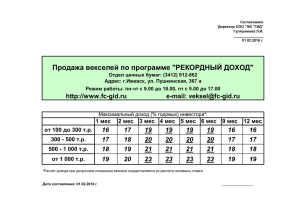
Молекулярный мониторинг хронического миелолейкоза
реклама

ОНКО Г Е М А ТО Л О Г И Я ISSN 1818-8346 Н А У Ч Н О - П Р А К Т И Ч Е С К И Й Е Ж Е К В А Р Т А Л Ь Н Ы Й Р Е Ц Е Н З И Р У Е М Ы Й Ж У Р Н А Л Молекулярный мониторинг хронического миелолейкоза Характеристики острого миелоидного лейкоза у детей первого года жизни Современные подходы к лечению цитомегаловирусной инфекции у онкогематологических больных 1 2013 Уверенность в результате при профилактике и лечении инванзивных микозов • • • • Доказанное превосходство в профилактике инвазивных грибковых инфекций у пациентов с нейтропенией1 Защита от инвазивных микозов пациентов после ТГСКb на фоне РТПХc2 Наиболее широкий спектр активности – от Aspergillus до Zygomycetes3 Проверенная эффективность в лечении рефрактерных микозов4 Международные руководства рекомендуют использование НОКСАФИЛА для профилактики ИГИa у пациентов группы высокого риска их развития Профилактика НОКСАФИЛОМ ECIL-3A NCCNB IDSAC AGIOH/DGHOD Пациенты с ОМЛd/МДСe в период нейтропении Al 1 Al Al Пациенты после ТГСК на фоне РТПХ Al 1 Al Al ECIL – Европейская конференция по инфекциям у больных лейкозами NCCN – Национальная онкологическая сеть США C IDSA – Американское общество инфекционных болезней D AGIOH/DGHO – Рекомендации рабочей группы по инфекционным заболеваниям Немецкого общества гематологов и онкологов A Краткая информация о препарате НОКСАФИЛ® (ПОЗАКОНАЗОЛ) Показания: Профилактика инвазивных грибковых инфекций у пациентов из группы высокого риска. Лечение инвазивных грибковых инфекций: инвазивный аспергиллёз, инвазивный кандидоз или кандидоз пищевода, рефрактерные к амфотерицину В или итраконазолу, а также при их непереносимости. Зигомикоз (мукормикоз), криптококкоз, фузариоз, хромомикоз и мицетома, кокцидиоидоз, рефрактерные к другим противогрибковым лекарственным средствам. Лечение орофарингеального кандидоза. Противопоказания: Повышенная чувствительность к какому-либо из компонентов препарата. Совместное применение с алкалоидами спорыньи, субстратами CYP3A4 – терфенадином, астемизолом, цизапридом, пимозидом, галофантрином или хинидином, ингибиторами ГМГ-КоА-редуктазы – симвастатином, ловастатином B и аторвастатином. С осторожностью: При повышенной чувствительности к азольным соединениям в анамнезе. При тяжёлом нарушении функции печени. При врождённом или приобретённом удлинении интервала Q-Tc; при кардиомиопатии; при других аритмиях; при совместном приёме с лекарственными средствами, удлиняющими интервал Q-Tc. Подробную информацию о дозировании препарата смотрите в инструкции по медицинскому применению. Побочные явления: Частые (>1 %, но <10 %): тошнота, головная боль, повышение АСТ, АЛТ, сыпь, лихорадка, астения, нарушение электролитного баланса. Нечастые (>0,1 %, но <1 %): тромбоцитопения, лейкопения, аллергическая реакция, гипергликемия, судороги, невропатия, расплывчатое зрение, удлинение интервала Q-T, изменения АД, панкреатит, повреждение гепатоцитов, боль в спине, нарушение функции почек, слабость, изменение сывороточных концентраций других лекарственных средств. Серьёзные нежелательные явления, зарегистрированные (с частотой 1 % каждое) у пациентов с инвазивными микозами, включали изменение концентрации других лекарственных средств, повышение печёночных ферментов, тошноту, сыпь и рвоту. Взаимодействие с другими лекарственными средствами: Позаконазол является ингибитором CYP3A4. Следует соблюдать осторожность при совместном назначении позаконазола и субстратов изофермента CYP3A4, поскольку такое применение приводит к повышению плазменных концентраций последних, что может вызывать нежелательные явления.Использование позаконазола во время беременности противопоказано, если преимущества лечения для женщины значительно не перевешивают потенциальный риск для плода. При назначении позаконазола кормление грудью следует прекратить. Литература: 1. Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs. fluconazole prophilaxis in patients with neutropenia. N Engl J Med. 2007;356:348–359. 2. Ullmann AJ, Lipton JH, Vesole DH, et al. Posaconazole or fluconazole for prophylaxis in severe graft versus host disease. N Engl J Med. 2007;356:335–347. 3. Lehrnbecher T. Current practice of antifungal prophylaxis and treatment in immunocompromised children and adults with malignancies: a single centre approach. Mycoses 52; 107-117. 4. Walsh TJ, Raad I, Patterson TF, et al. Treatment of invasive aspergillosis with posaconazole in patients who are refractory to or intolerant of conventional therapy: an externally controlled trial. Clin Infect Dis. 2007;44:2–12. 5. Инструкция по медицинскому препарату Ноксафил®. Перед назначением препарата, пожалуйста, ознакомьтесь с инструкцией по его применению. Компания MSD не рекомендует применять препараты компании способами, отличными от описанных в инструкции по применению. ООО «МСД Фармасьютикалс» 115093, г. Москва, ул. Павловская, д. 7, стр. 1 Tел.: +7 (495) 916 71 00, Факс: +7 (495) 916 70 94. www.msd.ru 07-2013-NOX-07-2011-RUS-012-EP Список сокращений: a ИГИ – инвазивные грибковые инфекции; b ТГСК – трансплантация гемопоэтических стволовых клеток; c РТПХ – реакция «трансплантант против хозяина»; d ОМЛ – острый миелолейкоз; e МДС – миелодиспластический синдром Журнал включен в Перечень ведущих рецензируемых научных журналов, в которых публикуются основные научные результаты диссертаций на соискание ученой степени доктора и кандидата наук ГЛАВНЫЙ РЕДАКТОР проф., д.м.н. Е.В. Самочатова Заместители главного редактора проф., д.м.н. В.В. Птушкин, проф., д.м.н. Б.В. Афанасьев Ответственный секретарь д.м.н. Ю.В. Румянцева РЕДАКЦИОННАЯ КОЛЛЕГИЯ проф., д.м.н. О.В. Алейникова (Минск) проф., д.м.н. А.К. Голенков (Москва) проф., д.м.н. А.И. Карачунский (Москва) д.м.н. Е.Н. Паровичникова (Москва) проф., д.м.н. Ю.А. Криволапов (С.-Петербург) доц., д.м.н. М.Л. Минков (Австрия) д.м.н. Н.В. Мякова (Москва) к.м.н. Е.А. Никитин (Москва) проф., д.м.н. О.А. Рукавицын (Москва) проф., д.м.н. С.А. Румянцев (Москва) д.м.н. Л.П. Менделеева (Москва) к.м.н. Л.Г. Фечина (Екатеринбург) д.м.н. А.Л. Усс (Минск) РЕДАКЦИОННЫЙ СОВЕТ проф., д.м.н. Е.А. Лукина (Москва) чл.-корр. РАМН И.В. Поддубная (Москва) чл.-корр. РАМН А.Г. Румянцев (Москва) к.м.н. В.А. Россиев (Самара) проф., д.м.н. А.Г. Талалаев (Москва) Адрес редакции: 115478, Москва, Каширское шоссе, д. 24, стр. 15, НИИ канцерогенеза, 3-й этаж. Тел./факс: +7 (499) 929-96-19 www.abvpress.ru e-mail: [email protected] Заведующая редакцией Т.В. Клюковкина Корректор В.В. Калинина Дизайн Е.В. Степанова Верстка О.В. Гончарук Служба подписки и распространения И.В. Шургаева, +7 (499) 929-96-19 e-mail: [email protected] Служба рекламы В.А. Клюковкин, +7 (499) 929-96-19 e-mail: [email protected] Журнал зарегистрирован в Федеральной службе по надзору в сфере связи, информационных технологий и массовых коммуникаций (Роскомнадзор) ПИ № ФС77-36928 от 21 июля 2009 г. EDITOR-IN-CHIEF Prof. Ye.V. Samochatova Deputy Editors Prof. V.V. Ptushkin, Prof. B.V. Afanasiev Executive Secretary D. Sci. Yu.V. Rumyantseva EDITORIAL BOARD Prof. O.V. Aleynikova (Minsk) Prof. A.K. Golenkov (Moscow) Prof. A.I. Karachunskiy (Moscow) D. Sci. Ye.N. Parovichnikova (Moscow) Prof. Yu.A. Krivolapov (St.-Petersburg) D. Sci. M.L. Minkov (Austria) D. Sci. N.V. Myakova (Moscow) PhD Ye.A. Nikitin (Moscow) Prof. O.A. Rukavitsyn (Moscow) Prof. S.A. Rumyantsev (Moscow) D. Sci. L.P. Mendeleeva (Moscow) PhD L.G. Fechina (Yekaterinburg) D. Sci. A.L. Uss (Minsk) EDITORIAL COUNCIL Prof. Ye.A. Lukina (Moscow) Prof. I.V. Poddubnaya (Moscow) Prof. A.G. Rumyantsev (Moscow) PhD V.A. Rossiyev (Samara) Prof. A.G. Talalayev (Moscow) При полной или частичной перепечатке материалов ссылка на журнал «Онкогематология» обязательна. Редакция не несет ответственности за содержание публикуемых рекламных материалов. В статьях представлена точка зрения авторов, которая может не совпадать с мнением редакции. ISSN 1818-8346 Онкогематология. 2013. № 1. 1–72 © ООО «ИД «АБВ-пресс», 2013 Подписной индекс в каталоге «Пресса России» — 42167 Отпечатано в типографии ООО «Тверская Городская Типография» Тираж 3000 экз. 1 2013 СОДЕРЖАНИЕ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Н.Р. Рябчикова, И.Р. Минниахметов, Г.Ш. Сафуанова, Д.В. Исламгулов, А.С. Карунас, Э.К. Хуснутдинова Хронический миелолейкоз: молекулярный мониторинг в клинической практике . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 Г.А. Цаур, А.М. Попов, О.М. Плеханова, А.М. Кустанович, О.В. Алейникова, Т.Л. Гиндина, А.С. Демина, А.Е. Друй, С.Ю. Ковалев, К.Л. Кондратчик, А.В. Мисюрин, Н.В. Мякова, Т.О. Ригер, Л.И. Савельев, О.И. Сокова, О.В. Стренева, М.В. Сучкова, Ю.П. Финашутина, Е.В. Флейшман, Е.В. Шориков, Р.И. Юцкевич, C. Meyer, R. Marschalek, Л.Г. Фечина Транслокация t(1;11)(p32;q23) с образованием химерного гена MLL-EPS15 при острых лейкозах: обзор литературы и описание 6 новых случаев. Подходы к мониторированию минимальной остаточной болезни . . . . . . . . . . . . . . . . . . . . 17 А.М. Попов, Г.А. Цаур, Т.Ю. Вержбицкая, О.В. Стренева, Е.В. Шориков, Л.И. Савельев, Л.Г. Фечина Иммунофенотипическая характеристика острого миелоидного лейкоза у детей первого года жизни . . . . . . . . . . . . . 33 Л.М. Мещерякова, Л.Г. Ковалева, С.Р. Карагюлян, Л.Ю. Колосова, О.В. Пороткова, Е.А. Семенова Отдаленные результаты спленэктомии при первичном миелофиброзе . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40 Д.Н. Балашов Современные подходы к лечению цитомегаловирусной инфекции у онкогематологических больных . . . . . . . . . . . . . . . 46 Е.А. Никитина, Н.Н. Климко, А.С. Колбин, Э.Г. Бойченко, И.А. Гарбузова, М.В. Голубева, Т.С. Богомолова Случай успешного лечения мукормикоза легких у ребенка с апластической анемией . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 ФАРМАКОТЕРАПИЯ В.В. Птушкин Современное представление о биоаналогах в гематологии и онкологии . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ От редакции . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 М.А. Орлова, Т.П. Трофимова, А.П. Орлов, О.А. Шаталов, Ю.К. Наполов, А.А. Свистунов, В.П. Чехонин Фуллерены и апоптоз . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65 КОНФЕРЕНЦИИ, СИМПОЗИУМЫ, СОВЕЩАНИЯ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72 CONTENTS HEMATOLOGIC MALIGNANCIES: DIAGNOSIS, TREATMENT, SUPPORTIVE CARE N.R. Ryabchikova, I.R. Minniakhmetov, G.Sh. Safuanova, D.V. Islamgulov, A.S. Karunas, E.K. Khusnutdinova Chronic myelogenous leukemia: molecular monitoring in clinical practice . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 G.A. Tsaur, A.M. Popov, O.M. Plekhanova, A.M. Kustanovich, O.V. Aleynikova, T.L. Gindina, A.S. Demina, A.Ye. Druy, S.Yu. Kovalev, K.L. Kondratchik, A.V. Misyurin, N.V. Myakova, T.O. Riger, L.I. Savelyev, O.I. Sokova, O.V. Streneva, M.V. Suchkova, Yu.P. Finashutina, Ye.V. Fleyshman, Ye.V. Shorikov, R.I. Yutskevich, C. Meyer, R. Marschalek, L.G. Fechina Translocation t(1;11)(p32;q23) with MLL-EPS15 fusion gene formation in acute leukemias: a review and 6 new case reports. Approaches to minimal residual disease monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 A.M. Popov, G.A. Tsaur, T.Yu. Verzhbitskaya, O.V. Streneva, E.V. Shorikov, L.I. Saveliev, L.G. Fechina Immunophenotypic investigation of infant acute myeloid leukemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 L.M. Meshcheryakova, L.G. Kovaleva, S.R. Karagyulyan, L.Yu. Kolosova, O.V. Porotkova, Ye.A. Semenova Long-term results of splenectomy in primary myelofibrosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40 D.N. Balashov Trends in the treatment of cytomegalovirus infection in oncohematological patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46 E.A. Nikitina, N.N. Klimko, A.S. Kolbin, E.G. Boychenko, I.A. Garbuzova, M.V. Golubeva, T.S. Bogomolova Successful treatment of pulmonary mucormycosis in a child with aplastic anemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 PHARMACOTHERAPY V.V. Ptushkin Modern concepts of biosimilars in hematology and oncology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 NEW TRENDS IN MEDICAL SCIENCE From edition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 M.A. Orlova, T.P. Trofimova, A.P. Orlov, O.A. Shatalov, Yu.K. Napolov, A.A. Svistunov, V.P. Chekhonin Fullerene and apoptosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65 CONFERENCES, SYMPOSIUMS, MEETINGS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72 Издательский дом «АБВ-пресс» специализируется на выпуске периодической научной медицинской литературы, книгопечатной продукции и создании сайтов медицинского направления НАШИ ЖУРНАЛЫ и ГАЗЕТЫ НАШИ КНИГИ Книги и наши издания можно заказать и приобрести в редакции по адресу: г. Москва, Каширское ш., д. 24, стр. 15 и по телефону: +7 (499) 929-96-19. Адрес электронной почты: [email protected] НАШИ САЙТЫ www.oncoproct.ru www.roou.ru www.netoncology.ru Москва, 2013 www.hnonco.ru www.urotoday.ru www.neuromuscular.ru 1 Хронический миелолейкоз: молекулярный мониторинг в клинической практике 7 ’2013 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Н.Р. Рябчикова1, И.Р. Минниахметов2, Г.Ш. Сафуанова1, Д.В. Исламгулов2, А.С. Карунас2, Э.К. Хуснутдинова2 1 ГБОУ ВПО «Башкирский государственный медицинский университет» Минздрава России, Уфа; 2 ФГБУН «Институт биохимии и генетики Уфимского научного центра» РАН Контакты: Гузель Шагбановна Сафуанова [email protected] Применение ингибитора тирозинкиназ (ИТК) иматиниба у пациентов с хроническим миелолейкозом (ХМЛ) привело к быстрому и значительному прогрессу в лечении данного заболевания. На сегодняшний день мониторинг терапии препаратами ИТК с помощью генетических технологий является обязательным атрибутом контроля эффективности лечения ХМЛ. Целью исследования было проведение комплексного молекулярно-генетического мониторинга с использованием методов стандартного цитогенетического исследования, полимеразной цепной реакции в режиме реального времени и мутационного анализа для оценки эффективности лечения пациентов с ХМЛ, получающих терапию иматинибом. Показана корреляция цитогенетического и молекулярного ответов и неоднородность структуры молекулярного ответа в каждой группе больных. Анализ характера мутаций гена BCR-ABL показал, что мутации киназного домена были выявлены у 32 % больных ХМЛ, резистентных к иматинибу. Ключевые слова: хронический миелолейкоз, молекулярно-генетический мониторинг, мутации в Рh-позитивных клетках Chronic myelogenous leukemia: molecular monitoring in clinical practice N.R. Ryabchikova1, I.R. Minniakhmetov2, G.Sh. Safuanova1, D.V. Islamgulov2, A.S. Karunas2, E.K. Khusnutdinova2 1 Bashkortostan State Medical University, Ministry of Health of Russia, Ufa; Institute of Biochemistry and Genetics, Ufa Scientific Center, Russian Academy of Sciences 2 Use of tyrosine kinase inhibitor imatinib has led to significant progress in chronic myeloid leukemia (CML) treatment. To date, genetic monitoring is a mandatory attribute of therapy with tyrosine kinase inhibitors. The purpose of this study was to access the imatinib therapy efficacy in CML patients using complete molecular genetic monitoring by standard cytogenetics, real-time polymerase chain reaction and mutational analysis. Correlation between cytogenetic and molecular response was shown. Heterogeneity of molecular response in each patient group was revealed by expression of BCR-ABL. Kinase domain mutations were detected in 32 % of CML patients resistant to imatinib. Key words: chronic myeloid leukemia, molecular genetic monitoring, Ph-positive cells mutations Введение Хронический миелолейкоз (ХМЛ) является формой лейкоза, которая характеризуется усиленным и нерегулируемым ростом преимущественно миелоидных клеток в костном мозге с их накоплением в крови. Основной причиной ХМЛ является реципрокная транслокация t(9;22),(q34;q11) с образованием филадельфийской хромосомы (Ph-хромосома) [1], обнаруживаемой у 90–95 % больных. В результате транслокации образуется химерный ген BCR-ABL, продуктом которого является патологический белок, обладающий выраженной тирозинкиназной активностью. Именно белок BCR-ABL, продуцируемый химерным геном, имеет ключевое значение в патогенезе ХМЛ [2]. Исследования молекулярных основ патогенеза ХМЛ подготовили почву для развития таргетной (целенаправленной) терапии этого заболевания, заключающейся в применении специфических ингибиторов BCR-ABL-тирозинкиназы (ИТК). Создание первого из них – иматиниба (STI571), стало революционным событием в области целенаправленной противоопухолевой терапии [3]. Иматиниб селективно ингибирует BCR-ABL-тирозинкиназу, встраиваясь в аденозин- трифосфат (АТФ)-связывающий карман киназного домена белка. Благодаря этому АТФ не может встроиться в АТФ-связывающий карман, BCR-ABL-тирозинкиназа остается в неактивном состоянии и тем самым блокируется сигнальный путь пролиферации опухолевых клеток [3, 4]. Несмотря на высокую эффективность препарата, у некоторых больных в 20–30 % случаев наблюдается первичная (рефрактерность) или вторичная (приобретенная) резистентность к проводимой терапии [5]. Возникновение устойчивости к ИТК является следствием взаимодействия многих факторов. Эти факторы включают в себя схему лечения, фармакодинамику ИТК, генетические изменения, мутации BCR-ABL киназного домена [5]. Наиболее широко обсуждается влияние мутаций химерного гена BCR-ABL на развитие резистентности. По современным данным, частота мутаций в гене BCR-ABL в разных группах больных колеблется от 15 до 90 % и зависит от критериев определения резистентности, методов обнаружения мутаций и фазы течения ХМЛ [6]. Эти мутации делают тирозинкиназу нечувствительной к действию иматиниба, при этом сохраняя трансформирующую актив- 1 ’2013 8 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ность фермента. Предполагается, что возникновение мутаций вызывает изменение конформации химерного белка тирозинкиназы, что затрудняет связывание иматиниба с аденозинтрифосфатным карманом белка [7, 8]. На сегодняшний день разработаны ИТК 2-го поколения, более эффективные по сравнению с иматинибом, 2 из которых уже зарегистрированы в России в начале 2008 г. – нилотиниб и дазатиниб. Применение ИТК 2-го поколения позволяет преодолеть проблемы с резистентностью и непереносимостью иматиниба у ряда больных, однако в некоторых случаях даже новые препараты не улучшают ответ на лечение и являются неэффективными [9]. В настоящее время основными методами диагностики и мониторинга заболевания при ХМЛ являются общий анализ крови, цитогенетическое определение содержания Ph-позитивных клеток в костном мозге и измерение уровня транскрипта гена BCR-ABL. Гематологический подсчет клеток является наименее чувствительным методом и используется для определения гематологического ответа (ГО) на лечение. Если анализ показывает отсутствие опухолевых клеток, ГО является полным; при наличии лейкозных клеток – частичным ГО [10]. Для определения цитогенетического ответа (ЦО) оценивается количество Ph-позитивных клеток по отношению к числу клеток, не несущих Ph-хромосомы [11]. Стандартное цитогенетическое исследование (СЦИ) клеток является единственным методом, позволяющим анализировать весь хромосомный набор клетки. Посредством СЦИ можно обнаружить дополнительные хромосомные аберрации, которые в ряде случаев свидетельствуют о неблагоприятном прогнозе заболевания. При этом необходимо понимать, что разрешающая способность этого метода относительно низкая и составляет 1–5 % клеток. ЦО на лечение определяется по относительному содержанию Ph+-клеток в костном мозге: полный – 0 %, частичный – 1–35 %, малый – 36–65 %, минимальный – 66–95 %, нет ответа – более 95 %. Большой ЦО (БЦО) определяется при достижении полного (ПЦО) или частичного ЦО при наличии в костном мозге 0–35 % Ph+-клеток [12]. Высокочувствительный молекулярно-цитогенетический метод флуоресцентной in situ гибридизации (FISH) хромосом позволяет обнаружить химерный ген BCR-ABL у Ph-позитивных больных ХМЛ не только в метафазе, но и в интерфазе митотического деления клетки [13], с частотой 1 опухолевая клетка на 200–500 клеток костного мозга. В соответствии с рекомендациями European Leukemia Net (ELN) контрольное СЦИ или FISH-анализ (в случае отсутствия митозов клеток костного мозга) необходимо проводить на 3 и 6 мес терапии ХМЛ ИТК, затем каждые 6 мес до достижения и подтверждения ПЦО, далее – каждые 12 мес в случаях, когда регулярная оценка молекулярных изменений неосуществима, и всегда при признаках миелодисплазии, субоптимальном ответе или неудаче лечения [12]. Кариотипирование с окрашиванием хромосом методом G-banding в клетках костного мозга, находящихся в стадии метафазы, необходимо проводить на 3 и 6 мес, а затем каждые 6 мес до достижения и подтверждения ПЦО, далее – каждые 12 мес в случаях, когда регулярная оценка молекулярных изменений неосуществима, и всегда при признаках миелодисплазии, субоптимальном ответе или неудаче лечения [12]. Наиболее чувствительным методом оценки молекулярного ответа (МО) на лечение является количественная полимеразная цепная реакция (ПЦР). Большой МО определяется как тысячекратное снижение количества транскриптов BCR-ABL по сравнению с исходным (до начала терапии). Полным МО (ПМО) считают случаи, когда BCR-ABL-транскрипты не выявляются [14]. Метод ПЦР применяется как для диагностики ХМЛ, так и для мониторирования минимальной остаточной болезни в процессе терапии [15, 16]. Чувствительность этого метода высока и позволяет обнаружить одну единственную клетку со специфической ДНК или РНК среди 104–106 клеток. В пределах чувствительности цитогенетического метода (10-2) оба метода (цитогенетический и молекулярный) равноценны для количественной оценки лейкемических клеток. В исследовании S. Branford et al. показано, что уровень экспрессии транскрипта гена BCR-ABL в периферической крови коррелирует с числом Ph-позитивных лейкемических клеток костного мозга (рис. 1) [17]. Молекулярный мониторинг уровня BCR-ABLтранскрипта при помощи количественной ПЦР в реальном времени (ПЦР-РВ) используется в качестве оценки ответа на лечение у пациентов с ХМЛ [18, 19]. Этот метод особенно важен при терапии ХМЛ ИТК, когда резидуальное количество лейкемических клеток ниже уровня чувствительности цитогенетического исследования [19–21]. Любая положительная динамика соотношения BCR-ABL/ABL является МО, так как отражает динамику соотношения нормальных и лейкемических клеток. Следовательно, количественный метод ПЦР можно использовать и на начальных этапах терапии ИТК [18, 20, 21]. Количественная ПЦР-РВ должна проводиться на цельных клетках из лейкоконцентрата, а результаты должны быть представлены в виде отношения BCRABL/ABL (или другим конститутивным генам), выраженного в процентах [12]. Целью нашего исследования было проведение комплексного молекулярно-генетического мониторинга с использованием методов СЦИ, ПЦР-РВ и мутационного анализа для оценки эффективности лечения пациентов с ХМЛ, получающих терапию иматинибом. 