kT
⎦ ⎣
⎣
⎦
Зная ориентационную поляризацию раствора P0, можно вычислить
ориентационную поляризацию растворенного вещества Р02, если известна (например,
вычислена по формуле (1.108)) ориентационная поляризация полярного растворителя
Р01:
P − P01
P02 = 0
+ P01 .
(1.110)
x2
29
1.11. Дальнейшее развитие теории диэлектрической поляризации
Дальнейшее уточнение теории поляризации жидких полярных диэлектриков [34,
35] связано с учетом анизотропии поляризуемости и несферичности
(эллипсоидальной формы) молекулы [36 — 40]. Ранее мы рассматривали молекулы как
точечные диполи с изотропной поляризуемостью, и считали, что они погружены в
сферу из изотропного поляризуемого вещества.
Впоследствии Шолте и другие распространили подход Онзагера на
эллипсоидальные молекулы с отношением значений главных осей тензора
поляризации а: b: с. В уравнении [16, 36, 40 — 42]
μ2 =
3ε 0 kTM (ε − ε ∞ )(2ε + 1)
[ε + (ε − ε ∞ )]A1
⋅
⋅
. (1.111)
Nρ
ε (2ε + ε ∞ )
[ε − (ε − 1) A1 ] ⋅ [1 + (ε − 1) A1 ]
где A1 — фактор деполяризации. Для сферической молекулы с а = b = с этот фактор равен
A1 = 1/3 и уравнение (1.111) переходит в уравнение Онзагера.
Другой путь уточнения выражений для диэлектрической проницаемости и
поляризуемости молекул - учет ассоциации молекул в жидкости. Значения дипольного
момента, рассчитанные по (1.111), нередко оказываются ближе к значениям,
полученным для газовой фазы, чем вычисленные по уравнениям Онзагера. Однако
имеется ряд исключений. Ассоциированные жидкости могут характеризоваться
значениями дипольных моментов, которые намного отличаются от измеренных в
газовой фазе, причем эти значения могут быть выше (ассоциация приводит к усилению
ориентации диполей в одном направлении) или ниже (ассоциация приводит к
антипараллельной ориентации диполей).
Из нижеследующего уравнения [43—45] определяют дипольный момент и число
ассоциированных молекул полярной жидкости в единице объема:
2
ε (ε − n 1 )
2ε + n 1
2
2
N
N
ε (ε − n 1 )
ϕ1 (1 − γ 2 ) +
⋅ γ ( 2 ϕ1 + ϕ 2 ) =
2
N1
N1
2ε + n 1
2
2
2
2
2
2
⎫
⎧⎪
1
2 ⎡ ε( n x + 2) ⎤ ⎪
2 ⎡ ε( n 2 + 2) ⎤
2 ⎡ ε( n 1 + 2) ⎤
N
(
1
)
=
⋅ ⎨( N 1 − γN 2 )μ 1 ⎢
N
+
−
γ
μ
+
γ
μ
2 x⎢
2
2⎢
2 ⎥ ⎬
2 ⎥
2 ⎥
9ε 0 kT ⎪
⎣ 2ε + n 1 ⎦
⎣ 2ε + n 2 ⎦
⎣ 2ε + n x ⎦ ⎪⎭
⎩
(1.112)
где степень ассоциации γ = N 2 x / N x - число ассоциированных молекул в единице
объема раствора; N2x, μ2x и n2x — соответственно их дипольный момент и показатель
преломления.
Заметим, что в уравнении (1.112) измеряемые величины суть: ε, x1, x2, φ1, φ2 . При
этом nх = φ1n1+ φ2n2, μ1 и μ2 измеряются заранее и неизвестными остаются собственно
интересующие нас величины μx и γ.
Постоянная равновесия реакции ассоциации К связана с γ соотношением
K=
γN
( N1 − γN 2 )(1 − γ )
. (1.113)
Значение постоянной равновесия реакции может быть определено различными
методами. Подставляя выражение (1.113) в формулу (1.112), можно вычислить
дипольный момент ассоциированных молекул μх для различных концентраций и
температур [46,47, 48].
30
ГЛАВА 2
РЕЛАКСАЦИОННЫЕ СВОЙСТВА
ЖИДКИХ ДИЭЛЕКТРИКОВ
В данной главе будут рассмотрены свойства жидких диэлектриков, обладающих по
преимуществу ориентационным механизмом поляризации (имеющих "жесткие"
диполи), а также свойства, характеризующие поведение диэлектриков в
переменных электромагнитных полях.
Очень медленно меняющиеся поля (колебания низкой частоты), в сущности,
ничем не отличаются по описанию взаимодействий с диэлектриком от статических
полей. Однако в области дисперсии, когда время установления равновесной
ориентации, называемое также временем релаксации τ (или временем корреляции),
становится сравнимым по порядку величины с периодом колебаний напряженности
поля, возникают существенные отличия от статического случая, связанные с рассеянием
энергии колебаний, затрачиваемой на периодическую переориентацию диполей в
диэлектрике. Эта энергия определяется необходимостью преодолеть силу трения,
которая и обусловливает конечное время установления в образце равновесного
распределения диполей по ориентациям, равное времени релаксации.
В свою очередь, в области более высоких частот, когда выступают на первый
план деформационные механизмы поляризации (отчасти — атомная при частотах
ИК-диапазона и ниже, а в основном — электронная в области оптических частот),
ориентационными явлениями можно пренебречь.
2.1. Дипольная релаксация
При наложении внешнего переменного электрического поля в полярной жидкости
в некоторых диапазонах частот и температур диполи полярных молекул способны
следовать за изменениями поля. В таком диэлектрике ток смещения опережает
приложенное напряжение точно на 90° (рис. 6,а). Ток является чисто реактивным и не
имеет составляющей, изменяющейся в фазе с электрическим полем, которая привела
бы к диссипации энергии колебаний с выделением тепла.
С увеличением частоты внешнего переменного поля (а, в известной мере, также и
при понижении температуры диэлектрика) диполи молекул не успевают следовать за
изменениями поля, и у тока смещения появляется активная составляющая (рис. 6,
б), что соответствует потере энергии на преодоление трения.
Рис. 6. Соотношение между фазами тока смещения и напряжения, приложенного к
диэлектрику для гармонического колебания: а — без потерь, б — с потерями.
31
Наличие отличной от 90° разности фаз между током смещения и приложенным
электрическим полем приводит к рассеянию энергии в виде тепла (диэлектрические
потери). При дальнейшем возрастании частоты колебаний ориентация молекул уже не
может следовать за приложенным полем, поляризация все более уменьшается и,
наконец, падает до величины, обусловленной атомной и электронной деформационной
поляризацией. Такая зависимость диэлектрической проницаемости от частоты
называется дисперсией проницаемости.
Оба явления (диссипация энергии и резкое уменьшение проницаемости)
обусловлены релаксацией дипольных моментов и могут служить для определения τ
косвенным методом при изучении относительной диэлектрической проницаемости с
изменением частоты приложенного внешнего гармонического электрического поля.
При невысоких частотах (достаточно далеких от области дисперсии) поляризация
P(t) меняется в переменном поле со временем по тому же закону, что и электрическое
поле E(t):
P (t ) = (ε − 1)ε 0 E (t ) ,
(2.1)
что аналогично (1.15). Однако, вообще говоря, уже при таких частотах
диэлектрическая проницаемость ε в (2.1) является комплексной величиной:
ε * = ε '−iε " ,
(2.2)
где ε' — диэлектрическая проницаемость вещества, пропорциональная изменению
свободной энергии диэлектрика, накопленной диэлектриком за период колебания поля,
а ε" — фактор, пропорциональный поглощаемой за период колебаний поля энергии.
Отношение мнимой части комплексной диэлектрической проницаемости к ее
действительной части называется тангенсом угла потерь:
tgδ =
ε"
,
ε'
(2.3)
где δ — угол, дополняющий до π/2 сдвиг фазы между приложенным напряжением и
током через диэлектрик (см. рис.6, б).
Комплексная диэлектрическая проницаемость применяется в уравнениях,
описывающих зависимость эффектов электрического поля от также комплексной
величины электрического поля E0 e iωt . При этом мы примем, что электрическое поле
на образце можно записать в виде
E( t ) = E 0 sin ωt ,
(2.4)
а так как
e iωt = cos ωt + i sin ωt ,
(2.5)
используя Im для обозначения мнимой части электрического поля, уравнение (2.4)
перепишем в виде
E( t ) = Im( E 0 e iωt ) . (2.6)
Уравнение (1.25) можно записать следующим образом:
r
r
D = ε 0εE ,
32
а согласно (1.25) с учетом (2.2), (2.6) имеем
r
r
r
D( t ) = ε 0 Im E( ε ' −iε" )e iωt = ε 0 Im E( ε ' 2 +ε" 2 )1 / 2 e i( ωt −δ ) ,
где tgδ =
(2.7)
ε"
.
ε'
Таким образом, комплексная диэлектрическая проницаемость (или отличное
r
r
от нуля значение ε") указывает на наличие разности фаз между D и E . В конечном
счете, именно это и обусловливает поглощение энергии системой.
2.2. Дисперсионные уравнения
Частотные зависимости ε' и ε" могут быть определены с использованием хорошо
известного принципа суперпозиции напряженностей электрических полей.
Принцип суперпозиции позволяет исследовать изменения поляризации в
зависимости от времени в тех случаях, когда напряженность электрического поля
изменяется со временем по сложному закону.
Допустим, что в момент времени t=0 релаксационная поляризация равна Р(0). В
момент t=0 отключается электрическое поле и релаксационная поляризация
начинает спадать, и в некоторый момент времени t она становится равной Р(t).
Функция спадания поляризации со временем определяется выражением
r
P( t )
ϕ( t ) = r
.
P( 0 )
Целью диэлектрической радиоспектроскопии является определение точных
количественных значений следующих величин:
а) среднего квадратичного значения сигнала шума электрической поляризации
r
P = ⟨ P 2 ( t )⟩ 1 / 2 ,
(2.8)
среднее значение поляризации при тепловом равновесии равно нулю
⟨ P( t )⟩ = 0 ;
(2.9)
б) нормализованной автокорреляционной функции, которая определяется
следующим образом [20,9,16]:
Ф( t ) =
⟨ P( t ) ⋅ P( 0 )⟩
.
⟨ P( 0 ) ⋅ P( 0 )⟩
(2.10)
Ф(t) также называется функцией отклика поляризации или функцией спадания
поляризации. При тепловом равновесии, однако, на сигнал, обусловленный
флуктуациями поляризации жидкости, накладываются также шумы измерительной
аппаратуры. Таким образом, нельзя с достаточной точностью определить
флуктуационную поляризацию образца вещества в равновесном состоянии.
Чтобы увеличить чувствительность измерения, на образец жидкости обычно
действуют монохроматическим электрическим полем E(f) с малой напряженностью:
μE0 « kT, где E0 — амплитуда.
С изменением частоты f поляризация P(f) измеряется как функция частоты f и
33
диэлектрические свойства жидкости выражаются
проницаемостью, определяемой равенством
ε * ( f ) = ε ' ( f ) − iε " ( f ) =
комплексной
1 P( f )
+1 .
ε 0 E( f )
электрической
(2.11)
Действительная часть диэлектрической проницаемости ε'(f) представляет
компоненту поляризации, изменяющуюся в фазе с полем E(f), в то время как мнимая
часть ε"(f) представляет собой вклад в P(f) составляющей со сдвигом фаз π/2
относительно
поля
E(f).
Следовательно,
использование
комплексной
диэлектрической проницаемости позволяет учитывать сдвиг фаз между
поляризацией и полем, являющийся результатом молекулярного взаимодействия,
которое не позволяет P(f) синфазно следовать за E(f).
Фазовый сдвиг между P(f) и E(f) означает, что электрическая энергия
рассеивается в виде тепла внутри образца жидкости.
Если построить график зависимости ε"(f) от f то получим кривую, схожую с
кривой поглощения (рис. 7). Как следует из флуктуационно-диссипационной теоремы
действительная часть ε'(f) определяет дисперсионные характеристики вещества.
Вернемся к рассмотрению временной спектроскопии. Межмолекулярные силы
проявляются в виде шумового сигнала поляризации и, таким образом, мы имеем
нетривиальную диэлектрическую функцию спадания.
Рис.7. Зависимости действительной части ε'(f) и мнимой части комплексной
диэлектрической проницаемости от частоты, где ε0 — диэлектрическая проницаемость
вакуума, п — показатель преломления.
Как следует из теории линейных систем, комплексная диэлектрическая
проницаемость ε*(f) и автокорреляционная функция шумового сигнала
поляризации Ф(t) связаны преобразованием Лапласа:
ε(f)=
1 Ps
ε0 Es
∞
∞
⎡
∫ ⎢⎣ −
0
d Ф ( t ) ⎤ − 2 ft
e
+1=
dt ⎥⎦
⎡ d Ф ( t ) ⎤ − 2 ft
e
+1
= [ε ( 0 ) − 1 ]∫ ⎢ −
dt ⎥⎦
0 ⎣
,
(2.12)
где Ps , ES, и ε(0) — статические значения этих величин (f→0).
34
Рис.8. Зависимость диэлектрической функции спадания от времени
Зависимость функции спадания от времени приведена на рис.8. Как видно из
графика, автокорреляционная функция резко падает при малых значениях времени t.
Такое поведение наблюдается для всех жидкостей. Это связано с тем, что механизм
поляризации смещения дает слишком быстрый спад (<1пс), который не может быть
разрешен с помощью микроволновой спектроскопии и значение Ф(t) соответствует
частоте 160 ГГц.
Медленно спадающая часть Ф r (t) функции Ф(t) представляет
релаксационные свойства вещества. Для многих полярных жидкостей при
комнатной температуре функция Ф r (t) может быть представлена экспоненциальной
зависимостью [16, 18, 19]:
t
Фr (t ) = Фr (0) exp( − ) .
τ
(2.13)
Время спадания этой функции τ называется временем диэлектрической
релаксации. При t = τ релаксационная поляризация уменьшается в е раз. Таким
образом, τ есть время, необходимое, чтобы поляризация уменьшилась в е раз
относительно первоначального значения под воздействием теплового движения
окружающих молекул.
Исходя из молекулярной модели жидкостей, желательно обсуждать
макроскопический процесс релаксации поляризации в понятиях дипольного времени
автокорреляции τμ.
Однако не существует применимого в общем случае соотношения между временем
спада функции Ф(t) и нормализованной дипольной автокорреляционной функцией ψ μ (t ) ,
определяемой соотношением
ψ μ (t ) =
⟨ μ (t ) ⋅ μ (0)⟩
.
⟨ μ (0) ⋅ μ (0)⟩
(2.14)
Сложность проблемы можно понять, используя в уравнении (2.10) определение
поляризации как полного диэлектрического момента в образце, отнесенного к объему
образца:
1
P(t ) =
V
Nν
∑ μ (t ) .
i =1
i
(2.15)
В этом уравнении Nν - число молекулярных диполей в объеме образца. Подстановка
суммы (2.15) в уравнение (2.10) указывает на два, по-существу, различных вклада в
P(t): результат взаимодействия между идентичными "собственными" дипольными
моментами и между "различными" дипольными моментами. Следовательно, помимо
35
дипольной автокорреляционной функции ψ μ (t ) также присутствует функция взаимной
корреляции
ψ μμ (t ) =
⟨ μ i (t ) ⋅ ∑ μ j ( 0 )⟩
i≠ j
,
⟨ μ ( 0 ) ⋅ μ ( 0 )⟩
(2.16)
которая может оказывать влияние на Ф(t). Таким образом, это следует иметь в
виду, когда макроскопически измеренное время релаксации интерпретируется как
молекулярное автокорреляционное время релаксации. Показано, однако, что в случае
воды
τμ ≈τ
.
(2.17)
При этих условиях τμ относится к слабо меняющейся части автокорреляционной
функции [16, 22, 23,21].
В соответствии с уравнением (2.12) экспоненциальная функция спадания
трансформируется в частотную зависимость.
r
Связь между электрической индукцией D и напряженностью электрического
r
r
r
r
r
поля E в случае сдвига фаз между D и E и между поляризацией P и E при
использовании уравнения (2.12) приводит к следующему выражению для комплексной
диэлектрической проницаемости [20, 8, 14, 16]:
ε* = ε∞ +
ε −ε∞
,
1 + iωτ
(2.18)
где ε∞ - предельная высокочастотная диэлектрическая проницаемость; ε - предельная
низкочастотная диэлектрическая проницаемость; ω - круговая частота; τ макроскопическое время релаксации.
Выделив в выражении (2.18) действительную и мнимую части, получим:
ε '= ε∞ +
ε "=
ε − ε∞
,
1 + ω 2τ 2
(ε − ε ∞ )ωτ
1 + ω 2τ 2
tgδ =
,
(2.19)
(2.20)
ε" (ε − ε ∞ )ωτ
=
. (2.21)
ε' ε + ε ∞ ω 2 τ 2
Выражения (2.19), (2.20) и (2.21) называются уравнениями Дебая.
Дебай [7] на основе формулы Лоренца для внутреннего поля предложил
следующее выражение для комплексной диэлектрической проницаемости:
ε* = ε∞ +
ε − ε∞
,
ε +2
)τ
1 + iω (
ε∞ + 2 μ
(2.22)
где τμ называется молекулярным или микроскопическим временем релаксации.
36
При выводе этой формулы Дебай предполагал, что полярная молекула является
сферической частицей, вращающейся в вязкой среде с коэффициентом η.
Разделяя вещественную и мнимую части в соотношении (2.22), находим
ε '= ε∞ +
где
ε − ε∞
1 + x2
,
(2.23)
ε "=
(ε − ε ∞ ) x
, (2.24)
1+ x2
x=
ε +2
ωτ .
ε∞ + 2 μ
(2.25)
Хотя зависимости (2.19, 2.20) и (2.23, 2.24) сходны по форме (различаются только
множителем (ε + 2)/(ε∞+2)), они в сущности разные, так как первые выведены для
макроскопического процесса релаксации, а вторые — для молекулярного процесса,
зависящего от внутреннего поля.
Частотные зависимости ε', а также ε", определяемые формулами (2.19), (2.20),
(2.23) и (2.24), одинаковы и различить их на эксперименте невозможно. Зависимости ε' и
ε" от частоты по формулам (2.19) и (2.20) приведены на рис.9
Рис.9. Зависимости ε' и ε" от частоты, вычисленные по уравнениям (2.19) и (2.20) при ε = 10,
ε ∞ = 2 и τ =10-10с
Уравнения (2.20) указывают, что комплексная часть диэлектрической
восприимчивости ε" стремится к нулю как при малых, так и при больших значениях ωτ и
достигает максимума при
ω mτ = 1 .
(2.26)
Значению ω mτ соответствуют ε'm и ε"m:
ε m′′ =
ε ′m =
ε − ε∞
2
ε + ε∞
.
2
,
(2.27)
(2.28)
37
Вместо ω m можно использовать критическое значение частоты fm, или
критическую длину волны λm:
ω
1
fm = m =
;
(2.29)
2π 2πτ
λm =
c
,
fm
(2.30)
где с — скорость света.
Исключив параметр ωτ из выражений для ε' и ε" в равенствах (2.19) и (2.20),
Коул [69] получил следующее соотношение:
(ε '−
ε + ε∞
2
) 2 + ε "2 = (
ε −ε∞
2
)2 .
(2.31)
Это уравнение представляет собой уравнение окружности, но поскольку все
входящие в него величины должны быть положительными, график зависимости ε" от ε'
фактически имеет вид полуокружности с центром на оси абсцисс на расстоянии
(ε+ε∞)/2 от начала координат и с радиусом (ε-ε∞)/2. Пересечение полуокружности с осью
абсцисс (ω=0) справа от центра дает значение ε, а слева от центра соответствует ε∞(ω→∞).
Максимум зависимости ε" от ε' достигается при ωτ =1 (рис. 10). Определение искомой
зависимости требует измерений в значительном частотном диапазоне [50].
Рис.10. Зависимость ε" от ε' при ε = 10, ε ∞= 2
Диэлектрическая проницаемость при предельно высоких частотах ε∞ может быть
вычислена по измеренному значению показателя преломления по формуле работы [51]:
ε∞ −1
n2 − 1
,
=A 2
ε∞ + 2
n +2
(2.32)
где А равно 1,05 — 1,15 (с учетом вклада атомной поляризации).
Построение
зависимости мнимой
части поляризуемости
а" от
действительной части лучше описывает отклонение свойств диэлектриков от
уравнения Дебая, чем зависимость ε" от ε' [52,53].
Комплексная поляризуемость связана с комплексной диэлектрической
проницаемостью следующим соотношением:
a* =
ε * −1
.
ε * +2
(2.33)
38
Подстановка ε* в уравнение (2.18) из формулы (2.33) приводит к выражению
(ε − 1) + iωτ (ε − 1)
.
(ε + 2) + iωτ (ε + 2)
a* =
(2.34)
Разделив мнимую и действительную части в выражении (2.34), получим
a' =
(ε '−1)(ε '+2) + ε "2
,
(ε '+2) 2 + ε "2
a" =
(2.35)
3ε "
.
(ε '+2) 2 + ε "2
(2.36)
Из формул (2.35) и (2.36) можно получить следующее уравнение:
(a '+
где a =
a + a∞ 2
a − a∞ 2
) + a"2 = (
) ,
2
2
(2.37)
ε −1
ε −1
и a∞ = ∞
ε∞ + 2
ε +2
Рис.11. Зависимость а' от а"
Время релаксации определяется из соотношения:
V
1− a
=
⋅ ωτ ,
U 1 − a∞
(2.38)
где V и U —- расстояния от экспериментальной точки на полуокружности до точек а’=а
и а" = a∞ соответственно (рис.11).
2.3. Линейные соотношения по Дебаю
Из уравнений (2.19), (2.20) был получен ряд выражений для графического
представления релаксационных процессов, описываемых теорией Дебая [50,54].
Умножив уравнение (2.18) на (1+iωτ) и выделив действительную и мнимую части,
39
придем к соотношениям [55]:
ε ' = ε − τ (ωε " ) ,
1 ε"
ε '= ε ∞ + ( ) .
τ ω
(2.39)
(2.40)
Эти уравнения линейно связывают измеряемые величины ε', ω ε", ε"/ω друг с
другом. Значения ε и ε∞ могут быть определены как пересечение прямых,
полученных из уравнений (2.39) и (2.40) с осью ε', а время релаксации τ по их наклону
к оси ε'. Экстраполяция зависимости ε' от ε"/ω прямой линией дает в этом случае
значение высокочастотного предела диэлектрической проницаемости ε∞.
Рис. 12. Зависимости ε' от ωε" и ε' от ε"/ω для 1-бутанола спирта: а — при t = -76°С;
б — при t = -106°С
На рис. 12 приведены зависимости (2.39) и (2.40) по данным измерений для нбутилового спирта при температурах -76 и -106°С [50]. Как видно из рисунка, при
t = -76°С получается прямая линия для широкого диапазона частот от 15 Гц до 3 МГц.
При t = -106°С наблюдается отклонение от линейности при частотах выше 100 кГц, что
указывает на наличие дополнительной области дисперсии.
Коул [50] и Брот [54] предложили следующие функции:
ε ' /(ε − n 2 ) = x /(1 + x 2 ),
(ε '−n 2 ) /(ε − n 2 ) = 1 /(1 + x 2 ) , (2.41)
x = ε " /(ε '− n 2 ) = (ε − ε ' ) / ε "
где ωτ = λc / λ ;
λc = 2πcτ ; λ = 2πc / ω .
Графики зависимостей:
ε"/x как функции от ε",
ε"x как функции от ε ',
(2.42)
40
являются линейными, и величины n2 и ε могут быть определены как пересечение этих
прямых с осью ε', а τ (или λc) по наклону соответствующих прямых.
Хилл [56] предложила следующие линейные функции:
a).x / ε " = x 2 /(ε − n 2 ) + 1 /(ε − n 2 ),
б ).1 / ε " x = 1 /(ε − n 2 ) x 2 + 1 /(ε − n 2 ),
в ).1 /(ε '− n 2 ) = x 2 /(ε − n 2 ) + 1 /(ε − n 2 ),
(2.43)
г ).1 /(ε − ε ' ) = 1 /(ε − n 2 ) x 2 + 1 /(ε − n 2 ).
Каждое уравнение включает в себя экспериментально измеряемые ε' и ε", но так
как частота входит в уравнения квадратично, частотная шкала искажена. Кроме того, в
уравнения б) и г) входят величины ε или n2, которые должны быть получены из
независимых экспериментов.
Различные методы определения времени релаксации с помощью уравнений Дебая
даны в литературе [50, 142, 143].
Для разбавленных растворов полярных жидкостей в неполярных растворителях
уравнение Дебая можно записать в виде [16]
ε "=
(ε '+2) 2 πNμ 2 C
ωτ
⋅
,
6750kT
1 + ω 2τ 2
(2.44)
где С - концентрация полярной жидкости в моль/л, μ - дипольный момент полярной
молекулы.
При ε" « ε' уравнение (2.44) имеет вид
1
ω
= ( A + ω 2τ ) ,
ε"
τ
где
A = 6750kT /(ε '+2) 2 Nπμ 2 C
Пользуясь графиком зависимости ω/ε" от ω2 можно определить τ и μ.
Результаты измерений показывают, что уравнения Дебая применимы для процесса
релаксации полярных молекул в бесконечно разведенных растворах полярных
жидкостей в неполярных растворителях, если молекулы полярных жидкостей велики
по сравнению с молекулами растворителя. Эти уравнения также применимы к полярным
жидкостям, молекулы которых имеют почти сферическую форму и для которых
релаксационные явления характеризуются одним временем релаксации [57—59].
С точностью до ошибки эксперимента уравнения Дебая описывают с хорошим
приближением диэлектрическую дисперсию воды [60] и большого числа одноатомных
спиртов [61]. В этих жидкостях релаксационные процессы могут быть
охарактеризованы одним временем релаксации. Однако большинство полярных
жидкостей имеют широкую область дисперсии с несколькими временами
релаксации, и для описания дисперсии в таких случаях уравнения Дебая
непосредственно неприменимы.
Такая ограниченная применимость уравнений Дебая объясняется тем, что при их
выводе использовано выражение для внутреннего поля, в котором предполагается,
что диполи молекул, окружающих данную молекулу, расположены совершенно
хаотично, а их ориентации не зависят от ориентации рассматриваемой молекулы. В этом
41
случае не учитывается ближний порядок ни в положениях, ни в ориентациях молекул.
2.4. Макроскопическое и микроскопическое времена релаксации
Определение эффективного внутреннего (локального) поля, действующего на
молекулу внутри диэлектрика, расположенного в электрическом поле, имеет
большое значение, так как дает возможность связать макроскопическую
диэлектрическую проницаемость со свойствами молекул и с характеристиками
межмолекулярных взаимодействий.
Связь релаксационного процесса с межмолекулярными взаимодействиями удобнее
обсуждать с применением представления о микроскопическом или молекулярном
времени релаксации.
Как уже говорилось (2.22), согласно Дебаю макроскопическое время релаксации
τ и молекулярное время релаксации τ μ связаны между собой следующим образом:
τ=
ε +2
τ .
ε∞ + 2 μ
(2.46)
Так как ε >ε∞ очевидно, что τ > τμ . При этом различие в величинах τ и τμ по (2.46)
весьма значительно. Например, для воды макроскопическое время релаксации в
двадцать раз больше вычисленного молекулярной, времени релаксации. Это
различие обусловлено использованием при выводе (2.46) выражения для
внутреннего поля по Лорентцу (отношения (ε+2)/(ε∞+2)). Отсюда и неприменимость
теории Дебая к сильно полярным жидкостям.
В работе [62] согласие с экспериментом попытались улучшить, используя
выражение для внутреннего поля по Онзагеру [24]. Для соотношения τ и τμ получено
τ=
2ε ∞ + 1
τμ .
2ε + 1
(2.47)
Паулс [63] нашел приближенное выражение для внутреннего поля, которое
приводит к соотношениям:
3ε
τ=
τ μ , (2.48)
2ε + ε ∞
⎡
ε −ε∞ ⎤
или τ = ⎢1 +
⎥τ μ .
⎣ 2ε + ε ∞ ⎦
(2.49)
Согласно [64] τ и τ μ связаны выражениями:
⎡
3x 2
⎤
τ = ⎢1 + x +
τμ
(1 − 2 x)(1 + x) ⎥
⎣
или τ =
⎦
2ε 2 + ε ∞3
τμ ,
3ε ∞ε (2ε + ε ∞ )
,
(2.50)
(2.51)
42
где x =
ε − ε∞
.
2ε + ε ∞
(2.52)
При малых значениях х можно пренебречь третьим слагаемым в (2.50) и тогда
(2.50) переходит в (2.48).
Различные соотношения между макроскопическим τ и молекулярным τμ
временами релаксации сопоставляются на рис.13.
Рис.13. Зависимость τ/τμ от статической диэлектрической проницаемости ε (при ε∞=2)
для различных представлений внутреннего поля: а — Лорентца—Дебая; б — Онзагера—Коула;
в — Паулса.
Сопоставление уравнений показывает, что согласно (2.50) и (2.46) отношение
τ/τ μ стремится к бесконечности при ε→∞, тогда как в соответствие с уравнениями (2.47) и
(2.48) отношение τ/τ μ в этом пределе стремится к значению 1,5.
Экспериментальные исследования веществ с большой и малой
поляризуемостью показали, что наилучшим выражением для связи между τ и τμ
является соотношение (2.48).
Применимость указанных уравнений была исследована Миллером и Смитом
[65] в предположении, что время релаксации слабополярной жидкости или
разбавленного раствора сильнополярных молекул в неполярной жидкости
совпадает с микроскопическим временем релаксации τ μ , которое сравнивалось с
непосредственно измеряемым макроскопическим временем релаксации чистой
полярной жидкости τ. Например, дипольный момент молекул толуола настолько мал
(0,4D), что при 20°С ε-ε∞=0,11 и уравнение (2.45) дает τ/τ μ =1,025; уравнение же
(2.48) — τ/τ μ =1,016, а уравнение (2.50) — τ/τ μ =1,005. Это означает, что вычисленное
микроскопическое или молекулярное время τ μ в данном случае неотличимо от τ, т.е.
τ μ = τ. Молекула п-пиколина имеет почти такие же размеры и форму, как и молекула
толуола, и вполне разумно предположить, что ее молекулярное время релаксации
должно быть почти таким же, как у толуола. Эти жидкости различаются вязкостями и,
безусловно, значениями внутренних полей (дипольный момент п-пиколина 2,6 D). По
уравнению (2.48) τ μ для п-пиколина на одну треть больше, чем у толуола. Согласно же
(2.50) время релаксации τ μ для п-пиколина меньше, чем у толуола. Такой результат
показывает, что уравнение (2.50) неприменимо при наличии дипольного
взаимодействия, а уравнение (2.46) приводит к еще большим противоречиям.
Для многих жидкостей с несимметричными молекулами установлена
приблизительная пропорциональность между временем релаксации и вязкостью.
Поэтому, возвращаясь к приведенному ранее сравнению значений τμ для п-пиколина
43
и толуола, чтобы свести к минимуму влияние вязкости, время релаксации для одной
жидкости помножим на обратное отношение вязкостей двух жидкостей η1 и η2:
η
τμ =τμ ( 2 ) ,
η1
2
1
(2.53)
где индекс (1) относится к слабополярной жидкости (например, к толуолу), а (2) к
сильнополярной (например, к п-пиколину). Обозначив τ μ1 = τ 1 , получим
τ
μ2
= τ1(
η2
).
η1
(2.53а)
Исследованные уравнения не учитывают эффектов, связанных с различиями в
форме молекул или в расположении диполей в молекулах. Предположение о
пропорциональности времени релаксации и вязкости жидкости является, повидимому, грубым приближением и серьезным источником ошибок при оценке
отношения времен релаксации. Так, оно приблизительно выполняется лишь при
условии, что полярные молекулы, по крайней мере, имеют втрое больший радиус, чем
окружающие их молекулы растворителя.
2.5. Распределение времен релаксации
Молекулы в жидкостях в каждый данный момент времени находятся в различных
условиях, так как величина сил взаимодействия между молекулами и внутреннее поле
(а иногда, и температура) различны в разных точках и изменяются во времени. Если
различия названных параметров меняются медленнее, чем ориентация дипольных
моментов, жидкость распадается на области, каждой из которых следует приписать свое
значение времени релаксации. Различия в окружении молекул могут быть как
макроскопическими (градиент температур или концентраций), так и микроскопическими
(образование в растворе разных долгоживущих ассоциаций, включающих дипольные
молекулы и другие различия микроструктуры). Для каждой из таких областей время
релаксации на ту или иную величину отклоняется от наиболее вероятного значения, т.е.
имеется распределение числа молекул по значениям времен релаксации. Это
распределение описывается некоторой функцией, которую обозначим G(τ). Тогда
вместо уравнений (2.19) и (2.20) получим:
ε '−ε ∞ ∞ G (τ )dτ
=
,
ε − ε ∞ ∫0 1 + ω 2τ 2
(2.54)
∞
G (τ)ωτdτ
ε"
. (2.55)
=∫
ε − ε ∞ 0 1 + ω2 τ 2
Величина G(τ)dτ характеризует вклад в диэлектрическую проницаемость группы
диполей, имеющих индивидуальные времена релаксации в интервале от τ до τ+dτ, а
амплитуду дисперсии диэлектрической проницаемости (как и все другие однотипные
молекулы) ε—ε∞.
С помощью соотношения (2.54), (2.55) при определенном выборе функции
распределения времен релаксации G(τ) можно получить множество дисперсионных
формул.
Знание функции G(τ) дает возможность описать экспериментальную зависимость
44
ε' и ε" от частоты. Егер [66] показал, что многие экспериментальные результаты
хорошо описываются, если в качестве функции G(τ) ввести распределение Гаусса:
F ( z) =
b
π
2 2
e −b z ,
(2.56)
F ( z ) = τY (τ ) , z = ln(τ / τ 0 ) , (2.57)
где
b — постоянная, характеризующая ширину распределения времен релаксации, τ0 —
наиболее вероятное время релаксации.
2.6. Функция распределения Фрелиха
В модели Фрелиха [9] предполагается, что в жидкости вследствие
неэквивалентности положений диполей должно существовать распределение числа
диполей по значениям энергетических барьеров переориентации диполей, причем это
распределение существует в некотором интервале значений энергии барьеров, которому
соответствуют определенные значения времен релаксации: от τ0 (минимального) до τ1
(максимального). Принимая распределение высот барьеров равномерным в заданном
интервале от Н0 до Н0+ VО, Фрелих получает следующие выражения для диэлектрической
проницаемости ε' и диэлектрических потерь ε":
ε '− ε ∞
⎡
1 + (ωτ 0 ) 2 e 2ν 0 / kT ⎤
kT
ln(
)⎥ ,
= ε − ε ∞ ⎢1 −
1 + (ωτ 0 ) 2
2ν 0
⎣
⎦
ε " = (ε − ε ∞ )
kT
ν0
(2.58)
⋅ [arctg (ωτ 0 exp(ν 0 / kT ) − arctg (ωτ 0 )] .
(2.59)
Эти формулы соответствуют функции распределения времен релаксации
⎧ (ε − ε 0 )kT
,τ 0 ≤ τ ≤ τ 1
⎪
G (τ ) = ⎨ 2τν 0
⎪⎩
0, τ p τ 0 , τ f τ 1
где
τ1 = τ 0eν
0
(2.60)
/ kT
. (2.61)
При такой функции распределения отношение значения диэлектрических потерь
к их максимальному значению εm" определяется как
ε"
=
ε "m
arctg (
ω τ1
ω τ0
) − arctg (
)
ωm τ 0
ωm τ 1
τ
τ
arctg ( 1 ) − arctg ( 0 )
τ0
τ1
.
(2.62)
45
Из последнего соотношения видно, что чем меньше величина τ0/τ1 т.е. чем больше
ширина области распределения потенциальных барьеров vО, тем больше отклонение от
дебаевской кривой. Если растет параметр
β=
τ1
f1 ,
τ0
(2.63)
кривая, описывающая функциональную зависимость ε" от частоты, обладает все более
уширенным и пологим максимумом. Зависимость ε"/ε"m от частоты приводится на
рис.14.
Рис. 14. Зависимость диэлектрических потерь ε"(ω)/ε"(ωm) при τ 1 τ 0 , равных 1; 5; 10.
В работе [67] исследовались растворы бензофенона в парафине. Наблюдалась
кривая поглощения с широким максимумом, которую можно описать набором времен
релаксации. На рассчитанную по (2.62) с β = 5 теоретическую кривую хорошо ложатся
экспериментальные точки. Однако при частотах, далеких от критической частоты
ω=1/τ0, имеют место заметные отклонения.
Этого можно было ожидать, так как функция распределения (2.60) может
характеризовать наиболее вероятное значение времени релаксации и ширину кривой,
описывающей распределение, но не дает сведения о более тонких деталях.
Подобная (2.60) функция распределения предлагается в работе [68]:
⎧1 Aτ ,τ 1 p τ p τ 2
G (t ) = ⎨
⎩ 0,τ p τ 1 ;τ f τ 2
,
(2.64)
где А является параметром, определяющим распределение времен релаксации.
Для некоторых веществ экспериментальные результаты согласуются с функцией
распределения (2.64) при значении А=4. Зависимость ε" от ε' описывается кривой,
очень близкой к эллипсу с полуосями:
a = ε "m ; b =
ε − ε∞
2
.
В пределах экспериментальной точности данные также могут ложиться на дугу
46
окружности при значении А=4,5. В общем случае отношение полуосей
рассматриваемого эллипса следующим образом выражается через параметр A:
a ⎛2⎞
= ⎜ ⎟arctg ( sh( A / 2)) .
b ⎝ A⎠
(2.65)
Для описания релаксационных процессов в алкилбромидах было использовано
распределение времени релаксации между двумя предельными значениями τ1 и τ2
[68]. В этом случае можно допустить, что нижний предел значений времен
релаксации τ1 есть время релаксации вращения малых полярных единиц, т.е. СН2Вrгрупп, расположенных на конце молекулярной цепи, а верхний предел τ2 соответствует
вращению всей молекулы в целом. С увеличением размеров молекулы первая
величина возрастает медленно, а вторая — быстро. Это качественно согласуется с
высказанной выше гипотезой о том, что τ2 и есть время ориентации молекулы в целом.
Результаты измерений хорошо описываются функцией распределения (2.64).
2.7. Распределение Коула—Коула
Для случая, когда имеется распределение времен релаксации Коул и Коул [69,70],
предложили взамен уравнения (2.18) эмпирическую формулу
ε * −ε ∞ =
ε − ε∞
,
1 + (iωτ 0 ) 1− α
(2.66)
где α называется коэффициентом распределения времен релаксации, причем
(0< α<1), τ0— наиболее вероятное значение времени релаксации.
Как вытекает из уравнения (2.18), диэлектрическая проницаемость на частоте
ω = 0 равна ε, а при ω→∞ стремится к ε∞, причем мнимая часть проницаемости снова
становится равной нулю.
Разделяя в (2.66) вещественную и мнимую части, находим соотношения:
ε ' = ε ∞ + (ε − ε ∞ )
ε " = (ε − ε ∞ )
1 + (ωτ 0 )1−α sin
1−α
1 + 2(ωτ 0 )
sin
πα
(ωτ 0 )1−α cos
1−α
1 + 2(ωτ 0 )
sin
πα
2
2
πα
2
+ (ωτ 0 )
2 (1−α )
,
(2.67)
πα
2
+ (ωτ 0 )
2 (1−α )
.
(2.68)
При α = 0 эти уравнения переходят в уравнения Дебая.
При ω = 0 максимальное значение ε' равно ε, минимальное значение ε" равно ε∞
при ω→∞. Дифференцируя ε" по ωτ0 и приравнивая производную нулю, получаем, что
ε" проходит через максимум при ωτ0 = 1, достигая в максимуме величины
47
πα ⎤
⎡
ε − ε ∞ ⎢ cos 2 ⎥ ε − ε ∞
ε "m =
⋅⎢
⎥=
πα
2
2
⎥
⎢
1 + sin
2 ⎦
⎣
πα
⎡
⎢1 − sin 2
⋅⎢
πα
⎢ cos
2
⎣
⎤
⎥
⎥.
⎥
⎦
(2.69)
Функция распределения времен релаксации, соответствующая уравнениям (2.67)
и (2.68), имеет вид
F ( z )dz =
1
sin πα ⋅ dz
⋅
.
2π cos(1 − α ) z − cos πα
(2.70)
Исключение параметра ωτ0 из уравнений (2.67) и (2.68) приводит к уравнению
окружности вида
2
2
⎤
1
⎡1
⎤ ⎡1
⎛ πα ⎞
2
2 ⎛ πα ⎞
⎢⎣ 2 (ε − ε ∞ ) − ε '⎥⎦ + ⎢ 2 (ε − ε ∞ ) tan⎜⎝ 2 ⎟⎠ − ε ′′⎥ = 4 (ε − ε ∞ ) sec ⎜⎝ 2 ⎟⎠ . (2.71)
⎣
⎦
При этом центр окружности определяется соотношениями:
⎡ε − ε ∞ ⎤
⎢ 2 ⎥ = ε',
⎣
⎦
а радиус окружности равен
⎡
⎛ πα ⎞ ⎤
⎢ ( ε − ε ∞ ) tan ⎜ 2 ⎟ ⎥
⎠ ⎥ = ε " , (2.72)
⎝
⎢
2
⎢
⎥
⎢
⎥
⎣
⎦
⎡
⎛ πα ⎞ ⎤
⎢ (ε − ε ∞ ) sec⎜ 2 ⎟ ⎥
⎝
⎠⎥ .
⎢
2
⎥
⎢
⎥
⎢
⎦
⎣
(2.73)
Время релаксации τ0 или соответствующая ему критическая длина волны λm = 2πnτ0
могут быть определены из соотношения:
V
= (ωτ 0 )1−α ,
U
(2.74)
где V и U— расстояния от экспериментальной точки на полуокружности до точек ε' = ε и
ε' = ε ∞ соответственно:
V = (ε − ε ' ) 2 + ε "2 ,
U = (ε '−ε ∞ ) 2 + ε "2 .
(2.75)
Зная угол ψ, образованный осью абсцисс и линией, соединяющей центр окружности с
точкой ε' = ε∞, можно определить параметр распределения времен релаксации α, так как
48
ψ =α
π
2
. (2.76)
Геометрический смысл уравнений (2.74), (2.75) и (2.76) можно понять из рис.15.
Рис. 15. Геометрическая интерпретация параметров уравнений (2.74), (2.75) и (2.76)
Эмпирическая формула Коула—Коула получила теоретическое обоснование
в работе [71].
Результаты измерения диэлектрической проницаемости ε' и потерь ε" многих
жидкостей показывают, что экспериментальные точки хорошо ложатся на дугу
полуокружности, описывающей зависимость ε" от ε'.
2.8. Распределение Девидсона—Коула
Диаграмма Коула—Коула является симметричной относительно линии,
проходящей через центр и параллельной оси ε".
Экспериментальные результаты, полученные при исследовании глицерина и
некоторых других жидкостей, показали, что зависимость ε" от ε' не является
симметричной, а соответствует скошенной дуге.
Девидсон и Коул [72, 73] предложили следующую эмпирическую формулу:
ε * −ε ∞ =
ε − ε∞
,
(1 + iωτ ) β
(2.77)
где β - эмпирический параметр, причем 0 < β < 1.
Уравнение (2.77) переходит в уравнение Дебая при β=1.
После разделения действительной и мнимой частей (2.77) имеем:
ε '−ε ∞ = (ε − ε ∞ )(cosϕ ) β cos βϕ ,
ε " = (ε − ε ∞ )(cosϕ ) β sin βϕ ,
(2.78)
(2.79)
где tgϕ = ωτ .
Функция распределения, соответствующая уравнению (2.77), имеет вид:
49
⎧ sin βπ
⎪
G (τ ) = ⎨ π
⎪
⎩
⎛ τ ⎞
⎜⎜
⎟⎟,τ p τ 0
τ
τ
−
0
⎝
⎠
0,τ f τ 0
(2.80)
Зависимость функции (2.78) от частоты показывает, что расхождение между
этими функциями и уравнением Дебая велико при высоких частотах. Дисперсионная
кривая более пологая, а абсорбционная кривая имеет в максимуме меньшее значение,
чем в случае уравнения Дебая (рис.16).
Рис.16. Зависимости ε" и ε' (в относительных единицах) от ωτ, полученные по уравнениям
(2.78) и (2.79) при β=0,67 (сплошная линия) и по уравнению Дебая при β=1,0 (пунктирная
линия).
Кривая зависимости ε" от ε' дана на рис.17.
Рис.17. Зависимости ε" и ε' от ωτ для разных β по уравнению (2.77) (стрелки показывают
частоту, при которой ωτ = 1).
Зависимость мнимой части диэлектрической проницаемости от действительной
части представляет собой асимметричную кривую, которая близка к окружности при
низких частотах и ассимптотически приближается к линии, образующей угол (βπ)/2 с
действительной осью для высоких частот.
Соответствующее значение β может быть найдено из предельного наклона
экспериментальной кривой на высоких частотах. Так как при ωτ = 1,
50
ε"
βπ
= tg
,
4
ε '−ε ∞
τ может быть найдено как обратная величина круговой частоты, которая
соответствует точке пересечения с биссектрисой предельного угла. Эта частота ниже
частоты, при которой ε" достигает максимального значения.
Другим методом определения τ, не зависящим от количества экспериментальных
точек, является построение зависимости:
⎛θ
tan ⎜⎜
⎝β
⎞
⎟⎟ = ωτ ,
⎠
(2.81)
⎛ ε" ⎞
⎟⎟ .
где θ = arctan⎜⎜
−
ε
'
ε
∞ ⎠
⎝
(2.82)
Частота, для которой эта функция равна единице, определяет время релаксации τ
(рис. 18). Значения β и τ могут быть определены по данным измерения на одной частоте,
но при различных температурах. При этом с изменением температуры значение ε"
должно проходить через максимум. Тогда
ωτ =
⎡ π
⎤
ε '−ε ∞
= tan ⎢
⎥.
ε"
⎣ 2(1 + β ) ⎦
(2.83)
Рис.18. Графическое представление значения времени релаксации, полученное согласно
уравнению (2.81). Кривая I - глицерин при - 50°С (β = 0.6), кривая II - пропилен гликол при
- 64°С (β= 0.66), кривая III - 1 - пропанол при - 113°С (β= 1.0)
2.9. Уравнение Фаусса - Кирквуда
Уравнение Фаусса-Кирквуда [74] связывает параметр β с максимальным значением
εm" и имеет вид:
ε " = ε "m sec h( β ln
ω
) ,
ωm
51
где 0<β<1.
ch −1 (
ε "m
) = 2,3β (lg f m − lg f )
ε"
(2.84)
Построение этой зависимости от lgf позволяет определить 2,3β по наклону линии,
а пересечение ее с осью абсцисс дает значения lgfт, где fт - частота,
соответствующая максимуму поглощения.
Параметр β уравнения Фаусса-Кирквуда связан с коэффициентом распределения
времен релаксации α Коула-Коула следующим соотношением:
β 2=
1−α
1−α
cos(
)π
4
.
(2.85)
Зависимость α от температуры выражается формулой [75]:
α=
A BT
,
e
T
(2.86)
где А и В - постоянные.
Более сложную функциональную зависимость предложили Гаврильяк и Негами
[76]:
ε − ε∞
ε * (ω ) − ε ∞ =
,
(2.87)
(1− β )
1 + (iωτ )1−α
[
]
где 0≤ α ≤1 и 0≤β≤1.
При β=1 (2.87) переходит в формулу Коула-Коула и при α=1, β=1 - в уравнение
Дебая. Уравнение (2.87) в ряде случаев удовлетворительно описывает частотные
свойства полимеров.
Уравнения, описывающие распределение времен релаксации, даны также в
[26,68,77].
2.10. Определение времени релаксации по данным одночастотных
измерений
Метод Гопала-Кришны [78-80] позволяет определить τ и μ индивидуальных
молекул и растворов из измерений на одной частоте.
Вводятся величины X и Y, определяемые соотношениями:
X=
ε '+ε "2 +ε '−2
,
ε "2 +(ε '+2) 2
Y=
3ε "
.
ε " +(ε '+2) 2
2
(2.88)
Эти величины связаны линейной зависимостью
52
X = P + (1 / ωτ )Y ,
где
P=
(2.89)
ε ∞ −1
.
ε∞ + 2
(2.90)
Из зависимости Y от X определяется время релаксации τ :
τ = (λ / 2πс)
dY
,
dX
(2.91)
где λ - длина волны в свободном пространстве.