12 10 1 1011 2 10 Заболевание в момент диагностики Полный ГО 109 4 108 5 7 10 6 106 10 % 1 10 3 100 ’2013 Log ПЦО Большой МО 0,1 0,01 0,001 0,0001 ПМО 0% Log 3 = 0,1 % (большой МО) Рис. 1. Оценка ответа на терапию молекулярным методом [18] Материалы и методы Пациенты. Материалом исследования послужили образцы РНК и кДНК, выделенные из периферической крови 114 больных (55 мужчин и 59 женщин, в возрасте от 14 до 76 лет, медиана – 43 года) с клинически и цитогенетически установленным диагнозом ХМЛ, обследовавшихся в Республиканской клинической больнице им. Г.Г. Куватова г. Уфы. Все больные получали лечение препаратом ИТК иматинибом в дозе 400, 600 или 800 мг/сут (согласно рекомендациям ELN) [12]. Максимальная длительность терапии составила 205 мес, минимальная – 6 мес; медиана длительности терапии – 65,5 мес. Исследование одобрено Этическим комитетом Института биохимии и генетики Уфимского научного центра Российской академии наук. Все больные ХМЛ дали информированное согласие на участие в исследовании. Выделение РНК. РНК выделяли из 5–7 мл венозной крови реагентом TriZol (Invitrogene) в соответствии с рекомендациями изготовителя. Эффективность выделения мРНК определяли с использованием спектрофотометра NanoDrop. Качество препаратов РНК оценивали по соотношению оптических плотностей А260/A280 и электрофорезом в 1 % агарозном геле по соотношению количества 28S и 18S рРНК [16]. Выделенную РНК обрабатывали ДНКазой I, свободной от РНКазы (Sigma). Наличие в образце РНК примесей ДНК определяли с помощью ПЦР со специфическими праймерами к гену «домашнего хозяйства» GAPDH (Fermentas). Обратная транскрипция проводилась с использованием набора “RevertAid™ First Strand cDNA” (Fermentas) с помощью гекса- и дексануклеотидных праймеров согласно рекомендациям производителя. 9 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Цитогенетические исследования костного мозга. Цитогенетическое исследование костного мозга на этапе постановки диагноза ХМЛ выполнено у 101 больного. Контрольные цитогенетические исследования проведены через 6, 12 и 18 мес терапии иматинибом. Образцы костного мозга транспортировали в лабораторию при температуре 4–8 °С в термоконтейнере в течение 1–24 ч. В работе использовали общепринятый прямой метод и метод культивирования костного мозга в течение 24 ч. Для СЦИ препараты окрашивались методом GTG (дифференциальная окраска G). Результаты кариотипирования описывали в соответствии с требованиями Международной цитогенетической номенклатуры ISCN 2009. ЦО, а также критерии отсутствия ответа определяли в соответствии с рекомендациями ELN [12]. ПЦР-РВ осуществляли с помощью системы детекции продуктов ПЦР-РВ CFX96™ (BioRad). Для анализа экспрессии гена BCR-ABL использовали набор реагентов Онкоскрин-1-1-Q (ООО «ГеноТехнология»). Определение интенсивности экспрессии химерного онкогена BCR-ABL и контрольного гена ABL проводили согласно протоколу производителя. Метод не стандартизирован по международной шкале (IS), результаты анализа уровня экспрессии представлены в процентах. Исследование мутаций в гене BCR-ABL. ПЦР проводили в 2 этапа с целью амплификации мутантного транскрипта ABL, согласно рекомендациям Soverini et al. [22]. На первом этапе амплифицировали участок гена BCR-ABL длиной в 1,2 тыс. пар оснований (п. о.) с использованием следующих праймеров: F-BCR-A и R-ABL-A (таблица). На 2-м этапе проводили реамплификацию, используя в качестве матрицы амплификат от первого этапа с использованием 2 пар внутренних праймеров (F-ABL-B/R-ABL-B и F-ABL-С/R-ABL-С) 1 ’2013 10 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Праймеры для ПЦР Название праймера Последовательность, 5'–3' F-BCR-A A* [1475 п. о. GAG CAG CAG AAG BCR (b3a2) или 1401 AAG TGT TTC AGA (3075–3098) п. о. (b2a2)] R-ABL-A CTC TAG CAG CTC ATA CAC CTG GG ABL (1484–1506) F-ABL-B CAT CAT TCA ACG TGT GCC GAC GG ABL (748–770) R-ABL-B GTT GCA CTC CCT CAG GTA GTC ABL (1120–1140) F-ABL-C GAA GAA ATA CAG CCT GAC GGT G ABL (930–951) R-ABL-C CGT CGG ACT TGA TGG AGA A ABL (1393–1411) Позиция Длина ампликона Статистическая обработка данных осуществлялась с использованием программы GraphPad Prism 3.0, GraphPad Software. Результаты и обсуждение B (393 п. о.) C (482 п. о.) Примечание. * – длина ампликона после первого этапа ПЦР варьирует в зависимости от типа транскрипта гена BCR-ABL (b3a2 или b2a2). и образованием 2 фрагментов длиной 393 п. о. и 482 п. о. соответственно. Для амплификации использовали реакционную смесь объемом 25 мкл, содержащую 2,5 мкл 10 × Taqбуфера, 0,5 единиц Taq полимеразы, смесь dNTP (dATP, dGTP, dCTP, dTTP по 200 мкМ каждого), 2,5 mM MgCl2 и по 5 пM каждого праймера. В реакцию брали 1–3 мкг кДНК. В первом раунде ПЦР проводилась при следующих условиях: после денатурации (94 °С, 5 мин) проводили 30 циклов амплификации в режиме 94 °С / 40 с; 60 °С / 60 с; 72 °С / 60 с; финальная достройка 72 °С / 7 мин. Реамплификация проводилась при следующих условиях: после первичной денатурации (94 °С, 5 мин) проводили 35 циклов амплификации в режиме 94 °С / 30 с; 55 °С / 40 с; 72 °С / 40 с; финальная достройка 72 °С / 7 мин. Определение нуклеотидной последовательности кДНК гена BCR-ABL выполняли с помощью автоматического секвенатора ABI PRISM модель 310 (“Applied Biosystems”) с использованием набора для флуоресцентного мечения DYEnamicTM ET, согласно протоколу фирмы-производителя (“Amersham Pharmacia Biotech” DYEnamicTM ET Terminator Cycle Sequencing Kit). Наличие мутаций подтверждали с помощью анализа полиморфизма длины рестрикционных фрагментов (ПДРФ). Фрагменты гена BCR-ABL подвергали рестрикции специфическими эндонуклеазами DdeI, Ncol. Реакцию проводили согласно протоколу фирмыпроизводителя (Fermentas). Продукты амплификации и рестрикции анализировали в 7 % полиакриламидном геле с последующей окраской в растворе бромистого этидия (конечная концентрация 0,1 мкг/мл) и визуализацией в проходящем УФ-свете при длине волны 312 нм. Цитогенетический мониторинг терапии хронического миелолейкоза иматинибом Цитогенетическое исследование клеток костного мозга позволяет наиболее точно определить размер опухолевого клона клеток и оценить эффективность терапии иматинибом в течение первых 12 мес лечения [23]. Одним из самых важных критериев прогноза ответа на терапию иматинибом является результат цитогенетического исследования клеток костного мозга после 6 мес лечения (рекомендации ELN 2006, 2009). В соответствии с данными рекомендациями в эти сроки больные должны достичь БЦО (содержание Ph-позитивных клеток менее 35 %) [12, 23]. В нашем исследовании эффективность лечения иматинибом оценивалась после 6 мес лечения у 101 пациента с ХМЛ, из них в хронической фазе были 82 (81,2 %) пациента, из которых иматиниб в дозе 400 мг/сут получали 76 (92,7 %) больных и 600 мг/сут – 6 (7,3 %) пациентов. В фазе акселерации находились 16 больных ХМЛ, из которых 1 (6,25 %) пациент получал терапию иматинибом в дозе 400 мг/сут, 12 (75 %) – в дозе 600 мг/сут, 3 (18,75 %) больных – в дозе 800 мг/сут. В бластном кризе – 3 пациента, которые получали иматиниб в дозировке 400, 600 и 800 мг/сут соответственно. Следует отметить, что достижение оптимального ответа на терапию иматинибом у пациентов в хронической фазе и особенно в фазе акселерации напрямую зависит от назначенной дозы иматиниба. Больные ХМЛ в хронической фазе должны получать иматиниб в дозе не менее 400 мг/сут, в фазе акселерации – не менее 600 мг/сут. Поэтому в нашем исследовании пациенты, получавшие иматиниб в фазе акселерации и бластного криза в дозе 400 мг/сут, были исключены из дальнейшего анализа. Результаты проведенного анализа показали, что после 6 мес лечения из 82 пациентов в хронической фазе ХМЛ, получавших иматиниб в дозе 400–600 мг/сут, 43 (52,4 %) пациента достигли ПЦО и 6 (7,3 %) больных достигли частичного ЦО. Таким образом, БЦО выявлен у 49 (59,7 %) пациентов, что в соответствии с рекомендациями ELN (2009) рассматривается как оптимальный ответ. Из 82 обследованных больных ХМЛ в хронической фазе после 6 мес лечения 9 (11 %) достигли малого ЦО и 4 (4,9 %) пациента имели минимальный ЦО. Таким образом, малый или минимальный ЦО, рассматриваемый в рекомендациях ELN (2009) как субоптимальный ответ, был достигнут у 13 пациентов (15,9 % случаев). Из 15 больных ХМЛ, находившихся в фазе акселерации и получавших иматиниб в дозе 600–800 мг/сут, Анализ экспрессии гена BCR-ABL у больных хроническим миелолейкозом Для количественной оценки транскриптов BCRABL у 114 больных ХМЛ был проведен анализ экспрессии методом ПЦР-РВ. Относительная экспрессия гена BCR-ABL определялась как отношение среднего числа копий гена BCR-ABL к среднему числу копий гена ABL, умноженное на 100 %. Согласно рекомендациям ELN, обнаружение гена BCR-ABL более чем в 1 % клеток свидетельствует о выраженной экспрессии BCR-ABL, оценить которую можно с помощью СЦИ. Экспрессия гена BCR-ABL в 0,1–1 % клеток характерна для умеренного уровня экспрессии, который уже невозможно оценить с помощью СЦИ. Экспрессия гена BCR-ABL на уровне менее 0,1 % свидетельствует о достижении БМО [23]. Для удобства представления данных по изменению уровня экспрессии BCR-ABL в процессе лечения была также использована логарифмическая шкала измерений. Результаты выражали в виде десятичного логарифма (lg) снижения экспрессии по отношению к базовому уровню. Снижение экспрессии на 1 lg означало снижение уровня BCR-ABL-транскрипта в 10 раз, на 2 lg – в 100 раз, на 3 lg – в 1000 раз, на 4 lg – в 10 000 раз. В результате проведенного нами анализа было выявлено, что медиана экспрессии гена BCR-ABL в общей выборке пациентов с ХМЛ составила 23,77 % (10–90-й процентиль, 0–316,8 %), или 1,5 lg. У 32 (28 %) больных ХМЛ был выявлен ПМО на терапию ИТК (0 % гена BCR-ABL). Медиана снижения уровня экспрессии BCR-ABL составила 4,2 lg. Умеренный уровень экспрессии был выявлен в 10 (9 %) случаях. Медиана экспрессии гена BCR-ABL составила 0,15 % (10–90-й процентиль, 0,05–0,93 %), или 2,5 lg. В группе больных ХМЛ с высоким уровнем экспрессии гена BCR-ABL (n = 72) медиана экспрессии составила 79,9 %, или 1,2 lg. Сравнение значений цитогенетического и молекулярного ответа Сравнение данных МО и ЦО проводилось на 77 образцах крови пациентов. Образцы были разделены на группы, соответствующие уровню ЦО. Разброс значений экспрессии BCR-ABL в каждой группе был очень широким, но с уменьшением процентного содержания Ph-позитивных клеток увеличивалась доля образцов с более слабым уровнем экспрессии. Сравнительный анализ данных ЦО и МО подтвердил наличие положительной корреляции между процентным содержанием Ph-позитивных клеток и уровнем экспрессии BCR-ABL. Коэффициент ранговой корреляции Спирмена для этих параметров составил 0,822 (p < 0,0001). В группе пациентов с ПЦО в 43,8 % (14 из 32) образцов транскрипты гена BCR-ABL не были выявлены; в 15,6 % (5 образцов) экспрессия BCR-ABL составляла от 0 до 0,1 %, в 40,6 % (13 образцов) – выше 1 %. В группе с частичным ЦО в 28,6 % (2 из 7) образцов экспрессия гена BCR-ABL не выявлялась, в 71,4 % (5 из 7) образцов экспрессия была выше 1 %. В группе с малым ЦО 14,3 % (1 из 7) образцов экспрессия была равна нулю, в остальных 85,7 % (6 из 7) наблюдалась экспрессия свыше 1 %. В группе с минимальным ЦО транскрипты BCR-ABL не выявлялись в 28,6 % (2 из 7) случаев, экспрессия выше 1 % наблюдалась в 71,4 % (5 из 7). В группе с отсутствием ЦО в 100 % (24 образца) случаев экспрессия была свыше 1 %. Таким образом, в нашем исследовании высокий уровень экспрессии гена BCR-ABL (> 1 %) наблюдался во всех группах больных, в том числе и в группе пациентов с ПЦО. Пациенты с низким уровнем экспрессии (< 0,1 %) имеются в группах с малым и частичным ЦО, в группе с частичным ЦО есть также пациенты с отсутствием экспрессии BCR-ABL. Доля образцов пациентов с большим и ПМО значительно возрастает в группе с ПЦО. Анализ мутаций в гене BCR-ABL Для проведения анализа мутаций мы сформировали группу из 50 больных, соответствующих критериям резистентности к иматинибу. В этой группе пациентов медиана экспрессии гена BCR-ABL составила 141,6 %. В состав группы входили 22 мужчины и 28 женщин с медианой возраста 46 лет. С целью идентификации мутаций в гене BCR-ABL у 50 пациентов с ХМЛ проведен анализ изменений нуклеотидной последовательности тирозинкиназного домена BCR-ABL путем прямого секвенирования кДНК (рис. 2а и б). В результате проведенного нами исследования у пациентов с резистентностью к иматинибу выявлены 4 миссенс-мутации (M244V, T315I, M351T и H396R). У 7 (14 %) больных с ХМЛ при секвенировании кДНК обнаружена замена цитозина на тимин в 944-м положении, приводящая к замене треонина на изолейцин в 315-м положении (T315I). Наличие мутации у всех больных с выявленными при секвенировании изменениями последовательности нуклеотидов подтверждено методом ПДРФ-анализа с помощью эндонуклеазы рестрикции DdeI (рис. 2в и г). Анализ про- ’2013 ПЦО после 6 мес лечения достиг только 1 (6,7 %) пациент, частичного ответа – 2 (13,3 %) пациента, минимальный ЦО был достигнут у 2 (13,3 %) пациентов. Таким образом, в группе больных в фазе акселерации оптимальный ответ был выявлен у 3 (20 %) пациентов, субоптимальный – у 2 (13,3 %) пациентов. У 20 (24,4 %) больных ХМЛ в хронической фазе, у 10 (66,7 %) пациентов с ХМЛ в фазе акселерации и у всех больных, находящихся в бластном кризе, ЦО отсутствовал, что определяется как «неудача терапии». Отсутствие ГО после 3 мес и любого ЦО после 6 мес терапии, а также потеря уже достигнутого ГО, ЦО и МО рассматриваются как проявления резистентности к терапии ИТК. 11 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ а A T C A 130 C б Т G A A 130 T C A C Т G A 1 ’2013 12 в г 489 404 * 5΄ 3΄ 162 + 35 190 147 110 197 162 116 114 67 55 55 116 114 197 35 34 1 2 3 4 5 6 7 Рис. 2. Секвенирование 2 образцов кДНК с мутацией T315I (а, б) с последующим ПДРФ-анализом (в, г): в – электрофореграмма ПДРФ-анализа гена BCR-ABL1 с помощью эндонуклеазы рестрикции DdeI. Дорожка 1 – маркер молекулярного веса pUC19/MspI; 2 – контрольный образец, не обработанный рестриктазой; 3, 4, 5, 7 – образцы кДНК с мутацией T315I; 6 – образец кДНК без мутации T315I; г – рестрикционная карта фрагмента кДНК длиной 482 п. о. для эндонуклеазы DdeI. Сайты рестрикции показаны стрелками. При мутации T315I пропадает один из сайтов узнавания для рестриктазы DdeI (отмечен *), с образованием фрагмента длиной 197 п. о. водили после амплификации фрагмента кДНК длиной 482 п. о., который в норме содержит 4 сайта рестрикции для эндонуклеазы DdeI, гидролизующей кДНК на 5 фрагментов длиной 35, 55, 114, 116, 162 п. о. При возникновении замены цитозина на тимин пропадает один из сайтов рестрикции DdeI (отмечен * на рис. 2г), и у мутантного варианта ABL-транскрипта появляется дополнительный негидролизованный фрагмент длиной 197 п. о. В образцах с мутацией также были выявлены фрагменты длиной 162 п. о. и 35 п. о., что свидетельствует о клоновом характере мутаций, т. е. мутантные образцы представлены разным соотношением транскриптов дикого и мутантного типов. Мутация T315I в гене BCR-ABL является, по данным литературы, наиболее распространенной мутацией, вызывающей резистентность ко всем известным ИТК [4, 5]. Треонин в положении 315 находится на периферии нуклеотид-связывающего сайта ABL1 и формирует ключевое взаимодействие через водородную связь с иматинибом [24]. Мутация T315I разрывает это взаимодействие, ослабляет связывание иматиниба и приводит к полной нечувствительности к нему. Резистентность к терапии при этой мутации гена BCRABL невозможно преодолеть увеличением дозы има- тиниба, так же как и терапией ИТК 2-го поколения, поэтому при наличии совместимого донора пациентам с мутацией T315I показана аллогенная трансплантация костного мозга [3]. В исследовании итальянской группы GIMEMA (Italian Group for Hematologic Malignancies of the Adult) [25] частота данной мутации у больных ХМЛ составила 12 %, в исследовании Catherine Roche-Lestienne у больных ХМЛ из Франции мутация T315I была выявлена с частотой 12,5 % [26], что коррелирует с результатами нашего исследования. При исследовании мутаций у больных из г. Ростова было обнаружено, что частота мутации T315I составляет 4 % [4]. Достаточно низкая частота этой мутации, возможно, связана с тем, что авторами в исследование были отобраны только больные с первичной резистентностью к иматинибу (рефрактерность). У 10 пациентов с ХМЛ при секвенировании кДНК гена BCR-ABL нами была обнаружена замена тимина на цитозин в 1052-м положении, приводящая к замене метионина в положении 351 на треонин (M351T) (рис. 3а и б). Для подтверждения наличия мутации M351T проводился ПДРФ-анализ (рис. 3в и г) с использованием специфической эндонуклеазы Ncol. ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ C A C G 240 G б A G C 240 A C C G G 1 C ’2013 а 13 в г * 489 5΄ 434 404 331 242 3΄ 48 219 + 215 434 219 215 190 Рис. 3. Секвенирование 2 образцов кДНК с мутацией M351T (а, б) с последующим ПДРФ-анализом (в, г): в – электрофореграмма ПДРФ-анализа гена BCR-ABL1 с помощью эндонуклеазы рестрикции Ncol. Дорожка 1 – маркер молекулярного веса pUC19/MspI; 2, 3 – контрольный образец, не обработанный рестриктазой; 4, 6, 7, 8 – образцы кДНК с мутацией M351T; 5 – образец кДНК без мутации M351T; г – рестрикционная карта фрагмента кДНК длиной 482 п. о. для эндонуклеазы Ncol. Сайты рестрикции показаны стрелками. При мутации M351T пропадает один из сайтов узнавания для рестриктазы Ncol (отмечен *) и образуется дополнительный фрагмент длиной 434 п. о. Гидролиз амплифицированного фрагмента кДНК без мутации приводит к образованию 3 фрагментов длиной 219, 215 и 48 п. о. При наличии мутации M351T пропадает один из двух сайтов узнавания эндонуклеазы Ncol и образуются 2 фрагмента длиной 434 п. о. и 48 п. о. Мутация M351T в гене BCR-ABL является одной из часто встречаемых при ХМЛ мутаций, связанных с развитием резистентности к терапии [3]. Данная мутация приводит к умеренному снижению чувствительности к иматинибу в тестах in vitro, которая восстанавливается при концентрации препарата 1–4 мкмоль (концентрация 4 мкмоль соответствует суточной дозе иматиниба 800 мг/сут) [3, 27]. Предполагается, что в основе развития резистентности при этой мутации лежит нарушение связи между SH2- и киназным доменами ABL-киназы. В норме метионин в положении 351 связывается с SH2-доменом и стабилизирует неактивную конформацию ABL-киназы. Показано, что терапия высокими дозами иматиниба может привести к преодолению резистентности, однако более эффективный результат достигается при применении ИТК 2-го поколения [23]. По литературным данным, частота мутации M351T у больных ХМЛ из Германии составляет 6 % [28], в Италии и Австралии мутация M351T определяется с частотой 11 % [25, 29]. У больных ХМЛ из Республики Башкортостан мутация M351T встречается с более высокой частотой – 20 %, и является самой распространенной мутацией. Мутация H396R в гене BCR-ABL, возникающая в результате замены аденина на гуанин в 1187-м положении, обнаружена у 1 (2 %) больного (рис. 4а). В исследовании Hochhaus et al. (Германия, 2002) частота мутации H396R составила 1,5 % [28], в то время как в исследовании итальянской группы GIMEMA (Italian Group for Hematologic Malignancies of the Adult) ее частота была 0,7 % [25]. Показано, что наличие данной мутации может являться причиной резистентности к терапии иматини- ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ а C 410 C б C G T T 110 G G C C C A T G C T G 110 G A A 1 ’2013 14 в C C G г A A C C A T Рис. 4. Результаты секвенирования образцов кДНК с мутациями H396R (а), M244V (в) и в норме (б, г) бом. Мутация H396R дестабилизирует расположение активационной (А) петли так, что домен киназы не может перейти в неактивную пространственную организацию, необходимую для связывания с иматинибом. Обнаружение данной мутации является основанием для повышения дозы иматиниба. В случае отсутствия положительного результата необходимо перейти на терапию препаратами 2-го поколения (нилотиниб и дазатиниб) [3]. У 1 больного ХМЛ нами выявлена замена аденина на гуанин в 730-й позиции, приводящая к замене метионина в 244-м положении на валин (M244V) (рис. 4в). Несмотря на то, что фосфорилирующая активность ABL-киназы с данной мутацией незначительно отличается от активности ABL-киназы дикого типа, в работах A. Corbin и T. O’Hare et al. была доказана роль мутации M244V в развитии резистентности при лечении иматинибом у больных ХМЛ. По мнению авторов, при обнаружении данной мутации у пациентов необходимо увеличить дозу иматиниба или назначить ИТК 2-го поколения [7, 27]. Частота мутации M244V в исследовании группы GIMEMA достигла 4,4 % [25]. По данным Hochhaus et al., мутация M244V определяется в 1,5 % случаев [28]. В выборке больных ХМЛ из Республики Башкортостан частота мутации M244V составила 2 %. В целом, в результате проведенного исследования мутаций в гене BCR-ABL у 16 резистентных к иматинибу пациентов из 50 (32 %) были выявлены 4 мутации в киназном домене. У 7 пациентов с ХМЛ была обна- ружена мутация M351T, у 4 пациентов – мутация T315I, у 3 больных было выявлено по 2 мутации – T315I и M351T, и у 2 пациентов было определено по 1 мутации – M244V и H396R. Частота встречаемости обнаруженных нами в гене BCR-ABL мутаций варьировала при различных фазах заболевания. У большинства резистентных пациентов была хроническая фаза заболевания. У 7 (17 %) из них были обнаружены мутации в гене BCR-ABL: M351T (n = 4), T315I (n = 1), M244V (n = 1) и H396R (n = 1). У 6 пациентов, находившихся в фазе акселерации, были выявлены мутации: у 3 больных – мутация M351T, у 2 – мутация T315I, у 1 – 2 мутации, T315I и M351T. Из 3 пациентов, у которых наблюдался бластный криз, у 1 была определена мутация T315I, у 2 – по 2 мутации, T315I и M351T. Нами проведен сравнительный анализ уровня экспрессии химерного гена у больных с различными мутациями в киназном домене BCR-ABL. Установлено, что наиболее высокий средний уровень экспрессии химерного гена наблюдается у пациентов с компаундмутацией T315I + M351T (медиана экспрессии – 442,3 %), а также у пациентов с мутацией M351T (медиана экспрессии – 94,6 %). Таким образом, в результате проведенного анализа мутаций в гене BCR-ABL у обследованной группы больных ХМЛ было установлено, что мутации киназного домена встречаются у 32 % больных ХМЛ, резистентных к терапии иматинибом, что свидетельствует о необходимости проведения своевременной Обсуждение В результате проведенного исследования была показана корреляция ЦО и МО, проявляющаяся в том, что с уменьшением процентного содержания Phпозитивных клеток увеличивалась доля образцов с более слабым уровнем экспрессии. В то же время мониторинг эффективности лечения иматинибом в группе больных с определенным уровнем ЦО показал неоднородность в структуре МО: разброс значений экспрессии гена BCR-ABL в каждой группе пациентов был очень широким. Пациенты с низким уровнем экспрессии (< 0,1 %) имеются в группах с малым и частичным ЦО, в группе с частичным ЦО есть также пациенты с отсутствием экспрессии гена BCR-ABL. Тем не менее, с учетом низкой разрешающей способности цитогенетического метода (10-2) и в особенности после достижения цитогенетической ремиссии, оценивать дальнейшую редукцию лейкозного клона клеток можно только с помощью молекулярного метода ПЦР. Мониторинг лечения ХМЛ в настоящее время должен включать как цитогенетические, так и молекулярные исследования (определение уровня экспрессии BCR-ABL, поиск мутаций в гене BCR-ABL) и проводиться регулярно. Такой подход позволяет оценить ди- намику терапии и предсказать возможность развития рецидива. Значимость молекулярного анализа определяется также и тем, что уровень МО служит предиктором безрецидивной выживаемости. Повышение уровня экспрессии BCR-ABL-транскрипта во время терапии иматинибом косвенно может указывать на наличие мутаций в гене BCR-ABL, являющихся ведущей причиной резистентности к препарату. Как следствие, молекулярный мониторинг целесообразно использовать в качестве скрининговой стратегии для мутационного анализа. Заключение Мутации киназного домена гена BCR-ABL являются одной из основных причин резистентности к терапии иматинибом у некоторых пациентов с ХМЛ, что подтверждается данными, полученными в нашем исследовании. Мутационный анализ необходим для выяснения причин резистентности к иматинибу и поиска путей ее преодоления, определения прогноза течения заболевания, особенно при продолжении терапии иматинибом. В 14 % случаев у больных ХМЛ встречается мутация T315I, приводящая к резистентности к лечению всеми видами ИТК, что свидетельствует о необходимости проведения скрининга данной мутации у всех пациентов с ХМЛ, имеющих признаки резистентности. Раннее и своевременное обнаружение мутации T315I позволит изменить тактику лечения в пользу трансплантации костного мозга. Л И Т Е Р А Т У Р А 1. Savona M., Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008;8:341–50. 2. Kurzrock R., Kantarjian H., Druker B. et al. Philadelphia chromosome–positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med 2003;138:819–30. 3. Куцев С.И., Вельченко М.В. Значение анализа мутаций гена BCR-ABL в оптимизации таргетной терапии хронического миелолейкоза. Клин онкогематол 2008;1(3):190–9. 4. Куцев С.И., Вельченко М.В., Морданов С.В. Роль мутаций гена BCR-ABL в развитии рефрактерности к иматинибу у пациентов с хроническим миелолейкозом. Клин онкогематол 2008;1(4):303–9. 5. Quintas-Cardama A., Kantarjian H., Cortes J. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control 2009;16(2):122–31. 6. Barnes D.J., Palaiologou D., Panousopoulou E. et al. BCR-ABL expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res 2005;65(19):8912–9. 7. Corbin A., La Rose P., Stoffregen E. et al. Several BCR-ABL kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood 2003;101(11):4611–4. 8. Druker B.J., Sawyers C.L., Kantarjian H. et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344(14):1038–42. 9. Виноградова О.Ю., Туркина А.Г., Хорошко Н.Д. Организация терапии хронического миелолейкоза. Первый общероссийский регистр больных хроническим миелолейкозом: анализ и перспективы. Гематол и трансфузиол 2008;53(5):54–8. 10. Sessions J. Chronic myeloid leukemia in 2007. J Manag Care Pharm 2007;13(Suppl S-a):S4–S7. 11. Аксенова Е.В., Крутов А.А., Солдатова И.Н. и др. Стандартизация молекулярной диагностики хронического миелолейкоза. Клин онкогематол 2010;3(2):160–5. 12. Baccarani M., Cortes J., Pane F. et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009;27:6041–51. 13. Jha C.B., Kucheria K., Choudhary V.P. Diagnostic role of conventional cytogenetics and fluorescence in situ hybridization (FISH) in chronic myeloid leukemia patients. KUMJ 2006;4(2):171–5. 14. Gabert J., Beillard E., van der Velden V.H. et al. Standartisation and quality control studies of «real-time» quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia. A Europe Against Cancer program. Leukemia 2003;17:2318–57. 15. Туркина А.Г. Ингибитор сигнальных путей STI 571 (Signal Transductor Inhibitor) – новое направление в лечении хронического миелолейкоза. Совр онкол 2001;3(2):46–8. 16. Мисюрин А.В., Аксенова Е.В., Крутов А.А. и др. Молекулярная ’2013 диагностики данных мутаций и коррекции тактики лечения. 15 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 16 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ диагностика хронического миелолейкоза. Гематол и трансфузиол 2007;2:35–40. 17. Branford S., Hughes T.P., Rudzki Z. Monitoring chronic myeloid leukaemia therapy by real-time quantitative PCR in blood is a reliable alternative to bone marrow cytogenetics. Br J Haematol 1999;107:587–99. 18. Дубина М.В., Куевда Д.А., Хомякова Т.Е. и др. Молекулярный мониторинг эффективности терапии больных хроническим миелолейкозом в России. Совр онкол 2010;4(12):11–7. 19. Челышева Е.Ю., Туркина А.Г., Мисюрин А.В., Захарова А.В. Раннее выявление цитогенетического рецидива при динамическом исследовании уровня BCR-ABL-транскрипта у больного хроническим миелолейкозом. Гематол и трансфузиол 2007;2:50–1. 20. Kaeda S.A., Chase A., Goldman J.M. Cytogenetic and molecular monitoring of residual desease in chronic myeloid leukemia. Acta Hematol 2002;107:64–75. 21. Goldman J., Gordon M. Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matter? Leuk Lymphoma 2006;47:1–7. 22. Soverini S., Martinelli G., Amabile M. et al. Denaturing-HPLC-based assay for detection of ABL mutations in chronic myeloid leukemia patients resistant to Imatinib. Clin Chem 2004;50:1205–13. 23. Куцев С.И., Вельченко М.В., Зельцер А.Н. Молекулярно-генетический мониторинг терапии хронического миелолейкоза ингибиторами тирозинкиназ. Онкогематол 2008;4:17–25. 24. Nagar B., Bornmann W.G., Pellicena P. et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 2002;62(15):4236–43. 25. Soverini S., Colarossi S., Gnan A. et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA working party on chronic myeloid leukemia. Clin Cancer Res 2006;12(24):7374–9. 26. Roche-Lestienne C., Soenen-Cornu V., Grardel-Duflos N. at al. Several types of mutations of the ABL gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 2002;100:1014–8. 27. O'Hare T., Eide C.A., Deininger M. BCR-ABL kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007;110(7):2242–9. 28. Hochhaus A., Kreil S., Corbin A.S. et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002;16(11):2190–6. 29. Branford S., Rudzki Z., Walshet S. et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002;99:3472–5. Г.А. Цаур1, 2, А.М. Попов1, 2, О.М. Плеханова1, А.М. Кустанович3, О.В. Алейникова3, Т.Л. Гиндина4, А.С. Демина1, 2, А.Е. Друй1, 2, 5, С.Ю. Ковалев6, К.Л. Кондратчик7, А.В. Мисюрин8, Н.В. Мякова8, Т.О. Ригер1, 2, Л.И. Савельев1, 2, 5, О.И. Сокова9, О.В. Стренева1, 2, М.В. Сучкова10, Ю.П. Финашутина8, Е.В. Флейшман9, Е.В. Шориков1, 2, Р.И. Юцкевич3, C. Meyer11, R. Marschalek11, Л.Г. Фечина1, 2 1 ГБУЗ CO «Областная детская клиническая больница № 1», Екатеринбург; ГБУЗ СО «Институт медицинских клеточных технологий», Екатеринбург; 3 ГУ «Республиканский научно-практический центр детской онкологии, гематологии и иммунологии», Минск, Республика Беларусь; 4 Институт детской гематологии и трансплантологии им. Р.М. Горбачевой ГБОУ ВПО «Санкт-Петербургский государственный медицинский университет им. акад. И.П. Павлова» Минздрава России; 5 ГБОУ ВПО «Уральская государственная медицинская академия», Екатеринбург; 6 ФГАОУ ВПО «Уральский федеральный университет им. первого Президента России Б.Н. Ельцина», Екатеринбург; 7 Морозовская детская городская клиническая больница, Москва; 8 ФГБУ «Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Д. Рогачева» Минздрава России, Москва; 9 ФГБУ «Российский онкологический научный центр им. Н.Н. Блохина» РАМН, Москва; 10 ФГБУ «Гематологический научный центр» Минздрава России, Москва; 11 Diagnostic Center of Acute Leukemia, Institute of Pharmaceutical Biology / ZAFES, Goethe-University of Frankfurt, Франкфурт-на-Майне, Германия 2 Контакты: Григорий Анатольевич Цаур [email protected] На основании собственных исследований (6 пациентов) и данных литературы (27 случаев) представлена клинико-лабораторная характеристика острых лейкозов с транслокацией t(1;11)(p32;q23)/MLL-EPS15. Установлено, что наиболее часто транслокация t(1;11)(p32;q23) обнаруживается у детей первого года жизни (медиана возраста составляет 8 мес). При остром лимфобластном лейкозе лица мужского пола болеют реже (соотношение 1:3), при остром миелоидном лейкозе соотношение полов 1:1. Хромосомные аномалии, дополнительные к t(1;11)(p32;q23), выявлены в 38 % случаев. Наиболее часто зоной разрывов в ДНК гена EPS15 является интрон 1. Описано 4 типа химерных транскриптов MLL-EPS15. Создана и успешно применена комбинация праймеров, флуоресцентных зондов и плазмиды для мониторирования химерного транскрипта MLL-EPS15 методом полимеразной цепной реакции в режиме реального времени. Подобраны условия и проведен мониторинг минимальной остаточной болезни (МОБ) по индивидуальным точкам разрыва в геномной ДНК у пациентов с наличием химерного гена MLL-EPS15. Сравнение данных определения МОБ в геномной ДНК и кДНК показало высокую качественную сопоставимость результатов (92 %). Ключевые слова: острый лейкоз, дети первого года жизни, перестройки гена MLL, транслокация t(1;11)(p32;q23), химерный ген MLL-EPS15, минимальная остаточная болезнь Translocation t(1;11)(p32;q23) with MLL-EPS15 fusion gene formation in acute leukemias: a review and 6 new case reports. Approaches to minimal residual disease monitoring G.A. Tsaur1, 2, A.M. Popov1, 2, O.M. Plekhanova1, A.M. Kustanovich3, O.V. Aleynikova3, T.L. Gindina4, A.S. Demina1, 2, A.Ye. Druy1, 2, 5, S.Yu. Kovalev6, K.L. Kondratchik7, A.V. Misyurin8, N.V. Myakova8, T.O. Riger1, 2, L.I. Savelyev1, 2, 5, O.I. Sokova9, O.V. Streneva1, 2, M.V. Suchkova10, Yu.P. Finashutina8, Ye.V. Fleyshman9, Ye.V. Shorikov1, 2, R.I. Yutskevich3, C. Meyer11, R. Marschalek11, L.G. Fechina1, 2 1 Regional Childrenʼs Clinical Hospital №1, Yekaterinburg; Research Institute of Medical Cell Technologies, Yekaterinburg; 3 Belarusian Research Center for Pediatric Hematology, Oncology and Immunology, Minsk, Republic of Belarus; 4 Raisa Gorbacheva Memorial Institute of Children Hematology and Transplantation, St.-Petersburg I.P. Pavlov State Medical University, Ministry of Health of Russia; 5 Ural State Medical Academy, Yekaterinburg; 6 The first President of Russia Boris Yeltsin Ural Federal University, Yekaterinburg; 7 Morozov Children Municipal Clinical Hospital, Moscow; 8 Dmitriy Rogachev Federal Research Center of Pediatric Hematology, Oncology and Immunology, Ministry of Health of Russia, Moscow; 9 N.N. Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Moscow; 10 Hematology Research Center, Ministry of Health of Russia, Moscow; 11 Diagnostic Center of Acute Leukemia, Institute of Pharmaceutical Biology / ZAFES, Goethe-University of Frankfurt, Frankfurt on the Main, Germany 2 ’2013 Транслокация t(1;11)(p32;q23) с образованием химерного гена MLL-EPS15 при острых лейкозах: обзор литературы и описание 6 новых случаев. Подходы к мониторированию минимальной остаточной болезни 17 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 18 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ We performed clinical and laboratory characterization of patients with rare translocation t(1;11)(p32;q23) leading to MLL-EPS15 fusion gene formation. Study cohort consisted of 33 primary acute leukemia (AL) cases including 6 newly diagnosed and 27 patients previously described in literature. Among study group patients t(1;11)(p32;q23) was found most frequently in infant AL cases (median age 8 months). In acute lymphoblastic leukemia (ALL) male/female ratio was 1:3, in acute myeloid leukemia (AML) it was 1:1. Additional cytogenetic aberrations in 38 % of patients were revealed. The most frequent breakpoint position in EPS15 gene was intron 1. Four different types of MLLEPS15 fusion gene transcripts were detected. Primers-probe-plasmid combination for MLL-EPS15 fusion gene transcript monitoring by real-time quantitative polymerase chain reaction (RQ-PCR) was developed and successfully applied. In 3 patients RQ-PCR was done on genomic DNA for absolute quantification of MLL-EPS15 fusion gene. High qualitative concordance rate (92 %) was noted between minimal residual disease data obtained in cDNA and genomic DNA for MLL-EPS15 fusion detection. Key words: acute leukemia, infants, MLL rearrangements, translocation t(1;11)(p32;q23), MLL-EPS15 fusion gene, minimal residual disease Введение Перестройки 11q23/MLL встречаются примерно в 10 % всех случаев острого лимфобластного лейкоза (ОЛЛ) и в 3 % случаев острого миелоидного лейкоза (ОМЛ) [1, 2]. На сегодняшний день на молекулярном уровне охарактеризовано 64 различных перестройки хромосомного района 11q23 с участием гена MLL, встречающихся при острых лейкозах (ОЛ) и при миелодиспластическом синдроме [3]. Наиболее частыми партнерами MLL являются гены AFF1 (AF4), MLLT3 (AF9), MLLT1 (ENL), MLLT10 (AF10), MLLT4 (AF6), ELL, на долю которых суммарно приходится около 85 % всех случаев MLL-позитивных ОЛ, как у детей, так и у взрослых. Среди оставшихся 15 % случаев наиболее часто обнаруживается транслокация t(1;11) (p32;q23) с образованием химерного гена MLL-EPS15, частота выявления которой составляет примерно 3 % от общего числа перестроек гена MLL [3, 4]. Данная транслокация встречается у детей и у взрослых как при de novo, так и при вторичных ОЛ [4–22]. Также описаны 2 случая миелодиспластического синдрома с наличием аберраций хромосомного района 1p32 у лиц, переживших атомную бомбардировку в Японии: в одном случае была выявлена делеция del(1)(p22p32), во втором транслокация t(1;11)(p32;q23) [23]. Ген EPS15 (AF1P, AF-1P, MLLT5), располагающийся в хромосомном регионе 1р32, кодирует один из рецепторов эпидермального фактора роста, отвечающий за эндоцитоз данного белка. Ген имеет протяженность 165 т. н., состоит из 25 экзонов и кодирует белок, состоящий из 896 аминокислот с молекулярным весом 98 656 Да [24]. Транскрипция гена осуществляется в теломерном направлении. Описано 10 транскриптных вариантов, образующихся вследствие альтернативного сплайсинга, однако только 4 из них кодируют белок [25]. ЕН1 ЕН2 ЕН3 У человека наиболее высокая экспрессия гена EPS15 выявлена в Т- и В-лимфоцитах, моноцитах и лимфобластах [26]. Белок EPS15, как участник сигнального пути рецептора эпидермального фактора роста (EGFR), регулирует рост и пролиферацию клеток. Структура белка EPS15 приведена на рис. 1. Химерный ген MLL-EPS15 впервые описан O. Bernard et al. [13]. В основе образования данного химерного гена лежит реципрокная транслокация t(1;11)(p32;q23) (рис. 2). Известно, что существует 2 альтернативных механизма активации онкогенного потенциала белка MLL [29, 30]. Первый, более универсальный механизм, связан с воздействием на мотив белка-партнера MLL различных внешних факторов, что ведет к последующему неконтролируемому синтезу белка. Данный механизм