Дипольный момент определяется по наклону прямой, соответствующей
зависимости X от концентрации:
X = P + KC ,
(2.92)
где
K=
1
Nμ 2 ⎛
⎞
⎜
2 2 ⎟
9kTε 0 ⎝ 1 + ω τ ⎠
(2.93)
и С — концентрация в моль/см3.
Метод Кришны позволяет также определить τ разбавленных растворов:
τ=
1
a"
ω a'−a
⋅
Значения а', а" и а∞ определяются по наклону линейных зависимостей от
концентраций следующих величин:
ε12 = ε1 + αX 2 ,
ε "12 = a" X 2 ,
ε '12 = ε '1 + a' X 2 ,
(ε ∞ )12 = (ε ∞ )1 + a∞ X 2 ,
(2.95)
где индекс "1" относится к растворителю, а "2" — к полярному компоненту. В этом
случае ε∞= n2, концентрация выражается в мольных долях.
Уравнение для определения времени релаксации τ чистых жидкостей и их
растворов, по результатам измерения на одной частоте приведены также в работах
[81,82]:
⎛ 1 ⎞
ε ' = ε ∞ + ⎜ ⎟ε " . (2.96)
⎝ ωτ ⎠
В случае растворов предполагается, что ε∞ является постоянной величиной.
Зависимость ε' от ε" представляет собой прямую линию, и τ рассчитывается по ее наклону.
В работе [83] для времени релаксации разбавленных растворов полярных жидкостей
и их растворов в неполярных растворителях предложено выражение вида:
53
1
τ
=∑
Ai
τi
,
(2.97)
где А - параметр, зависящий от влияния на данную молекулу окружающих молекул.
При допущении, что доля каждого релаксационного процесса пропорциональна
квадрату дипольного момента и в пренебрежении эффектом растворителя, параметр Аi
можно записать как:
Ci μ i2
,
(2.98)
Ai =
∑ Ci μ i2
где Сi — параметр, описывающий влияние эффектов, отличающихся от влияния
дипольного момента.
Для бинарных систем (2.97) принимает форму:
⎤ 1 ⎡
⎤
C1μ12
C 2 μ 22
1 ⎡
= ⋅⎢
+ ⋅⎢
=
2
2⎥
2
2⎥
τ τ 1 ⎣ (C1 μ1 + C2 μ 2 ⎦ τ 2 ⎣ (C1μ1 + C2 μ 2 ⎦
1
1 ⎡ Cμ12 ⎤ 1
= ⋅⎢
+
τ 1 ⎣ (Cμ12 + μ 22 ⎥⎦ τ 2
⎡
⎤
μ 22
⋅⎢
.
2
2⎥
⎣ (Cμ1 + μ 2 ⎦
(2.99)
При С1/С2=С можно предположить, что С приблизительно равен молекулярному
объему растворенного вещества.
В системе, состоящей из молекул одинаковой формы и размеров можно принять
С = 1 и (2.99) приводится к виду
1
τ
=
μ12 / τ 1 + μ 22 / τ 2
.
μ12 + μ 22
(2.100)
Уравнения (2.99) и (2.100) могут быть применены для простых систем в
разбавленных растворах. Вышеуказанные допущения справедливы при измерениях на
одной частоте.
Исследование релаксационных процессов жестких молекул и их бинарных
растворов показывают, что экспериментальные значения времени релаксации хорошо
согласуются с расчетными, полученными из вышеуказанных уравнений.
Время релаксации τ и коэффициент распределения времени релаксации α
разбавленных растворов могут быть определены по данным измерения на одной частоте
по формуле[84-86]:
1 / 2 (1−α )
1 ⎛ A2 + B 2 ⎞
⎟
τ = ⋅ ⎜⎜
ω ⎝ C 2 ⎟⎠
где
,
(2.101)
A = a" (a − a∞ ) ,
B = (a − a ' )(a"−a) = a"2 ,
C = (a '−a∞ ) + a"2 .
54
2.11. Диэлектрическая релаксация с двумя временами релаксации
Релаксационный процесс в некоторых жидкостях может быть охарактеризован с
помощью нескольких типов независимых релаксационных процессов. В этом случае
диэлектрическая релаксация описывается уравнениями [87, 88]:
Ck
ε '−ε ∞
,
=∑
2
ε − ε∞
k 1 + (ωτ k )
C k ωτ k
ε"
=∑
2
ε − ε∞
k 1 + (ωτ k )
(2.102)
.
(2.103)
Параметры Сk характеризуют вклад, вносимый
релаксационным процессом, и удовлетворяют соотношению:
∑С
k
каждым
независимым
= 1.
k
В случае двух независимых релаксационных процессов уравнения (2.102) и (2.103)
можно представить в виде:
ε '−ε ∞
C1
C2
,
=
+
2
ε − ε ∞ 1 + (ωτ 1 ) 1 + (ωτ 2 ) 2
(2.104)
C1ωτ 1
C2ωτ 2
ε"
,
=
+
2
ε − ε ∞ 1 + (ωτ 1 ) 1 + (ωτ 2 ) 2
(2.105)
где С1 и С2 — вклад каждого релаксационного процесса, причем
С1 + С2 = 1 .
(2.106)
Для определения τ1 , τ2 , С1, С2 обычно пользуются графическим методом.
Уравнения (2.104) и (2.105) можно записать в следующем виде:
X = C1 X 1 + C 2 X 2 ,
Y = C1Y1 + C 2Y2 ,
X1 =
1
1
, X2 =
2
1 + Z1
1 + Z 22
Y1 =
Z1
Z2
, Y2 =
2
1 + Z1
1 + Z 22
Z 1 = ωτ 1 ,
Z 2 = ωτ 2 .
,
,
(2.107)
(2.108)
(2.109)
(2.110)
Если релаксационный процесс описывается одним временем релаксации,
экспериментальная точка лежит на полуокружности приведенной диаграммы
Коула—Коула. При наличии двух времен релаксации экспериментальная точка с
55
координатами (X, У) лежит на хорде полуокружности между точками (X1, У1) и (X2, У2).
Эта точка делит хорду в отношении С1/С2 (рис.19).
Рис. 19. Графический метод определения диэлектрической релаксации
независимыми релаксационными процессами по уравнениям (2.102), (2.103).
двумя
Меняя положение хорд, проходящих через экспериментальные точки, необходимо
получить одинаковые значения τ1 и τ2 для всех экспериментальных точек, измеренных на
различных частотах.
Метод решения уравнений (2.105—2.110) с помощью вычислительных машин и
упрощенные методы расчета τ1 и τ2 приведены в работах [89—94].
В случае чистых жидкостей весовые факторы С1 и С2 в равенстве (2.104) и (2.105)
связаны с составляющей дипольного момента, направленной вдоль оси вращения молекулы
μ║ и составляющей, направленной перпендикулярно к оси вращения μ┴ , соотношением:
С1 μ ∏
.
=
С2 μ ⊥
(2.111)
Для растворов эта связь имеет вид:
С1 μ12 x1
=
С 2 μ 22 x2
,
(2.112)
где μ1, μ2, x1 и х2 — дипольные моменты и концентрации компонентов соответственно.
Использование функции дипольной автокорреляции и преобразования Лапласа для
определения частотной зависимости ε* приводит к уравнениям (2.104), (2.105) [95-98].
Экспериментальные результаты показывают, что диэлектрические свойства некоторых
молекул можно описать с помощью двух времен релаксации. Большее время релаксации
соответствует ориентации полярных молекул в целом, а меньшее — внутримолекулярному
вращению.
Вторая область дисперсии обнаружена для таких тяжелых молекул, как йодбензол,
нитробензол и бензонитрил на волне 0,802 см [99].
Дополнительные области дисперсии можно ожидать и для других монозамещенных
бензолов при расширении области измерения на более короткие длины волн.
Результаты исследования многих жидкостей хорошо представляются дугой
полуокружности на плоскости ε" от ε', но значение ε∞, определяемое по точке пересечения
дуги с осью ε' со стороны высоких частот, значительно больше квадрата показателя
преломления п2, вычисленного по оптическим измерениям. Это расхождение
указывает на наличие дополнительной области дисперсии [99].
Наличие нескольких областей дисперсии может быть определено также
56
построением зависимости ε' от ε"/λ. В этом случае каждой области поглощения
соответствует прямая линия с определенным наклоном (рис.20).
Рис. 20. Зависимость ε' от ε"/Х для 1-гептанола при 0 и 20°С по данным Магат [100].
Как видно, три прямых отрезка на рисунке соответствуют трем областям
дисперсии, которые обычно называют метровой (I), дециметровой (II) и
сантиметровой (III). В конце третьей дисперсионной области ε∞3 равен 2,28, в то время
как п2=2,02. Такая разница указывает на существование других, не учтенных,
областей дисперсии.
Данные высокочастотных измерений некоторых жидкостей, имеющих несколько
областей дисперсии, не дают отдельных максимумов поглощения при построении
зависимости ε" от ε'. Однако в тех случаях, когда времена релаксации,
соответствующие этим областям поглощения, различаются в 10-15 раз, возможно
разделение максимумов потерь для отдельных областей [101-105]. На примере 1пропанола проиллюстрируем метод разделения отдельных областей поглощения [101].
Предположим, что дисперсия аддитивно складывается из вкладов их областей
дисперсии (рис.21). Первая область поглощения количественно описывается
уравнением Дебая:
ε1 * −ε ∞ = (ε '1 −ε ∞ ) − iε " =
ε − ε∞
f
1+ i
fm
,
(2.113)
где индекс "1" относится к первой области дисперсии.
Кривая ε* является полуокружностью, которая пересекает ось ε' в точках ε1 = ε и
ε' =ε∞1. Этой области дисперсии соответствует предельная частота fm1.
Для того чтобы определить вторую область дисперсии, необходимо из
экспериментальных данных ε1* вычесть δε1*= ε1*- ε∞ , значение которой получено из
уравнения (2.113). При частоте измерения f>10f m1 , диэлектрическая
проницаемость ε'2 и потери ε"2 для второй области дисперсии вычисляются по
формулам:
2
⎛f ⎞
ε 2′ = ε '−(ε − ε ∞ )⎜⎜ m1 ⎟⎟ ,
⎝ f ⎠
(2.114)
57
2
⎛f ⎞
ε 2′′ = ε "−(ε − ε ∞ )⎜⎜ m1 ⎟⎟ .
⎝ f ⎠
(2.115)
Значения ε'3 и ε"3 для третьей области дисперсии могут быть получены из
экспериментальных данных вышеописанным методом. Центр дуги полуокружности на
плоскости ε" от ε' для этой области выбирается между ε∞2 и ε∞3.
Измерения нормальных спиртов (от С4 до С12) в диапазоне длин волн от 30 до
0,22см и в широком интервале температур показали, что все эти спирты имеют три
области дисперсии, критическая частота которых зависит от температуры и длины
углеродной цепочки [106-109].
По Магат [100], время релаксации, соответствующее метровой области
дисперсии,
определяется
продолжительностью
жизни
ассоциированных
комплексов. Время релаксации молекул меньше, чем продолжительность жизни
комплексов. Например, для 1-гептанола при 20°С продолжительность жизни
комплексов 3,2 · 10-12 с, время релаксации 4 · 10-12 с.
Вторая
область
дисперсии
обусловлена
двумя
связанными
релаксационными процессами, которые не протекают параллельно, а развитие одного
процесса влияет на развитие другого. Первый релаксационный процесс составляет
переход диполей из положения I в положение II, а второй релаксационный процесс
соответствует ориентации диполей в положении II при допущении, что их ориентация
в положении I была закреплена. В этом случае течение второго процесса в некоторой
степени зависит от первого процесса. Для спирта за положение I можно принять
положение диполей в средней части цепи, где их ориентация практически закреплена, а
положение II есть положение тех же диполей, когда они оказываются близкими к концу
цепи, и переход I→II состоит в обрыве близких водородных связей. В положении II
диполи могут ориентироваться путем поворота сегмента цепи.
Третья
область
дисперсии
объясняется
релаксацией
быстро
ориентирующихся молекул, т.е. свободных молекул и молекул, расположенных в
конце цепи и имеющих несвязанный диполь ОН. Такое предположение
подтверждается исследованиями растворов спиртов в неполярных растворителях.
Например, раствор 1 объемного процента гептанола в гептане дает только одну
сантиметровую область дисперсии, в то время как для чистого гептанола
наблюдается три области дисперсии.
Результаты исследования галоидалкилов, спиртов и их растворов в неполярных
растворителях подтверждают вышеуказанные предположения [110— 113].
Рис. 21. Микроволновая часть зависимости ε" от ε' 1-пропанола. Сплошная линия экспериментальные точки, пунктирные линии — полученные из уравнения (2.113)
58
2.12. Связь между диэлектрической релаксацией, вязкостью и
структурой жидкости
Соотношение, связывающее время релаксации с молекулярными постоянными
и предложенное Дебаем [7, 65, 114, 115], имеет вид
4πa 3η
τμ =
.
kT
(2.116)
При выводе этой формулы молекула рассматривается как сфера с радиусом а,
вращающаяся в непрерывной вязкой среде с коэффициентом внутреннего трения η.
Микроскопическое время релаксации может быть вычислено из измеренного
макроскопического времени τ с помощью уравнения (2.48).
Радиусы молекул, вычисленные по формуле (2.116), оказываются намного меньше
величин, получающихся из других измерений. Согласно выражению (2.116) при
постоянстве размеров молекул, время релаксации должно быть пропорциональным
вязкости.
Результаты исследования времени релаксации жидкостей с различной вязкостью
показывают, что макроскопическая вязкость существенно не влияет на время
релаксации в тех случаях, когда полярные молекулы имеют сферическую форму и,
следовательно, могут вращаться, не смещая соседние молекулы. При возрастании
молекулярной асимметрии и размеров молекул вращение полярной молекулы вокруг
какой-нибудь оси вызывает смещение соседних молекул, и в этом случае время
релаксации сильно зависит от вязкости. Формула (2.116) дает удовлетворительные
результаты для очень ограниченного числа жидкостей, а в большинстве случаев имеет
место значительное расхождение между расчетными и опытными данными. Это
объясняется тем, что в соотношении (2.116) используется макроскопическая вязкость, в
то время как в релаксационных процессах должна рассматриваться микроскопическая
вязкость.
Многие экспериментальные данные хорошо совпадают с расчетными, если вместо
η в уравнении (2.116) принять 0,3η.
В работе [116] для времени релаксации получено теоретическое уравнение,
которое включает в себя такие параметры, как средняя вязкость, момент инерции
полярной молекулы, среднее расстояние между соседними полярными молекулами и
некоторые другие. Указанные величины не всегда можно быстро определить или
измерить, что сильно затрудняет пользование этим уравнением. Оно лучше
подтверждается экспериментом, чем уравнением Дебая.
Большая разница между экспериментальными значениями и вычисленными по
формуле (2.116) возникает потому, что при выводе этой формулы использовано
уравнение Стокса для вращения жесткой сферы в вязкой жидкости [117].
Уравнение предполагает, что момент сил трения создается исключительно
вязкостью жидкости, молекула имеет сферическую форму, среда является
непрерывной.
Для установления связи между временем релаксации и вязкостью необходимо
детально рассмотреть природу молекулярного взаимодействия.
Было сделано несколько попыток улучшить формулу Дебая [116, 118—126]. В
работах [123—125] рассматривали молекулу как эллипсоид, разлагая дипольный
момент на составляющие вдоль его трех главных осей. Такой подход оказался
полезным при изучении растворов больших молекул (белков), когда молекула
растворенного вещества настолько велика, что дискретным строением окружающего
растворителя вполне можно пренебречь.
59
Среднее время релаксации
соотношениями [126, 127]:
концентрированных
τ e = C1τ 1 + C2τ 2 ; τ e = τ
η1
,
η2
растворов
определяется
(2.117)
где τ1 — время релаксации всей молекулы, τ2 — время релаксации групп, С1 и С2 —
вклад каждого типа релаксации, τ — время релаксации, определяемое из уравнения
Коула—Коула, η1 — вязкость раствора, η2— вязкость растворителя.
Связь τ с η определяется соотношением [128-131]:
τ =
β
T
η
x
,
(2.118)
где β — постоянная, х — параметр (х < 1).
В случае вращения молекулы необходимо учитывать влияние плотности жидкости
на время релаксации. Для установления связи τ с η чистых жидкостей и растворов
предложены также следующие уравнения [117, 132, 133]:
τ=
3Mη (ε − 1)(2ε + 1)
⋅
,
RTρ
9ε 2
(2.119)
где М, ρ — молекулярный вес и плотность полярной жидкости, η и ε — вязкость и
диэлектрическая проницаемость растворителя, и
τ=
4πa 2
(η + C ρ ) ,
kT
(2.120)
где ρ - плотность жидкости, а - радиус молекулы, С - постоянная, k - постоянная
Больцмана.
Для разбавленных растворов:
τ=
Mη
( ε − 1) .
RT
(2.121)
С целью экспериментальной проверки уравнения (2.120) измеряли τ для
бензофенола в 17 неполярных растворителях с различными вязкостями и
плотностями. Плотность жидкости менялась от 0,6 до 1,6 г/см3, а измерения
проводились на частотах от 1 МГц до 24 ГГц. Значения вязкости, вычисленные по
формуле (2.120), совпадали с измеренными с точностью до 3 сПз, что указывает на
хорошее совпадение расчетных и опытных данных.
Динамическая вязкость связана со статической вязкостью выражением [130]:
η d = η (1 + ω 2τ 2 ) −1 .
(2.122)
2.13. Диэлектрическая релаксация и термодинамические функции
В жидкостях, состоящих из дипольных молекул, процесс диэлектрической
релаксации представляет собой сочетание вращательных качаний молекул около
60
некоторых временных положений равновесия и последующих быстрых, скачкообразных
переориентации, приводящих к переходу молекулы в новое положение равновесия. Такой
переход может сопровождаться как изменением положения диполя молекулы по
отношению к окружающим молекулам, так и изменением числа окружающих молекул.
Таким образом, переориентация молекул имеет характер активированных скачков через
потенциальный барьер. Следовательно, процесс диэлектрической релаксации можно
обсуждать в понятиях теории абсолютных скоростей реакций. Процесс перехода молекулы
из одного положения равновесия в другое требует свободную энергию активации для
преодоления энергетического барьера, определяющего эти положения равновесия. Частота
такого процесса перехода определяется величиной 1/τ, а время релаксации τ связано с
изменением свободной энергии активации дипольной релаксации ΔF следующим
соотношением [134,135]:
τ=
hN
⎡ ΔF ⎤
exp ⎢
,
RT
⎣ RT ⎥⎦
(2.123)
где h — постоянная Планка; R — газовая постоянная; N — число Авогадро. Так как ΔF
связана с теплотой активации дипольной релаксации ΔH и энтропией активации дипольной
релаксации соотношением
ΔF = ΔH − TΔS ,
(2.124)
то уравнение (2.123) может быть записано в виде:
τ=
hN
⎡ ΔH − TΔS ⎤
exp ⎢
⎥⎦ .
RT
⎣ RT
(2.125)
Значения ΔF вычисляется из наклона зависимости lnτ от 1/T, так как
дифференцирование выражения (2.123) по 1/T приводит к уравнению:
ΔH =
1 dτ
Rd (lnτ )
− RT = − RT 2 ⋅
− RT .
τ dT
⎛1⎞
d⎜ ⎟
⎝T ⎠
(2.126)
Величина ΔS определяется из выражения (2.124). Экспериментальные данные
показывают линейную зависимость lnτ от 1/Т для отдельных жидкостей, но в
некоторых случаях наблюдаются отклонения от линейности.
Для улучшения соответствия теории и эксперимента уравнение (2.123)
приведено к виду [136]:
⎡ ΔF ⎤
hN
(2.127)
exp ⎢
τ=
⎥,
RT
⎣ R(T − T0 ) ⎦
где Т0 = (0,25 — 0,35)Tкип (температура кипения); h — постоянная Планка. Это уравнение
приводит к линейной зависимости ln(τT) от 1/(T—T0) для многих полярных
жидкостей. Аналогичное с (2.125) выражение получено для вязкости жидкости [137]:
61
η=
hN
exp(ΔFη / RT ) ,
V
(2.128)
где V - молярный объем, ΔFη - изменение свободной энергии активации вязкостного
процесса:
ΔFη = ΔH η − TΔSη ,
(2.129)
где ΔHη и ΔSη — теплота и энтропия активации вязкостного процесса.
Хилл [138] указал, что поскольку процесс вязкого течения не идентичен
диэлектрической релаксации, величины, входящие в оба эти уравнения, не обязательно
должны иметь одинаковые значения.
Процессы диэлектрической и вязкостной релаксаций связаны соотношением
[139-141]:
ln(τT ) = ln B + x ln η ,
(2.130)
где
B=
x
1 ⎡⎛ V ⎞ (1− x ) ⎤
⎡1
⎤
⎢⎜ ⎟ h
⎥ ⋅ exp ⎢ ( xΔSη − ΔS )⎥
k ⎢⎣⎝ N ⎠
⎣R
⎦
⎥⎦
ΔH
x=
ΔH η
, (2.131)
Зависимость ln(τT) от lnη — прямая линия, по наклону которой можно определить х
и по пересечению с осью ординат — В.
62
ГЛАВА 3
МЕТОДЫ ИЗМЕРЕНИЯ ДИЭЛЕКТРИЧЕСКОЙ
ПРОНИЦАЕМОСТИ И ДИЭЛЕКТРИЧЕСКИХ ПОТЕРЬ
Подробное описание различных методов измерения диэлектрической
проницаемости ε' и диэлектрических потерь ε" приведено в нескольких
монографиях и обзорных статьях [5,13,15,19,160-174]. В этой главе кратко
рассмотрены методы измерения и даны соответствующие рекомендации для
получения наилучшей точности при их практическом применении.
Выбор метода измерения зависит от величины диэлектрической
проницаемости ε', диэлектрических потерь ε", диапазона температур и частот, в
котором производятся измерения. Измерения параметров цепей и сигналов
достигаются с помощью емкостных мостов, Q-метров, измерителей коэффициента
стоячей волны, анализаторов спектров частот, измерительных генераторов,
измерителей импедансов и т.п.
Все известные методы измерения ε' и ε" можно разделить условно на следующие
группы:
1) резонансные методы;
2) коаксиальные и волноводные методы;
3) широкодиапазонные и высокоточные методы измерения диэлектрической
проницаемости;
4) методы, использующие волны в свободном пространстве;
5) методы измерения во временной области.
3.1. Резонансные методы
Для измерения ε' и ε" на частотах от долей кГц до 50МГц применяются
колебательные контуры с сосредоточенными параметрами.
Исследуемая жидкость заливается в измерительный конденсатор. В качестве
измерительных конденсаторов используются плоские, дисковые, цилиндрические и
сферические конденсаторы. Наиболее широко распространены цилиндрические
конденсаторы. Обычно они состоят из трех коаксиальных цилиндров. Наружный и
внутренний цилиндры соединены между собой и при подключении к измерительному
прибору заземляются. Средний цилиндр для уменьшения краевых эффектов делается на
4-6 мм короче наружного и внутреннего цилиндров.
В
последние
годы
широко
используются
четырехэлектродные
конденсаторы, где до минимума сведены паразитные емкости и достигается
повышенная точность измерений [175—181].
Конденсаторы различных конструкций описаны в литературе [182—186].
При изготовлении конденсаторов в качестве конструкционных материалов
используются материалы, не взаимодействующие с исследуемым веществом:
посеребренная латунь, золото, платина, титан.
Определение диэлектрической проницаемости сводится к измерению изменения
емкости измерительного конденсатора при заполнении его жидкостью.
Так как
63
С = εС 0 + С n , (3.1)
то x =
C − C0
, (3.2)
C0
где С0 - емкость пустого конденсатора; С - емкость заполненного жидкостью
измерительного конденсатора; Сn - паразитная емкость, χ - диэлектрическая
восприимчивость.
Для определения рабочей емкости пустого измерительного конденсатора и
паразитной емкости монтажных проводников необходимо откалибровать конденсатор
по эталонным жидкостям.
Измерительный конденсатор калибруется измерением ε жидкостей с различной
диэлектрической проницаемостью. Необходимо, чтобы измерения проводились с одним и
тем же конденсатором.
При калибровке с двумя жидкостями можно записать:
С1 = ε 1С 0 + С n ,
(3.3)
С 2 = ε 2 С0 + Сn ,
(3.4)
где С1 и С 2 — измеренная емкость конденсатора с жидкостью; ε 1 и ε 2 диэлектрические проницаемости эталонных жидкостей; С0 - рабочая емкость пустого
конденсатора. Тогда:
С0 =
С1 − С 2
,
ε1 − ε 2
С n = С1 − ε 1С 0 .