реализуется в том случае, если белок-партнер локализуется в ядре клетки. Второй механизм, предложенный для MLL-EPS15 и ряда других химерных белков с участием MLL, локализующихся в цитоплазме, – двуспиральная олигомеризация функциональных доменов белка EPS15, приводящая к лейкозогенной трансформации белка MLL, что, в свою очередь, ведет к увеличенной способности клеток к самообновлению. Было показано, что при ОЛ действие химерного белка опосредуется через гены семейства HOX [31–34], однако по отношению к ОЛЛ данная точка зрения разделяется не всеми авторами [35]. В доступной нам литературе встретилось описание всего 25 случаев de novo ОЛ, а также 2 рецидивов ОЛ с наличием t(1;11)(p32;q23) [4–22]. Структура химерного гена MLL-EPS15 была описана всего в 2 случаях, а структура химерного транскрипта – в 5. Целью данной работы была клинико-лабораторная характеристика пациентов с транслокацией t(1;11)(p32;q23)/ ССD DPF UIM1 UIM2 Рис. 1. Структура EPS15. В структуре данного белка выделяют: 1. Три N-концевых EPS15-гомологичных домена (EH1, EH2, EH3), которые необходимы для присоединения к цитоплазматической мембране путем взаимодействия с белками и прямого связывания с фосфолипидами; 2. Биспиральный домен (ССD), ответственный за гомо- и гетеродимеризацию; 3. Аспартат-пролин-фенилаланиновые повторы (DPF), которые могут связываться с α-субъединицей адапторного белка AP-2; С-концевой фрагмент представлен 4 убиквитин-связывающими мотивами (UIM), которые необходимы для убиквитинилирования EGFR [27, 28] 11 (N) Der(1) Der(11) MLL-EPS15 EPS15-MLL Рис. 2. Основной механизм образования химерного гена у пациентов с наличием MLL-EPS15 – реципрокная транслокация MLL-EPS15, а также разработка методики мониторирования минимальной остаточной болезни (МОБ) у пациентов с этой транслокацией методом полимеразной цепной реакции (ПЦР). В отличие от других, более часто встречающихся перестроек гена MLL, для которых описаны различные комбинации праймеров и зондов [36, 37], нам не встретилось методики мониторирования МОБ у пациентов с наличием химерного гена MLL-EPS15 или его транскрипта. Материалы и методы За период с февраля 2000 по апрель 2012 г. было обследовано 186 детей в возрасте от 1 до 365 дней с ОЛ, включая 120 пациентов с ОЛЛ, 58 – с ОМЛ, 3 – с острым недифференцированным лейкозом, по 2 – с острым бифенотипическим и острым билинейным лейкозами, еще в 1 случае тип ОЛ не был определен. Транслокация t(1;11)(p32;q23) и/или химерный ген MLL-EPS15 были выявлены нами в 6 случаях. Пациенты, у которых была обнаружена транслокация t(1;11) (p32;q23), получали терапию по протоколам MLL-Baby [38] или ALL IC-BFM 2002 в детских онкогематологических клиниках Российской Федерации и Республики Беларусь. Информированное согласие на проведение диагностических и лечебных процедур было получено во всех случаях. Для цитогенетического исследования использовали клетки костного мозга, взятые до начала терапии. Применялась техника краткосрочного культивирования клеток в течение 24 ч. Методом окраски хромосомных препаратов был GTG-вариант. В большинстве случаев анализировали не менее 20 метафазных пластинок. Кариотипирование проводили в соответствии с международной номенклатурой хромосом человека, принятой на момент выполнения стандартного цитогенетического исследования [39–41]. В ходе данной работы все кариотипы были повторно оценены с учетом рекомендаций ISCN 2009 [41]. Дополнительными хромосомными аберрациями считали все клональные аномалии, сочетавшиеся с t(1;11)(p32;q23) [42]. Кариотип считали комплексным, если каждая клетка лейкозного клона у пациентов с ОМЛ содержала не менее 3 хромосомных изменений, включая перестройки района 11q23 [43, 44]. Флуоресцентную гибридизацию in situ (FISH) проводили с использованием гибридизационной системы TermoBrite (StatSpin, США) на микроскопе DM 4000 (Leica Microsystems Wetzlar GmbH, Германия) при помощи программного обеспечения CW4000 FISH (Leica Microsystems Wetzlar GmbH, Германия). Применяли локус-специфичный зонд LSI MLL Dual Color Rearrangement Probe 11q23 (Abbott Molecular, США), а также цельнохромосомные зонды для полного окрашивания 1-й и 11-й хромосом, меченные FITC и TexasRed соответственно (Metasystems, Германия). Длинную инвертированную ПЦР (long-distance inverse PCR-LDI-PCR) выполняли по описанной ранее методике [45]. В качестве исходного материала использовали геномную ДНК в количестве 1 мкг, выделенную из лейкоцитов и бластных клеток костного мозга, взятого в момент установления диагноза. ДНК обрабатывали экзонуклеазами рестрикции BamHI и BglII, а затем лигировали. Полученный продукт использовали для проведения нескольких инвертированных ПЦР-реакций. Ампликоны секвенировали с целью определения индивидуальной для каждого пациента точки разрыва в MLL и гене-партнере. Выделение РНК и обратную транскрипцию проводили по ранее описанной методике [46]. Качество полученной РНК оценивали методом капиллярного электрофореза на биоанализаторе Agilent 2100 (Agilent, США). В дальнейший анализ брали образцы с показателем целостности РНК более 4,2, который, как было доказано ранее, является достаточным для эффективного проведения ПЦР в режиме реального времени (ПЦР-РВ) [47]. Обратно-транскриптазную ПЦР (ОТ-ПЦР) проводили с использованием набора HemaVision (DNAtechnology A/S, Дания) и TaqF ДНК-полимеразы (ЦНИИ эпидемиологии, Россия) в 2 этапа, согласно инструкции производителя. На первом этапе ставили 8 мультиплексных ПЦР-реакций. При получении положительного результата в одной из пробирок на втором этапе проводили несколько моноплексных реакций. Результат расценивали как позитивный, если на обоих этапах ПЦР обнаруживалось наличие специфического ПЦР-продукта, совпадающего по размеру с ожидаемым. Результат расценивали как отрицательный, если в ходе первого этапа ПЦР не было выявлено продуктов амплификации и во всех пробирках присутствовала полоса внутреннего контроля размером 911 п. н. Детекцию проводили методом горизонтального электрофореза в 2 % агарозном геле. Дополнительно ’2013 1 (N) 19 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ж ж м ж ж ж ж ж м м ж ж ж ж 1 2 3 4 5 6 7 8 9 10 11 12 13 14 № паПол циента 8 мес 5 мес 1 год 43 года 4 года НД 35 лет 3,3 года 1 год 2 мес < 1 года 4 мес 8 мес НД** Возраст НД НД НД НД 465 НД НД 153 НД 260 НД 104 425 НД Лейкоциты (× 109/л) 46,XX,t(1;11)(p32;q23) 46,XX,t(1;11)(p32;q23) ВI-ОЛЛ ВI-ОЛЛ ОЛЛ ОМЛ M5a 46,XX,t(1;11)(p32;q23),+der(1)t(1;12) (p12;q12),-12[5]/46,XX[4] 46,XX,t(1;11)(p32;q23) ОМЛ М0 46,XY,t(1;11)(p32;q23)[11]/46,XY[1] ОЛЛ ОЛЛ 46,XX,t(X;5)(q26;p15),t(1;11)(p32;q23),i(9) (q10),del(17)(p11)/46,XX,i(9)(q10),add(12) (p13),del(17)(p11) 50,XY,t(1;11)(p32;q23),+?7,+21,+2mar** ВI-ОЛЛ 46,XX,t(1;11)(p32;q23)[5]/46,XX[18] BI-ОЛЛ ОМЛ 46,XX,del(1)(p32),del(11)(q13q23),der(4) t(1;11;4)(1pter 1p32::11q23 11q13::4p16 4qter)[29]/44,XX,-14,-21[1] 46,XX,t(1;11)(p32;q23) НД BIV-ОЛЛ BI-ОЛЛ ВП-ОЛЛ Вид ОЛ 46,XX,t(1;11)(p32;q23) 46,XY,t(1;11)(p32;q23)[17]/46,XY[20] 46,XX,t(1;11)(p32;q23)[6] 46,XX,t(1;11)(p32;q23) Кариотип — — — — — — — — — — — — — — FISH — — — — — — — — — — — — — — Химерный ген Таблица 1. Клинико-лабораторная характеристика пациентов с ОЛ и наличием t(1;11)(p32;q23)/MLL-EPS15 (начало) НД MLL экзон 9 – EPS15 экзон 2 — — Событие через 21 мес от начала терапии, смерть через 32 мес Пациент жив в ППР после проведения ТГСК. Срок наблюдения 54 мес Событие* через 20 мес от начала терапии, смерть через 24 мес НД MLL экзон 9 – EPS15 экзон 2 — НД C. Harrison et al., 1998 [4] O. Bernard et al., 1994 [13] T. Abshire et al., 1992 [12] C. Shippey et al., 1989 [11] S. Raimondi et al., 1989 [10] Смерть от рецидива через 5 мес от начала терапии НД A. Hagemeijer et al., 1987 [9] A. Selypes et al., 1987 [8] M. Gregoire et al., 1987 [7] Y. Kaneko et al., 1986 [6] D. Williams et al., 1984 [5] Источник ’2013 НД НД НД Смерть в ремиссии от инфекции через 9 мес от начала терапии Пациент жив в ППР со сроком наблюдения 19 мес НД Исход терапии — — — — — — — — — Химерный транскрипт 1 20 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ж ж ж м м ж м м ж м м ж ж ж м 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 № паПол циента 156 2 мес# 7 мес НД 430 102 2 мес# 5 мес 2 37 87 187 189 89 НД НД НД НД НД Лейкоциты (× 109/л) 76 лет 62 года 2 года 6 мес 9 мес 6 мес 59 лет 3 мес 1 год 67 лет 2 мес Возраст ОМЛ М5 ОМЛ М5 46,XY,t(1;4)(q21;p15),−8,del(9) (p11),der(11)t(1;11)(p32;q23),+13,del(15) (q22) 46,XY,der(1)t(1;11)(p32;q23),der(10) t(1;11;10)(p32;q23q14;p11),del(10) (q22q25),r(11)(p11q14) 47,XY,t(1;11)(p32;q23),+8[11] НД BI-ОЛЛ BI-ОЛЛ 46,XX,t(1;11)(p32;q23) [8]/47,idem,+X[4]/46,XX[8] 46,XX,t(1;11)(p32;q23)[11] BI-ОЛЛ ОМЛ М5 55,XY,t(1;11)(p32;q23),+der(1)t(1;11) x2,add(3)(p11)+6,+8x3,+21[] 46,XX,t(1;11)(p32;q23)[13]/46,XX[7] ОМЛ М4 45,XY,t(1;11)(p32;q23),-7[9]/46 XY[4] ОМЛ М5 ОЛЛ 46,XX,der(1)t(1;11)(p32;q23),inv(1) (p13q21),der(11)t(1;11)(p32;q23) 46,XX,t(1;11)(p32;q23)[17]/46,XX[3] ОЛЛ ОБфЛ ОМЛ М4 ОМЛ М5 ОМЛ М0 Вид ОЛ 46,XY,t(1;11)(p32;q23)[8]/47,idem,+18[2] 46,XY,t(1;11)(p32;q23)[16]/46,XY[4]** 46,XX,t(1;11)(p32;q23) 46,XX,t(1;11)(p32;q23)/47,idem,+8 46,XX,t(1;11)(p32;q23) Кариотип — MLL R — — MLL R MLL R — — — Отсутствие ответа на терапию, смерть через 2 мес от начала лечения Пациент жив в ППР со сроком наблюдения 39 мес MLL экзон 9 – ЦНС-рецидив на сроке 18 мес EPS15 экзон после ТГСК. Пациент жив 10 Смерть от рецидива через 13 мес от начала терапии MLL экзон 10 – EPS15 интрон 9 — K. Park et al., 2001 [17] и H. Kim et al., 2002 [18] J. Chessells et al., 2002 [16] A. von Bergh et al., 2000 [15] C. Felix et al., 1998 [14] C. Harrison et al., 1998 [4] Источник 1 ’2013 Настоящая работа R. Kotecha et al., 2012 [21, 22] M. Sagawa et al., 2006 [20] Рецидив через 16 мес от начала N. Douet-Guilbert терапии. Смерть от рецидива et al., 2005 [19] через 1 нед НД Время наблюдения до события 6 мес Пациент находится в ППР со сроком наблюдения 140 мес Время наблюдения до события 31 мес НД Смерть от лейкоза через 9,7 мес от начала терапии Пациент жив в ППР. Срок наблюдения 37 мес Пациент жив в ППР. Срок наблюдения 5 мес Событие через 4 мес от начала терапии, смерть через 6 мес Исход терапии MLL экзон 9 – Смерть от веноокклюзионной EPS15 экзон болезни на день +8 после 10 ТГСК MLL экзон 9 – EPS15 экзон 2 — — — — — — НД — — — Химерный транскрипт MLL экзон 10 – EPS15 интрон 9 — — MLL R MLL R — — НД НД — — MLL R НД НД НД — — MLL R — — Химерный ген — FISH ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 21 Примечание. FISH – результаты флуоресцентной гибридизации in situ c локус-специфичным зондом; «НД» – нет данных; «—» – исследование не проведено; * – вид неблагоприятного события не был указан авторами; # – монозиготные близнецы; ОБфЛ – острый бифенотипический лейкоз; ТГСК – трансплантация гемопоэтических стволовых клеток; ППР – полная продолжающаяся ремиссия; ** – образцы № 9 и 18 взяты от пациентов при рецидиве ОЛ, инициально у пациента № 18 определялся нормальный кариотип, у пациента № 9 – случайная транслокация 46,XY,t(1;12)(p32;q24). Пациент жив в ППР со сроком наблюдения 20 мес MLL экзон 10 – EPS15 экзон 2 MLL интрон 10 – 1p32 MLL R ж 33 4 мес 300 46,XX,t(1;11)(p32;q23)[11] BI-ОЛЛ Рецидив на сроке 7 мес, смерть от рецидива через 9 мес от начала терапии MLL экзон 11 – EPS15 экзон 2 MLL интрон 11 – EPS15 интрон 1 MLL R BI-ОЛЛ — 49 ж 32 3 мес Рецидив на сроке 18 мес, смерть от рецидива через 28 мес от начала терапии MLL экзон 11 – EPS15 экзон 2 MLL интрон 11 – EPS15 интрон 1 MLL R BI-ОЛЛ 46,XY,t(1;11)(p32;q23)[13]/46,XY[1] 212 м 31 1 день Пациент жив в ППР со сроком наблюдения 64 мес MLL экзон 10 – EPS15 экзон 2 MLL интрон 10 – EPS15 интрон 1 MLL R BI-ОЛЛ 46,XX,t(1;11)(p32;q23)[11] 91 9 мес ж 30 Вид ОЛ Кариотип Лейкоциты (× 109/л) Возраст № паПол циента Таблица 1. Клинико-лабораторная характеристика пациентов с ОЛ и наличием t(1;11)(p32;q23)/MLL-EPS15 (окончание) Исход терапии Химерный транскрипт Химерный ген FISH 1 Источник ’2013 Настоящая работа ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 22 всех пациентов с выявленным химерным геном MLLEPS15 тестировали в гнездной ОТ-ПЦР с праймерами, предложенными N. Palisgaard et al. [36]. Полученные в ходе гнездной ОТ-ПЦР ампликоны очищали при помощи «Wizard SV Gel and PCR CleanUp System» (Promega, Германия). Секвенирующую реакцию ставили с применением праймеров, использовавшихся во втором раунде гнездной ОТ-ПЦР, и набора BigDye Terminator 3.1. (Applied Biosystems, США) согласно инструкции производителя. Секвенирование проводили в 2 направлениях на генетическом анализаторе ABI Prism 3130 (Applied Biosystems, США). Для конструирования калибратора, используемого для количественной оценки величины химерного транскрипта MLL-EPS15 в ходе ПЦР-РВ, была использована РНК пациента № 30 (табл. 1). После проведения гнездной ОТ-ПЦР по описанной ранее методике [36] визуализацию ампликонов проводили методом электрофореза в 6 % полиакриламидном геле. Выделение ДНК-фрагмента из геля выполняли путем элюирования TE-буфером с последующим осаждением 70 % этиловым спиртом по общепринятой методике. Перед процедурой лигирования проводили наращивание «тупых» концов с использованием Taq ДНК-полимеразы (ООО «Генотехнология») в присутствии дезоксиаденозинтрифосфата. Полученный ампликон лигировали в плазмидный вектор «pGEM-T Еasy» (Promega, Германия) с последующей его трансформацией в бактериальную культуру Е. coli. Из отобранных положительных клонов E. coli выделяли плазмидную ДНК методом щелочного лизиса. Концентрацию ДНК определяли спектрофотометрически. Для использования в качестве калибратора для ПЦР-РВ сделали 5 разведений (101, 102, 103, 105, 106 копий/5 мкл), аналогичных использованным в международном проекте «Европа против рака» [48]. ПЦР-РВ для количественной оценки химерного транскрипта выполняли в соответствии с ранее описанными протоколами [48, 49]. В качестве калибраторов применяли плазмиды, несущие фрагменты химерного транскрипта MLL-EPS15 и контрольного гена ABL1 (Ipsogen, Франция). Нуклеотидная последовательность использовавшихся праймеров и зондов для определения транскрипта MLL-EPS15 приведена в табл. 2. Для количественной оценки величины МОБ на основании данных, полученных путем ПЦР-РВ, использовалась методика расчета, рекомендованная консорциумом «Европа против рака» [49]. ПЦР-РВ для определения количества химерного гена MLL-EPS15 в геномной ДНК проводили по ранее описанному протоколу [50] с рядом модификаций. ПЦР выполняли в мультиплексном формате: реакционная смесь включала 2 пациент-специфичных праймера и зонд, меченный карбоксифлуоресцеином (FAM), комплементарных зоне слияния генов MLL и EPS15, а также 2 праймера и зонд, меченный карбокси-Х-родамином (ROX), для амплификации контрольного гена по изучению МОБ при ОЛЛ ESG-MRD-ALL [51] c дополнениями T. Burmeister et al. [50]. Величина МОБ была рассчитана как среднее значение числа копий 2 повторов каждого образца при следующих условиях. 1. Отсутствие амплификации в отрицательном контроле (смесь ДНК, использованная для создания разведений) или превышение значения порогового цикла в образце пациента более чем на 3,0 от величины порогового цикла отрицательного контроля. 2. Разброс значений порогового цикла контрольного гена NAGK в 2 повторах 1 образца не превышает 1,5. 3. Коэффициент корреляции (R2) выше 0,95. 4. Угол наклона калибровочной кривой укладывается в диапазон от –2,5 до –4,5. В каждом образце рассчитывался количественный диапазон согласно рекомендациям ESG-MRD-ALL [51]. Таблица 2. Использованные праймеры и зонды Нуклеотидная последовательность 5’-3’ Ген Экзон Локализация MLL 10 4135-4155 EPS15 3 201-182 MLL 10 4157-4185 Химерный транскрипт MLL-EPS15 (кДНК) Прямой праймер AGGAGAATGCAGGCACTTTGA Обратный праймер AAGCCAACACCCTTCCAGTA Зонд ROX-CATCCTCAGCACTCTCTCCAATGGCAATA-BHQ2 Контрольный ген ABL (кДНК) Прямой праймер AGCTCCGGGTCTTAGGCTAT ABL 2 263-282 Обратный праймер TAGTTGCTTGGGACCCAGCC ABL 3 357-328 Зонд FAM-CCATTTTTGGTTTGGGCTTCACACCATT-BHQ1 ABL 3 323-296 Химерный ген MLL-EPS15 у пациента № 30 (ДНК) Прямой праймер GCAGCAGTTATTTTTGGACTCATTGA MLL Обратный праймер GATCTTTGTGACAGAGCAAGTCTTTA EPS15 Зонд FAM-TGATTACGCTTACAGTTACATGAACCCACACAT-BHQ1 EPS15 Химерный ген MLL-EPS15 у пациента № 31 (ДНК) Прямой праймер TCCCTGTTTAAACCAGCTAAAGAAATGT Обратный праймер TCAAGTGTAATTTAAAACAAACACCATTTCC Зонд FAM-TGCTACTCTAATAGCAGATTCCTTCCTAAAATCT-BHQ1 MLL EPS15 MLL Химерный ген MLL-EPS15 у пациента № 32 (ДНК) MLL Прямой праймер AGTGGGCATGTAGAGGTAAG Обратный праймер GCAGGAGGATTGGTTGAG EPS15 Зонд FAM-TGCACTCTAGCCTGGGCAACAGAGTGAGA-BHQ1 EPS15 Контрольный ген NAGK (ДНК) Прямой праймер TGGGCAGACACATCGTAGCA NAGK Обратный праймер CACCTTCACTCCCACCTCAAC NAGK Зонд ROX-TGTTGCCCGAGATTGACCCGGT-BHQ-2 NAGK Примечание. Нуклеотидная последовательность и нумерация экзонов гена MLL дана I. Nilson et al. [52], EPS15 – согласно NM_001981.2, ABL – согласно NM_005157. ’2013 N-ацетилглюкозамин киназы (NAGK), располагающегося на коротком плече хромосомы 2 в регионе 2p12. Ген NAGK использовался для исключения случайной ошибки. Нуклеотидные последовательности праймеров и зондов приведены в табл 2. В ПЦР-смесь вносили 600 нг геномной ДНК. Амплификацию проводили с использованием TaqF-полимеразы (ЦНИИ эпидемиологии, Россия) по следующей программе: 95 °С 15 мин, 45 циклов: 95 °С 15 с – 60 °С 60 с. Для создания калибровочной кривой проводили амплификацию 10-кратных разведений ДНК пациента в смеси ДНК, полученной от 5 пациентов без онкогематологических заболеваний, взятых в общем количестве 600 нг в соотношениях от 1:10 до 1:100 000. Все образцы тестировали в 2 повторах. Интерпретация результатов ПЦР-РВ проводилась исходя из рекомендаций Европейской рабочей группы 23 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 24 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Результаты терапии оценивались по кривым общей (ОВ) и бессобытийной выживаемости (БСВ), построенным по методу Каплана–Майера [52]. Для сравнения кривых выживаемости использовался непараметрический log-rank критерий. Различия считались достоверными при р < 0,05. Результаты Данные о 27 пациентах, представленных в литературе, а также 6 вновь описываемых нами случаев, приведены в табл. 1. Возраст на момент установления диагноза был известен у 31 пациента. Среди них было 19 (62 %) детей первого года жизни, 6 (19 %) детей в возрасте от 1 года до 4 лет и 6 (19 %) взрослых в возрасте от 35 до 76 лет. При этом медиана возраста во всей исследуемой группе составила 8 мес (диапазон 1 день – 76 лет). У лиц женского пола транслокация t(1;11)(p32;q23) выявлялась в 2 раза чаще по сравнению с мужчинами: 22 и 11 человек соответственно. В первую очередь это было связано с преобладанием лиц женского пола среди пациентов с ОЛЛ, где соотношение женщин и мужчин составило 3:1 (15 и 5 человек соответственно). Это же соотношение сохранялось и у детей первого года жизни с ОЛЛ (10 девочек и 3 мальчика). Для пациентов с ОМЛ вне зависимости от возраста соотношение лиц женского и мужского пола было близко к 1:1. У 20 (63 %) пациентов диагностирован ОЛЛ, у 11 (34 %) – ОМЛ, в 1 (3 %) случае – острый бифенотипический лейкоз. У 1 пациента (№ 4 в табл. 1) вариант ОЛ не был указан авторами. У детей первого года жизни ОЛЛ встречался еще чаще. Из 18 пациентов с известным типом ОЛ у 13 (72 %) был ОЛЛ, у 4 (22 %) – ОМЛ и у 1 (6 %) – острый бифенотипический лейкоз. Иммунофенотип бластных клеток был известен в 15 случаях ОЛЛ, в том числе полностью в 13 и частично в 2 (№ 1 и 9 в табл. 1). У всех описанных пациентов выявлен B-линейный иммунофенотип бластных клеток, в том числе 14 случаев ОЛЛ из В-линейных предшественников, в 1 случае (№ 3) нельзя исключить зрелый B-ОЛЛ. BI-ОЛЛ выявлен у 10 (77 %) из 13 пациентов первого года жизни с ОЛЛ. FAB-вариант описан у 10 пациентов с ОМЛ. В эту группу вошли 2 пациента с ОМЛ с минимальной дифференцировкой (М0 морфологический вариант по FAB-классификации), 2 – с острым миеломонобластным лейкозом (М4) и 6 – с острым монобластным лейкозом (М5). У детей первого года жизни с ОМЛ в 2 случаях выявлен ОМЛ М5, в 1 – ОМЛ М0. Данные об инициальном уровне лейкоцитов приведены для 18 пациентов. У них превалировал инициальный гиперлейкоцитоз: уровень лейкоцитов выше 100 × 109/л на момент установления диагноза выявлен в 12 (67 %) случаях. Медиана количества лейкоцитов составила 156 × 109/л (диапазон 2–465). Дети первого года жизни имели