(3.5)
(3.6)
В результате измерения емкости конденсатора необходимо внести поправку на
паразитную емкость, а так же вычислить диэлектрическую проницаемость [174, 175].
Измерение комплексной диэлектрической проницаемости часто проводится с
использованием резонансных цепей. Основными составляющими такой цепи являются
генератор, работающий на фиксированной стабильной частоте, связанный (в
большинстве случаев индуктивно) с резонансной цепью LС, которая содержит
измерительную ячейку с жидкостью.
При измерении емкости конденсатора используются различные
электрические схемы. Наиболее часто встречаются схемы биений и мостовые
схемы.
3.1.1. Метод биений
В этом методе сравниваются резонансные частоты двух генераторов. Первый
генератор является опорным и работает на фиксированной частоте, а в
колебательный контур второго генератора включается эталонный переменный
конденсатор. Колебания от двух генераторов подаются на смеситель, позволяющий
получить разностную частоту (биения), которая регистрируется выходным
индикатором. Изменяя переменную емкость второго генератора, добиваются
равенства частот генераторов (нулевые биения). После получения нулевых биений
64
параллельно к эталонному конденсатору подключается измерительный конденсатор с
исследуемой жидкостью. При этом появляется разностная частота, которая может быть
сведена к нулю изменением емкости эталонного конденсатора. Разность значений
емкости эталонного конденсатора до подключения измерительного конденсатора С и
его емкости С после подключения его и получения нулевых биений равна емкости
измерительного конденсатора, а диэлектрическая проницаемость жидкости
определяется по формуле:
ε=
С1 − С 2
,
С0
(3.7)
где С0 — емкость пустого измерительного конденсатора.
Большая чувствительность этого метода позволяет использовать его для
измерения малых изменений емкости (диэлектрической проницаемости) при
изменении температуры, давления и т.д. В этом случае определяется изменение
емкости в зависимости от внешнего фактора. Расчетная формула имеет вид:
ΔС = 2С
Δf
f
,
(3.8)
где ∆f - разностная частота; f - частота опорного сигнала; С - емкость эталонного
конденсатора. Так как разностная частота может быть измерена с большой точностью,
достигается высокая точность измерения 8 (порядка 106).
Метод биений не позволяет измерять диэлектрические потери жидкости и
определять ε в частотном диапазоне.
3.1.2. Метод расстройки контуров
Метод расстройки контуров не имеет вышеуказанных недостатков. Схема
измерения этим методом приведена на рис. 22.
Рис. 22. Схема измерения диэлектрической проницаемости и диэлектрических потерь
методом расстройки контуров.
Резонанс в контуре достигается изменением эталонной емкости С при отключенном
и подключенном измерительном конденсаторе. Достижение резонанса в контуре
фиксируется индикаторным прибором ln, индуктивно связанным с контуром.
Резонансные значения эталонной емкости при измерениях с измерительным конденсатором
С1 и без него C2 используются для расчета емкости измерительного конденсатора с
исследуемой жидкостью:
С = С1 − С 2 .
(3.9)
65
Диэлектрическая проницаемость определяется по известной емкости С0 пустого
измерительного конденсатора:
С
ε=
.
(3.10)
С0
При измерении диэлектрических потерь с подключенным измерительным
конденсатором достигается резонанс, и эталонную емкость изменяют до тех пор, пока
значение тока в контуре не уменьшится до значения 1 2 относительно его значения
при резонансе. Тогда измерение емкости определяется выражением:
ΔС =
1
2
( С 2′′ − C 2′ ) ,
(3.11)
где С2" и С2' — емкости эталонного конденсатора при резонансе и при расстройке от
резонанса.
Диэлектрические потери вычисляются по следующей формуле:
tgδ =
ΔC
C
⋅
A1 − A2
A1
,
(3.12)
где А1 и А2 — показания индикаторного прибора при резонансе с измерительным
конденсатором и без него.
При этой схеме можно использовать другой вариант расчета.
Когда измерительный конденсатор с жидкостью подключен к цепи
эталонного конденсатора, резонанс достигается изменением емкости эталонного
конденсатора и при этом ток в контуре имеет максимальное значение. При резонансе
фиксируется значение частоты fr и тока Ir.
Затем эталонная емкость изменяется до тех пор, пока значение тока не
уменьшается в 1 2 раз и фиксируются значения частоты как с левой, так и с
правой стороны от резонансной кривой. Разность этих частот и есть ширина
резонансной кривой Δf.
Добротность контура определяется отношением:
Q=
fr
.
Δf
(3.13)
Добротность контура связана с tgδ соотношением:
Q=
ε'
1
=
.
ε" tgδ
(3.14)
Погрешность вышеописанного метода составляет 1% для ε' и 20—25 % для tgδ .
Подробное описание метода, анализ ошибок и техника эксперимента приведены в
работах [191, 192, 193, 194, 195].
66
3.1.3. Метод куметра
Величины ε и tgδ часто измеряются куметром. В этом случае расчетные формулы
имеют следующий вид:
С = С1 − С 2 ,
(3.15)
С
,
С0
(3.16)
ε=
tgδ =
( Q1 − Q2 )C1
,
Q1Q2 ( C1 − C 2 )
(3.17)
где С1 , С2 ,Q1 , Q2 — емкости эталонного конденсатора и добротности контура до и после
подключения измерительного конденсатора.
Погрешность измерения куметром составляет 5—10% для ε' и 10—15% для tgδ
[190, 196—198].
Для измерения ε' и ε" в метровом диапазоне широко применяются приборы,
выпускаемые промышленностью.
3.1.4. Методы измерения в объемном резонаторе
Измерения ε' и ε" в сантиметровом и миллиметровом диапазонах дают важную
информацию о дополнительных областях дисперсии, релаксационных процессах и других
явлениях, связанных со структурой жидкости. В связи с этим некоторые методы,
используемые в этих диапазонах, будут в дальнейшем рассмотрены более подробно.
В сантиметровом диапазоне чаще всего используются круглый или прямоугольный
объемный резонатор.
Исследуемая жидкость в кювете помещается в резонатор, ε' и tgδ жидкости
определяются по измеренным значениям резонансной длины волны и добротности
резонатора при отсутствии и наличии исследуемой жидкости в кювете. Резонаторы без
диэлектрика и частично заполненный диэлектриком изображены на рис. 23.
Рис. 23. Схемы полого резонатора без диэлектрика и частично заполненного диэлектриком.
Диэлектрическая проницаемость определяется из выражения [199]:
67
ε' =
1 + ( λ c a / 2πd ) 2
1 + ( λc / λ p )2
.
(3.18)
Причем а определяется из следующего уравнения:
tga λ c 2π ( Z см + d )
=
⋅
,
a
λp
2πd
(3.19)
где λρ — резонансная длина волны резонатора; λс — критическая длина волны
резонатора; Zсм — смещение безконтактного поршня при настройке в резонанс с
жидкостью в резонаторе; d — высота столба жидкости. При известных значениях λρ,
tga
Zсм, и d величина а определяется по таблицам функции
[160].
a
Добротность
цилиндрического
резонатора,
частично
заполненного
диэлектриком с потерями, в котором возбуждаются колебания типа Нои,
определяется формулой [200]:
Q=
где D = 2d −
Δ⎛
1
⎜⎜ 2
r ⎝β +k2
1
β
1
PD + L
3
[
]
⎞ 2
⎟⎟ k ( PD + L ) + 2r( β 2 P + β 0 2 ) + PDtgδ
⎠
sin βd ,
L = 2l 0 −
1
β0
sin β 0 l 0 ,
P=
sin β 0 l 0
,
sin βl
,
(3.20)
k=
2π
λc
,
r — радиус резонатора, l и l0 — длины частей резонатора, заполненных диэлектриком
и воздухом соответственно.
Добротность резонатора с диэлектриком без потерь определяется выражением
Qm′ =
PD +
Δ⎛
1
⎜⎜ 2
r ⎝β + k2
[
1
L
3
]
⎞ 2
⎟⎟ k ( PD + L ) + 2r( β 2 P + β 0 2 )
⎠
.
(3.21)
Глубина скин-слоя Δ может быть вычислена по экспериментально определяемой
добротности резонатора Qт, заполненного воздухом:
Qm =
r( k 2 − β 2 )
2
⎛
2 rβ 0
Δ⎜⎜ k 2 +
l+d
⎝
⎞
⎟
⎟
⎠
,
(3.22)
tgδ вычисляется по измеренной добротности резонатора с диэлектриком и без него по
формуле
68
⎛
L ⎞⎛ 1
1
⎟⎟⎜⎜ −
tgδ = ⎜⎜1 +
Pl 0 D ⎠⎝ Q Qm
⎝
⎞
⎟⎟ .
⎠
(3.23)
Добротность резонатора может быть определена методами, которые указаны
в литературе [198, 199].
При колебаниях в цилиндрическом резонаторе волн типа Н01n или Е010 кювета с
жидкостью устанавливается на оси резонатора. Кювета должна иметь
цилиндрическую форму, тонкие стенки и малый диаметр. Расчетные формулы,
метод измерения и анализ ошибок даны в работах [201—203].
В прямоугольном резонаторе тонкостенная капсула с жидкостью
помещается через отверстие в широкой стенке резонатора в пучности
электрического поля [204]. Тогда при измеренной резонансной частоте fr , для
добротности резонатора имеем:
Δf r = ( ε ' −1 )
2 fr S
l ,
V
(3.24)
1
S
1
= 4ε " l +
, d/a≤0,1 , (3.25)
Q1
V
Q0
где S, l — поперечное сечение жидкости в капсуле и длина столба жидкости, Q1 и Q0 —
добротности резонатора с образцом и без него, V — внутренний диаметр капсулы, а
— размеры широкой стенки волновода.
По наклону зависимости Δfr от l и 1/Q1 от l определяются ε' и ε". В этом случае
отпадает необходимость определения Q0. Погрешность этого метода порядка 1%
для ε' и 3% для ε".
Достоинством резонаторных методов являются простота измерений, малые
габариты установки, приемлемые погрешности ε' порядка 1,5%, а ε" 3—5%, малое
количество жидкости, необходимое для измерения [205—210].
К недостаткам этих методов можно отнести механическую трудность
изготовления резонаторов с большой добротностью, необходимость обеспечения
большой стабильности генератора колебаний и невозможность измерения ε' и tgδ
жидкостей с большими потерями.
3.2. Коаксиальные методы
Микроволновую область часто делят на отдельные области: дециметровые волны
(0,3—3 ГГц), сантиметровые волны (3—30 ГГц) и миллиметровые волны (30— 300
ГГц).
Традиционные методы определения диэлектрической проницаемости проводятся
в частотной области. Во всех этих методах жидкость помещается в измерительную
ячейку, и диэлектрическая проницаемость измеряется в дискретных частотных точках.
Однако нет экспериментальной установки или метода, который мог бы перекрыть весь
диапазон микроволн.
Мы рассмотрим микроволновые методы, в которых используются такие линии
передачи, как коаксиальные системы и волноводы.
Коаксиальные системы могут использоваться в частотном диапазоне 50МГц—
18 ГГц.
Наиболее привлекательными для применения в микроволновой области являются
69
коаксиальные линии [216—224]. Коаксиальные линии используются при частотах,
когда распространяются поперечные электромагнитные волны типа ТЕМ.
Электрические и магнитные поля направлены перпендикулярно друг другу, и оба поля
перпендикулярны к направлению распространения волны.
Теория таких систем требует решений уравнений Максвелла с
соответствующими краевыми условиями [211] и приводит к концепции
комплексной постоянной распространения γ. Если для коаксиальной линии в точке x = 0
электрическая компонента поля равна Е0, тогда в точке х = d при отсутствии отражения
поле может быть представлено как:
E = E 0 l − γd
где
,
(3.26)
γ = α + jβ ,
(3.27)
α — коэффициент поглощения, β — фазовая постоянная.
Величина α и β определяются с помощью измерительной линии, в которой
изменения мощности связано с показаниями индикаторного прибора при двух
положениях зонда. Тогда коэффициент поглощения определяется формулой:
α=
⎛V
1
20 log 10 ⎜⎜ 1
x
⎝ V2
⎞
⎟⎟
⎠ ,
(3.28)
x = x 2 − x1
где х — расстояние между точками, где определяется V1 и V2, V1 - показания
индикатора в положении зонда х1, V2 - показания прибора при х2. Единица измерения
α — дБ·см-1.
Расстояние между двумя максимумами или минимумами показаний индикатора
равно половине длине волны в волноводе. Для коаксиальных линий, кроме частоты,
используется понятие длины волны излучения. Соответственно длина волны в
свободном пространстве обозначается λ0. Фазовая постоянная связана с длиной волны
в веществе соотношением:
2π
.
(3.29)
β=
λd
Следовательно, техника коаксиальной линии описывает измерение постоянной
распространения γ, т.е. (α , β) или (α , λd ).
Это позволяет получить выражение для комплексной диэлектрической
проницаемости из равенства:
2π ⎡⎛ λ 0
⎢⎜
γ=
λ 0 ⎢⎜⎝ λ с
⎣
2
⎤
⎞
⎟⎟ − ε * ⎥
⎥⎦
⎠
1
2
,
(3.30)
где λс называется критической длиной волны.
Для основной моды в коаксиальной линии λс равна бесконечности и уравнение
(3.30) приводится к виду:
70
γ=
2π
λ0
[− ε * ] 12
.
(3.31)
Это уравнение является комплексным. Разделяя его на действительную и мнимые
части, получим:
ε' =
λ0
[β 2 − α 2 ] ,
2
4π
ε" =
αβλ 0 2
.
2π 2
(3.32)
(3.33)
Отсюда видно, что ε' и ε" являются функциями α и β, хотя ε' зависит сильно от β, ε" от α.
Для жидкостей с малыми диэлектрическими потерями (α = 0) из уравнений (3.32) и
(3.29) имеем
λd =
λ0
ε'
.
(3.34)
Это равенство хорошо известно из оптической теории.
В коаксиальных, а также в волноводных линиях измерительная ячейка с образцом
может быть введена в различные области линии (рис. 24).
Рис. 24. Секции образца, используемые в частотной и временной диэлектрической
спектроскопии:
а — волновые сопротивления линии и образца согласованы, b — образец бесконечной
длины, с — модель сосредоточенной емкости, d и е — за образцом линия закорочена, f —
образец переменной толщины с подвижным короткозамыкателем [213].
Эти варианты расположения образца проанализированы в [213]. Теория метода и
конкретные измерительные системы в большинстве случаев автоматизированы и
описаны в работах [214—224].
3.3. Волноводные методы
В сантиметровом и миллиметровом диапазонах широко используются волноводные
методы для измерения ε' и ε" жидкостей с большими, средними и малыми потерями.
Имеются несколько разновидностей волноводных методов. Рассмотрим наиболее
применяемые варианты.
71
3.3.1. Метод короткого замыкания
Если на один из концов волновода поместить источник электромагнитных волн, а
второй конец замкнуть металлической пластинкой, то образуется стоячая волна.
Расстояние между узлами этой волны равно половине длины волны в волноводе.
Пусть на некотором участке волновода помещен исследуемый диэлектрик, а за
диэлектриком волновод закорочен с помощью короткозамыкающего поршня. При
наличии диэлектрика в волноводе положение минимума стоячей волны сдвигается, часть
падающей волны поглощается диэлектриком, а значение напряженности электрического
поля в минимуме стоячей волны отличается от нуля.
Соотношение, связывающее параметры отраженной волны с диэлектрическими
свойствами диэлектрика и его толщиной, для такого случая имеет вид [225]:
− jλв 1 − jηtg ( β 0 x0 )
th( γ 1 d )
=−
⋅
, (3.35)
γ 1d
2πd η − jtg ( β 0 x0 )
Emax
2π
— коэффициент стоячей волны (КСВ), β 0 =
— фазовая постоянная,
Emin
λв
λв — длина волны в волноводе, x0 — расстояние от диэлектрика до первого узла
стоячей волны, d — толщина жидкости, γ1 — постоянная распространения в жидкости.
С помощью измерительной линии можно определить λв , х0, η, а постоянная γ1
thγd
определяется по таблицам функции
[160]. По этим данным вычисляются значения
γd
ε' и ε".
Если короткозамыкающий поршень находится на расстоянии λв/4 за
диэлектриком, то уравнение имеет вид:
где η =
λ 1 − jηtg( β 0 x0 )
cth( γ 1d )
=−j в ⋅
. (3.36)
γ 1d
2πd η − jtg( β 0 x0 )
Трансцендентные уравнения (3.35) и (3.36) решаются графическими методами
или методами последовательных приближений [226—228].
Погрешность определения комплексной диэлектрической проницаемости
ухудшается за счет погрешностей определения этих величин по графикам, а метод
последовательных приближений является очень громоздким. Кроме того, эти
уравнения не имеют однозначного решения и для определения ε' и ε" необходимо
заранее знать их приблизительные значения или измерять их при различных
толщинах столба жидкости.
Существует несколько частных случаев, когда исключается решение этих
трансцендентных уравнений и получаются более простые выражения для
определения постоянной распространения.
1. С помощью измерительной линии измеряется входное сопротивление
волновода один раз при положении короткозамыкающего поршня непосредственно за
диэлектриком (Zвх)1, а другой — на расстоянии λв/4 от диэлектрика (Zвх)2. Эти величины
входят в формулу для расчета γ1:
Z0 β0
γ1 =
,
( Z вх )1 ( Z вх )2
2
2
(3.37)
72
где Z0 — входное сопротивление волновода без диэлектрика.
2. Если потери в диэлектрике малы (γd ≤ 0,3) и КСВ мал (η<5), то уравнение
(3.35) упрощается:
− λ ⎛ 2π ⎞
th( β1d )
x0 ⎟ .
= − в tg ⎜⎜
β1 d
2πd ⎝ λв ⎟⎠
(3.38)
3. Входное сопротивление волновода с жидкостью измеряется один раз при
толщине d , а другой раз при толщине 2d [229, 230].
Согласно [229], в этом случае расчетная формула принимает вид:
⎛λ
ε * = ε ' − jε " = ⎜⎜ 0
⎝ λc
где
A=
2
⎡ ⎛λ
⎞
⎟⎟ + ⎢1 − ⎜⎜ 0
⎢⎣ ⎝ λ d
⎠
1 − jη1tg ( β 0 x01 )
,
η1 − jη1tg ( β 0 x01 )
B=
⎤⎛ 2
1⎞
⎥⎜
− ⎟ ,
⎥⎦⎝ AB A ⎠
(3.39)
1 − jη 2 tg ( β 0 x02 )
,
η 2 − jη 2 tg ( β 0 x02 )
(3.40)
⎞
⎟⎟
⎠
2
η 1 и η 2 - коэффициенты стоячей волны при толщине d и 2d, х 01 , х 02 соответствующие сдвиги минимума стоячей волны при толщине столба жидкости d и 2d.
Для получения максимальной точности измерения ε' и ε" толщина жидкости
должна удовлетворять соотношению:
d = ( 2n + 1 )
λd
4
ε'
для n = 0 и 1. (3.41)
При больших значениях коэффициента стоячей волны η интенсивность сигнала
изменяется значительно при переходе от минимума к максимуму напряжения
вдоль линии. Такая перегрузка вызывает отклонения от квадратичности
характеристики кристаллического детектора и нелинейные искажения в усилителе. В
этом случае значения η определяются из зависимости от х напряженности
электростатического поля по формуле:
η=
где θ =
Emax
E min
⎡ E 2
⎤
⎢ x − cos 2 θ ⎥
⎢ E min
⎥⎦
=⎣
sin θ
1
2
,
(3.42)
πΔx
, E x = 2Emin , λв — длина волны в волноводе, Δх — расстояние между
λв
точками по обе стороны от минимума, в которых напряженность поля в 2 раза
больше его минимального значения.
Если показания индикатора пропорциональны Е2 и значение Δх малы, можно
положить соs2θ=1 и sinθ=θ. В этом случае равенство (3.42) можно записать в виде:
73
η=
λв
.
πΔ x
(3.43)
Эти предположения верны при Δх < 6—7% от λ0 .
Несоблюдение этого условия приводит к ошибке 1—2% для ε' ≤ 5 и tgδ = 0,1.
Для сильно полярных жидкостей с большими потерями ошибка за счет
неоптимальности толщины жидкости может быть значительной [231]. Эти методы
позволяют определить ε' с погрешностью 0,5—2%, а ε" примерно с погрешностью 2—
4%, в зависимости от величины диэлектрической проницаемости и потерь.
Блок-схема установки для измерения ε' и ε" методом короткого замыкания
приведена на рис. 25.
Рис. 25. Схема экспериментальной установки для измерения ε' и ε" методом
короткого замыкания:
1 — модулятор, 2 — стабилизатор напряжения, 3 — клистрон, 4 — ферритовый вентиль,
5 — направленный ответвитель, 6 — волномер, 7 — аттенюатор, 8 — измерительная линия,
9 — измерительный усилитель, 10 — измерительная ячейка, 11 — термостат, 12 —
микрометрический винт.
3.3.2. Балансные методы
Рассмотрим балансный метод для измерения ε' и ε" жидкостей, как с малыми
потерями, так и с большими потерями [232, 233]. Блок-схема установки дана на рис. 26.
Клистронный генератор моделируется частотой в несколько килогерц. Ферритовый
вентиль или аттенюатор развязывает клистрон от основного тракта.
Длина волны непосредственно измеряется волномером. Высокочастотная мощность
подается на плечо а первого двойного волноводного тройника и делится на две равные
части. Одна часть из плеча моста С через вентиль и согласующий трансформатор попадает
в ячейку с жидкостью. С выхода ячейки волна через согласующий трансформатор и
вентиль попадает в плечо С' второго волноводного двойного тройника. Другая часть
высокочастотной мощности из плеча моста d через вентиль, аттенюатор и фазовращатель
попадает в плечо d' моста. Сигнал модуляции, выделяющийся на детекторе, подается на
вход усилителя, к которому подключен индикатор.
В другом варианте схемы установки модулированный высокочастотный сигнал
подается на смеситель, к которому подается также сигнал гетеродина. Разностная частота
со смесителя подается на усилитель. В этом случае создается возможность производить
измерения в широком диапазоне частот.
Измерительная ячейка состоит из двух круглых или прямоугольных волноводов.
Один волновод имеет чуть меньшие размеры и может перемещаться внутри другого.
Ячейка закрыта с двух сторон с помощью герметических окон из слюды толщиной 0,010,02 мм или тефлона. На стенке волновода ячейки сделаны маленькие отверстия, через
которые жидкость может поступать из резервуара или вытесняться в резервуар при
перемещении подвижного волновода. Перемещение волновода отсчитывается с помощью
74
микрометрической системы. Ячейка с резервуаром для жидкости окружена
термостатирующей рубашкой. Температура контролируется термометром или термопарой.
Процедура измерения заключается в определении изменения фазы волны при
прохождении через жидкость и поглощенной энергии.
Если векторная сумма волн, поступающих в плечо С' и d' двойного тройника,
равна нулю, т. е. волны поступают в противофазе, то сигнал в плече а' моста и показания
индикатора равны нулю, мост сбалансирован. Баланс моста достигается с помощью
прецизионного фазовращателя и аттенюатора.
Рис. 26. Схема установки для измерений балансным методом:
1 — блок питания, 2 — модулятор, 3 — клистрон, 4 — волномер, 5 — двойной тройник, 6
— нагрузка, 7 — согласующий трансформатор, 8 — измерительная ячейка, 9 — индикатор
перемещения, 10 — согласующий трансформатор, 11 — двойной тройник, 12 — нагрузка, 13
— переменный аттенюатор, 14 — фазовращатель, 15 — вентиль, 16 — гетеродин, 17 —
смеситель, 18—детектор, 19 — усилитель, 20 — индикатор.
Микрометрическая система перемещает подвижной волновод до установления
нулевой толщины жидкости, и достигается баланс моста. Таким образам, микрометр
перемещается на несколько миллиметров в зависимости от величины поглощения.
Зависимость поглощения в неперах от толщины столба жидкости строится по
показанию прецизионного аттенюатора. Наклон этой зависимости дает нам
величину коэффициента поглощения α. По наклону зависимости сдвига фазы от
толщины жидкости определяется фазовая постоянная β.
То, что фазовращатель вносит незначительное затухание, зависит от величины
изменения фазы. Поэтому затухание фазовращателя необходимо калибровать.
Аттенюатор также дает незначительный сдвиг фазы. Для исключения фазового
сдвига нужно измерить фазовую постоянную βвоз при отсутствии жидкости в ячейке.