большую опухолевую массу: из 13 человек только трое имели уровень лейкоцитов менее 100 × 109/л, а медиана количества лейкоцитов в этой возрастной группе составила 187 × 109/л (диапазон 49–425 × 109/л). Цитогенетическое исследование показало, что транслокация t(1;11)(p32;q23) как единственная аномалия выявлена в 20 (59 %) из 32 случаев. У 1 пациента (случай № 5) найдена комплексная транслокация с вовлечением хромосомных районов 1p32, 11q23 и 4p16. Дополнительные хромосомные аномалии (ДХА) в опухолевых клетках пациентов с наличием t(1;11)(p32;q23) и информативным цитогенетическим исследованием выявлены у 12 (38 %) из 32 пациентов. В 3 случаях (№ 8, 20, 22 в табл. 1) обнаружены только количественные ДХА, в 6 (№ 16, 19, 24, 25, 27, 29) – только структурные; в 3 случаях – сочетание количественных и структурных (№ 9, 11, 24). Наиболее частой количественной ДХА являлась трисомия 8 (3 случая). Комплексный кариотип обнаружен у 4 (36 %) из 11 пациентов с ОМЛ (№ 11, 21, 22, 25) (рис. 3). Молекулярно-генетические исследования выполнены 15 пациентам, в том числе исследование методом FISH с локус-специфичным зондом проведено у 11 больных, и во всех случаях была выявлена перестройка гена MLL. В 3 случаях (№ 30, 32, 33) нами проведен дополнительный анализ методом FISH c использованием цельнохромосомных зондов для хромосом 1 и 11, который также подтвердил присутствие t(1;11) (рис. 4). У пациентов № 14 и 18 перестройки MLL подтверждены методом гибридизации по Саузерну. Структура химерного транскрипта определена у 9 пациентов, химерного гена – у 6. Выявлено по 2 случая локализации точек разрыва в экзоне 10 (№ 26 и 27), интроне 10 (№ 30 и 33) и интроне 11 (№ 31 и 32) ДХА (n = 12) Несбалансированный кариотип (n = 12) Сбалансированный кариотип (n = 12) Комплексный кариотип* (n = 4) < 3 аберраций* (n = 4) Количественные (n = 3) Количественные и структурные (n = 3) Структурные (n = 3) Рис. 3. Типы ДХА у пациентов с t(1;11)(p32;q23). Цифры соответствуют числу пациентов; * – разделение по количеству ДХА на более 3 (комплексный кариотип) и менее 3 проведено только для пациентов с ОМЛ а б 1 der(1) (p32) 25 ’2013 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 11 1 der(11) (q23) в г Рис. 4. Результаты цитогенетического и молекулярно-генетических методов исследования: а – частичная кариограмма пациента с наличием t(1;11)(p32;q23); б – результаты скрининговой (слева) и подтверждающей (справа) ОТ-ПЦР с использованием набора HemaVision (DNA-technology A/S, Дания) у пациента с наличием химерного транскрипта MLL-EPS15. K- – негативный контроль. Также на каждой электрофореграмме присутствует ДНК-маркер размерности ПЦР-продуктов; в – результат исследования методом FISH в интерфазных ядрах, характерный для перестройки гена MLL; г – данные FISH c применением цельнохромосомных зондов к 1-й и 11-й хромосомам, показывающие наличие t(1;11) а б Рис. 5. а – нуклеотидная последовательность зоны слияния гена MLL (выделен черным цветом) и хромосомного района 1p32 (выделен красным цветом), полученные при проведении LDI-PCR у пациента № 33. Жирным шрифтом отмечен экзон 10 гена MLL. Синим цветом выделена тринуклеотидная вставка; б – результат секвенирования химерного транскрипта у этого же пациента выявил слияние экзона 10 гена MLL c экзоном 2 гена EPS15 в ДНК гена MLL. В гене EPS15 в 3 случаях точка разрыва находилась в пределах интрона 1 (№ 30–32), в 1 случае в регионе 1p32 на расстоянии 1580 нуклеотидов от экзона 1 гена EPS15 (№ 33) (рис. 5a). Интересно отметить, что в этом случае точка слияния в химерном транскрипте располагалась во 2-м экзоне гена EPS15 (рис. 5б). Еще в 2 случаях у монозиготных близнецов точка разрыва в гене EPS15 локализовалась в относительно нетипичном месте – интроне 9 (№ 26 и 27). Выявлены следующие типы химерных транскриптов MLL-EPS15: e9e2 – 3 случая, и по 2 случая e9e10, e10e2, e11e2 (рис. 6). Для проведения количественного мониторинга МОБ нами была создана комбинация флуоресцентных ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ зондов, праймеров и плазмиды, несущей фрагмент химерного транскрипта MLL-EPS15. На основании 10-кратных разведений плазмиды (рис. 7а) была получена калибровочная кривая (рис. 7б и табл. 3). Специфичность комбинации зондов и праймеров была проверена исследованием 15 пациентов с ОЛ и нормальным кариотипом и 15 пациентов с ОЛ и другими химерными транскриптами с участием MLL, в том числе 6 – с MLLAF4, 5 – с MLL-MLLT1, 2 – с MLL-MLLT4, 1 – с MLLMLLT3, 1 – с MLL-MLLT10. Ни в одном случае неспецифической амплификации не выявлено. При разведении РНК, выделенной из исходного образца костного мозга пациента № 30, смесью РНК 10 пациентов без онкогематологических заболеваний была определена чувствительность системы, созданной для выявления MLLEPS15 методом ПЦР-РВ. Она составила 1 × 10-5 (рис. 7в). Мониторинг МОБ путем качественного и количественного определения химерного транскрипта MLLEPS15 проводили пациентам № 29–33. Во время проведения первого курса консолидации ремиссии пациенты № 30, 31, 32 (рис. 8а) достигли МОБ-негативности, однако 2 из них позднее рецидивировали (№ 31 и 32) (рис. 8б). Пациент № 33 достиг МОБ-негативности во время проведения 3-го курса консолидации ремиссии и находится в полной продолжающейся ремиссии (ППР) со сроком наблюдения 20 мес (рис. 8в). У пациента № 29 мониторинг МОБ стал проводиться через 5,5 мес от начала терапии. Для исключения случайных ошибок одновременно с ПЦР в этом случае проводилось исследование методом FISH. Химерный транскрипт MLL-EPS15 методом ПЦР и перестройки гена MLL методом FISH ни разу не определялись. MLL Количество образцов EPS15 NM_001981.2 3 2 2(2*) 2(2*) 3593 3658 4036 4110 4242 4164-4187 4356 4205-4228 126 168 258 306 273-294 Всего: 9 256-276 Рис. 6. Типы химерных транскриптов MLL-EPS15. Белые прямоугольники – экзоны гена MLL, серые – экзоны гена EPS15. Нумерация экзонов гена MLL дана по работе I. Nilson et al. [53], нумерация экзонов EPS15 – согласно референсной последовательности NM_001981.2. Праймеры для проведения гнездной ОТ-ПЦР схематически представлены в виде черных прямоугольников. Цифры внутри черных прямоугольников соответствуют местам начала и конца праймера по отношению к нормальной мРНК соответствующего транскрипта. Цифры под схематическим изображением экзонов соответствуют началу экзона. * – цифры в скобках соответствуют числу пациентов, впервые описанных в данной работе б в 104 Интенсивность флуоресценции, rfu а Интенсивность флуоресценции, rfu 103 Пороговый цикл 1 ’2013 26 102 101 R squared = 0,999 Slope = –3,356 Y-intercept = 38,790 Цикл ПЦР-РВ 10-кратные разведения плазмиды 100 0 5 10 15 20 25 30 35 40 45 50 Цикл ПЦР-РВ Рис. 7. Получение и оценка калибровочной кривой для последующего проведения количественного анализа химерного транскрипта MLL-EPS15 в кДНК пациентов: а – 10-кратные разведения плазмиды, несущей фрагмент транскрипта MLL-EPS15; б – калибровочная кривая, полученная на основании 10-кратных разведений плазмиды, с аналитическими характеристиками; в – чувствительность созданной комбинации праймеров и зонда методом 10-кратных лимитирующих разведений составила 1 × 10-5. Одно из разведений 1 × 10 -6 лежит за пределом линейности, во втором – амплификация отсутствует День 15 День 36 День 43 Консолидация I Консолидация II Консолидация III Поддерж. терапия День 0 День 15 День 36 День 43 Консолидация I Рецидив МОБ, % Чувствительность,% б День 0 День 36 День 43 Консолидация I Консолидация II Консолидация III Поддерж. терапия Таблица 3. Аналитические характеристики полученных калибровочных кривых МОБ, % Чувствительность,% в Для пациентов № 30, 31 и 33 проведено также определение МОБ путем количественной оценки химерного гена в геномной ДНК. Параметры калибровочных кривых, полученные при использовании индивидуально подобранных комбинаций зондов и праймеров, приведены в табл. 3. Все значения укладываются в диапазоны, рекомендованные рабочей группой ESGMRD-ALL [51] с дополнениями T. Burmeister et al. [50] (рис. 9). Пример определения абсолютного количества MLL-EPS15 в геномной ДНК у пациента № 31 приведен на рис. 10. Сравнение результатов качественного определения МОБ в кДНК и геномной ДНК у пациентов с наличием MLL-EPS15 проведено в 23 образцах. В 21 (92 %) из них были получены идентичные результаты. В 2 случаях МОБ была выявлена только в кДНК, но не в геномной ДНК, что, скорее всего, связано с более высокой чувствительностью методики по определению химерного транскрипта. Несмотря на различные величины МОБ, получаемые при использовании различных подходов для мониторирования MLL-EPS15, нами была выявлена сходная кинетика МОБ при определении в геномной ДНК и кДНК (рис. 11). Прогностическое значение транслокации t(1;11) (p32;q23) в настоящее время трудно оценить, посколь- Коэффициент корреляции (R2) Угол наклона кривой (Slope) Точка пересечения с осью ординат (Y-intercept) ПЦР-РВ химерного транскрипта 0,999 –3,356 38,790 ПЦР-РВ химерного гена в ДНК Рис. 8. Результаты мониторинга МОБ в кДНК пациентов № 30 (а), № 32 (б), № 33 (в). На всех графиках синим цветом показана величина МОБ, красным – чувствительность (нижний предел детекции), рассчитанные согласно рекомендациям консорциума «Европа против рака» [49]. В тех точках, где кривые чувствительности и МОБ сходятся, пациент становился МОБ-негативным Пациент № 30 0,990 –3,323 25,602 Пациент № 31 0,992 –2,987 21,987 Пациент № 32 0,996 –3,244 21,491 Пороговый цикл Интенсивность флуоресценции, rfu в R squared = 0,992 Slope = –2,987 Y-intercept = 21,987 Цикл ПЦР-РВ 10-кратные разведения ДНК пациента до лечения Интенсивность флуоресценции, rfu б а Цикл ПЦР-РВ Рис. 9. Получение и оценка калибровочной кривой для последующего проведения количественного анализа химерного гена MLL-EPS15 в геномной ДНК пациента № 31: а – 10-кратные разведения ДНК пациента, взятой до лечения в смеси ДНК от 5-го пациента без онкогематологических заболеваний; б – калибровочная кривая, полученная на основании 10-кратных разведений с аналитическими характеристиками; в – амплификация контрольного гена NAGK во всех образцах, использовавшихся для получения калибровочной кривой ’2013 День 0 МОБ, % Чувствительность,% а 27 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 28 День 8 День 15 День 36 День 43 Консолидация I Консолидация II Рецидив Величина МОБ 1 ’2013 День 0 Предел чувствительности ДНК а негативно позитивно Рис. 10. Определение МОБ в геномной ДНК. Динамика количества химерного гена MLL-EPS15 в геномной ДНК у пациента № 31. Красной горизонтальной линией обозначен предел чувствительности, рассчитанный исходя из рекомендаций ESG-MRD-ALL [51]. Два образца, значения которых лежат ниже уровня чувствительности, являются негативными негативно позитивно кДНК б День 0 День 15 День 36 День 43 Консолидация I Рецидив Предел чувствительности Рис. 11. Сравнение результатов определения МОБ в кДНК и геномной ДНК: а – сходимость в 23 образцах пациентов составила 92 %; б – кинетика МОБ, определенной в кДНК (синий цвет) и ДНК (красный цвет) у пациента № 32. Синей и красной горизонтальными линиями указаны пределы чувствительности в кДНК и ДНК соответственно ку эта аномалия является весьма редкой. Из доступной литературы мы выбрали данные о выживаемости пациентов, начиная с середины 1980-х годов. За это время терапевтические подходы претерпели значительные изменения, и, соответственно, улучшилась эффективность лечения как ОЛЛ, так и ОМЛ. В анализ ОВ было включено 12 пациентов с ОЛЛ и 6 больных с ОМЛ. Внутри этой группы ОВ составила 0,37 ± 0,12 с медианой наблюдения 38 мес (диапазон 5–140 мес), а БСВ – 0,33 ± 0,11. ОВ и БСВ в исследуемой группе не зависели от типа ОЛ и возраста пациентов (рис. 12а и 12б). Однако при более детальном разделении на группы было показано, что ОВ и БСВ в группе мальчиков с ОЛЛ младше 1 года были достоверно ниже, чем у девочек с ОЛЛ этой же возрастной группы (0 и 0,64 ± 0,21, p = 0,033; 0 и 0,46 ± 0,18, p = 0,049 соответственно) (рис. 12в и 12г). Обсуждение Транслокация t(1;11)(p32;q23) – относительно редкая генетическая перестройка, возможно, именно с этим связано небольшое количество работ, посвященных этой хромосомной аномалии. Так, на крупнейшем онлайн-ресурсе – Базе данных хромосомных аберраций и химерных генов F. Mitelman приводятся данные всего о 32 пациентах с транслокацией t(1;11) (p32;q23) [54]. Такие же цифры представлены в обзорной статье J.-L. Huret [55]. К сожалению, в этих источниках не разделены сведения о пациентах с первичными (n = 20) и вторичными ОЛ (n = 12), что затрудняет детальную оценку случаев ОЛ de novo. Участники Европейской рабочей группы по изучению перестроек 11q23 собрали информацию о 7 пациентах с транслокацией t(1;11)(p32;q23), включая 1 пациента со вторичным ОМЛ [4]. Мы в своей работе не касались случаев вторичных ОЛ из-за совершенно особой биологии опухолевого процесса у данной категории пациентов. Мы подтвердили ранее опубликованные данные о более высокой частоте t(1;11)(p32;q23) у лиц женского пола [4, 55]. Также необходимо отметить, что для пациентов с наличием t(1;11)(p32;q23) характерны общие черты всех MLL-позитивных ОЛ: преимущественное выявление у детей первого года жизни, доминирование CD10-негативного иммунофенотипа при ОЛЛ и М5 морфологического варианта при ОМЛ, частый инициальный гиперлейкоцитоз [56–58]. Согласно ранее опубликованным данным транслокация t(1;11)(p32;q23) наблюдается примерно с одинаковой частотой как при ОЛЛ, так и при ОМЛ [3, 4, 55]. Интересно, что нами ранее не выявлено ни одного случая ОМЛ с данной хромосомной аномалией. Отчасти это можно объяснить тем, что мы ограничили свой выбор изучением только детей первого года жизни, хотя и в этой возрастной группе встречается ОМЛ с t(1;11). Возможно, что отсутствие случаев с относительно редкой транслокацией t(1;11) объясняется малочисленностью (58 человек) нашей группы больных с ОМЛ, среди которой перестройки 11q23/MLL выявлены в половине случаев [59]. В то же время частота обнаружения t(1;11)(p32;q23) среди всех перестроек 11q23/MLL у детей первого года жизни с ОЛЛ по нашим наблюдениям достигает 6 % [60], что значительно выше, чем по литературным данным. Возможно, этот вопрос требует отдельного исследования на большем числе пациентов. Прогностическое значение t(1;11)(p32;q23)/MLLEPS15 трудно оценить, как в силу небольшого числа на- ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 0,9 0,9 0,8 0,8 0,7 0,7 0,6 0,5 0,4 0,3 0,6 0,5 0,4 0,3 0,2 0,2 0,1 0,1 0,0 0 20 40 60 80 100 120 Время наблюдения, мес 0,0 140 1 1,0 ’2013 б 1,0 Вероятность ОВ Вероятность БСВ а 0 Вся группа; n = 20; в ППР – 8; БСВ 0,33 ± 0,11 ОЛЛ; n = 14; в ППР – 5; БСВ 0,29 ± 0,13 ОМЛ; n = 6; в ППР – 3; БСВ 0,41 ± 0,22 ОЛЛ младше 1 года; n = 12; в ППР – 5; БСВ 0,35 ± 0,15 20 40 60 80 100 120 Время наблюдения, мес 140 Вся группа; n = 18; живы – 8; ОВ 0,37 ± 0,12 ОЛЛ; n = 12; живы – 5; ОВ 0,33 ± 0,15 ОМЛ; n = 6; живы – 3; ОВ 0,40 ± 0,21 ОЛЛ младше 1 года; n = 10; живы – 5; ОВ 0,42 ± 0,17 г 1,0 1,0 0,9 0,9 0,8 0,8 0,7 0,7 Вероятность ОВ Вероятность БСВ в 0,6 0,5 0,4 0,3 0,6 0,5 0,4 0,3 0,2 0,2 0,1 0,1 р = 0,049 0,0 0 10 20 30 40 50 Время наблюдения, мес 60 70 ОЛЛ < 1 года мальчики; n = 3; в ППР – 0; БСВ 0 ОЛЛ < 1 года девочки; n = 9; в ППР – 5; ОВ 0,46 ± 0,18 0,0 0 р = 0,033 20 40 60 80 100 120 Время наблюдения, мес 140 ОЛЛ < 1 года мальчики; n = 3; живы – 0; ОВ 0 ОЛЛ < 1 года девочки; n = 7; живы – 5; ОВ 0,64 ± 0,21 Рис. 12. Оценка прогностического значения t(1;11)(p32;q23) / MLL-EPS15: а – показатели БСВ в общей группе пациентов, а также пациентов с ОЛЛ, ОМЛ и ОЛЛ в возрасте младше 1 года; б – показатели ОВ в вышеупомянутых группах; в и г – БСВ и ОВ соответственно у пациентов первого года жизни с ОЛЛ в зависимости от пола. ППР — полная продолжающаяся ремиссия блюдений, так и гетерогенности терапевтических подходов, использовавшихся у данной группы пациентов. Мы, так же как и J.-L. Huret [55], при ОЛЛ выявили достоверно более низкие показатели БСВ и ОВ у пациентов мужского пола в возрасте младше 1 года по сравнению с девочками данной возрастной группы, но следует принимать во внимание, что в исследуемой группе было всего 12 пациентов (9 девочек и 3 мальчика). Более того, из-за отсутствия данных о том, живы ли 2 из 9 девочек, они были исключены из расчета вероятности ОВ. Целенаправленное исследование структуры химерного гена MLL-EPS15 было выполнено лишь однажды: 29 C. Meyer et al. приводят собственные данные о 13 пациентах, включая 12 случаев ОЛ у детей [3]. Однако в этой работе отсутствуют клинико-лабораторные характеристики пациентов с MLL-EPS15 при ОЛ. Точка разрыва в гене EPS15 у обследованных нами пациентов локализовалась либо в интроне 1, на долю которого приходится около 30 % всех случаев формирования химерного гена MLL-EPS15 [3], либо в некодирующей последовательности ДНК в хромосомном районе 1p32 в непосредственной близости от 5’-конца гена MLL. В гене MLL точки разрыва у обследованных нами пациентов располагаются в 10-м 1 ’2013 30 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ и 11-м интронах, что хорошо согласуется с ранее опубликованными данными [3, 50, 61]. Несмотря на относительно небольшую частоту встречаемости транслокации t(1;11)(p32;q23), важно проводить ее молекулярный мониторинг, который, как было показано ранее, позволяет прогнозировать исходы терапии [62]. Технически определение МОБ может быть проведено как методом FISH, так и ПЦР. Первый метод более универсален, но обладает низкой чувствительностью: в среднем он способен выявлять 1 бластную клетку среди 500–1000 нормальных. Довольно часто для оценки МОБ используется количественное определение индивидуальных клональных перестроек генов Т-клеточных рецепторов (TCR) и тяжелых цепей иммуноглобулинов (IgH) методом количественной ПЦР-РВ со специфичными для каждого пациента праймерами и зондом. Однако ранее было показано, что по сравнению со старшими детьми у детей первого года этот метод имеет ограниченное применение вследствие того, что в данной группе пациентов перестройки IgH/TCR встречаются реже и часто являются олигоклональными [63, 64]. Применение проточной цитометрии, еще одного метода, широко используемого для мониторинга МОБ у детей и взрослых, также может быть затруднено в данной возрастной группе вследствие совершенно особого иммунофенотипа опухолевых клеток [65–67] и нестабильной экспрессии антигенов под действием химиопрепаратов [68, 69]. Альтернативой может служить мониторинг МОБ путем определения химерного гена в геномной ДНК методом ПЦР-РВ. Для этого необходимо предварительно определить индивидуальную для каждого пациента точку слияния гена MLL и гена-партнера и, исходя из сиквенса зоны слияния 2 генов, подобрать комбинацию праймеров и зонда [51, 70]. Проведенное сопоставление показало хорошее совпадение результатов с данными МОБ, получаемыми при использовании IgH/TCR [70]. Наконец, в качестве мишени для оценки МОБ можно использовать химерный транскрипт в мРНК/кДНК, определяемый методом ПЦР-РВ [48, 71]. Нами в качестве основного метода для мониторирования МОБ была выбрана ПЦР с определением химерного гена MLL-EPS15 в геномной ДНК и химерного транскрипта в РНК/кДНК. Наш выбор связан с тем, что и химерные гены, и химерные транскрипты представляют собой стабильные мишени для определения МОБ [37, 48, 72], патогенетически связанные с развитием ОЛ, так как они выявляются только в опухолевых клетках. Подобранные условия ПЦР позволили нам успешно провести мониторинг МОБ во всех доступных образцах пациентов, что дало возможность оценить ответ опухоли на химиотерапию. На основании данных, получаемых при мониторинге МОБ, уже сегодня проводится стратификация пациентов с ОЛЛ, получающих терапию по большинству современных протоколов. Заключение Транслокация t(1;11)(p32;q23) при ОЛ наиболее часто выявляется у детей первого года жизни. Медиана возраста составляет 8 мес. Соотношение девочек и мальчиков 2:1. В 38 % случаев у пациентов с транслокацией t(1;11)(p32;q23) обнаружены ДХА. Наиболее часто зоной разрывов в гене EPS15 является интрон 1, в гене MLL – интроны 10 и 11. Описано 4 типа химерных транскриптов MLL-EPS15. Создана и успешно применена комбинация праймеров, флуоресцентных зондов и плазмиды для мониторирования химерного транскрипта MLLEPS15 методом ПЦР-РВ. Подобраны условия и проведен мониторинг МОБ по индивидуальным точкам разрыва в геномной ДНК у пациентов с наличием химерного гена MLL-EPS15. Сравнение результатов определения МОБ в геномной ДНК и кДНК показало высокую качественную сопоставимость (92 %). Благодарность. Авторы выражают искреннюю благодарность врачам-гематологам, детским онкологам из Областной детской клинической больницы № 1 (Екатеринбург), Республиканского научно-практического центра детской онкологии, гематологии и иммунологии (Минск), Российской детской клинической больницы (Москва), Морозовской детской городской клинической больницы (Москва), предоставившим данные о пациентах, описанных в этой статье. Л И Т Е Р А Т У Р А 1. Armstrong S., Look A. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 2005;23:6306–15. 2. Schoch C., Schnittger S., Klaus M. et al. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003;102:2395–402. 3. Meyer С., Kowarz E., Hofmann J. et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009;23:1490–9. 4. Harrison C., Cuneo A., Clark R. et al. Ten novel 11q23 chromosomal partner sites. Leukemia 1998;12:811–22. 5. Williams D., Look A., Melvin S. et al. New chromosomal translocations correlate with specific lmmunophenotypes of childhood acute lymphoblastic leukemia. Cell 1984;36:101–9. 6. Kaneko Y., Maseki N., Takasaki N. et al. Clinical and hematologic characteristics in acute leukemia with 11q23 translocations. Blood 1986;67:484–91. 7. Gregoire M., Peeters M., Bene M. et al. Karyotype, immunophenotype, and clinical outcome: correlations in childhood acute lymphoblastic leukemia. Haematol Blood Transfus 1987;30:504–8. 8. Selypes A., László A. A new translocation t(1;4;11) in congenital acute nonlymphocytic 21. Kotecha R., Ford J., Beesley A. et al. Molecular characterization of identical, novel MLL-EPS15 translocation and individual genomic copy number alterations in monozygotic infant twins with acute lymphoblastic leukemia. Haematologica 2012;97:1447–50. 22. Kotecha R., Murch A. Kees U., Cole C. Pre-natal, clonal origin of t(1;11)(p32;q23) acute lymphoblastic leukemia in monozygotic twins. Leuk Res 2012;36:46–50. 23. Nakamura H., Hata T., Tagawa M. et al. Chromosome 1 abnormalities at band 1p32 in two atomic bomb survivors with myelodysplastic syndrome. Rinsho Ketsueki 2000;41:152–8. 24. http://www.genecards.org/cgi-bin/ carddisp.pl?gene=EPS15. 25. http://www.ensembl.org/Homo_sapiens/ Gene/Summary?g=ENSG00000085832; r=1:51819935-51985000. 26. Su A., Wiltshire T., Batalov S. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 2004;101:6062–7. 27. Salcini A., Chen H., Iannolo G. et al. Epidermal growth factor pathway substrate 15, Eps15. Int J Biochem Cell Biol 1999;31:805–9. 28. Parachoniak C., Park M. Distinct recruitment of Eps15 via its coiled-coil domain is required for efficient down-regulation of the MET receptor tyrosine kinase. J Biol Chem 2009;284(13):8382–94. 29. Hess J. MLL: a histone methyltransferase disrupted in leukemia. Trends in Molecular Medicine 2004;10(10):500–7. 30. Slany R. The molecular biology of mixed lineage leukemia. Haematol 2009;94:984–93. 31. Armstrong S., Staunton J., Silverman L. et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7. 32. Ferrando A., Armstrong S., Neuberg D. et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood 2003;102:262–8. 33. Yeoh E., Ross M., Shurtleff S. et al. Classification, subtype discovery, and prediction of out-come in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002;1:133–43. 34. Ayton P., Cleary M. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes and Development 2003;17:2298–307. 35. Trentin L., Giordan M., Dingermann Th. et al. Two independent gene signatures in pediatric t(4;11) acute lymphoblastic leukemia patients. Euro J Haematol 2009;83:406–19. 36. Pallisgaard N., Hokland P., Riishoj D. et al. Multiplex reverse transcriptionpolymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood 1998;92:574–88. 37. Dongen van J., Macintyre E., Gabert J. et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Leukemia 1999;13:1901–18. 38. Fechina L., Shorikov E., Tsaur G. et al. Contribution of all-trans retinoic acid to improved early relapse-free outcome in infant acute lymphoblastic leukemia comparing to the chemotherapy alone. Blood 2007;110(11):832А; abstr. 2828. 39. ISCN 1995: An International System for Human Cytogenetic Nomenclature (1995). Ed. Mitelman F. Basel: S. Karger, 1995. 40. ISCN 2005: An International System For Human Cytogenetic Nomenclature (2005). Eds.: Shaffer L., Tommerup N. Basel: S. Karger, 2005. 41. ISCN 2009: An International System For Human Cytogenetic Nomenclature (2009). Eds.: Schaffer L., Slovak M., Campbell L. Basel: S. Karger, 2009. 42. Coenen E., Raimondi S., Harbott J. et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL -rearranged AML patients: results of an international study. Blood 2011;117:7102–11. 43. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Eds.: Swerdlow S., Campo E., Harris N. et al. Lyon, France: IARC, 2008. 44. Betts D., Ammann R., Hirt A. et al. The prognostic significance of cytogenetic aberrations in childhood acute myeloid leukaemia. A study of the Swiss Paediatric Oncology Group (SPOG). Eur J Haematol 2007;78(6):468–76. 45. Meyer C., Schneider B., Reichel M. et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci USA 2005;102(2):449–54. 46. Цаур Г.А., Наседкина Т.В., Попов А.М. и др. Время достижения молекулярной ремиссии как фактор прогноза у детей первого года жизни острым лимфобластным лейкозом. Онкогематол 2010;2:46–54. 47. Цаур Г.А., Друй А.Е., Попов А.М. и др. Возможность использования микроструйных биочипов для оценки качества и количества РНК у пациентов с онкологическими и онкогематологическими заболеваниями. Вестн Урал мед акад науки 2011;4:107–11. 48. Gabert J., Beillard E., Velden van der V. et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a Europe Against Cancer Program. Leukemia 2003;17:2318–57. 49. Beillard E., Pallisgaard N., Velden van der V. et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ ’2013 leukemia (acute myeloblastic leukemia). Hum Genet 1987;76:106–8. 9. Hagemeijer A., van Dongen J., Slater R. et al. Characterization of the blast cells in acute leukemia with translocation (4;11): report of eight additional cases and of one case with a variant translocation. Leukemia 1987;1:24–31. 10. Raimondi S., Peiper S., Kitchingman G. et al. Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 1989;73:1627–34. 11. Shippey C., Lawlor E., Secker-Walker L. Isochromosome 9q in acute lymphoblastic leukemia: a new non-random finding. Leukemia 1989;3:195–9. 12. Abshire T., Buchanan G., Jackson J. et al. Morphologic, immunologic and cytogenetic studies in children with acute lymphoblastic leukemia at diagnosis and relapse: a Pediatric Oncology Group study. Leukemia 1992;6:357–62. 13. Bernard O., Mauchauffe M., Mecucci C. et al. A novel gene, AF-1p, fused to HRX in t(1;11)(p32;q23), is not related to AF-4, AF-9 nor ENL. Oncogene 1994;9: 1039–45. 14. Felix C., Hosler M., Slater D. et al. MLL genomic breakpoint distribution within the breakpoint cluster region in de novo leukemia in children. J Pediatr Hematol Oncol 1998;20:299–308. 15. Bergh von A., Emanuel B., ZelderenBhola van S. et al. A DNA probe combination for improved detection of MLL/11q23 breakpoints by double-color interphase-FISH in acute leukemias. Genes Chromosomes and Cancer 2000;28:14–22. 16. Chessells J., Harrison C., Kempski H. et al. Clinical features, cytogenetics and outcome in acute lymphoblastic and myeloid leukaemia of infancy: report from the MRC Childhood Leukaemia working party. Leukemia 2002;16:776–84. 17. Park K., Lee D., Lee H. et al. Granulocytic Sarcoma in MLL-Positive Infant Acute Myelogenous Leukemia: Fluorescence in Situ Hybridization Study of Childhood Acute Myelogenous Leukemia for Detecting MLL Rearrangement. Am J Pathol 2001;159(6):2011–6. 18. Kim H., Cho H., Kim E. et al. A study on 289 consecutive Korean patients with acute leukaemias revealed fluorescence in situ hybridization detects the MLL translocation without cytogenetic evidence both initially and during follow-up. Br J Haematol 2002;119(4):930–9. 19. Douet-Guilbert N., Morel F., Le Bris M. et al. Rearrangement of the MLL gene in acute myeloblastic leukemia: report of two rare translocations. Cancer Genet Cytogenet 2005;157:169–74. 20. Sagawa M., Shimizu T., Shimizu T. et al. Establishment of a new human acute monocytic leukemia cell line TZ-1 with t(1;11)(p32;q23) and fusion gene MLLEPS15. Leukemia 2006;20:1566–71. 31 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 32 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) – a Europe Against Cancer Program. Leukemia 2003;17:2474–86. 50. Burmeister T., Marschalek R., Schneider B. et al. Monitoring minimal residual disease by quantification of genomic chromosomal breakpoint sequences in acute leukemias with MLL aberrations. Leukemia 2006;20:451–7. 51. Velden van der V., Cazzaniga G., Schrauder A. et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007;21:604–11. 52. Kaplan E., Meier P. Non-parametric estimation from incomplete observations. J Am Stat Assoc 1958;53:457–81. 53. Nilson I., Loechner K., Siegler G. et al. Exon/intron structure of ALL1 (MLL) gene involved in translocations to chromosomal region 11q23 and acute leukemias. Br J Haematol 1996;94(4):966–72. 54. Mitelman database of chromosome aberrations and gene fusions in cancer (2012). Mitelman F., Johansson B. Mertens F. (eds.). http://cgap.nci.nih.gov/ Chromosomes/Mitelman. 55. Huret J.-L. t(1;11)(p32;q23). Atlas Genet Cytogenet Oncol Haematol (September 2010). http:// AtlasGeneticsOncology.org/Anomalies/ t0111p32q23ID1046.html. 56. Chowdhury T., Brady H. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol Dis 2008;40:192–9. 57. Forestier E., Schmiegelow K. The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol 2006;28:486–95. 58. Zweidler-McKay P., Hilden J. The ABCs of Infant Leukemia. Curr Probl Pediatr Adolesc Health Care 2008;38(3):78–94. 59. Цаур Г.А., Флейшман Е.В., Гиндина Т.Л. и др. Характеристика перестроек 11q23/MLL при остром миелоидном лейкозе у детей первого года жизни. Клин онкогематол 2012;5(4):365–71. 60. Цаур Г.А., Попов А.М., Алейникова О.В. и др. Характеристика перестроек 11q23 (MLL) у детей первого года жизни с острым лимфобластным лейкозом. Онкогематол 2011;3:57–64. 61. Meyer C., Schneider B., Jakob S. et al. The MLL recombinome of acute leukemias. Leukemia 2006;20:777–84. 62. Цаур Г.А., Попов А.М., Наседкина Т.В. и др. Прогностическое значение минимальной остаточной болезни, определенной путем выявления химерных транскриптов у детей первого года жизни, больных острым лимфобластным лейкозом, получающих терапию по протоколу MLL-BABY. Гематол и трансфузиол 2012;57(4):12–22. 63. Jansen M., Corral L., Velden van der V. et al. Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia 2007;21:633–41. 64. Peham M., Panzer S., Fasching K. et al. Low frequency of clonotypic Ig and T-cell receptor gene rearrangements in t(4;11) infant acute lymphoblastic leukaemia and its implication for the detection of minimal residual disease. Br J Haematol 2002;117:315–21. 65. De Zen L., Bicciato S., te Kronnie G., Basso G. Computational analysis of flowcytometry antigen expression profiles in childhood acute lymphoblastic leukemia: an MLL/AF4 identification. Leukemia 2003;17:1557–65. 66. Schwartz S., Rieder H., Schlaeger B. et al. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/ CD15(+) B-cell phenotype. Leukemia 2003;17:1589–95. 67. Attarbaschi A., Mann G., König M. et al. Mixed lineage leukemia-rearranged childhood pro-B and CD10-negative pre-B acute lymphoblastic leukemia constitute a distinct clinical entity. Clin Cancer Res 2006;12:2988–94. 68. Попов А.М., Вержбицкая Т.Ю., Цаур Г.А. и др. Особенности мониторинга минимальной остаточной болезни при B-линейных острых лимфобластных лейкозах методом проточной цитометрии у детей первого года жизни. Дет онкол 2008;2:32–5. 69. Попов А.М., Вержбицкая Т.Ю., Цаур Г.А. и др. Возможности применения NG2 для мониторинга минимальной остаточной болезни методом проточной цитометрии у детей первого года жизни с острым лимфобластным лейкозом, ассоциированным с реарранжировками гена MLL. Гематол и трансфузиол 2009;6:19–22. 70. Velden van der V., Сorral L. Valsecchi M. et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia 2009;23:1073–9. 71. Jansen M., Velden van der V., Dongen van J. Efficient and easy detection of MLL-AF4, MLL-AF9 and MLL-ENL fusion gene transcripts by multiplex real-time quantitative RT-PCR in TaqMan and LightCycler. Leukemia 2005;19(11):2016–18. 72. Szczepanski T. Why and how to quantify minimal residual disease in acute lymphoblastic leukaemia. Leukemia 2007;21:622–6. 1 Иммунофенотипическая характеристика острого миелоидного лейкоза у детей первого года жизни 33 ’2013 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ А.М. Попов1, 2, Г.А. Цаур1, 2, Т.Ю. Вержбицкая1, 2, О.В. Стренева1, 2, Е.В. Шориков1, 2, Л.И. Савельев1, 2, 3, Л.Г. Фечина1, 2 1 ГБУЗ СО ОДКБ № 1, Екатеринбург; ГБУЗ СО «Институт медицинских клеточных технологий», Екатеринбург; 3 ГБОУ ВПО УГМА, Екатеринбург 2 Контакты: Александр Михайлович Попов [email protected] Цель исследования – характеристика иммунофенотипа острого миелоидного лейкоза (ОМЛ) у детей первого года жизни. В исследование было включено 90 пациентов (40 мальчиков и 50 девочек) с острым лейкозом (ОЛ) в возрасте от 0 до 11 месяцев включительно. Частота выявления ОМЛ составила 26,67 % от всех ОЛ у детей первого года жизни, что было значительно чаще, чем у более старших детей (10,83 %; р = 0,0002). Отсутствие экспрессии CD61, а также высокая экспрессия CD99, CD15, CD133 и NG2 характеризовали иммунофенотип опухолевых клеток при наличии перестроек гена MLL. Диагностическая эффективность выявления экспрессии CD99 и NG2 для прогнозирования наличия перестроек гена MLL оказалась наиболее высокой. Таким образом, иммунофенотип ОМЛ у детей младше 1 года существенно различается в зависимости от наличия или отсутствия перестроек гена MLL. Описанные особенности экспрессии антигенов позволяют прогнозировать наличие какой-либо перестройки гена MLL при ОМЛ у детей первого года жизни, при этом наибольшей диагностической эффективностью обладает наличие экспрессии CD99 или NG2. Ключевые слова: острый миелоидный лейкоз, дети первого года жизни, проточная цитометрия Immunophenotypic investigation of infant acute myeloid leukemia 1, 2 A.M. Popov , G.A. Tsaur1, 2, T.Yu. Verzhbitskaya1, 2, O.V. Streneva1, 2, E.V. Shorikov1 ,2, L.I. Saveliev1, 2, 3, L.G. Fechina1, 2 1 2 Regional Childrenʼs Clinical Hospital №1, Yekaterinburg; Research Institute of Medical Cell Technologies, Yekaterinburg; 3 Ural State Medical Academy, Yekaterinburg Aim of the study – characterization of immunophenotype in infant acute myeloid leukemia (AML). 90 patients (40 boys and 50 girls) with acute leukemia (AL) aged up to 365 days were included in the current study. AML was found more frequently in infants than in older children (26.67 % and 10.83 % respectively; p = 0.0002). Significant immunophenotypic differences were observed in patients with and without MLL gene rearrangements. Number of cases in those tumor cells expressed CD99, CD61, CD133, CD15, NG2 varied between MLL-positive and MLL-negative groups. CD61-negativity, high CD99, CD15, CD133 and NG2 expression were immunophenotypic signatures of MLLrearranged infant AML, although CD99 and NG2 had the highest diagnostic efficacy. Thus infants’ AML immunophenotype differs significantly due to the presence of MLL gene rearrangements. Diagnostic immunophenotyping of infants’ AML allows predicting presence of MLL rearrangements by either CD99 or NG2 expression. Key words: acute myeloid leukemia, infants, flow cytometry Острый миелоидный лейкоз (ОМЛ) представляет собой гетерогенную группу заболеваний, отличающихся друг от друга по морфологическим, иммунофенотипическим, цитогенетическим и молекулярногенетическим характеристикам [1, 2]. У детей пик заболеваемости ОМЛ приходится на первый год жизни. Доля пациентов этого возраста составляет по разным источникам до 20 % всех случаев ОМЛ у детей [3–5]. Среди характерных признаков ОМЛ у детей первого года жизни следует отметить относительно высокий инициальный лейкоцитоз (более 100 × 109/л) [6–9], гепатоспленомегалию, инициальный нейролейкоз [6, 9, 10–12], частые случаи хлоромы [9, 13]. Среди всех морфологических вариантов по франко-американо-британской (FAB) классификации у детей первого года жиз- ни выявляются острый миеломонобластный (ОМЛ М4), острый монобластный (ОМЛ М5) [6, 9, 14] и острый мегакариобластный лейкозы (ОМЛ М7) [6, 10, 14, 15]. Примерно в половине случаев ОМЛ у детей первого года жизни чаще всего выявляются перестройки 11q23/ MLL [4, 11, 12, 16–19]. Методами молекулярной биологии охарактеризована структура 64 химерных генов с участием MLL [20], наиболее частыми из которых при ОМЛ у детей являются MLL-MLLT3, MLL-MLLT10, MLL-ELL, MLL-MLLT11, MLL-SEPT6 (даны в порядке убывания частоты встречаемости) [16–20]. В работе S. Armstrong et al. было впервые показано, что опухолевые клетки при острых лейкозах (ОЛ), ассоциированных с перестройками гена MLL, по профилю экспрессии генов отличаются от бластов при типичных ОМЛ и остром лимфобластном лейкозе 1 ’2013 34 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ (ОЛЛ) [21]. В дальнейшем было установлено, что по профилю экспрессии генов ОЛЛ и ОМЛ, ассоциированные с перестройками гена MLL, ближе друг к другу по сравнению с ОЛЛ и ОМЛ без данных перестроек соответственно [22–24]. Таким образом, наличие перестроек гена MLL обусловливает развитие биологически совершенно особенной опухоли [21–24]. Иммунофенотип опухолевых клеток при ОЛЛ с наличием перестроек гена MLL описан достаточно детально [25–31], в то время как при ОМЛ чаще всего в качестве типичной для наличия перестроек гена MLL аберрации иммунофенотипа упоминается только экспрессия нейрогликана NG2, считающегося крайне специфичным MLL-ассоциированным антигеном [28, 32–34]. При этом комплексное описание иммунофенотипа ОМЛ у детей первого года жизни как с наличием перестроек гена MLL, так и с их отсутствием, в литературе не встречается. Цель исследования – характеристика иммунофенотипа ОМЛ у детей первого года жизни. Материалы и методы исследования Исследование проводилось в Лаборатории иммунофенотипирования гемобластозов Отдела детской онкологии и гематологии ОДКБ № 1 г. Екатеринбурга с октября 2005 по ноябрь 2012 г. Из 441 ребенка, у которых было проведено исследование первичного иммунофенотипа, 90 детей (40 мальчиков и 50 девочек) были в возрасте от 0 до 11 месяцев включительно. Эти пациенты составили исследуемую группу. Пациентам проводились: стандартное цитоморфологическое исследование, иммунофенотипирование, цитогенетическое исследование, включающее исследование методом флуоресцентной гибридизации in situ (FISH), и молекулярно-генетические исследования. Иммунофенотипирование опухолевых клеток в костном мозге производилось методом 4–6-цветной проточной цитометрии на приборах “FACS Canto” и “FACS Canto II” (Becton & Dickinson (BD), США). Настройка проточных цитометров производилась с использованием калибровочной системы “7-color Setup Beads” (BD). Мониторинг стабильности работы приборов осуществлялся при помощи калибровочных систем “Cytometer Setup and Tracking” (BD) и «DAKO Fluorospheres» (Dako, Дания). Использовались моноклональные антитела (МКАТ), меченные флуоресцеинизотиоцианатом (FITC), R-фикоэритрином (PE), перидининхлорофилл протеином (PerCP), аллофикоцианином (АРС), а также тандемными конъюгатами PE с цианином 7 (Cy7), PerCP с цианином 5.5 (Cy5.5) и APC с Су7. Для иммунофенотипирования применялись МКАТ, представленные в табл. 1. Окрашивание первично меченными МКАТ производилось согласно инструкции производителя. Результаты иммунофенотипирования оценивались при помощи программного обеспечения FACS Diva 4.0-6.1 (BD). Анализировали не менее 10 000 ядро- Таблица 1. Моноклональные антитела, применявшиеся для первичного иммунофенотипирования ОЛ Флуорохром Моноклональные антитела FITC CD58, CD45, CD99, CD7, CD7*, CD65*, CD15, CD33, CD10, CD19, CD4, CD3, MPO, CD64, CD66b, CD61, CD5, CD71 PE CD10, CD7, CD34, NG2*, CD1a, CD45, CD22, CD133**, CD13, CD8, CD58, CD20, CD79a, TdT, CD38, CD11a, CD11b, CD11c, CD2, CD235a, CD99, MPO