Истинная фазовая постоянная в жидкости определяется из равенства:
β = β изм − β воз , (3.44)
где
β воз =
2π
λв
,
(3.45)
λв — длина волны в волноводе без жидкости.
ε' и ε" определяются из следующих соотношений:
2
⎛λ ⎞
ε " = ⎜ 0 ⎟ 2αβ ,
⎝ 2π ⎠
(3.47)
75
β =
2π
λ
.
(3.48)
d
При отладке балансных схем необходимо принять ряд мер.
1. Рассогласование в волноводном тракте свести до минимума. Особое
внимание следует уделить тем узлам, электрическая длина которых меняется в
процессе измерения.
2. Степень баланса необходимо довести до достаточно высокой. Для этого все
регулирующие элементы должны допускать плавную регулировку.
В балансных методах точность измерения ограничена точностью аттенюатора,
фазовращателя и микрометрической системы, используемой для отсчета приращения
толщины жидкости. Точность измерения ε' в этих методах порядка 0,3—0,6%, а ε"
порядка 1%.
В [234] описана измерительная установка с двухканальной системой, позволяющая
с высокой точностью определять комплексную диэлектрическую проницаемость
жидкостей с высокими потерями. Установка работает в диапазоне 9 ГГц и позволяет
проводить измерения абсолютных значений комплексной диэлектрической
проницаемости воды, ε' с точностью 0,06% и ε" с точностью 0,15%.
Как видно из вышесказанного, резонаторные методы измерения диэлектрической
проницаемости имеют высокую точность. Вполне оправдано их применение на частотах
свыше 5 ГГц. Их применение на более низких частотах связано с созданием громоздких
объемных резонаторов, что не удобно как с точки зрения геометрических размеров, так и
с необходимостью иметь для измерения большие количества образца. Выход из этой
ситуации предложен в работе [306] путем использования резонаторов со спиральной
линией задержки. Подобный подход позволяет решить сразу две проблемы: уменьшить
геометрический размер резонатора и минимизировать количество измеряемого вещества.
Кратко рассмотрим устройства и методику измерений диэлектрической проницаемости с
помощью спиральных резонаторов.
3.3.3. Диапазон частот 250 ÷ 3000 МГц
Имеющиеся методы измерения диэлектрических параметров в данном диапазоне
частот являются менее совершенными и менее освоенными по сравнению c
имеющимися метрологическими устройствами в области низких и сверхвысоких
частот.
Ввиду ряда технических трудностей, устройства для измерения диэлектрических
параметров диэлектриков в дециметровой области длин волн (250 ÷ 3000 МГц)
отличаются большой ошибкой измерений (до 40%), громоздкостью измерительных
систем, требованием значительных количеств исследуемого вещества, сложностью
настройки, производства измерений и др.
Используя замедляющие системы и компенсационный принцип измерений, было
сконструировано устройство для определения диэлектрических параметров как низко-,
так и высокопотерных веществ в диапазоне от 250 МГц до 3000 МГц.
Резонансный метод определения диэлектрических констант
В качестве датчика используется четвертьволновый коаксиальный резонатор со
спиральным внутренним проводником, играющим роль замедляющей системы.
Резонатор в этом случае образуется тогда, когда спираль, экранированную кожухом,
закоротить с двух сторон проводящими плоскостями (рис. 27). При подводе к
резонатору высокочастотной энергии в нем будут наблюдаться резонансы всякий раз,
76
когда вдоль спирали будет укладываться целое число четвертей волн.
Резонансная длина волны λР и добротность Q такой сиcтемы выражаются
следующим образом:
⎛ D
λ Р = 4πnlD⎜⎜
⎝ DЭ
1
⎞2
⎟⎟ ,
⎠
⎡ ⎛ D ⎞2 1 ⎤
⎟⎟ ⋅ f 02 ⎥
0.024πD ⎢1 − ⎜⎜
D
⎢⎣ ⎝ Э ⎠
⎥⎦
Q=
3
⎡
⎛ D ⎞ ⎤
⎟⎟ ⎥
⎢(πnd )−1 + ⎜⎜
⎢⎣
⎝ D Э ⎠ ⎥⎦
(3.49)
,
(3.50)
где f 0 - резонансная частота, n - число витков в замедляющей системе на 1 см длины,
d - диаметр провода внутреннего спирального проводника.
Рис. 27. Четвертьволновый коаксиальный резонатор.
l - длина спирали, lЭ - длина экрана, D - диаметр спирали, DЭ - диаметр экрана.
Наличие спирали в системе обеспечивает существование волн медленного типа,
т.е. спираль действует как микроволновая линия задержки, уменьшая скорость
распространения электромагнитной волны. Так как спираль является широкополосной
системой, то в резонаторе со спиральным внутренним проводником постоянной длины
резонансы будут наблюдаться в широкой полосе частот.
При измерении диэлектрических констант исследуемый образец помещается в
максимум электрического поля, т.е. у разомкнутого конца спирали. Так как между
верхней гранью и разомкнутым концом спирали имеется небольшая емкость, то,
используя специальный подвижный плунжер, можно подстраивать резонансную
полость. Для того, чтобы не происходило искажения силовых линий
электромагнитного поля, приводящих к потерям, в нижней части резонатора имеется
D
зазор Э . Связь выхода и входа передающих линий с резонатором, т.е. возбуждение
4
резонатора и отвод энергии, осуществляется с помощью зонда и петли, вводимых
внутрь полости резонатора. Из рис.27 видно, что внутренняя длина экрана
⎛ D ⎞
l Э = ⎜ l + Э ⎟ , т.е. для определения l Э необходимо знать длину спирали, которая в
2 ⎠
⎝
свою очередь определяется общим числом витков и её шагом. Число витков на 1 см
l
длины внутреннего спирального проводника при D = 0,55 и
= 1.0 (отношения,
D
DЭ
выполнение которых ведет к максимальной добротности резонатора) можно оценить по
формуле:
77
n≈
4800
.
ν 0 DЭ
(3.51)
Шаг намотки:
τН =
где ν 0 =
ν 0D2
(3.52)
5800
1
.
f
f0 =
Резонансная частота:
c
λ0
.
Дальнейший расчет четвертьволнового коаксиального резонатора (ЧКР) со
спиральным внутренним проводником с заданной добротностью Q0 на резонансную
частоту f 0 производится следующим образом. Зная f 0 , выбираются значения D , l ,
DЭ из условия, что
D
l
0.3 <
0.6 и
> 1.0 .
DЭ
D
Тем самым для резонансной частоты f 0 определяется размер экрана резонатора.
По формулам (3.51) и (3.52) находится число витков спирали и шаг намотки. По
величине Q0 и выбранным параметрам определяется диаметр провода спирали d и
находится l Э . Для облегчения расчета параметров резонатора составляется номограмма
параметров ЧКР со спиральным внутренним проводником.
Достоинства четвертьволновых коаксиальных резонаторов прежде всего
заключаются в том, что за счет широкополосности спирали и наличия настроечных
плунжеров, резонансные частоты таких резонаторов могут перестраиваться в широком
диапазоне частот Δf = 40% .
В таблице 3 приведены параметры изготовленных ЧКР с внутренними
спиральными проводниками, которые обеспечивают перекрытие частотного диапазона
от 2,5·108Гц до 3·109Гц. Изготовленные резонаторы были покрыты изнутри слоем
серебра толщиной 0,02 мм и отшлифованы с чистотой 8 класса.
Таблица 3
Параметры изготовленных четвертьволновых коаксиальных резонаторов (ЧКР)
№
резонатора
1
2
3
4
5
Интервал
частот, МГц
250-700
700-1200
1200-1800
1800-2300
2300-2800
D , мм
Dэ , мм
l , мм
d , мм
Q
8,0
6,0
4,0
3,0
2,5
24,0
23,0
18,8
16,5
10,0
14,0
11,0
10,0
9,0
4,3
1,0
0,9
0,8
0,7
0,5
1000
1300
1740
1600
1900
При помещении диэлектрика в полость резонатора сдвиг резонансной частоты
будет пропорционален диэлектрической проницаемости, а изменение добротности
системы позволит определить величину tgδ исследуемого вещества.
Для вывода аналитических выражений для ε ′ и tgδ можно воспользоваться
методом малых возмущений. Относительное смещение частоты резонатора Δf от
внесения в него образца имеет вид:
78
rr
Δf ∫ PEdV
=
. (3.53)
f
4W
Если образец помещен вдоль оси спирали и площадь поперечного сечения его
мала, можно считать, что все компоненты поля в области, занимаемой образцом,
значительно меньше продольной компоненты и поэтому ими можно пренебречь. С
учетом того, что
r
r (ε − 1)E
,
P=
4π
и тангенциальная составляющая поля на границе раздела непрерывна, т.е. поле внутри
образца E Z′ равно полю, внешнему относительно образца E Z , выражение для
относительного смещения частоты приводится к виду:
l
Δf
f
E
(
ε − 1)S ∫
=
⋅
2
Z
dz
0
16π
W
,
(3.54)
где S - площадь поперечного сечения образца. Для получения окончательного
l
выражения для ε необходимо определить ∫ E Z dz / W .
2
0
Поля находятся методом, общепринятым в решении задач о распространении
волн в анизотропно-проводящем волноводе (рис.28).
Рис. 28. Спиральная линия задержки в коаксиальной линии.
Система разбивается на две области: внутри спирали 0 ≤ r ≤ d и вне спирали
d ≤ r ≤ R и для каждой из областей записываются решения уравнений Максвелла.
Для достаточно густой намотки спирали (ctg ϕ > 10 ) справедливо равенство
радиального волнового вектора продольному волновому вектору k 1 ≈ k 2 ≈ k 3 ≈ 2 π .
λg
При этом поля в каждой из областей имеют вид:
I область:
E z(1) = A1 J 0 ( k 3 ) e i ( ω t − k 3 z ) ,
E r(1) = iA1 J 1 ( k 3 ) e i (ω t − k 3 z ) ,
Eϕ(1) = −
ik
B1 J 1 ( k 3 r ) e i (ωt − k 3 z ) ,
k3
(3.55)
H z(1) = B1 J 0 ( k 3 r ) e i ( ω t − k 3 z ) ,
79
H r(1) = iB1 J 1 ( k 3 r )e i (ωt − k3 z ) ,
H ϕ(1) =
II область:
ik
A1 J 1 ( k 3 r )e i (ωt − k3 z ) ,
k3
E z( 2 ) = [A2 J 0 ( k 3 r ) + B 2 K 0 ( k 3 r ) ]e i ( ω t − k 3 z ) ,
E r( 2 ) = i[A2 J 1 ( k 3 r ) + B2 K 1 ( k 3 r ) ]e i (ωt − k 3 z ) ,
Eϕ( 2 ) = −
ik
[CJ 1 (k3 r ) − DK 1 (k3 r )]e i (ωt − k3 z ) ,
k3
(3.56)
H z( 2 ) = [CJ 0 ( k 3 r ) + DK 0 ( k 3 r ) ]e i (ωt − k3 z ) ,
H r( 2 ) = i [CJ 1 ( k 3 r ) − DK 0 ( k 3 r ) ]e i ( ω t − k 3 z ) ,
H ϕ( 2 ) =
ik
[A2 J 1 ( k 3 r ) − B2 K 1 ( k 3 r ) ]e i (ωt − k3 z ) .
k3
Произвольные постоянные А1, А2, B1, B2, С, D определяются из граничных
условий. В приведенном случае граничные условия записываются в общепринятом
виде при r = d и при r = R :
E z( 2 ) = 0
Eϕ( 2 ) = 0
После подстановки в граничные условия значений полей из (3.55) и (3.56)
получаются выражения для постоянных коэффициентов интегрирования:
A1 = E 0 ,
−1
⎛ ik
⎞
B1 = E0 J ⎜⎜ J1a ctgϕ ⎟⎟ ,
⎝ k3
⎠
a
0
A2 = E0 J 0a K 0R J 0a K 0R − J 0a K 0R
(
)
,
(
)
,
B2 = E0 J 0a K 0R J 0a K 0a − J 0a K 0R
⎛ ik
⎞
C = E0 J K ⎜⎜ ctgϕ ⎟⎟
⎝ k3
⎠
a
0
−1
R
1
⎛ ik
⎞
D = E0 J J ⎜⎜ ctgϕ ⎟⎟
⎝ k3
⎠
a a
0 1
−1
(J
(J
−1
−1
(3.57)
a
1
K1R − J1R K1a
)
,
a
1
K1R − J1R K1a
)
,
−1
−1
где J 0 , J1 , K 0 , K1 - функции Бесселя. Постоянная часть аргументов функций Бесселя
80
записывается в виде индекса сверху, а множитель 2π опускается.
λg
Энергия системы записывается как сумма энергий в первой и второй областях
W = W1 + W2 , где:
W1 =
1
W2 =
8π
2
l
1 ⎛ (1) 2
⎜ E z + Er(1) ⎞⎟dV = E02 a J 0a J1a ,
∫
⎠
8π V ⎝
4k3
R
{[
l
(2) 2
(2) 2
r
r
∫V ⎛⎜⎝ E r + E z ⎞⎟⎠ dV = 4 ∫0 A2 J 0 + B2 K 0
] + [A J
2
2
r
1
− B2 K 1r
] }rdr .
2
(3.58)
В результате дальнейших подстановок в уравнение (3.54) и с учетом того, что
σ , где σ - проводимость образца, получаем выражения для диэлектрических
tg δ =
ε ′ω
констант:
ε ′ = 1+
ε ′′ =
16Δf
,
2
f 0 (k3d ) μ 0a − μ 0R
(
8
(k3d ) (μ
2
a
0
−μ
R
0
)
)
⎛1 1 ⎞
⎜⎜ − ⎟⎟ ,
⎝ Q Q0 ⎠
(3.59)
(3.60)
где Q0 - добротность пустого резонатора, Q - добротность резонатора с внесенным в
K 0a ,R
- параметр, являющийся отношением бесселевых
J 0a ,R
функций первого рода нулевых порядков, при численных значениях a и R .
него диэлектриком, μ0a ,R =
На рис. 29 представлена блок-схема измерительного устройства.
Рис. 29. Блок-схема измерительного устройства.
1 - генератор, 2 - развязка, 3 - аттенюатор, 4 - 2Т-мост, 5 - прецизионный аттенюатор, 6 фазовращатель, 7 - частотомер, 8 - стабилизированный источник питания, 9 - согласователь,
10 - резонатор, 11 - смеситель, 12 - усилитель, 13 - вольтметр ВК7-10А.
Рабочий генератор модулируется меандром (Г3-35). Для определения
генерируемой частоты используется волномер типа Ч4-3. Поступающая от генератора
мощность регулируется по амплитуде аттенюатором типа Д2-20. Далее, энергия от
генератора через разветвитель мощности поступает в два канала, один из которых
содержит измерительный резонатор, а второй - компенсационные устройства.
Компенсационный канал состоит из аттенюатора, трансформатора полных
81
сопротивлений (ТПС) и согласующих элементов, работающих в диапазоне 200 3000МГц. Компенсационный или шунтирующий канал позволяет улучшить
метрологические параметры устройства. Мощность, поступающая в шунтирующее
плечо, возвращается в основной канал, содержащий резонатор на участке между
рабочим резонатором и детектором. С помощью ТПС можно отрегулировать фазу
волны, вновь попадающей в основной канал, таким образом, чтобы либо усиливалась,
либо ослаблялась мощность волны, проходящей по главному волноводному тракту.
Аттенюатор А2 и ТПС позволяют так отрегулировать сигнал, возвращающийся в
основной канал, чтобы получить нужный уровень мощности на детекторе.
Скомпенсировав оба плеча по нулевой мощности на детекторе, помещаем в резонатор
исследуемый образец. Возникающий при этом сигнал разбаланса не будет
скомпенсирован и пройдет на детектор. В дальнейшем сигнал усиливается и
регистрируется
цифровым
вольтметром
ВК7-10А.
Благодаря
введению
компенсационной схемы, получена возможность измерять как низко-, так и
высокопотерные диэлектрики (см. табл. 4, 5). Вся схема имеет характер моста, что
существенно улучшает чувствительность устройства.
Процесс измерений и результаты калибровки измерительного устройства
Исследуемый раствор наливается в стеклянный капилляр диаметром 1,0-1,5 мм и
длиной 4-5 см с толщиной стенок 0,15-0,2 мм и помещается вдоль оси резонатора.
Резонансная частота резонатора f P определяется волномером. Добротность резонатора
f
определяется как Q = P , где Δf = f1 − f 2 - полуширина резонансной кривой.
2Δf
Полученные результаты измерений на данной установке приведены в таблицах 4 и 5.
Таблица 4
Диэлектрические характеристики веществ, измеренных на установке при f = 700 МГц
в сравнении с литературными данными при близких частотах; t°=25°С.
Вещество
ε ′ изм
ε ′′ изм
ε ′ [87,100]
ε ′′ [87,100]
Нитробензол
32.21
4.44
32.5
4.5
Глицерин
13.74
10.7
12.79
9.69
Метиловый спирт
30.02
7.6
30.00
7.9
Этиловый спирт
18.5
9.6
18.5
9.7
Вода
80.10
2.80
80.81
2.75
В таблице 5 представлены результаты измерений диэлектрических параметров
воды и водных растворов спиртов при разных частотах и температурах для сравнения с
литературными данными.
Из таблиц 4 и 5 видно, что данным методом хорошо измеряются как низко-, так и
высокопотерные вещества.
82
Таблица 5
Диэлектрические параметры веществ в диапазоне частот 300 - 2800 МГц
Частота,
МГц
Вещество
Измеряемые
величины
300
Вода би-дист.
600
Температура, °С
10
20
30
40
50
ε ′ изм.
ε ′′ изм.
84,01
1,87
80,52
1,32
76,87
0,96
73,60
0,73
69,98
0,57
Метиловый
спирт
ε ′ изм.
ε ′′ изм.
32,1
5,68
31,8
5,43
29,8
4,9
27,3
4,2
25,8
3,9
1762
Метиловый
спирт
ε ′ изм.
ε ′′ изм.
11,6
26,3
10,9
9,8
8,4
7,9
1762
Этиловый
спирт
ε ′ изм.
ε ′′ изм.
19,9
12,8
19,4
11,6
18,7
10,5
17,8
9,3
16,4
8,1
2800
Этиловый
спирт
ε ′ изм.
ε ′′ изм.
16,5
7,8
15,8
7,3
15,4
6,8
15,0
6,0
14,5
5,4
Относительные погрешности δε ′ и δε ′′ оценивались по следующим формулам:
δε ′ =
δε ′′ =
16Δf 0δf p
32Δf 0δα
16δ (Δf )
+ 2
+
,
2
2
a
R
2 3
a
R
a
R
f R (k3 d ) (μ 0 − μ 0 ) f p (k 3 d ) (μ 0 − μ 0 ) f p k 3 d (μ 0 − μ 0 )
⎛ δf εp δf ⎞
16δ (Δf ) ⎛⎜ 1
1 ⎞ 16(Δf εp − Δf )Δf ⎛⎜ 1
1 ⎞
32δα
⎜
− ⎟+
− 2⎟+ 2 3 a
− ⎟.
a
R ⎜
a
R
R
2
k3d (μ0 − μ0 ) ⎝ f εp f p ⎟⎠ (k3d )(μ0 − μ0 ) ⎜⎝ f εp f p ⎟⎠ k3 d (μ0 − μ0 ) ⎜⎝ fεp f p ⎟⎠
Погрешности измерений частоты составляли ±0,005%, амплитуды тока детектора
±0,5%, диаметра резонатора и образца ±10-3 см. Рассчитанные погрешности δε ′ , δε ′′
составили:
δε ′ =±(0.5÷0.8)%, δε ′′ =±(1.2÷2.0)%.
Метод "тонкого" образца для измерения диэлектрических констант
Вследствие отсутствия широкого набора калибровочных веществ в диапазоне от
200 до 3000 МГц для сравнительной оценки полученных результатов была собрана
установка с применением коаксиальной линии по методу тонкого образца.
Суть метода сводится к раздельному измерению диэлектрической проницаемости
ε ′ и коэффициента потерь ε ′′ . Измеряемый образец помещается в максимум
электрического поля на расстоянии четверти длины волны от поршня. Далее находится
положение пучности напряжения в линии без образца l и с образцом lε . Разность
положений l − lε = Δl . Расчет ε ′ и ε ′′ производится по формулам:
83
ε′ =
λ
π
tg
,
πα λ (d + Δl )
⎛ 1
ε ′′ = ⎜⎜
⎝ Kc
(3.61)
⎞
λ
⎟⎟
,
⎠ изм 2πd
где K c - коэффициент стоячей волны.
Оригинальным местом в данной установке является измерительная ячейка,
которая была собрана в лаборатории диэлектрической спектроскопии (рис. 30).
Рис. 30. Измерительная ячейка
Установка была проградуирована эталонными веществами. Данные по
измерению диэлектрических характеристик воды на разных частотах в сравнении с
результатами, полученными методом ЧКР со спиральным внутренним проводником,
приведены в таблице 6.
Погрешности измерений оценивались по формулам:
δε ′ =
1
πα
tg
π
λ ⎤ 1
⎡
⋅
δλ − δd ⎥ +
⎢
λ (d + Δl ) ⎣
d ⎦ πα
1 ⎤
⎡
⎢⎣δΔl + λδd − λ dλ ⎥⎦ ,
⎡
⎤
π
1+ ⎢
⎥
⎣ λ (d + Δl ) ⎦
1
2
⎛
1 ⎞ ⎛
λ
λ ⎞
⎟⎟ ⋅ ⎜⎜ δλ −
δK c − δd ⎟⎟ ,
Kc
d ⎠
⎝ K c 2πα ⎠ ⎝
δε ′′ = ⎜⎜
где точность измерений Δl - 0,001 мм, d - 0,01 мм, погрешность определения длины
волны λв по внутреннему волномеру - 0,25%. Полученная погрешность для ε ′ - 5-6%,
для ε ′′ - 8-10%. Причем, метод "тонкого" образца позволяет измерять только
высокопотерные вещества. Следовательно, мы еще раз убеждаемся в преимуществах
предложенного метода с использованием ЧКР со спиральным внутренним
проводником.
84
Таблица 6
Таблица сравнения диэлектрических констант воды, измеренных разными методами
f , МГц
250
500
900
1200
1500
1700
2000
2500
2800
λ , см
-
60,0
33,3
25,0
20,0
17,65
15,0
12,0
-
Резонаторный
метод
80,38
80,33
80,19
80,02
79,81
79,64
79,36
78,78
78,09
0,79
2,23
4,01
5,33
6,67
7,51
8,82
10,91
12,97
Метод тонкого
образца
81,62
80,31
80,30
80,10
79,72
79,0
78,5
77,3
77,5
0,67
2,30
3,10
5,71
5,32
8,3
7,72
10,1
11,3
На основе вышеизложенного можно сделать следующие заключения:
1) Предложенный резонансный метод измерения диэлектрических констант с
использованием набора изготовленных четвертьволновых резонаторов со спиральными
внутренними проводниками можно использовать для измерения диэлектрических
параметров диэлектриков в диапазоне 200 - 2800 МГц.
2) Применение компенсационного принципа измерений дает возможность
измерять не только низкопотерные, но также и высокопотерные вещества.
3.3.4. Измерения диэлектрической проницаемости на частотах:
9584,6 МГц, 163,93 МГц, 37037,0 МГц
Для измерения диэлектрических характеристик диэлектриков на СВЧ существует
довольно большое количество методов, каждый из которых имеет свои преимущества и
недостатки. Основным недостатком почти всех методов является возможность только
дискретных измерений диэлектрической проницаемости относительно tgδ. Чтобы
избавиться от этого недостатка, была разработана схема установки, позволяющая
измерять как низко-, так и высокопотерные диэлектрики в области СВЧ.
Резонансный метод ( f = 9584.6 МГц)
Введение в известной резонансной схеме дополнительного приема независимой
регулировки мощности в резонаторе и на детекторе позволяет проводить измерения
веществ, tgδ которых может меняться в широких пределах. Блок-схема рабочей
установки на этой частоте аналогична схеме, приведенной на рис.29.