PerCP CD19, CD20, CD8, CD45, CD34 PerCP-Cy5.5 CD20, CD33, CD38, CD19, CD79a, CD5 PE-Cy7 CD34, CD13, CD3, CD56, CD10, CD38, CD22*, CD33* APC CD19, CD117, CD3, CD133**, CD56, CD2, CD79a, CD79a*, CD10, CD11c, CD38, CD41a, TdT, IgM APC-Cy7 CD45, CD20, CD14, CD3, CD4 Примечание. * – антитела производства Beckman Coulter (США); ** – антитела производства Miltenyi Biotec (Германия); остальные антитела произведены Becton & Dickinson (США). содержащих клеток. Опухолевые клетки выделяли на точечных графиках по экспрессии CD45, значениям параметра бокового светорассеяния и экспрессии линейно-ассоциированных маркеров (CD19, CD7, CD33). Дальнейший анализ проводили при помощи гистограмм. Согласно критериям группы EGIL [35], популяцию считали позитивной, если 20 % и более клеток экспрессировали исследуемый антиген. В качестве негативного контроля использовали сохранившиеся в образце нормальные клетки. Определение иммунологических вариантов производилось также согласно критериям EGIL [35]. Всем пациентам проводилось цитогенетическое исследование клеток костного мозга и/или периферической крови, взятых до начала терапии. Применялась техника приготовления «прямых препаратов» или краткосрочное культивирование клеток (24 ч, 48 ч). Методом окраски хромосомных препаратов был GTGвариант. В большинстве случаев анализировали не менее 20 метафазных пластинок. Кариотипирование проводили в соответствии с международной номенклатурой хромосом человека ISCN 2009 [36]. Для выявления перестроек гена MLL выполняли исследование методом FISH с использованием локус-специфичного зонда LSI MLL Dual Color Break Apart Rearrangement Probe (Abbott Molecular, США). Гнездная ПЦР с обратной транскрипцией (ОТ-ПЦР) выполнялась для выявления следующих типов химерных транскриптов с участием гена MLL: MLL-AF4, MLL-MLLT1, MLL-MLLT3, MLL-MLLT4, MLL-MLLT10 по ранее описанным методикам [37–39]. У пациентов с отсутствием указанных химерных транскриптов до- Результаты исследования Распределение вариантов ОЛ, по данным иммунофенотипирования, у пациентов первого года жизни (n = 90) и более старших детей (n = 351) показано на рис. 1. У детей первого года жизни ОЛЛ выявлялся достоверно реже (р < 0,0001), а ОМЛ и острый билинейный лейкоз (ОБлЛ) – достоверно чаще (р = 0,0002 и р = 0,0359 соответственно), чем у пациентов старше 1 года. Для острого бифенотипического лейкоза (ОБфЛ) и острого недифференцированного лейкоза (ОНдЛ) значимых различий в возрастных группах обнаружено не было (р = 0,7787 и р = 0,8717 соответственно). Среди 24 детей младше 1 года с ОМЛ данные цитоморфологического исследования были доступны у 23 (95,8 %). Наиболее частыми цитологическими вариантами (по FAB-классификации) были ОМЛ М5 и ОМЛ М7 – 7 (30,4 %) пациентов с каждым вариантом соответственно; кроме того, были выявлены 3 (13,0 %) случая ОМЛ М2; 2 (8,7 %) случая ОМЛ М4 и по 1 случаю ОМЛ М0 и ОМЛ М1 (по 4,3 %). У 1 (4,3 %) пациента были определены бласты 2 типов: ОМЛ М5 и ОМЛ М6. Еще у 1 (4,3 %) ребенка опухолевые клетки имели иммунофенотип ОМЛ, в то время как при цитоморфологическом исследовании был определен ОЛЛ L1. В 14 (58,3 %) из 24 случаев были выявлены различные перестройки гена MLL. Шесть (42,9 %) пациентов имели MLL-MLLT3, 4 (28,6 %) – MLL-MLLT10, 2 (14,3 %) – MLL-MLLT11, 1 (7,1 %) – MLL-MYO1F, а у 1 (7,1 %) пациента ген-партнер гена MLL идентифицировать не удалось. Данные цитоморфологического исследования существенно различались у пациентов с наличием перестроек гена MLL и пациентов, у которых эти перестройки выявлены не были. Так, у 6 из 9 MLL(–) пациентов был определен ОМЛ М7 (66,7 %), в то время как среди MLL(+) пациентов данный цитологический вариант был обнаружен лишь в 1 (7,1 %; р = 0,0050) случае. В то же время в MLL(+) группе монобласты определялись в 8 (57,1 %) случаях, а в MLL(–) группе – не были определены ни у одного ребенка (р = 0,0061). Иммунофенотип опухолевых клеток пациентов с MLL(+) ОМЛ также существенно отличался от фенотипа бластов пациентов MLL(–). Экспрессия антигенов опухолевыми клетками пациентов с MLL(+) и MLL(–) ОМЛ приведена в табл. 2. Наши данные позволяют утверждать, что экспрессия NG2, CD99, CD15 и CD133, а также отсутствие экспрессии CD61 могут прогнозировать наличие перестройки гена MLL. Типичные примеры экспрессии антигенов представлены на рис. 2. Характеристики параметров диагностической информативности определения экспрессии CD15, CD133, CD99, CD61 и NG2 а б ОНдЛ 0,57 % ОБлЛ 3,33 % ОМЛ 26,67 % ОБфЛ 2,22 % ОБлЛ 0,28 % ОБфЛ 1,14 % ОМЛ 10,83 % ОЛЛ 67,78 % ОЛЛ 87,18 % Рис. 1. Распределение вариантов ОЛ по данным иммунофенотипирования: а – у пациентов первого года жизни (n = 90); б – у более старших детей (n = 351) ’2013 полнительно оценивали экспрессию MLL-MLLT6, MLL-MLLT11, MLL-EPS15, MLL-ELL с использованием набора “HemaVision” (DNA-Technology A/S, Дания). Для статистической обработки результатов применяли программу Statistica 6.0. Статистическая значимость различий определялась при помощи точного критерия Фишера и другого непараметрического критерия χ2. Достоверными считались различия при р < 0,05. Для оценки возможности прогнозирования наличия перестроек гена MLL для каждого иммунологического маркера рассчитывали диагностическую чувствительность, специфичность, прогностическую ценность положительного и отрицательного результатов, а также общую диагностическую эффективность теста [40]. 35 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Таблица 2. Экспрессия антигенов опухолевыми клетками пациентов с MLL(+) и MLL(–) ВП-ОЛЛ для прогнозирования наличия перестроек гена MLL представлены в табл. 3. Экспрессия CD61 характерна для острого мегакариобластного лейкоза (ОМЛ М7 по FAB-классификации). Данный вариант ОМЛ редко ассоциирован с перестройками гена MLL [41, 42], однако в исследуемой группе среди MLL(+)-пациентов был выявлен 1 случай ОМЛ М7. Девочка, 6 месяцев, поступила с выраженными признаками интоксикации, анемией, геморрагическим синдромом, увеличением печени и селезенки, умеренным лейкоцитозом. В миелограмме MLL(+)* MLL(–)* p CD13 5/14 4/9 0,5046 CD33 12/14 7/10 0,3320 CD117 5/13 3/9 0,5836 CD45 14/14 10/10 0,9999 CD34 3/14 5/9 0,1102 CD133 5/12 0/9 0,0389 CD99 10/11 1/6 0,0054 CD7 2/14 5/9 0,0517 Таблица 3. Характеристики параметров диагностической информативности определения экспрессии антигенов для прогнозирования наличия перестроек гена MLL CD19/22 3/14 0/9 0,2055 ДЧ, % Сп, % ПЦПР, % ПЦОР, % ДЭТ, % CD15 12/14 3/9 0,0166 CD65 10/12 1/4 0,0632 CD61 1/14 6/10 0,0088 NG2 11/14 0/7 0,0010 CD235a 1/14 0/9 0,6087 CD14 1/12 2/8 0,3439 CD4 11/12 5/9 0,0805 CD56 11/13 4/9 0,0642 MPO 4/14 2/10 0,5057 1 ’2013 36 Примечание. Данные представлены в формате «число позитивных пациентов / общее число пациентов, которым проводилось определение экспрессии антигена». MLL(+)* – пациенты с наличием перестроек гена MLL; MLL(–)* – пациенты, у которых перестройка гена MLL выявлена не была. Экспрессия CD133 41,7 100 100 56,3 66,7 Экспрессия CD99 90,9 83,3 90,9 83,3 88,2 Экспрессия CD15 85,7 66,7 80,0 75,0 78,3 Отсутствие экспрессии CD61 92,9 60,0 76,5 85,7 79,2 Экспрессия NG2 78,6 100 100 70,0 85,7 Экспрессия NG2 или CD99 100 87,5 93,3 100,0 95,5 Примечание. ДЧ – диагностическая чувствительность, Сп – специфичность, ПЦПР – прогностическая ценность положительного результата, ПЦОР – прогностическая ценность отрицательного результата, ДЭТ – диагностическая эффективность теста. MLL – MLL + Количество клеток MLL + CD99 CD61 NG2 MLL – CD133 CD15 Рис. 2. Типичные примеры экспрессии антигенов при ОМЛ с наличием перестроек гена MLL и без них у детей первого года жизни Обсуждение полученных результатов ОМЛ у детей первого года жизни, как и ОЛЛ, характеризуется особенной биологией опухоли [21–24]. Эти особенности обусловлены наличием перестроек гена MLL, наиболее часто встречающихся именно у пациентов младше 1 года. Однако частота встречаемости перестроек гена MLL при ОМЛ у детей первого года жизни ниже таковой при ОЛЛ [4, 11, 12, 16–18]. На протяжении длительного времени ведутся работы по поиску иммунологических маркеров, позволяющих с достаточно высокой вероятностью прогнозировать наличие молекулярно-генетических аберраций с вовлечением региона 11q23. И если типичный иммунофенотип опухолевых клеток при ОЛЛ, ассоциированном с перестройками гена MLL, описан достаточно четко [25–31], то подобных данных относительно ОМЛ сравнительно немного. Чаще всего описывается только экспрессия опухолевыми клетками при MLL(+) ОМЛ маркера NG2, считающегося очень специфичным MLL-ассоциированным антигеном [28, 32–34]. В доступной нам литературе мы не встретили работ, посвященных иммунофенотипированию ОМЛ у детей первого года жизни, как имеющих MLL-перестройки, так и без них. Обычно описывается иммунофенотип только MLL-позитивных пациентов, вне зависимости от возраста [32–34]. В нашей популяции больных выявление ОМЛ составило 26,67 % всех ОЛ у детей первого года жизни, в то время как среди ОЛ у более старших детей ОМЛ встречался значительно реже (10,83 %). Цитоморфологические варианты и иммунофенотипы ОМЛ существенно отличались внутри исследуемой группы, в зависимости от наличия/отсутствия перестроек гена MLL. Отсутствие экспрессии CD61, а также высокая экспрессия CD99, CD15, CD133 и NG2 характеризовали иммунофенотип опухолевых клеток при наличии перестроек гена MLL. Диагностическая эффективность выявления экспрессии CD99 и NG2 для прогнозирования наличия перестроек гена MLL оказалась наиболее высокой, в то время как диагностическая эффективность остальных маркеров была значительно ниже. Одновременное же определение экспрессии NG2 и CD99 позволило по наличию на опухолевых клетках хотя бы одного из этих антигенов правильно прогнозировать перестройку гена MLL у 95,5 % пациентов. Ранее было показано, что экспрессия CD15, CD133 и NG2 характерна для наличия перестроек гена MLL и при ОЛЛ [26–31], в то время как для CD99 такой зависимости выявлено не было [31]. Таким образом, ОЛЛ и ОМЛ у детей первого года жизни с наличием перестроек гена MLL имеют ряд общих особенностей иммунофенотипа. Среди пациентов с ОМЛ без перестроек гена MLL преобладал острый мегакариобластный лейкоз с характерной экспрессией CD61. Однако и среди пациентов MLL(+)-группы был выявлен 1 случай ОМЛ М7. Этот описанный выше случай является нетипичным, поскольку при данном варианте ОМЛ перестройки гена MLL обнаруживаются редко [41, 42]. Однако и при ОМЛ М7 эффективность NG2 для прогнозирования наличия перестроек гена MLL оказалась очень высока. По данным литературы, при ОМЛ у детей первого года жизни самым частым партнером гена MLL является ген MLLT3 (32,0 % случаев), несколько реже выявляются MLLT10 (23,1 %), ELL (11,6 %), MLLT4 (7,5 %), MLLT11 (4,8 %), EPS15 и MLLT1 (по 3,4 %) [20]. На долю других химерных генов приходится менее 15 % всех случаев ОМЛ у детей, но именно за счет них достигается чрезвычайно высокая вариабельность типов перестроек гена MLL, которых описано более 60 [20]. Это тем более важно, потому что прогноз при ОМЛ во многом определяется геном-партнером [43–46]. Так, роль t(9;11)(p21;q23)/MLL-MLLT3 при ОМЛ – скорее благоприятная [45], хотя это показано не для всех протоколов терапии ОМЛ у детей младше 12 месяцев [44]. В то же время наличие t(6;11)(q27;q23)/MLL-MLLT4 и t(10;11)(p12;q23)/MLL-MLLT10 ассоциировано с неблагоприятным прогнозом [46]. Поскольку в рутинной лабораторной практике методом ОТ-ПЦР чаще всего проводится определение только MLL-MLLT3, MLLT-MLLT4 и реже MLL-MLLT10, MLL-ELL [38], то существует большая вероятность «пропустить» имеющуюся перестройку гена MLL. Важным исследованием, использующимся для скрининга наличия перестроек MLL, является метод FISH ’2013 при цитоморфологическом исследовании обнаружено 82 % бластных клеток, имеющих морфологические признаки мегакариоцитарной линии дифференцировки. Во всех опухолевых клетках выявлена цитохимическая реакция на кислую фосфатазу, хлорацетат эстеразу, неспецифическую α-нафтилацетат эстеразу, в части клеток – позитивная ШИК-реакция. Реакции на миелопероксидазу и липиды (окраска суданом черным В) была отрицательна. При иммунофенотипировании определено, что все опухолевые клетки экспрессировали CD45, однако экспрессия данного маркера была снижена по сравнению с лейкоцитами. Бласты также экспрессировали CD61, CD41a, CD7, CD117, CD4, CD56 и гликофорин А (CD235a), 23,5 % опухолевых клеток также слабо экспрессировали NG2. Экспрессия других миелоидных, В- и Т-линейных антигенов, а также маркеров клеток-предшественников (CD34 и CD133) не выявлена. По совокупности данных цитологического, цитохимического исследований и иммунофенотипирования был поставлен диагноз ОМЛ М7. При цитогенетическом исследовании была определена транслокация t(9;11), а при проведении ОТ-ПЦР – химерный транскрипт MLL-MLLT3. Наличие перестройки гена MLL было также подтверждено методом FISH. Таким образом, нами было выявлено достаточно редкое сочетание перестройки гена MLL с острым мегакариобластным лейкозом. 37 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 38 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ [47–51], однако данный диагностический подход доступен только в ограниченном числе лабораторий. Таким образом, при проведении обычного рутинного определения наиболее частых молекулярногенетических нарушений у пациентов младше 1 года с ОМЛ у существенного количества MLL(+)-больных перестройки данного гена выявлены не будут. В то же время эффективность прогнозирования наличия любых перестроек гена MLL с помощью определения экспрессии CD99 и NG2 в нашем исследовании составила 95,5 %, поэтому применение данных антигенов при первичном иммунофенотипировании ОМЛ у детей первого года жизни представляется крайне необходимым. Для максимально точной оценки прогноза у пациентов с ОМЛ при обнаружении экспрессии опухолевыми клетками CD99 или NG2 следует проводить более углубленный поиск перестроек гена MLL при помощи FISH, мультиплексной ОТ-ПЦР и длинной инвертированной ПЦР, а не ограничиваться стандартным определением наиболее частых химерных генов. Выводы Частота выявления ОМЛ у детей первого года жизни составила 26,67 % всех случаев ОЛ, что было значительно чаще, чем у старших детей (10,83 %). Иммунофенотип ОМЛ у детей младше 1 года существенно различается в зависимости от наличия или отсутствия перестроек гена MLL. У детей младше 1 года с ОМЛ экспрессия NG2, CD99, CD133, CD15, а также отсутствие экспрессии CD61 позволяют с уверенностью предполагать наличие какой-либо перестройки гена MLL, при этом диагностическая эффективность одновременного определения экспрессии CD99 и NG2 наиболее высока. Л И Т Е Р А Т У Р А 1. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Swerdlow S., Campo E., Harris N.L. et al. (eds.). Lyon, France: IARC, 2008. 2. Vardiman J., Thiele J., Arber D. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;111:937–51. 3. Ross J., Davies S., Potter J., Robison L. Epidemiology of childhood leukemia, with a focus on infants. Epidemiol Rev 1994;16:243–72. 4. Pui C.-H., Kane J., Crist W. Biology and treatment of infant leukemias. Leukemia 1995;9:762–9. 5. Biondi A., Cimino G., Pieters R., Pui C.H. Biological and therapeutic aspects of infant leukemia. Blood 2000;96:24–33. 6. Webb D., Harrison C., Stevens R. et al. Relationships between age at diagnosis, clinical features, and outcome of therapy in children treated in the Medical Research Council AML 10 and 12 trials for acute myeloid leukemia. Blood 2001;98:1714–20. 7. Pui C.-H., Kalwinsky D., Schell M. et al. Acute nonlymphoblastic leukemia in infants: clinical presentation and outcome. J Clin Oncol 1988;6:1008–13. 8. Chessells J., Harrison C., Kempski H. et al. Clinical features, cytogenetics and outcome in acute lymphoblastic and myeloid leukaemia of infancy: report from the MRC Childhood Leukaemia working party. Leukemia 2002;16:776–84. 9. van Wering E., Kamps W. Acute leukemia in infants. A unique pattern of acute nonlymphocytic leukemia. Am J Pediatr Hematol Oncol 1986;8:220–4. 10. Vormoor J., Ritter J., Creutzig U. et al. Acute myelogenous leukaemia in children under 2 years – experiences of the West German AML studies BFM-78, -83 and -87. AML-BFM Study Group. Br J Cancer 1992;18(Suppl.):63–7. 11. Pui C-H., Raimondi S., Srivastava D. et al. Prognostic factors in infants with acute myeloid leukemia. Leukemia 2000;14:684–7. 12. Sorensen P., Chen C., Smith F. et al. Molecular rearrangements of the MLL gene are present in most cases of infant acute myeloid leukemia and are strongly correlated with monocytic or myelomonocytic phenotypes. J Clin Invest 1994;93:429–37. 13. Park K., Lee D., Lee H. et al. Granulocytic sarcoma in MLL-positive infant acute myelogenous leukemia: fluorescence in situ hybridization study of childhood acute myelogenous leukemia for detecting MLL rearrangement. Am J Pathol 2001;159(6):2011–6. 14. Pieters R. Biology and treatment of infant leukemias. In: Pui С.-H. (editor), Treatment of Acute Leukemias: New Directions for Clinical Research. Totowa, USA: Humana Press, 2003. Pp. 61–73. 15. Felix C., Lange B. Leukemia in infants. Oncologist 1999;4:225–40. 16. Balgobind B., Raimondi S., Harbott J. et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 2009;114:2489–96. 17. Harrison C., Hills R., Moorman A. et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 2010;28(16):2674–81. 18. Neuhoff von C., Reinhardt D., Sander A. et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 2010;28(16):2682–9. 19. Chantrain C.F., Poirel H.A. The genetic signature of acute leukemia in infancy. Hem Oncol 2010;1:1–8. 20. Meyer С., Kowarz E., Hofmann J. et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009;23:1490–9. 21. Armstrong S.A., Staunton J.E., Silverman L.B. et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genetics 2002;30:41–7. 22. Zangrando A., Dell’Orto M.C., te Kronnie G., Basso G. MLL rearrangements in pediatric acute lymphoblastic and myeloblastic leukemias: MLL specific and lineage-specific signatures. BMC Med Genom 2009;23:2–36. 23. Stam R.W., Schneider P., Hagelstein J.A.P. et al. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010;115:2835–44. 24. Qazi S., Uckun F.M. Gene expression profiles of infant acute lymphoblastic leukemia and its prognostically distinct subsets. Br J Hematol 2010;149:865–73. 25. Szczepanski T., Harrison C.J., van Dongen J.J.M. Genetic aberrations in paediatric acute leukaemias and implication for management of patients. Lancet Oncol 2010;11:880–9. 26. Borkhardt A., Vuchter C., Viehmann S. et al. Infant acute lymphoblastic leukemia – combined cytogenetic, immunophenotypical and molecular analysis of 77 cases. Leukemia 2002;16:1685–90. 27. Hrusak O., Porwit-MacDonald A. Antigen expression patterns reflecting monoclonal antibody 7.1. Leukemia 2000;14:1232–8. 35. Bene M., Castoldi G., Knapp W. et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995; 9(10):1783–6. 36. ISCN 2009: An International System Human Cytogenetic Nomenclature (2009). Eds.: Shaffer L.G., Slovak M., Campbell L. Karger, Basel, Switzerland, 2009. 37. Borkhardt A., Repp R., Haupt E. et al. Molecular analysis of MLL/AF4 recombination in infant acute lymphoblastic leukemia. Leukemia 1994;8:549–53. 38. Pallisgaard N., Hokland P., Riishoj D. et al. Multiplex reverse transcriptionpolymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood 1998;92:574–88. 39. Цаур Г.А., Наседкина Т.В., Попов А.М. и др. Время достижения молекулярной ремиссии как фактор прогноза у детей первого года жизни с острым лимфобластным лейкозом. Онкогематол 2010;2:46–54. 40. Weinstein S., Obuchowski N.A., Lieber M.L. Clinical evaluation of diagnostic tests. Am J Roentgenol 2005;184:14–9. 41. Bernstein J., Dastugue N., Haas O. et al. Nineteen cases of the t(1;22)(p13;q13) acute megakaryoblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 2000;14:216–8. 42. Матвеева Е.А., Казакова А.Н., Калинина И.И. и др. Методы молекулярной цитогенетики для диагностики острого мегакариобластного лейкоза. Онкогематол 2012;2:51–6. 43. Hilden J., Smith F., Frestedt J. et al. MLL Gene Rearrangement, Cytogenetic 11q23 Abnormalities, and Expression of the NG2 Molecule in Infant Acute Myeloid Leukemia. Blood 1997;89(10):3801–5. 