Исследуемый образец, в виде стержня, помещается вдоль оси цилиндрического
резонатора (если образец жидкий, то он находится в стеклянном капилляре d = 1.5-2.5
мм с толщиной стенок 0.15±0.02 мм). Определение диэлектрических констант состоит в
измерении частотомером Ч2-32 резонансной частоты при помещении образца ( f 0 ) и
полуширины резонансной кривой (Δf ) пустого резонатора и нагруженного.
fP
. Диэлектрические
2Δf
параметры изучаемых образцов рассчитываются по следующим формулам:
Добротность резонатора определяется по формуле Q =
85
2
0.325 ⎛ a ⎞ f 0 − f1
,
ε ′ = 1+
⎜ ⎟
B ⎝b⎠
f0
ε ′′ =
(3.62)
0.325 ⎛ 1 1 ⎞
⎜ −
⎟,
B ⎜⎝ Q Q0 ⎟⎠
где a, b - размеры резонатора и образца диэлектрика, B - числовой коэффициент,
зависящий от отношения b .
a
Для иллюстрации предлагаемого метода в таблице 7 приведены измеренные
параметры образцов в сравнении с литературными.
Таблица 7
Диэлектрические характеристики воды и раствора NaCl на частоте 9584 МГц
Вещество
Вода
би-дист.
NaCl
C=5 М
Измеренные
величины
10°
20°
30°
40°
50°
60°
Примечание
ε′
ε ′′
54,0
61,69
65,08
65,69
64,80
63,21
Измер.
39,0
32,10
26,51
21,56
17,50
14,32
Измер.
ε′
ε ′′
-
63,5
65,6
66,2
65,2
63,4
[105]
-
31,1
25,7
26,0
16,7
13,3
[105]
ε′
ε ′′
-
63,0
65,8
66,3
65,5
63,5
[63]
-
31,5
26,0
20,5
16,05
13,5
[63]
ε′
ε ′′
-
57,8
59,7
59,6
58,3
56,4
Измер.
-
27,3
23,4
18,5
16,7
13,1
Измер.
ε′
ε ′′
-
57,9
60,0
59,8
58,6
55,3
[105]
-
21,1
22,6
19,1
15,3
12,3
[105]
ε′
ε ′′
-
57,5
59,5
59,5
58,2
55,5
[63]
-
27,5
22,5
19,7
15,0
12,3
[63]
Примечание: рабочий резонатор изнутри покрыт Au толщиной 0,002 мм и
отшлифован по 8 классу.
Введение схемы компенсации позволило увеличить чувствительность установки.
Для проверки достоверности полученных результатов была собрана широко известная
установка на частоте 9585 МГц, работающая по методу частичного заполнения сечения
волновода.
86
Метод частичного заполнения сечения волновода
( f = 9584.6 МГц и f = 16393.4 МГц)
Этот метод является одной из модификаций метода, основанного на измерении
импеданса образца, расположенного в измерительной линии.
Методика измерений состоит в следующем. Определяется величина ΔX
смещения поршня, фиксирующее положение минимума тока в линии без образца и с
образцом. При этом регистрируется величина тока в максимуме стоячей волны без
образца J max и в минимуме с образцом J min . Из этих величин, при условии
квадратичности характеристики детектора, рассчитываются ε ′ и ε ′′ по формулам:
⎡ ⎛ λl ⎞ 2
⎤ u
ε ′ = 1 + ⎢2⎜ ⎟ + 0.5⎥ 2 2 ,
⎢⎣ ⎝ π ⎠
⎥⎦ u − v
(3.63)
⎡ ⎛ λ0 ⎞
⎤ v
,
⎟ + 0.5⎥ 2
2
⎢⎣ ⎝ π ⎠
⎥⎦ u − v
2
ε ′′ = ⎢2⎜
где
u = 2 ∑ − 1 .75 ln
v=2
∑
λв
a
⋅
4a
πα
+2
λв
d
⋅
B
,
B +Y2
2
Y
,
B +Y2
2
⎡
⎢
∞
⎢
1
= ∑ ⎢
n =3,5, 7
⎢ n 2 + ⎛⎜ 2α
⎜λ
⎢
⎝ 0
⎣
⎤
⎥
1⎥ ,
− ⎥
⎞ n⎥
⎟⎟
⎥
⎠
⎦
a - размер широкой стенки волновода, d - диаметр образца.
Величины B и Y находятся непосредственно из измерений:
⎛ 2π
⎞
B = tg ⎜
Δx ⎟ ,
⎜λ
⎟
⎝ g
⎠
Y=
1
J max
−1
J min
.
Точность определяемых данным методом ε ′ и ε ′′ зависит от точности
измеряемых экспериментальных величин λ0 , λв , B, Y .
Измеряемые образцы помещались в стеклянные капилляры с толщиной стенок
0,1-0,15 мм и диаметром 0,8 мм (для f = 16393,4 МГц) и 1,2 мм (для f = 9584,6 МГц).
Результаты, полученные на описанных установках, на частотах 9581 МГц и 16393
МГц согласуются с литературными, что можно видеть из таблиц 8, 9.
87
Таблица 8
Результаты измерений ε ′ и ε ′′ чистых жидкостей на f = 9581 МГц при t 0 =24,5°С
Вещество
ε ′ изм.
ε ′ [87,105]
ε ′′ изм.
ε ′′ [87,105]
Вода
55,5
55,0
31,08
29,7
Метиловый спирт
8,36
8,35
7,39
7,20
Этиловый спирт
4,56
4,55
2,48
2,50
н-бутиловый спирт
3,12
3,16
0,76
0,74
Диэтиленгликоль
4,38
4,47
3,29
3,22
Пиридин
11,4
11,6
3,67
3,74
Зная погрешности измерений величин λ0 , λв , B, Y ,
относительные погрешности измерений δε ′ и δε ′′ :
нетрудно
рассчитать
⎡ ⎛ λ0 ⎞2
⎤
⎡ ⎛ λ ⎞2
⎤ 2 2
2
0
.
5
2
2⎜ ⎟ + 0.5⎥
v
u
δ
u
uv
+
−
⎟
⎜
⎢
⎢
⎥
4λ
4λ2
u
u
⎢⎣ ⎝ πα ⎠
⎥⎦
⎢ ⎝ πα ⎠
⎥⎦
δv ,
−
δε ′ = 0 ⋅ 2 2 δλ0 + 2 0 2 2 2 δα + ⎣
2
2
(πα ) u + v
π α u +v
u 2 + v2
u 2 + v2
(
(
)
)
(
)
⎡ ⎛ λ0 ⎞2
⎤
⎡ ⎛ λ ⎞2
⎤ 2 2
2
2⎜ ⎟ + 0.5⎥δu
uv
2
0
.
5
δ
u
v
v
+
−
⎢
⎜
⎟
⎢
⎥
4λ
4λ2
⎢⎣ ⎝ πα ⎠
⎥⎦
⎢ ⎝ πα ⎠
⎥⎦
v
u
δε ′′ = 0 2 ⋅ 2 2 δλ0 + 2 0 3 2 2 δα + ⎣
−
.
2
2
π α u +v
(πα) u + v
u 2 + v2
u 2 + v2
(
(
)
)
(
)
Полученные величины составляли 2% и 5% для ε ′ и ε ′′ соответственно, при
λ =1,83 см и 1,5% и 4,5% для ε ′ и ε ′′ при λ = 3,13 см.
Таблица 9
Результаты измерений ε ′ и ε ′′ чистых жидкостей на f = 16393 МГц при t°=20°С
Вещество
ε ′ изм.
ε ′ [87]
ε ′′ изм.
ε ′′ [87]
Вода
43,37
-
38,00
-
Изобутиловый
спирт
2,86
2,88
0,53
0,52
н-бутиловый
спирт
2,99
2,98
0,61
0,64
Изопропиловый
спирт
3,14
3,19
0,72
0,71
Пропиловый
спирт
3,30
3,33
0,86
0,88
Для измерения ε ′ и ε ′′ при λ = 0,81 см применяется метод вариации толщины
образца.
88
Метод вариации толщины образца ( f = 37037.0 МГц)
Рабочая установка состоит из стандартных узлов, за исключением измерительной
ячейки, которая является термостатируемым отрезком прямоугольного волновода
17x10 мм, закрытого фторопластовой пластиной снизу. Исследуемый образец
помещается в ячейку. Внутренняя часть ячейки и поршень покрыты Au толщиной 0,08
мм. Длина хода поршня и соответственно высота столба образца выбираются в
соответствия с длиной волны и коэффициентом поглощения. В непосредственной
близости от фторопластовой перегородки расположен направленный ответвитель,
регистрирующий только отраженную энергию.
Измерение производится следующим образом. Поршень из своего исходного
самого нижнего положения плавно поднимается вверх. В зависимости от толщины слоя
образца, находящегося между фторопластовой перегородкой и нижней поверхностью
поршня, показания индикатора тока проходят через максимум и минимум.
Максимальные показания соответствуют такой толщине слоя, при которой отражения
волн от нижней и верхней его поверхностей совпадают по фазе. При увеличении
толщины слоя амплитуда осцилляции уменьшается, стремясь к некоторому
постоянному значению, соответствующему слою бесконечной толщины, при котором
волна, отраженная от верхней границы, затухает, не доходя до нижней поверхности
раздела. В нашей установке использован оптический длинномер типа ИВВ-2,
позволяющий производить отсчет с точностью до 0,005 мм.
Расчет диэлектрических параметров ε ′ и ε ′′ производится по формулам
[109,110]:
⎛λ
ε ′ = ⎜⎜
⎝ λε
2
2
2
⎞ ⎛ λ0 ⎞ ⎛ αλ0 ⎞
⎟⎟ + ⎜⎜ ⎟⎟ − ⎜
⎟ ,
⎠ ⎝ λc ⎠ ⎝ 2π ⎠
ε ′′ =
(3.64)
αλ
,
πλε
2
0
где λε - длина волны в образце.
Постоянная затухания α определяется из соотношения:
1
αnλε =
2
1
⎛ J
th⎜⎜ max
⎝ J∞
⎞
⎟
⎟
⎠
,
где J max - показания индикаторного прибора в каком-либо максимуме, J ∞ - показания
прибора при «бесконечной» толщине образца и n - число полуволн, укладывающихся
между положениями поршня, соответствующим показаниям прибора J max и J ∞ . Для
учета влияния затухания на экспериментально наблюдаемую λε были использованы
поправочные графики.
В таблице 10 приведены результаты измеренных значений ε ′ и ε ′′ в сравнении с
литературными.
89
Таблица 10
Сравнение литературных данных со значениями ε ′ и ε ′′ , измеренными методом
«вариации толщины образца» при t° = 20°С
Вещество
ε ′ [100, 110, 112]
ε ′′ [100,110,112]
ε ′ изм.
ε ′′ изм.
Вода диcт.
20,5
29,4
20,3
29,5
Этиловый спирт
3,89
1,30
3,88
1,30
Метиловый спирт
5,68
3,23
5,7
3,22
Вода - метанол
10%
90%
5,45
4,17
5,48
4,19
30%
70%
5,95
6,60
5,98
6,54
50%
50%
7,07
10,00
7,01
10,11
70%
30%
9,50
15,10
9,44
15,00
90%
10%
14,00
23,9
13,91
24,00
величин,
вычисляются
Зная погрешности определяемых промежуточных
относительные погрешности измерений δε ′ и δε ′′ :
δε ′ = 2
λ0
λ2
λ
αλ
δλ0 + 2 30 δλ0 + 2 02 δλ0 + 2 02 (λ0δλ + αδλ0 ) ,
λε
λε
λс
4π
δε ′′ =
λ0
πλε
⎛
⎞
λ
⎜⎜ 2αλ0δ + λδλ0 + α 0 δλε ⎟⎟ .
λε
⎝
⎠
Оцененные погрешности составляли 2,5% для ε ′ и 4,5% для ε ′′ .
Следует отметить, что в определенные на ВЧ и СВЧ величины ε ′′ растворов
жидкостей необходимо вводить поправки на низкочастотную проводимость по
следующей формуле:
′′ = ε изм
′′ −
ε ист
2σ
f ,
(3.65)
где σ - низкочастотная проводимость (ом-1 см-1) раствора, определяемая на частоте
1кГц при каждой температуре производимых измерений. Проводимость воды
(растворителя) составляла σ = 10-4 ÷ 10-5 ом-1 см-1 при t=20°С.
Определение ε ∞ растворов жидкостей
Определение ε ∞ представляет собой сложную задачу. Существует ряд методов,
которые позволяют определить ε ∞ . Перечислим некоторые из них.
1) Измеряют ε S и плотности ρ при различных температурах и экстраполируют
90
T 0 C → ∞ [67].
2) Изучают поглощение и отражение света жидкостями.
3) Рассчитывают ε ∞ теоретически, зная молекулярные параметры исследуемого
объекта.
4) Для некоторых (неполярных) веществ выполняется соотношение Максвелла
ε ∞ ≅ n 2 что значительно упрощает расчеты.
5) Имея измеренные ε ′ и ε ′′ на разных частотах, строят диаграмму Коула-Коула
и, интерполируя кривую на ось абсцисс, находят ε ∞ .
Во всех перечисленных случаях экспериментальные методы имеют высокую
погрешность и некоторые недостатки, описанные в [67].
3.4. Методы, использующие волны в свободном пространстве
Рассмотренные выше методы широко применяются в сантиметровом диапазоне, но с
уменьшением длины волны до миллиметрового диапазона и, в особенности, на
коротковолновом участке миллиметрового диапазона распространены методы,
использующие
волны
в
свободном
пространстве.
Теория
метода,
экспериментальные системы подробно описаны в книге [235] и статьях [236—241]. В этих
методах для определения диэлектрической проницаемости используется оптическая
характеристика диэлектрика, т.е. комплексный показатель преломления:
n* = n − jk ,
(3.66)
где п* - показатель преломления диэлектрика с потерями; k - коэффициент
поглощения.
Для немагнитных веществ (μ =1) скорость распространения электромагнитной
волны в среде связана с комплексной диэлектрической проницаемостью среды
выражением:
v=
c
ε*
,
(3.67)
где с - скорость света в пустоте. Следовательно,
n* =
c
= ε* .
v
(3.68)
Так как ε * = ε ′ − jε ′′ , из уравнений (3.49) и (3.51) имеем:
ε ' = n 2 − k 2 , ε " = 2nk .
(3.69)
Блок-схема измерительной установки для определения ε' и ε" этим методом
приведена на рис. 31.
91
Рис. 31. Схема установки для измерения методом свободного пространства: 1 — блок
питания, 2 — генератор СВЧ, 3 — модулятор, 4 — полноводный ответвитель, 5 — волномер, 6 —
детектор, 7 — усилитель, 8 — переменный аттенюатор, 9 — передающая рупорная антенна,
10 — измерительная ячейка с микрометрическим устройством, 11 — приемная рупорная
антенна, 12 — детектор, 13 — усилитель, 14 — самопишущее устройство.
Высокочастотная энергия излучается на поверхность жидкости рупорной антенной с
диэлектрической линзой, и отраженный сигнал принимается приемной антенной.
Коэффициент отражения измеряется в зависимости от толщины слоя жидкости,
изменяемой с помощью микрометрической системы.
Зависимость коэффициента отражения от толщины жидкости имеет ряд
максимумов и минимумов. Расстояние между максимумом и минимумом равно λж/2.
Коэффициент поглощения α определяется из выражения:
exp( na λ ж ) =
1 − SL − ( 1 − L )( 1 − S )
L− S
,
(3.70)
где п — целое число, L и S — коэффициенты отражения в максимуме и минимуме.
Определив α и λж ε' и ε", вычислим по формулам:
⎛λ
ε ' = ⎜⎜ 0
⎝ λж
2
2
⎞ ⎡ ⎛ αλ ж ⎞ ⎤
⎟⎟ ⎢1 − ⎜
⎟ ⎥ + sin 2 θ ,
⎠ ⎢⎣ ⎝ 2π ⎠ ⎦⎥
(3.71)
2
⎛ λ ⎞ ⎛ αλ ж ⎞
⎟⎟ ⎜ −
⎟,
π ⎠
⎝ λж ⎠ ⎝
ε " = ⎜⎜
где θ — угол падения.
Рис. 32. Схема установки для измерения методом свободного пространства (нормальное
падение волны): 1 — блок питания, 2 — генератор, 3 — согласующий трансформатор, 4 —
гибридное соединение, 5 — рупорная антенна, 6 — диэлектрическая линза, 7 — измерительная
ячейка, 8 — индикатор перемещения, 9 — переменный аттенюатор, 10 — короткозамыкающий
поршень, 11 —детектор, 12 — индикатор.
92
Высокочастотная мощность поступает на вход гибридного соединения, где
разделяется на опорную и измерительную компоненты. С одного плеча гибридного
соединения с помощью рупорной антенны с диэлектрической линзой измерительный
сигнал падает на ячейку с жидкостью и после отражения попадает в гибридное соединение.
Опорный сигнал проходит через переменный аттенюатор и после отражения от
короткозамыкателя также поступает в гибридное соединение. Часть измерительного
сигнала, поступившего в гибридное соединение после отражения от жидкости, поступает
в выходное плечо и здесь интерферируется частью опорного сигнала, предварительно
прошедшего через аттенюатор и претерпевшего отражение от короткозамыкающего
поршня.
Перед началом измерений поршень измерительной ячейки опускается до плотного
соприкосновения с пластиной, герметизирующей ячейку. После этого с помощью
переменного аттенюатора и короткозамыкающего поршня добиваются, чтобы
выходной сигнал был равен нулю. Далее перемещают поршень и снимают зависимость
величины выходного сигнала от толщины столба жидкости. Эта зависимость
представляет собой осциллирующую кривую с минимумами и максимумами.
Расстояние между последующими минимумами и максимумами кривой равно
половине длины волны в жидкости.
По величине максимумов определяются ε' и ε". Погрешность измерения метода
порядка 1 - 2% для ε' и 2 - 4% для ε" в зависимости от величины потерь.
93
3.5. Временная спектроскопия жидкостей
Метод временной спектроскопии диэлектриков используется для измерения
диэлектрических свойств веществ в широком диапазоне частот от 10-4 до 1010 ГГц.
Основные соотношения, связывающие во временной спектроскопии сигнал отклика и
комплексные частотные зависимости свойств вещества, описаны в работах [285-290].
Развитие автоматизации измерений оказало значительное влияние на методику
временной спектроскопии, в особенности в случае применения современных
анализаторов параметров цепи [291, 292].
Рассмотрим связь между частотной и временной спектроскопией. При выключении
внешнего электрического поля функция спадания поляризации диэлектрической
релаксации имеет вид
α( t ) =
P( t )
,
P( 0 )
(3.72)
где P (t ) — вектор поляризации в момент времени t, P (0) — вектор поляризации в
момент времени t=0, т.е. перед выключением внешнего электрического поля.
Вектор смещения D (t ) и напряженность электрического поля E (t ) связаны
соотношением [9, 17, 22, 23, 290]
D( t ) = ε ∞ ( t ) +
t
∫ E ( t' )ϕ( t − t' )dt' ,
(3.73)
−∞
где производная φ обозначена точкой. Функция D ( t ) = ε 0 E ( t ) + P ( t ) диэлектрического
отклика φ(t) имеет вид
ϕ ( t ) = ε ∞ + Ф( t ) ,
(3.74)
Ф( t ) = ( ε − ε ∞ )[1 − α ( t )] ,
(3.75)
α(t) — функция релаксации или функция спадания диэлектрической поляризации.
Комплексная диэлектрическая проницаемость ε*(ω) является аналогом φ(t):
∞
ε * ( ω ) = iωL[ϕ ( t )] = iω ∫ ϕ ( t ) exp( −iωt )dt , (3.76)
0
где L — оператор преобразования Фурье-Лапласа.
Если
α(t)~exp(-t/τ) ,
(3.77)
где τ представляет время диэлектрической релаксации, то в этом случае удовлетворяется
соотношение Дебая для частотной области [9, 17, 22, 23, 290]:
ε * (ω ) − ε ∞
1
=
.
ε −ε∞
1 + iωτ
(3.78)
94
Методом получения информации о динамических молекулярных свойствах вещества
являются прямые измерения во временной области.
Соотношение (3.76) показывает, что эквивалентная информация о диэлектрических
свойствах вещества может быть получена по измерениям как в частотной, так и во
временной области.
Действительно, флуктуации поляризации, вызванные тепловым движением, в случае
линейного отклика имеют такой же характер, как и изменение поляризации под
действием макроскопического электрического поля [284, 293]. Это значит, что можно
приравнять функцию спадания диэлектрической поляризации α(t) к макроскопической
дипольной функции корреляции Г(t):
α( t ) = Г( t ) =
M(0)⋅ M(t )
M(0)⋅ M(0)
,
(3.79)
где М(t) - макроскопический флуктуационный дипольный момент единицы объема,
равный векторной сумме всех молекулярных диполей; символ < > означает усреднение по
ансамблю. Скорость и закон спадания функции Г(t) непосредственно связаны со
структурой и кинетическими свойствами изучаемого объекта и характеризуют
макроскопические системы.
Строго говоря, необходимо найти, как связаны между собой φ(t) и Г(t). Однако,
задача установления связи между макроскопической и молекулярной дипольными
корреляционными функциями до сих пор является одной из самых проблематичных в
теории жидких диэлектриков [23, 24, 284, 294].
Таким образом, экспериментальная функция φ(t) и, следовательно, α(t) и Г(t)
могут быть использованы для получения информации о динамических свойствах
диэлектриков в понятиях дипольной корреляционной функции [284, 294, 23, 24].
Метод временной спектроскопии базируется на рефлектометрии и в нем
неоднородность коаксиальной линии определяется по изменению формы
тестирующего импульса [285—289]. Пока линия однородна, импульс не
изменяется, но когда в линии введена неоднородность, например диэлектрик,
импульс частично отражается от поверхности воздух-диэлектрик, а часть сигнала
проходит через диэлектрик.
Качественно форма падающего и отраженного сигналов показаны на рис. 33.
Рис. 33. Характерная форма сигнала, наблюдаемого в эксперименте временной
диэлектрической спектроскопии; G — сигнал от образца с большой проводимостью.
Суммарный отраженный от образца сигнал V(t) складывается из двух
составляющих: отражения исходного быстро возрастающего скачка напряжения от
передней поверхности образца V0(t) и сигнала R(t), прошедшего в образец и
отраженного со сдвигом на интервал времени, соответствующий двойному
прохождению сигналом пути от входной поверхности образца до места отражения
сигнала.
Дальнейший анализ данных основан на рассмотрении связи между падающим
95
и отраженным сигналами [287, 290, 295, 296].
Напряжение, приложенное к образцу, можно представить в виде:
V ( t ) = V0 ( t ) + R( t ) ,
(3.80)
где V0(t) и R(t) — падающий и отраженный сигналы соответственно.
Ток, протекающий через диэлектрик [287, 290, 297, 298], определяется как:
I( t ) =
1
[V0 ( t ) − R( t )] ,
Z0
(3.81)
где Z0 — характеристический импеданс измерительной линии.
Общий ток, протекающий через диэлектрик, состоит из тока смещения IQ(t) и
низкочастотного тока проводимости IR(t).
Активное сопротивление измерительной ячейки, содержащей жидкость,
записывается в виде [287, 298]:
r=
V0 ( ∞ )
V ( ∞ ) + R( ∞ )
= Z0 0
.
I( ∞ )
V0 ( ∞ ) − R( ∞ )
(3.82)
Низкочастотный ток проводимости выражается как:
IR(t ) =
т.е.
IR =
V(t )
,
r
(3.83)
1 V0 ( ∞ ) − R( ∞ )
[V0 ( t ) + R( t )] .
Z 0 V0 ( ∞ ) + R( ∞ )
(3.84)
Таким образом, уравнение (3.81) может быть записано как
I( t ) = I Q ( t ) + I R ( t ) =
⎫
V0 ( ∞ ) − R( ∞ )
1 ⎧
[V0 ( t ) + R( t )]⎬ . (3.85)
⎨[V0 ( t ) − R( t )] −
Z0 ⎩
V0 ( ∞ ) + R( ∞ )
⎭
Соотношения (3.80) и (3.85) представляют собой основные уравнения,
связывающие ток I(t) и определяемый в процессе эксперимента сигнал V(t).
Выражение (3.85) показывает, что временная спектроскопия диэлектриков
позволяет определить низкочастотную проводимость образца:
σ=
ε 0 V0 ( ∞ ) − R( ∞ )
Z 0 C 0 V0 ( ∞ ) + R( ∞ )
,
(3.86)
где ε0 = 8,55·10-12 Ф/м, С0 — емкость частично заполненной образцом коаксиальной
линии.
Используя выражения для I(t), V(t) и их Фурье-образы, получаем
96
∞
V ( iω ) = ∫ V ( t ) exp( −iωt )dt ,
(3.87)
0
∞
I ( iω ) = ∫ I ( t ) exp( −iωt )dt .
(3.88)
0
Можно вывести соотношения, которые описывают диэлектрические
характеристики исследуемого образца как во временном, так и в частотном
представлении. Конкретные виды этих соотношений зависят от геометрической
конфигурации измерительной ячейки и ее эквивалентного представления [276, 287—
290,299].
Например, эквивалентная схема ячейки может быть представлена как
сосредоточенная емкость, присоединенная к линии (образец расположен на конце
линии). Электрический заряд Q(t) связан с диэлектрической функцией отклика Ф(t) и
приложенным напряжением V(t) следующим образом:
t
Q (t ) = C 0ε ∞V (t ) + ∫ Ф(t − t ' )V (t ' )dt ' ,
(3.89)
0
где С0 — емкость пустой измерительной ячейки.
t
Q( t ) = ∫ I ( t' )dt' ,
(3.90)
0
⎡ ε ( iω ) − ε ∞ ⎤
Ф( t ) = L−1 ⎢
⎥ , (3.91)
iω
⎣
⎦
здесь L — оператор обратного преобразования Фурье-Лапласа.
Соотношение (3.89) также справедливо, когда образец в линии рассматривается
как секция с распределенными параметрами. В этом случае С0 есть геометрическая
емкость единицы длины линии [256, 276, 288—290]. Если применять преобразования
Лапласа к обеим сторонам уравнения (3.89), получим спектр комплексной диэлектрической
проницаемости образца:
ε * (ω ) =
1 L[I ( t )]
,
iωZ L[V ( t )]
(3.92)
где I(t) и V(t) определяются соотношениями (3.80) и (3.85).
Если принять в расчет определение физической длины образца и многочисленные
отражения от поверхностей воздух-диэлектрик и диэлектрик-воздух, это соотношение
принимает вид:
ε * (ω ) =
L[I ( t )]
C
xctgx , (3.93)
iω( γd ) L[V ( t )]
где x = ωd ε * (ω ) / C , d— эффективная длина внутреннего проводника линии, γ отношение емкости на единицу длины ячейки, заполненной вакуумом, к погонной
97
емкости согласованного коаксиального кабеля [275, 287, 289, 290].
Соотношения (3.92) и (3.93) дают возможность определить диэлектрические
характеристики исследуемого образца в виде спектра комплексной диэлектрической
проницаемости ε*(ω).
Важной особенностью временной спектроскопии диэлектриков является
возможность определить релаксационные характеристики образца прямо во временной
области.
Таким образом, решая интегральное уравнение, находим величину функции
диэлектрического отклика Ф(t) для метода сосредоточенной емкости [23, 24, 289, 290, 299].
Функцию φ(t) = Ф(t) + ε∞ можно связать с функцией макроскопической дипольной
релаксации Г(t) [284, 290, 293], которая является основополагающей в теории линейного
отклика.
Экспериментальная реализация метода временной спектроскопии диэлектриков
состоит в применении рефлектометра, используемого для измерения неоднородности
коаксиальной линии. Рефлектометр содержит высокоскоростной генератор ступенчатого
напряжения и широкополосную регистрирующую систему с одно- или двухканальной
измерительной ячейкой. Для улучшения отношения сигнал/шум применяется метод
накопления. Кроме того, необходимо принять меры к стабилизации параметров системы в
процессе измерения [308].
На рис. 34 приведена принципиальная схема двухканальной системы для временной
спектроскопии диэлектриков, которая позволяет записывать сигналы от эталонного и
измеряемого образцов. Диапазон частот системы составляет 100 кГц—10 ГГц.
В работе [300] представлены методы увеличения полосы временной
диэлектрической
спектроскопии
с
использованием
метода
отбора
последовательных
значений
сигнала
переменной
временной
шкалой.
Последовательные сегменты переходных во времени процессов временной спектроскопии
отбираются с возрастающими временными интервалами, и весь переходный во времени
процесс преобразуется в частотном диапазоне с использованием преобразования Лапласа.
Приборные ошибки идентифицируются и контролируются исследованием паразитных
отраженных сигналов. Представлены результаты исследования водного раствора солей с
большой проводимостью. Получен непрерывный частотный спектр в диапазоне частот от
100 кГц до 5 ГГц и чтобы захватить весь отклик, шесть разрядов времени во
временной диэлектрической спектроскопии.
Рис. 34. Принципиальная схема двухканальной системы для временной спектроскопии:
1 — образец, 2 — нагрузка 50 Ом, А, В — узлы отбора канала испытания, 3 — делитель
мощности, 4 — тунельный диод, 5 — осциллограф, 6 — интерфейс, 7 — компьютер, 8 —
принтер, 9 — графопостроитель [303].
Точность измерения с помощью этого устройства составляет ±2% для ε' и ±5%
для ε" в диапазоне до 10 ГГц.
98
Ступенчатое напряжение распространяется вдоль линии с полной
проводимостью G и переходит в напряжение V(t) в конце образца.
В сочетании с отраженным r(t) это приводит к объединенному напряжению
V(t)+r(t) в образце. Трансформированное ступенчатое напряжение обусловливает
ступенчатый ток GV(t), который сочетается с отраженным током [-Gr(t)]. Знак (-)
показывает направление комбинированного ступенчатого тока в образце G[V(t)-r(t)].
Частотные компоненты напряжения и тока в образце представляются выражениями
V(ω)+r(ω) и G[V(ω)-r(ω)], соответственно, где V(ω) и r(ω) являются комплексными
преобразованиями Лапласа от V(t) и r(t). Зная частотные компоненты напряжения и
тока в образце, окончательную общую проводимость образца можно представить как:
Y(ω ) = G
V ( ω ) − r( ω )
.
V ( ω ) + r( ω )
(3.94)
Комплексная диэлектрическая проницаемость образца записывается в виде:
ε * (ω ) =
Y(ω )
,
iωC 0
(3.95)
где С0 — геометрическая емкость измерительной ячейки без образца.
Отраженный от образца сигнал rх(t) сравнивается с отраженным от
калиброванного эталонного образца rr(t) сигналом взамен на V(t). Основное
соотношение можно получить, написав уравнение (3.94) для стандартного и
неизвестного сигнала, в результате которого можно исключить V(ω). В результате
получим:
{
}
Yx ( ω ) Yr ( ω )
1 − [Y r ( ω ) / G ] ρ ( ω )
−
=
.
G
G
1 + [Y r ( ω ) / G ]ρ ( ω )
2
(3.96)
Коэффициент отражения можно получить из равенства:
ρ( ω ) =
rr ( ω ) − rx ( ω )
rr ( ω ) + rx ( ω )
(3.97)
где ε(ω) =Y(ω)/iωС0 заменяется на стандартную и неизвестную полные проводимости.
Рассмотренный подход позволяет решать поставленную задачу.
В заключение рассмотрим методику измерения параметров диэлектриков во
временной области на примере экспериментальной установки «Диполь», созданной под
руководством проф. Андрианова А.В.
Радиоволновые методы исследования веществ основаны на анализе процессов
взаимодействия исследуемого объекта и электромагнитного поля. Данное
взаимодействие формализуется материальными уравнениями электродинамики.
Мнимая
часть
диэлектрической
проницаемости
однозначно
определена
действительной частью, а действительная часть определяется по мнимой с точностью
до постоянного слагаемого, равного диэлектрической проницаемости постоянного
тока.
Главным достоинством методов измерения диэлектрической проницаемости во
временной области на основе использования сверхширокополосных импульсных
измерительных сигналов является высокая скорость измерений спектра
99
диэлектрической проницаемости, возможность работы в сверхшироком диапазоне
частот (от 100 кГц до 18 ГГц и более). Благодаря этому, импульсный метод является
незаменимым в задачах динамического анализа изменений диэлектрических свойств в
реальном времени. Он также находит широкое применение при измерении
диэлектрических свойств эмульсий, жидких кристаллов [308], органических и
биологических материалов. Зависимость диэлектрической проницаемости от частоты
получают путем цифровой обработки сигнала, рассматриваемого как зависимость
напряжения или напряженности поля от времени.
Комплексный коэффициент отражения от измерительной ячейки с веществом
определяется по временным сигналам, отраженным от измерительной ячейки и от
опорных объектов. При использовании в качестве опорных объектов
короткозамыкателя и согласованной нагрузки:
Г =−
F [и0 (t ) − иСН (t )]
F [и КЗ (t ) + иСН (t )]
где F[.] - преобразование Фурье; ио, икз и исн - сигналы, отраженные от измерительной
ячейки с диэлектриком, короткозамыкателем и согласованной нагрузки соответственно.
Согласованная нагрузка применяется для определения нулевой линии прибора, а
короткозамыкатель для нахождения падающей волны.
Связь диэлектрической проницаемости с коэффициентом отражения определяется
используемой измерительной ячейкой. В зависимости от используемой измерительной
ячейки различают следующие методы измерения диэлектрической проницаемости во
временной области: одного отражения, тонкого образца, концевой емкости. Приближенное
описание метода, вид ячейки и расчетные соотношения для случая, когда волновое
сопротивление измерительной ячейки W = Wo, где Wo - волновое сопротивление
подводящего тракта, приведены в таблице 11.
Таблица 11.
Ниже рассмотрены методы измерения диэлектрической проницаемости непроводящих
диэлектриков.
100
3.5.1. Метод одного отражения (толстого образца)
Метод справедлив при условии fmin>c/2Lε½ , L-длина измерительной ячейки.
Диэлектрическая
выражением:
проницаемость
непроводящих
диэлектриков
определяется
2
⎛ W (1 − Г ) ⎞
⎟⎟ ,
ε = ⎜⎜
⎝ W0 (1 + Г ) ⎠
ε = ε ′ − jε ′′ .
Здесь W и Wo - волновые сопротивления измерительной ячейки и подводящего тракта
(для коаксиальных линий W=601n(D/d), D и d - диаметры внешнего и внутреннего
проводников соответственно). Недостатком метода является необходимость использования
больших объемных образцов и длинных измерительных ячеек. Форма нормированного к
амплитуде сигнала, отраженного от измерительной ячейки, показана на рис. 35.
Рис. 35. Форма сигнала, отраженного от измерительной ячейки с толстым образцом, при
зондировании перепадом напряжения. Коэффициенты отражения ρо и ρ1 определяют
значения диэлектрической проницаемости εo и ε1 на высоких и низких частотах.
3.5.2. Метод тонкого образца
Метод справедлив при условии fmax<< c/Lε½ , где fmax - верхняя граничная частота
диапазона измерений, с — скорость света в вакууме, L — длина измерительной ячейки.
Коэффициент отражения ячейки при W = WO и малой толщине образца L, так, что
выполняется условие јωL/c « 1 может быть записан в виде:
Г = S11 =
jωL ⎛
4πσ
⎜⎜1 − ε −
2c ⎝
jω
⎞
⎟⎟ .
⎠
В случае если W ≠ Wo, диэлектрическая проницаемость непроводящих
диэлектриков (σ = 0) определяется выражением:
ε =
W
W
2
2
0
+ i
2 cW Г
.
ω LW 0
Метод удобен для реализации, однако имеет систематическую погрешность,
101
связанную с тем, что формула для расчета получена из точного выражения, путем
разложения в ряд Тейлора и учета только первых двух членов. Относительная
погрешность определения диэлектрической проницаемости приблизительно равна
значению модуля коэффициента отражения. Для получения погрешности порядка 10%
при больших значениях диэлектрической проницаемости, необходимо использование
тонких образцов - толщиной в доли миллиметра. Форма отраженного сигнала для
метода тонкого образца показана на рис. 36.
Использование тонкого образца в сочетании с однородной воздушной линией
позволяет реализовать измерительную ячейку с минимальными неоднородностями и
применять зондирующий сигнал, как в форме перепада напряжения, так и короткого
импульса.
Рис. 36. Форма сигнала, отраженного от измерительной ячейки с тонким образцом.
Точное решение по методу образца конечной длины L.
Диэлектрическая проницаемость ячейки с образцом длиной L связано на каждой
частоте с параметром S11 = Г измерительной ячейки трансцендентным уравнением:
S11 =
[1 − exp(−2γL)]ρ
1 − ρ 2 exp(1 − 2γL)
ρ=
1− ε
,
1+ ε
γ =
jω
ε.
c
,
Это уравнение может быть решено относительно ε с использованием итерационного
алгоритма. Для обеспечения правильного решения необходимо, чтобы переходный
процесс, возникающий вследствие переотражений волн от границ образца в отраженном
сигнале, установился в пределах регистрируемого временного окна. Другим условием,
ограничивающим применение метода тонкого образца, является возникновение волн
высшего типа в измерительной ячейке.
3.5.3. Метод сосредоточенной емкости
½
Метод справедлив при условии fmax<< c/lε’ , где l - наибольший из линейных
размеров неоднородности. Диэлектрическая проницаемость непроводящих диэлектриков
при использовании метода концевой емкости определяется формулой:
102
ε=
1
1− Г
,
iωC0W0 1 + Г
где Со - емкость измерительной ячейки, W0 - волновое сопротивление подводящей линии
передачи.
Использование перепада напряжения в качестве зондирующего сигнала при использовании
метода концевой емкости позволяет проводить измерения в широком частотном диапазоне,
начиная от нескольких мегагерц до десятков гигагерц.
Рис. 37. Форма сигнала, отраженного от измерительной ячейки с концевой емкостью.
При определении комплексного коэффициента отражения необходимо для
измерения падающей волны в точке подключения емкости организовать режим
короткого замыкания и измерить сигнал uкз(t). Однако, на результаты измерения влияют
неоднородности конструкции ячейки, поскольку в точке падения на диэлектрик падающая
волна может отличаться от точки подключения короткозамыкателя. Во избежание этого в
качестве опорного объекта используют измерительную ячейку, заполненную диэлектриком
с известной диэлектрической проницаемостью. В этом случае для определения
коэффициента отражения используется формула:
Г=
F [и0 (t ) − иСН (t )] (1 − iωC0W0ε оп )
,
F [и КЗ (t ) + иСН (t )] (1 + iωС0W0ε оп )
где Uоп(t) - сигнал, отраженный от емкости с опорным диэлектриком, εоп диэлектрической проницаемости опорного диэлектрика.
На практике измерительная ячейка не заполняется полностью диэлектриком и ее
эквивалентная схема представима в виде, показанном на рис.38. Здесь заполняемая
диэлектриком часть общей емкости представлена емкостью С1.
Рис. 38. Эквивалентная схема ячейки концевой емкости. C1, Се — емкости, заполняемая
и не заполняемая диэлектриком.
В данном случае общая емкость Со = C1 + Ce и расчетные соотношения для
определения диэлектрической проницаемости будут следующими:
103
1 1− Г
− Cе
jωW0 1 + Г
.
ε=
С0 − С е
В разностном методе концевой емкости коэффициент отражения определяется
соотношением
Г=
F [и0 (t ) − иСН (t )] (1 − jωW0 (Cе + (С0 − Се )ε оп ))
.
F [и КЗ (t ) + иСН (t )] (1 + jωW0 (Cе + (С0 − Се )ε оп ))
Емкости Со и Се определяются по результатам калибровки по образцам диэлектрика с
известной диэлектрической проницаемостью. В качестве таких опорных веществ могут
служить вещества с малым изменением диэлектрической проницаемости от температуры или
частоты, например, воздух, фторопласт или жидкие диэлектрики с известным значением
диэлектрической проницаемости. При этом, если в измерительной ячейке находится воздух,
то наибольшее влияние на результат измерений оказывает емкость Со, а если в ячейке находится
диэлектрик с большим значением диэлектрической проницаемости, то результат измерений
в большей степени зависит от емкости С1. Параметры ячейки С0, С1, Се определяются
экспериментальным путем при калибровке и для ячейки с заданной геометрией в
дальнейшем не изменяются.
Значение емкости измерительной ячейки в первом приближении можно рассчитать по
формуле емкости плоского конденсатора:
С=
А
[см] ≈ А [пФ] ,
4πd
14d
где A и d - площадь пластин и расстояние между ними в сантиметрах.
104
ЗАКЛЮЧЕНИЕ
В данном учебном пособии представлены сведения о теории диэлектрической
поляризации, методах ее наблюдения и возможных применениях для исследования
межмолекулярных взаимодействий в простых и сложных системах.
Представленные выше материалы будут полезны и востребованы студентами,
аспирантами и научными сотрудниками, работающими в области исследовании
структуры и свойств вещества.
105
Литература
1. Marriott A. A., Smith E.R., Table of Dielectric Constants of Pure Liquids, NBS, circular 514,
Washington (1951).
2. Timmermans I., The Physico-chemical Constants of Pure Liquids, New-York - London (1965).
3. Burckley F., Marriott A.A., Tables of Dielectric Dispersions Data for Pure Liquids and Dilute
Solutions, Nat Bur. Standarts circular 589 (1958).
4. Landolt-Bornstein, Static Dielectric Constants of Pure Liquids and Binary Liquids Mixtures, vol b,
Springen-Verlag, Berlin (1991).
5. Ахадов Я.Ю., Диэлектрические свойства чистых жидкостей, Москва, изд. Стандартов
(1972).
6. Ахадов Я.Ю., Диэлектрические свойства бинарных растворов, изд. Наука (1981).
7. Дебай П., Полярные молекулы, ГТТИ (1931).
8. Сканави Г.И., Физика диэлектриков, Гостехиздат (1949).
9. Фрелих X., Теория диэлектриков, изд. ИЛ (1960).
10.Хиппел А.Р., Диэлектрики и волны, изд. ИЛ (1962).
11.Броун В.Б., Диэлектрики, изд. ИЛ (1961).
12.Шахпаронов М.И., Методы исследования теплового движения молекул и строение
жидкостей, изд. МГУ (1963).
13.Богородицкий Н.П., Волакобинский Ю.М., Воробьев А.А., Тареев В.М., Теория
диэлектриков, Энергия (1965).
14.Smith C.P., Dielectric Behaviour and Structure, New-York (1955).
15.Hill N.E., Dielectric Properties and Molecular Behaviour, London (1969).
16.Böttcher C.J.F., Theory of Electric Polarization, 1, 2 nd. edn. Elsevier, Amsterdam (1973).
17.Böttcher C.J.F. and Bordewijk P., Theory of Electric Polarization, 2, 2 nd. edn. Elsevier,
Amsterdam (1978).
18.Гайдук В.И., Теория диэлектрической дисперсии полярных сред, изд. МИФИ (1980).
19.Grant E.H., Sheppard R.J. and South G.P., Dielectric Behaviour of Bialogical Molecules in
Solution, Oxford University Press, Oxford (1978).
20.Scaife B.K.P., Principles of Dielectrics, Oxford University Press, Oxford (1989).
21.Потапов А.А., Мецик M.C., Диэлектрическая поляризация, Иркутск (1986).
22.Kaatze U., Radiat. Phys. Chem., 45, 539 (1995).
23.Kaatze U., Radiat. Phys. Chem., 45, 549 (1995).
24.Onsager L., J. Amer. Chem. Soc, 58, 1486 (1936).
25.Kirkwood J.G., J.Chem. Phys., 7, 911 (1939).
26.Fröhlich H., Trans.Faraday Soc, 44, 238, (1948).
27.Chen Т., Dannhauser W., Jonori G.P., J.Chem. Phys., 50, 2046 (1969).
28.Loon R.V., Fuks S., Bellemans A., Bull. Soc. Chem. Belg., 76, 202 (1967).
29.Fouss R.M., Kirkwood 1С, J. Amer. Chem. Soc, 63, 385 (1941).
30.Cole R.H., J.Chem. Phys., 27, 33 (1957).
31.Сыркин В.К., ДАН, 35, 43 (1942).
32.Smith J.W., Walshaw S., J. Chem. Soc, 3217 (1957).
33.Smith J.W., Walshaw S., J. Chem. Soc, 3784 (1959).
34.Dashpaude D.K., Suryanaruyana Rao, Indian J. Pure Appl. Phys., 7, 439 (1969).
35.Ротайчак Г., Собчик Л., ЖСХ, 6, 262 (1965).
36.Scholte Th.G., Physica, 13, 437 (1949).
37.Poley J.P., J.Chem. Phys., 22, 1466 (1954).
38.Buckingham A.D., Trans. Faraday Soc. 49, 881 (1953).
39.Buckingham A.D., Auslr. J. Chem., 6, 93 (1953).
40.Wilson J.N., Chem. Rev., 25, 377 (1939).
41.Scholte Th.G., De Vos F.C., Rec. Trav. Chem.. 72, 625 (1953).
42.Klug D.D.,Waugham W.E., J.Chem. Phys., 56, 5005 (1972).
43.KharaB.N., J.Cfem. Phys., 47, 5173 (1967).
44.Loon R.V., Dauchot J.P., Bellemans A., Bull. Soc. Chem. Belg., 77, 397 (1968).
45.Weisbecker A., J.Chem. Phys., Biol., 66, 226 (1969).
46.Милаев С.М., ЖФХ, 56, 1816 (1982).
47.Nath J. and Dixit A.P., J. Chem. Soc. Faraday Trans. 2, 81, 111(1985).
106
48.Вилков Л.В., Пешин Ю.А., Физические методы исследования в химии, М, Высшая
школа(1987).
49.Вукс М.Ф., Электрические и оптические свойства молекул конденсированных сред, Л., изд.
ЛГУ (1984).
50.Cole R.H., J.Chem. Phys., 23,493 (1955).
51.Solderwood J.H., Smith С.Р., J.Amer. Chem. Soc., 78, 1295(1956).
52.Scaife B.K.P., Proc.Phys. Soc., 81,194 (1963).
53.Chandra S., Prakash J., Indian J. Pure Appl. Phys., 6, 116 (1968).
54.Brot C., Compt. Rend. Acad. Sci. Paris, 19, 397 (1959).
55.Petro A.J., Smith С.Р., J.Amer. Chem. Soc., 79, 6142 (1957).
56.Hill N.E., Proc. Phys. Soc., London, 67 B, 149 (1954).
57.Holland R.S, Robert G.N., J. Amer. Chem. Soc, 78, 20 (1956).
58.Henelly E.J., Heston W.M., Smith С.Р., J.Amer. Chem. Soc, 70, 4102 (1948).
59.Put D.A., Smith С.Р., J.Amer. Chem. Soc., 80, 1061 (1958).
60.Buchanan T.J. and Grant E.H., Brit. J. Appl. Phys., 6, 64 (1955).
61.Girard P. and Abadie P., Trans. Faraday Soc., 42A, 40 (1946).
62.Cole R.H., J.Cftem. Phys., 6, 385 (1938).
63.Powles J.G., J.Chem. Phys., 21, 633 (1953).
64.O'Dwyer J.J., Sack R.A., Austral. J. Sci. Res., A5, 647 (1952).
65.Miller R.C, Smyth С.Р., J. Amer. Chem. Soc., 79, 3310 (1957).
66.Yager W.A., Physica, 7, 434 (1936).
67.Jacson W., Powles J., Trans. Faraday Soc, 42A, 101 (1946).
68.Higasi K., Bergman K., Smyth С.Р., J. Chem. Phys., 64, 880 (1960).
69.Cole R.H. and Cole K.S., J. Chem. Phys., 9, 341 (1941).
70.Cole K.S. and Cole R.H., J. Chem. Phys., 10, 98 (1942).
71.Kastha G.S., J Phys. Soc. Jpn., 34, 699 (1973).
72.Dawidson D.W., Cole R.H., J. Chem. Phys., 19, 1484(1951).
73.Dawidsoji D.W., Canad. J. Chem., 39, 571 (1961).
74.Fuoss R.M., Kirkwood J.G., J. Amer. Chem. Soc., 63, 385 (1941).
75.Muralidhara Rao V., Indian J. Pure Appl. Phys., 1, 33 (1963).
76.Гаврильяк С., Негами С., в книге "Переходы и релаксационные явления в полимерах, М.,
"Мир", 99(1966).
77.Govande S.G., Garg S.K., Kadaba P.K., Mater. Sci. Engrs., 4, 206 (1969).
78.Gopala Krishna K.V., Trans. Faraday Soc, 53, 769 (1957).
79.Higasi K., Uchiyana K., Res. Inst. Appl. Elect., 17, 164(1965).
80.Prakashi J., J. Phys, Soc Jpn., 34,134 (1973).
81.Fmhlich H., Magan M.P., Canad. J, Phys., 58, 30 (1980).
82.Prakash Jai, Indian J. Pure Appl. Phys., 8,106(1970).
83.Gupta M., Chauhan M., Shukla J.P., Chem. Phys. Liq., 11, 241(1981).
84.Clark G.L., J. Chem. Phys., 25, 125(1956).
85.Higasi K., Bull. Chem. Soc. Jpn., 39, 2157 (1966).
86.Srivastava K.S.C., Nath D., Bull.Chem.Soc.Jpn., 43, 2809 (1970).
87.Budo H., Physik Z., 39, 706 (1938).
88.Beryman K., Roberty D. M, Smyth С.Р., J. Phys.Chem., 5, 665 (1960).
89.Mayee M.D., Walker S., Trans. Faraday Soc., 62, 3093 (1966).
90.Bhattachazyya J., Hasan R., Roy S.B., Kastha G.S., J. Phys. Soc. Jpn., 28, 204 (1970).
91.Barriol J., Boule. P., Diguet R, Соmр. Rend., Acad. Sci. Paris., 268C, 1977 (1969).
92.Boule P., J. Chim. Phys. France, 66, 1987 (1969).
93.Sabench J., Layer G., Reynier J., Champ. Ren. Acad. Sci. Paris, 274C, 973 (1972).
94.Higasi K., Koga Y., Makamura M., Bull. Chem. Soc. Jpn., 44, 988 (1971).
95.Turner E. M, Ehrhardt W. W., Leone G., Vaughan W. E., J. Phys Chem., 74, 3543 (1970).
96.Klanbuchl D. E., Kluy D.D., Vaughan W.E., J. Chem. Phys., 50, 5266 (1969).
97.Birnbaym G., Cohen E. R., J. Chem. Phys., 53, 2885 (1970).
98.Williams G., Chem. Rev., 72, 55 (1972).
99.Poley J.P., Appl. Sci. Res., B4, 337 (1965).
100.Magat M, Izv. A.N. SSSR, 24, 10 (1960).
101.Cole R.H., Davidson D. W., J. Chem. Phys., 20, 1388 (1952).
107
102.Dannhauser W.J., J. Chem. Phys., 55,629 (1971).
103.Danney D.Y., J. Chem. Phys., 30, 1019 (1959).
104.Dollert M., Mayat M, Surdut A., Bull. Soc. Chim. France, 7-8, D345 (1949).
105.Hassion F.X., Cole R.H., J. Chem. Phys., 13, 1766 (1955).
106.Dannhauser W.J., Cole R.H., J. Chem. Phys., 23, 1762 (1955).
107.Davidson D.W., Wheller J., J. Chem. Phys., 30, 1357 (1959).
108.Reinisch L., J. Chem. Phys., 51, 113 (1954).
109.Rompolla R.W., Miller R.S., Smyth СР., J. Chem. Phys., 30, 566 (1959).
110.Danney D.J., J. Chem. Phys., 30, 159 (1959).
111.Danney D.J., J. Chem. Phys., 30, 1010 (1959).
112.Danney D.J., Cole R.H., J. Chem. Phys.,23, 1767 (1955).
113.Sarojini V., Trans. Faraday Soc, 57, 425 (1961).
114.Meakins R.J., Trans. Faraday Soc, 54, 1160 (1958).
115.Pitt A.J., Smyth СР., J. Chem. Phys., 63, 582 (1959).
116.Нill N.E., Proc. Phys. Soc, 67B, 149 (1954).
117.Fairweather A., Frast E.J., Molecular Relaxation Processes, Chem. Society spec. pub. №20, 45
(1966).
118.Perrin F.J., Phys. Radium (Paris), 5, 497 (1934).
119.Fischer E., Phus. Z., 40, 645 (1939).
120.Gierer A., Wirtz K., Z. Naturf, 8a, 532 (1953).
121.Andrade E.N., Phil. Mag., 17, 497 (1934).
122.Hayghan W.E., Russell W.P., Smyth СР., J. Amer. Chem. Soc, 83, 571 (1961).
123.Perrin L., J. Amer. Chem. Soc, 58, 1486 (1936).
124.Budo A., Fisher E., Miyamoto S., Physik Z, 40, 337 (1939).
125.0ncley J.L., In: Proteins Amino Acids and Reptides (Ed. Cohn E.J.,) Reinhold, New York, (1943).
126.Magee M.D., Walker S., J. Chem. Phys., 50, 2580 (1969).
127.Magee M.D., Canad. J. Chem., 49, 1106 (1961).
128.Haigasi K., Dielectric relaxation and molecular structure, Spporo, Japan (1961).
129.Chitoku K., Higasi K., Bull. Chem. Soc. Jpn., 36, 1064 (1963).
130.Kalman O.F., Smyth СР., J. Amer. Chem. Soc, 82, 783 (1960).
131.Krishnaji, Srivastava S.L., Nath D., J. Chem. Phys., 52, 940 (1970).
132.Suamalamba K., Premaswarup D., Indian J. Pure Appl. Phys., 4, 84 (1966).
133.Despandl D.K., Suryanarayana Rao, Indian J. Pure Appl. Phys., 7, 71 (1969).
134.Glasstone S., Laider K.J., Eyring H., The Theory of Rate Processes, McGraw-Hill, N.Y.(1941).
135.Панченков Г.М., Лебедев В.П., Химическая кинетика и катализ, изд. МГУ (1964).
136.Bhandari R.C., SisodiaM.L., Indian J. Pure Appl. Phys., 2, 132 (1964).
137.Crossley J.,Smyth С.Р., J. Amer. Chem. Soc, 91, 2482 (1969).
138.Nelson R.D. and Smyth С.Р., J. Phys. Chem., 68, 2704 (1964).
139.Krishnaji, Mansingh A., Indian! Pure Appl. Phys., 2, 176 (1964).
140.Shukla J.P., Shukla D.D., Saxena M.C., J. Phys. Chem., 73, 2187 (1969).
141.SrivastsvaG.P., MatturP.C, Tripathui K.N., Indian J. Pure Appl. Phys., 6, 561(1968).
142.Williams G., J. Phys. Chem., 63, 537 (1959).
143.Davies M., J. Chem. Educ, 46, 17 (1969).
144.Roy S.K., SenguptaK. and Roy S.B., Bull. Chem. Soc. Jap., 49, 669 (1976).
145.Srivastava S.K. and Srivastava S.L., J. Phys. Soc. Jap., 41, 1310 (1976).
146.Roy S.K. and Roy S.B., Adv. Mol.Relax.Inter.Proc, W, 273 (1977).
147.Cole R.H., Molecular Liquids., ed. Barnes A.J., Dielectric Polarization and Relaxation , NATO
ASI, ser, ser C, 135, 59 (1984).
148.Williams G., Molecular Liquids , ed. Barnes A.J., Low frequency dielectric Spectroscopy , NATO
ASI, ser, ser C, 135, 239 (1984).
149.Smyth P.C., Dipole moment dielectric loss and intermolecular motion , in Internal Rotation in
Molecules , ed. Orwille-Thomas, N.Y. (1974).
150.Гайдук В.И., Калмыков Ю.П., Диэлектрическая релаксация и молекулярное движение в
полярных жидкостях, М., изд. МИФИ, 34, 1-222 (1987).
151.Cauhan M., Gupta M., Saxena S.K., Shukla J.P., Phys. Chem. Liq., 14, 335 (1981).
152.Evans M., Evans C.J., Coffey W.T., Crigolini P., Molecular Dinamic and Theory of Broad
Spectroscopy, N.Y., (1986).
108
153.Johry G.K., A new method of calculation of dipole moment and relaxation time for dilute solution
at microwave frequency , J. Phys. Soc. Jap., vol 2, 802 (1984).
154.McClellan A.L., Tables of Experimental Dipole Moments, Rahara Enterprises, vol 2 (1974).
155.Daubert Т.Е. and Danner R.P., Data Compilations, Tables of Properties of Pure Compounds ,
AICHE , N.Y., X (1985).
156.Hasted J.В.,Aqueous Dielectrics, (London) (1973).
157.Brot C, Dielectric and related molecular processes, Burlington House, London, 2,1 (1975).
158.Усманов СМ., Релаксационная поляризация диэлектриков, "Наука" (1996).
159.Минкин Ю.А., Осипов О.А., Дипольные моменты в органической химии, Л., изд.’'Химия"
(1968).
160.Hippel A.R., Dielectric Materials and Application, MIT Technology Press and Wiley , New York
(1954).
161.Брант А.А., Исследование диэлектриков на сверхвысоких частотах, Госиздат, М.(1963).
162.Chemberlain J. and Chantry G.W., High Frequency Dielectric Measurements, IPC Science and
Technology (1972).
163.Collier R.P., Transmission lines In Microwave Measurements (Editor by Baikay A.E.,London)
(1985a).
164,Gardolf F.E., Introduction to Microwaves (1984).
165.Smith B.L. and Carpenter M.H., The Microwave Engineering Handbook, Microwave Technology
Series vol. 1-3 (Chapman and Hall), London (1983).
166.Wayher F.L., Microwave network analyzers. In Microwave Mesurements, (Edited by Bailey
A.E.), London (1985).
167.Потапов А.А., Молекулярная диэлектрометрия, Наука, Новосибирск (1994).
168.Завьялов А.С., Дунаевский Г.Е., Измерение параметров материалов на сверхвысоких
частотах, Наука, Томск (1981).
169.Брагинский В.Б., Митрофанов В.П., Панков В.И., Системы с малой дисперсией, Наука
(1994).
170.Feldman Yu., Andrianov A., Polygalov E., Rev. Sci. Instrum (1996).
171.Chelkowski A., Dielectric Physics, Elsevier (1980).
172.Боровиков Ю.Я., Диэлектрометрия в органической химии, Химия (1987).
173.Усиков С.В., Электрометрия жидкостей, Химия (1974).
174.0chme W.F., Dielektrische Messmethoden (1965).
175.Якшин М.М., Езучевская К.М., ЖНХ, 6, 2425 (1961).
176.Birch J.R.,Clarke R.N., The radio and electronic Engineer, 52, 565 (1982).
177.Alder J.F. at. Al., The Application of microwave frequency spectrometry, permittivity and loss
measurements to chemical analysis , Trans. Inst. Meas. Contr., 5,99 (1983).
178.Sturdily M.A. and Sturchly S.S., Coaxial line reflection method for measuring dielectric
properties of biological substances at radio and microwave frequencies, IEEE Trans. Instr. Meas. IM29,176(1980).
179.Cook R.J. and Jones R.G., Measurement of electrical properties :Relative permittivity and loss
angle, Plastic and rubber materials and Appl.,1,216 (1976).
180.Pething R., Dielectric and Electronic Properties of Biological Materials,Willy, NewYork(1979).
181.Aminalbavi T.M.,Raikar S.K.,Aralaguppi M.l.Jndian J. Technol., 31, 739 (1993).
182.Steward J.W., J. Chem. Phys.,40, 3297 (1964).
183.Jezewski M., Ada. Phys. Polonica, 14, 483 (1955).
184.Hartshon L.,Parry J.V., Essen L., Proc. Phys. Soc, 68B, 422 (1955).
185.Mopsik F.I., J. Res. NBA, 71A, 287 (1967).
186.Hersch H.G.,Gen. Radio Exp., 36, 314 (1962).
187.Maczynskis., Hurwic J., Ada Phys. Polonica, 30, 519 (1966).
188.Nanassy A.J., Rev. Sci.Instr., 36, 766 (1065).
189.Vidulich G.A., Evans D.F., Kay R.L., J. Phys. Chem., 71, 636 (1967).
190.Hartshorn , Radio frequency measurements by bride and resonance Methods,London (1947).
191.Болулях К.С, Резонансные методы измерения, Энергия, 120 (1965).
192.Cachet H., Lestrade J.C. and Epelboin J., J. Chem. Phys., 62, 902 (1965).
193.Hill G.J., Proc. IEE, 140, 382 (1993).
194.Нill G.J. and Francis P.C., IEE Cont. Publ. (London IEE), 129, 135 (1975).
195.Kakimoto A., Ogova I. and Matsushita, Improvements on the measurements of dielectric constant
109
and dispersion factor in a wide frequency range, Rev. Sci.-Instr., 48, 1570(1977).
196.Иманов Л.М., Известия АН Азербайджанской ССР, 2, 29 (1959).
197.Subramanian V., Bellubi B.C., Sobnandry J-, Rev. Sci. Instrum., 64, 231 (1991).
198.Грохольский А.Л., Измерение добротности, кумметры, "Наука" (1966).
199.Укстин Е.Ф., Худяков Б. А.. Труды НИИ кабельной промышленности, 5, 72 (1960).
200.Horner F.J., Proc. IEE, 93, 21 (1954).
20l. Kraft W.D. and Gerdens E., Z Phys. Chem., 228, 5 (1965).
202.Ткач В.К., Степин А.Д., Казанский В.Б., Радиотехника и электроника, 12, 2009 (1960).
203.Roussi G., Boule P., J. Chim. Phys., 64, 529 (1965).
204.Бондаренко Р.Н., Гладышев Г.И., Радиотехника и электроника, 11, 149 (1966).
205.Cook R.J., Microwave cavity methods, (in [162]) (1973).
206.Sen A., Soha P.K. and Nag B.R., Rev. Sci. Instrum., 50,1594 (1979).
207.Ni E. And Stumper U., Proc. ЕЕ, pt H, 132, 27 (1985).
208.Margineda J., J. Phys. K; Sci. Instrum., 14, 961 (1981).
209.Ronssy G. and Falden M, IEEE Trans. Microwave and Technique, MTT-14, 196 (1966).
210.Stumper U.,Rev. Sci. Instrum., 44, 165 (1973).
21l.Braunt G.R.,Principles of Microwave Measuments, Peregrinus, London (1988).
212.Asfar M.N., Birch J.R., Clarke R.N., Proc. IEEE ,74, 187 (1986).
213.Ligthart L.P., IEEE Trans. Microw. Theory Tech., MMT-31, 249 (1983).
214.Sheppard P.J., J.Phys. D., Apple. Phys., 5, 1576 (1972).
215.Narayana Rao Daas , Prit. J. Appl. Phys., 17, 109 (1966).
216.Szwarnowski S. And Sheppard P.J., J. Phys. E.; Sci. Instrum., 12, 939 (1979).
217.Marcuvits N., Waveguide Handbook. MIT. Lab. Ser., vol 10, NewYork(1951).
218.Mathaci G.L., Young I. and Jones E.M.T., Microwave Filters, Impedance-Matching Networks
and Compling Structures, NewYork (1964).
219.Jones D.S.,Methods in Electromagnetic Wave Propagation , Oxford (1979).
220.Mayer E. And Pottel R., Physikalische Grundlayen der Hochfaequenz Tecknic (1969).
221.Warner F.L., Microwave network analysers. In Microwave Measuments (edited by Bailey A.E.),
London (1985).
222.Gardiol F.E., Introduction to Microwaves, Dedham (1984).
223.Collier R.J., Modern Lines. In Microwave Measuments (Edited by Bailley A.E.), London (1985b).
224.Helszajn J., Microwave Engineering : Passive, Active and Non-Receprocal Circuits, London
(1992).
225.Robert S. and HippelA., J. Appl. Phys., 17, 610 (1946).
226.Бурдун Г.Д., Журн. техн. физики, 26, 813 (1950).
227.Koizumi N. and Ikad E., Bull. Inst. Chem. Res. Kyoto Univ., 43, 392 (1961).
228.Nelson S.O., Stetson L.E., Schlaphof C.W., IEEE Trans. Instrum. Meas., TM-23, 455 (1974).
229.Mash D., Mayants A. And Fabelinskii, Zhur. Tekhn. Fiz., 19, 10 (1949).
230.Krishna K.V., Trans. Far. Sac., 52, 110 (1956).
231.Mendel M. And Marco C, Physica, 30, 597 (1964).
232.Hassel W.F., Mayel D., Tucker S.W. and Walkers S., Tetrahearon, 20, 2137, 2751 (1964).
233.Grant E.H. and Shack R., Brit. J. Appl. Phys., 18,1807 (1967).
234.McAvoy and Buckmaster H.A., J. Phys. E; Sci. Insrtum., 18, 244 (1985).
235.Musyl J., Microwave Measurements of Complex Parmittivity by Free Spiece Methods and Their
Application , Elsevie (1986).
236.Vaughan W.E., Beryman K. and Smyth СР., J. Phys. Chem., 65, 94 (1961).
237.Dagg I.D. and Reesor G.E., Canad. J. Phys., 41, 1314 (1963).
238.Schunzel M.Z., Angew. Phys., 21, 508 (1966).
239.Pottel R., Ber. Bungen. Phys. Chem., 69, 363 (1966).
240.Garg K.,Klip H. And Smyth СР., J. Chem. Phys.,43, 2341 (1965).
241.Catgookar D.K., Varadan V.V.,Varaden V.K., IEEE Trans, on Instrumentation and Measurement,
37, 789 (1989).
242.Gleadhikk S.J., Spectrum analysis. Microwave Measurements (Ed. by Bailey A.E.),
London (1985).
243.Friedson G.I., Biebl E.M., 1996 Conf. Precis. Electr. Meas.: IEEE, 210 (1996).
244.Asfar M.N., Button K.J., Proc.IEEE, 73, 131 (1985).
245.Alder J.F., et all., Trans. Inst. Meas. Contr., 5, 99 (1983).
110
246.Vincent D.,Jorat L. ,Menin J. and Noyel G., Meas. Sci. Technol., 5, 990 (1994).
247.Weir W.B., Proc.IEEE, 62, 33 (1974).
248.Baker-Jarvis J.,Vanzuza E.J. and Kissick W.A., IEEE Trans. Microwave Theory Tech.,38,
1096 (1990).
249.Yan-Zhen W. and Sridhar S., Rev. Sci. Instrum., 60, 3041 (1989).
250.HP 85070A Dielectric Probe Kit 200 Mhz to 20 GHz. Convenient non-destructive dielectric
measurement. (1990).
251.Fleming M.A., Plasted G.N., Urguhart A.J., Dielectric measurements techniques at microwave
frequencies (1989).
252.Davidson C.W., Transmission lines for Communication , Macmillan Press LTD (1978).
253.Measuring Dielectric Constant with the HP 8510 Network Analyser, HP Product Note 8510-3.
254.Hu Xi-Ping, Using Six-Port Reflectometer Measurement of Complex Dielectric Constant, IEEE
Trans. Instrum. Meas. 1M-36, 2, №2 (1987).
255.Mattahaei G.L., Young L. and Jones E.M.T., Microwave Filters, Impedans Matching, Networks
and Coupling Structures, McGrow-Hill (1964).
256.Kaatze U., Pottel R. and Wallusch H.,Meas. Sci. Technol., 6, 1201 (1995).
257.Buchmaster H.A., Hausen C.H. and Van Kalleveen T.H.T., IEEE Trans.
Instrum. Meas., 39, 964 (1990).
258.Kaatze U. and Giese K., Adv. Molec. Relaxation Processes, 36, 15 (1987).
259.Kaatze U., Complex Permittivity of Mater as a Function frequency and temperature, J. Chem.
Eng. Data., 34, 371 (1989).
260.Gottmann O., Kaatze U. and Petong P., Meas. Sci. Technol., 7, 525 (1996).
261.Bottrean A. and Merzouki, IEEE Trans. Instrum. Meas., 42, 899 (1993).
262.Colling R.E., Field Theory of Guided Waves, New-York (1960).
263.Davies M., Dielectric and Related Molecular Processes, vol. 1-3, London (1972,1975,1977).
264.Smith B.L.and Carpentier M-H.The Microwave Engineering Handbook,vol.3 Microwave Systems
and Applications, London (1993).
265.Alison J.M. and Sheppard R.J., Meas. Sci. Technol., 1, 1093 (1990).
266.Richards M.G.M. and Sheppard R.J., Meas. Sci. Tehnol., 2, 663 (1991).
267.Steel M.C. and Sheppard R.J., J. Phys. E: Sci. Instrum., 20, 872 (1987).
268.Szwarnowski S. and Sheppard R.J., J. Phys. EL: Sci. Instrum., 10, 1163 (1977).
269.Nightingal N.R.V., Szwarnowski S., J. Phys. D.:Appl. Phys., 6, 790 (1981).
270. Van Loon R. and Finsey R., Rev. Sci. Instrum., 45, 523 (1974).
271.Zanforlin L., IEEE Trans. Microwave Theory Tech. MTT-31, 417 (1983).
272.Sheppard R.J., J. Phys. E., Sci. Instrum., 14, 156 (1973).
273.Fulyero K., Triiso Т., Hilland T. and Tjumsland Y., Meas. Sci. Tehnol.,6, 995 (1995).
274.Jenkins S., Hodgetts Т.Е., Clarke R.N., Preece A.W., Meas. Sci.Tehnol., 1, 691 (1990).
275.Cole R.H., Mashrimo S. and Winsor IV. P., J. Phys. Chem., 84, 786 (1980).
276.Cole R.H., Berferian J.G., et. el., J. Appl. Phys., 66, 793 (1989).
277.Sakamoto Т., Nakamura H., et. al., J. Phys. Chem., 93, 357 (1989).
278.Gestblom B. and Sjoblom J., Acta. Chem. Scand., A 38, 47 (1984).
279. Gestblom B. and Elmyren H., Chem. Phys. Lett., 90, 412 (1982).
280.Somlo PL, IEEE Trans. Instrum. Meas., 42, 213 (1993).
281.Schwan H.P., Physical Techniques in biological Research. Determination of Biological
Impedance (ed. Oster G.) (1963).
282.Ramo J.R. and van Duzer Т., Fields and Waves in Communication Electronics, New York (1965).
283.Giacomo P., News from BIPM, Metrologia, 17, 69, (1981).
284.Cole R.H., Dielectric Polarization and Relaxation In: NATOASISer. C, 135, 59 (1984).
285.Глебович Г.В., Исследование объектов с помощью пикосекундных импульсов, М.,255р.
286.Suggers A., Time Domain Methods: In Dielectric and related molecular processes, 1, 100(1972).
287.Cule R.H., Winsor P., Fourier transform, dielectric spectroscopy Fourier, Hadamard and Hilber
transforms in Chemistry IV (ed. by Marshall A.J.), N.-Y. (1984).
288.Cole R.H., Evaluation of Dielectric Behavior by Time Doman Spectroscopy. 1. Dielectric
Response by Real Time Analyses, J. Phys. Chem., 79, 1459 (1975).
289.Cole R.H., J. Phys. Chem., 79, 1469 (1975).
290.Фельдман Ю.Д., Зуев Ю.Ф., Colloid Polym. Sci., 270, 768 (1992).
291.Chanine R., Rose Т.К., J. Chem.Phys., 65, 2211 (1976).
111
292.Hines M., Stinehelfer H.E., IEEE Trans. Microwave Theory Theck, MMT-22, 276 (1974).
293.Фельдман Ю.Д., Левин В.В., Chem. Phys. Lett., 85, 528 (1982).
294.Makamura H., Mashimo S., Wada A., Japan J. Appl. Phys., 21,1022 (1982).
295.Cole R.H., Time domain reflectometry, Ann. Rev. Phys. Chem., 28,283 (1977).
296.Kaatze U. and Giese K., J. Phys. E: Sci. Instrum., 13, 133 (1980a).
297.Nazaki R. and Bose Т.К., IEEE Trans. Instrum. Meas., 39, 945 (1990).
298. Фельдман Ю.Д., Гончаров В.А., Зуев Ю.Ф., Chem. Phys. Lett., 58, 304 (1978).
299.Фельдман Ю.Д., Зуев Ю.Ф., Валитов В.М., Приборы и техн. измерения, 3, 611 (1979).
300.Hayer N.E., Rev. Sci. Instrum., 65, 887 (1994).
301.Kaatze U. and Giese K., J. Molec. Liquids., 36, 15 (1987).
302.Rozaz M. And Davis J.B., IEEE Trans. Microwave Theory Techn., 27, 564 (1979).
303.Mashimo S., Umehara Т., Ota Т., et. all., J. Molec. Liquids, 36,135 (1987).
304.Bertolino D., Cassettari M., et. all., Rev. Sci. Instrum., 61, 450 (1990).
305.Bone S., Biochimica et Biophysica Acta., 967, 401 (1988).
306. Гусев Ю.А. Диэлектрическая релаксация в растворах некоторых аминокислот. Дисс.
канд. физ.-мат. наук, КГУ, Казань. 1975.
307. Р.Р. Нигматуллин, “Что такое диэлектрическая спектроскопия? Это спектроскопия
коллективных движений”. Научно-технический журнал “Георесурсы” 2(17) 2005, 7-14с.
308. Гончаров В.А. Флуктуационная и импульсная диэлектрометрия и молекулярное
движение в жидких кристаллах. Дисс. канд. физ.-мат. наук, КФТИ им. Е.К. Завойского КФ
АН СССР, 1986.
112