44. Kawasaki H., Isoyama K., Eguchi M. et al. Superior outcome of infant acute myeloid leukemia with intensive chemotherapy: results of the Japan Infant Leukemia Study Group. Blood 2001;98(13):3589–90. 45. Rubnitz J., Raimondi S., Tong X. et al. Favorable Impact of the t(9;11) in Childhood Acute Myeloid Leukemia. J Clin Oncol 2002;20:2302–9. 46. Balgobind B., Zwaan C., Pieters R., van den Heuvel-Eibrink M. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia 2011;25:1237–48. 47. Thirman M., Gill H., Burnett R. et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. New Eng J Med 1993;329:909–14. 48. Cuthbert G., Thompson K., Breese G. et al. Sensitivity of FISH in detection of MLL translocations. Genes Chromosomes Cancer 2000;29:180–5. 49. von Bergh A., Emanuel B., ZelderenBhola van S. et al. A DNA probe combination for improved detection of MLL/11q23 breakpoints by double-color interphaseFISH in acute leukemias. Genes Chromosomes Cancer 2000;28:14–22. 50. Dyson M., Talley P., Reilly J. et al. Detection of cryptic MLL insertions using a commercial dual-color fluorescence in situ hybridization probe. Cancer Genet Cytogenet 2003;147:81–3. 51. Цаур Г.А., Плеханова О.М., Гиндина Т.Л. и др. Применение метода флуоресцентной гибридизации in situ для выявления перестроек гена MLL при острых лейкозах у детей первого года жизни. Мед ген 2012;7:35–43. ’2013 genotype of acute leukemias. Leukemia 2002;16:1233–58. 28. De Zen L., Bicciato S., te Kronnie G., Basso G. Computational analysis of flow cytometry antigen expression profiles in childhood acute lymphoblastic leukemia: an MLL/AF4 identification. Leukemia 2003;17:1557–65. 29. Schwartz S., Reider H., Schlager B. et al. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangements and a CD10–/CD24–/CD65s+/CD15+ B-cell immunophenotype. Leukemia 2003;17:1589–95. 30. Attarbaschi A., Mann G., Konig M. et al. Mixed Lineage Leukemia – rearranged childhood pro-B and CD10-negative pre-B acute lymphoblastic leukemia constitute a distinct clinical entity. Clin Cancer Res 2006;15(10):2988–94. 31. Попов А.М., Цаур Г.А., Вержбицкая Т.Ю. и др. Иммунофенотипическая характеристика острого лимфобластного лейкоза у детей первого года жизни. Онкогематол 2012;2:16–23. 32. Smith F.O., Rauch C., Williams D.E. et al. The human homologue of rat NG2, a chondroitin sulphate chromosome band 11q23blasts from poor-prognosis patients with abnormalities of hematopoietic cells but is expressed by acute myeloid leukemia proteoglycan, is not expressed on the cell surface of normal. Blood 1996;87:1123–33. 33. Mauvieux L., Delabesse E., Bourquelot P. et al. NG2 expression in MLL rearranged acute myeloid leukaemia is restricted to monoblastic cases. Br J Haematol 1999;107:674–6. 34. Wuchter C., Harbott J., Schoch C. et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using 39 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Отдаленные результаты спленэктомии при первичном миелофиброзе 1 ’2013 40 Л.М. Мещерякова1, Л.Г. Ковалева1, С.Р. Карагюлян1, Л.Ю. Колосова1, О.В. Пороткова2, Е.А. Семенова1 1 ФГБУ ГНЦ Минздрава России, Москва; БУЗ ВО «Воронежская областная клиническая больница № 1» 2 Контакты: Людмила Михайловна Мещерякова [email protected] Приведена оценка ближайших и отдаленных результатов спленэктомии в зависимости от соблюдения показаний и противопоказаний у 18 больных первичным миелофиброзом (ПМФ). Отмечено уменьшение интра- и послеоперационных осложнений при соблюдении показаний и противопоказаний к спленэктомии у пациентов с ПМФ, а также при проведении профилактики тромботических осложнений гепарином. Продолжительность жизни больных после спленэктомии увеличена при проведении в последующем соответствующего специфического лечения. Ключевые слова: первичный миелофиброз, спленэктомия, результаты Long-term results of splenectomy in primary myelofibrosis 1 L.M. Meshcheryakova , L.G. Kovaleva1, S.R. Karagyulyan1, L.Yu. Kolosova1, O.V. Porotkova2, Ye.A. Semenova1 1 Hematologic Scientific Center, Ministry of Health of Russia, Moscow; 2 Voronezh Regional Clinical Hospital № 1 The estimation of immediate and long-term results after splenectomy depending on indications and contra-indications for 18 primary myelofibrosis (PMF) patients was shown. The reduction of intra- and postoperative complications in PMF patients was noted if the operation was performed under strict indications and also with prophylactic heparin therapy. Life expectancy is extended after splenectomy during subsequent relevant specific treatment. Key words: primary myelofibrosis, splenectomy, results Первичный миелофиброз (ПМФ) впервые был описан более 100 лет назад. К настоящему времени показано, что при ПМФ имеет место поражение стволовых полипотентных кроветворных клеток с дальнейшей неуправляемой пролиферацией клеток-предшественников миелопоэза с повышенной продукцией факторов, стимулирующих рост фибробластов и синтез коллагена, а именно пластиночный дериват фактора роста, который затем трансформируется в усиливающий рост фибробластов фактор b и эпидермальный ростовой фактор. Фактор b фибробластов усиливает синтез коллагенов I и III типов, фибронектина, секрецию компонентов экстрамедуллярного матрикса, который принимает непосредственное участие в образовании фиброзной ткани. Миелоидная пролиферация поддерживается за счет действия колониестимулирующего фактора-1 (КСФ-1) и гранулоцитарномакрофагального колониестимулирующего фактора. У больных ПМФ отмечается положительная корреляция содержания КСФ-1 с размером селезенки. У большинства больных ПМФ основными клиническими проявлениями являются спленомегалия, недостаточность костномозгового кроветворения с угнетением эритро- и тромбоцитопоэза. В течение болезни селезенка у 70 % пациентов достигает гигантских размеров и занимает более половины брюшной полости, что приводит к развитию компрессионного синдрома, инфарктов селезенки, периспленитов, спонтанных разрывов, синдрома мальабсорбции, катаболического синдрома, анемии и тромбоцитопении, как аутоиммунного, так и гиперспленического характера. Консервативная терапия становится неэффективной, проведение ее невозможно из-за цитопенического синдрома. Возникает вопрос о проведении спленэктомии, известной с 1871 г. [1–3]. Целью настоящего исследования явилась оценка ближайших и отдаленных результатов лечения больных ПМФ с включением спленэктомии в зависимости от соблюдения предложенных показаний и противопоказаний. Материалы и методы В поликлиническом и хирургическом отделениях Гематологического научного центра (ГНЦ) Минздрава России (Москва) в 2003 г. проанализировано 68 пациентов с ПМФ, которым произведена спленэктомия, которая не являлась основным методом лечения и применялась в программе терапии по следующим показаниям: спленомегалия с компрессионным синдромом при невозможности применения средств, сокращающих размеры селезенки, однако без признаков бластной трансформации; аутоиммунная гемолитическая анемия, рефрактерная к иммунодепрессивной терапии, или при значительной секвестрации эритро- ной рецидивирующее кровотечение из вен пищевода с последующей гастротомией и прошиванием вен пищевода, у 1 пациентки – варикозное расширение вен пищевода III степени и опасность кровотечения, у 2 – тромбоз v. lienalis и мезентериальных сосудов); спленомегалия, не купируемая цитостатической терапией, была показанием к спленэктомии у 9 больных; аутоиммунная анемия в сочетании с тромбоцитопенией – у 5 пациентов. К моменту операции давность заболевания до 1 года была у 2 больных, от 1 года до 7 лет – у 9, от 8 до 9 лет – у 7 пациентов. В предоперационном периоде селезенка была увеличена у всех больных; гигантская селезенка, занимавшая более половины брюшной полости и спускавшаяся в малый таз, отмечена у 70 % пациентов. Увеличение печени было отмечено у всех больных. Асцит был у 5 больных, варикозное расширение вен пищевода и желудка также у 5 больных. Результаты и обсуждение Трепанобиопсия в процессе предоперационного обследования была сделана в 14 случаях. По результатам гистологического исследования у 9 больных (1-я группа) показанием к спленэктомии была спленомегалия (спленомегалический вариант ПМФ); у 5 больных – цитопения (тромбоцитопения, анемия; цитопенический вариант). У 2 (25 %) больных 1-й группы была снижена клеточность костного мозга, у остальных, несмотря на диффузный фиброз, содержание элементов 3 ростков миелопоэза было близко к норме (рис. 1). У больных с цитопеническим вариантом определялся остеомиелосклероз с резким снижением клеточности костного мозга (рис. 2, 3). При гистологическом исследовании оперативно удаленных селезенок выявлена редукция белой пульпы, особенно у больных со спленомегалическим вариантом, инфильтрация элементами 3 ростков гемопоэза. Эти изменения имели разную степень выраженности: Рис. 1. Трепанобиоптат подвздошной кости. Объектив 200. ПМФ, клеточная стадия (наблюдается в первые годы развития заболевания) ’2013 цитов в селезенке, не сокращаемой цитостатической терапией; глубокая аутоиммунная тромбоцитопения (тромбоциты менее 30 × 109/л) с геморрагическим синдромом или без него, рефрактерная к глюкокортикоидам и препятствующая проведению адекватной цитостатической терапии. К противопоказаниям отнесены: бластная трансформация; быстрое прогрессирование процесса на стадии предвестников бластной трансформации, особенно с анемическим синдромом и выраженным фиброзом костного мозга, интоксикацией; наличие тяжелых некомпенсируемых сопутствующих заболеваний [4]. Лейкоцитоз и умеренный тромбоцитоз (менее 1000 × 109/л) не являются абсолютными противопоказаниями при условии адекватной цитостатической подготовки. В процессе выполнения работы мы анализировали послеоперационное течение ПМФ в зависимости от учета показаний и противопоказаний при выполнении спленэктомии. Тридцать четыре больных, оперированных в соответствии с выработанными показаниями, имели значительно более высокую выживаемость, чем те больные, которым спленэктомию проводили как экстренную диагностическую процедуру без учета вышеперечисленных противопоказаний. Медиана выживаемости пациентов, оперированных с соблюдением предложенных показаний и противопоказаний, составила 154 мес, без их учета – 50 мес. Медиана дожития после спленэктомии, выполненной с учетом показаний, составила 50 мес, а без учета – 8 мес. В раннем послеоперационном периоде умерло 7 человек. Причины смерти были следующие: аспирационная пневмония, абсцесс поджелудочной железы, инсульт (2 больных), тромбоэмболия легочной артерии (3 больных) [5]. В течение 6 мес после спленэктомии умерло еще 6 пациентов. Причины смерти: у 50 % больных прогрессирование заболевания в виде бластной трансформации с поражением клапанов сердца, перикарда, легких; печеночная недостаточность. В последующие годы: 60 % больных прожили более 5 лет, все умерли от бластного криза. От инфекционных осложнений, хронической печеночной недостаточности умерли 40 % больных. Продолжительность жизни после спленэктомии определялась вариантом заболевания. Так, у больных спленомегалическим вариантом показанием к операции был компрессионный синдром. Продолжительность жизни после операции составила 48 ± 4 мес. Показанием к спленэктомии у пациентов с тромбоцитопеническим и анемическим вариантами была цитопения. В данной группе продолжительность жизни после операции составила 68 ± 2 мес. Мы провели анализ историй болезни 18 больных ПМФ (12 женщин и 6 мужчин), которым была выполнена спленэктомия: в возрасте от 20 до 49 лет – 7 больных, от 50 до 70 лет – 11 пациентов. Показаниями к операции были: у 4 больных хирургические (у 1 боль- 41 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 42 Рис. 2. Трепанобиоптат подвздошной кости. Объектив 200. ПМФ, стадия фиброза во всех полях зрения обнаруживались наряду с лимфоидными и гистиоцитарными элементами мелкие группы эритрокариоцитов, отдельные мегакариоциты, уродливые и голоядерные, местами образующие кластеры. В части удаленных селезенок найдены склеропигментные узелки, свидетельствующие о длительности процесса, а также свежие и организующиеся инфаркты. Наблюдался фиброз ткани селезенки, причем при спленомегалическом варианте грубоволокнистый диффузный, а при цитопеническом варианте – очаговый (рис. 4, 5). У 6 больных в воротах удаленной селезенки обнаруживали лимфатические узлы. В большинстве наблюдений при обоих вариантах рисунок строения лимфатического узла был сохранен, в расширенных синусах выявлялась пролиферация кроветворных элементов на всех стадиях созревания, проникающих из синуса в ткань узла (рис. 6). Рис. 3. Трепанобиоптат подвздошной кости. ПМФ, стадия остеомиелосклероза Рис. 5. Фрагмент предшествующего препарата. Объектив 200 Рис. 4. Биоптат оперативно удаленной селезенки при ПМФ. Объектив 400 Рис. 6. Лимфатический узел ворот селезенки при ПМФ. Объектив 400 по 3 млн МЕ ежедневно в течение 2 нед, затем по 3 млн МЕ 3 раза в нед. У 1 пациентки указанная цитостатическая терапия была неэффективна, что потребовало назначения милерана по 4 мг в сутки до суммарной дозы 450 мг. Лейкоцитоз от 23 × 109/л до 12 × 109/л снижался под действием цитостатической терапии. Анемия оставалась у 3 больных, что потребовало трансфузий эритроцитарной массы в течение 4–6 нед после операции с одновременным продолжением терапии рекомбинантными эритропоэтинами (эпрекс, рекормон) в дозе 30–40 тыс. в неделю до 10–12 мес [7]. Продолжительность жизни больных, оперированных по хирургическим показаниям (тромбозы v. lienalis, инфаркты селезенки, разрывы селезенки), составила в среднем 78 ± 4 мес. Двум пациентам проводили адекватную предоперационную подготовку, осложнения в ранний послеоперационный период вовремя купировали и в дальнейшем назначали сочетанную терапию гидреа в дозе 1500 мг в сутки и реаферон в дозе 3 млн МЕ 3 раза в неделю, а также прямые антикоагулянты (гепарин 10 000 ЕД в сутки в течение 7 дней, затем фраксипарин по 0,3 мл 2 раза в сутки в течение 4 мес с последующим переводом на сулодексид по 1 капсуле (250 ЛЕ) 2 раза в сутки). Также назначали дезагреганты: тромбо асс 100 мг и/или плавикс 75 мг в сутки [8]. Продолжительность жизни больных после операций, проведенных по поводу спленомегалии с компрессионным синдромом, составила 46 ± 7 мес. До спленэктомии больным с тромбоцитозом проводили цитостатическую терапию до снижения уровня тромбоцитов до 300 × 109/л с целью уменьшения постоперационных тромботических осложнений. При этом в послеоперационный период цитостатическую терапию продолжали, к гидреа добавляли препараты α-интерферона в стандартных дозировках, с одновременной терапией антикоагулянтами и дезагрегантами. В настоящее время, спустя 36 мес, 7 больным продолжается сочетанная терапия, 2 пациентам проводится терапия только гидреа [9]. Больные, оперированные в связи с цитопеническим синдромом, прожили 37 ± 6 мес. До спленэктомии больным анемией проводили терапию преднизолоном, ретаболилом, рекомбинантным эритропоэтином в дозе 12 000–30 000 МЕ в нед, трансфузии эритроцитарной массы. При исследовании с радиоактивным хромом у 3 больных выявлено преимущественное разрушение эритроцитов в селезенке. Положительная проба Кумбса была у 2 больных. В послеоперационный период у 1 больной концентрация гемоглобина увеличилась до 87 г/л, противоанемическая терапия включала в себя витамины группы В и фолиевую кислоту. Остальным 4 больным в 1-й месяц после операции продолжали выполнять трансфузии эритроцитарной массы с одновременным введением рекомбинантных эритропоэтинов в дозе 30 000 ЕД в нед, 2 больным в сочетании с препаратами α-интерферона 3 млн МЕ 3 раза в нед с последующим назначением одному ’2013 Во всех биоптатах печени изменения были однотипными: при сохранной дольковой структуре, незначительная лимфоидная и гистиоцитарная инфильтрация в портальных трактах, умеренная дистрофия гепатоцитов. В синусоидных капиллярах обнаружены мелкие группы клеток эритро- и гранулоцитопоэза, отдельные мегакариоциты. Между выраженностью миелоинфильтрации селезенки и печени корреляции нет. В печени она очень умеренная. В предоперационный период в связи с тромбоцитозом и спленомегалией 12 больных принимали гидреа (1000–2000 мг в сутки). Препараты α-интерферона вводили 5 больным в дозе 3 млн МЕ 5 раз в нед (трем из них в сочетании с гидреа). Эритропоэтин получали 3 больных в дозе 30 000 ЕД в нед, преднизолон – 2 больных в дозе 40 мг в сутки в связи с тромбоцитопенией и аутоиммунной гемолитической анемией [6]. В связи с опасностью тромботических осложнений в ближайший и ранний послеоперационный период все больные профилактически получали терапию гепарином в дозе 10 000–12 000 ЕД в сутки и трансфузии свежезамороженной плазмы в первые 3–10 дней после операции (в среднем по 1000 мл). В раннем послеоперационном периоде у 40 % больных наблюдались следующие осложнения: рецидивирующее кровотечение из ложа удаленной селезенки, потребовавшее 2 повторных лапаротомий у 1 больной; у 3 пациентов – нижнедолевая левосторонняя пневмония и у 3 – реактивный панкреатит. У остальных пациентов ранних послеоперационных осложнений не было. В позднем послеоперационном периоде у всех больных была купирована тромбоцитопения, в отдельных случаях наблюдался тромбоцитоз до 2000 × 109/л. Отмечены также изменения функциональных свойств тромбоцитов в виде снижения агрегации с адреналином, появления макроагрегатов, повышения скорости агрегации тромбоцитов. В плазменном звене гемостаза выявлены: укорочение активированного частичного тромбопластинового времени, положительный этаноловый тест, замедление фибринолиза, которое сохранялось длительно и в отдаленном послеоперационном периоде. В связи с выраженными коагулологическими изменениями больным проводили исследование генетических факторов тромбофилии. У больного, у которого в постоперационном периоде развилась тромботическая окклюзия ствола портальной и верхней брыжеечной вен с асцитом, при исследовании маркеров тромбофилии выявлено гомозиготное наследование в 2 аллелях гена PAI-1 и гетерозиготная мутация в гене метилентетрагидрофолатредуктазы. В связи с гипертромбоцитозом у всех оперированных больных дозу гидреа сначала увеличивали до 3000 мг в сутки, но из-за развития токсических осложнений в виде анорексии, рвоты, диареи, повышения уровня трансаминаз в 1,5 раза дозу снижали до 2000 мг в сутки и сочетали с введением препаратов α-интерферона 43 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 44 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1500 мг гидреа ежедневно в связи с ростом тромбоцитоза. У 1 больного зависимость от гемотрансфузий значительно уменьшилась, но через год после спленэктомии имеются признаки бластной трансформации. После спленэктомии у больных наблюдался тромбоцитоз и гипертромбоцитоз, максимально до 5000 × 109/л. Тромбоцитопения является неблагоприятным признаком фазы бластной трансформации, которая является причиной смерти больных после спленэктомии в отдаленные сроки. В остальных случаях причинами смерти были: инфаркт миокарда, сепсис, не купируемая эритропоэтинами анемия. Таким образом, спленэктомия не излечивает ПМФ, но улучшает качество жизни больных, влияя на физическое, психологическое и социальное функционирование больного. В связи с ликвидацией компрессионного синдрома исчезают боли, приступы почечной колики, явления пиелонефрита, цистита, нормализуется работа кишечника, имеет место прибавка массы тела. Больные становятся более активными, исчезает чувство страха из-за угрозы разрыва селезенки. В исследовании влияния эритропоэтина на параметры качества жизни в ходе лечения отмечено уменьшение и исчезновение зависимости от гемотрансфузий, повышение активности повседневной жизни. В заключение следует отметить, что у всех анализируемых нами больных, которым спленэктомия была проведена по показаниям, не наблюдалось интраоперационных осложнений. Применение прямых антикоагулянтов в достаточных дозах и длительно, назначение цитостатической терапии в ранний послеоперационный период позволили избежать таких жизнеугрожающих осложнений, как тромбоэмболия легочной артерии и инфаркт миокарда. После спленэктомии возможна коррекция глубокой анемии, связанной с недостаточностью кроветворения вследствие остеомиелофиброза и остеомиелосклероза костного мозга при длительном применении рекомбинантных эритропоэтинов в адекватных дозах. В качестве примера адекватной тактики выполнения спленэктомии при ПМФ, проведенной по строгим показаниям, приводим историю болезни больного З., 64 лет, который обратился в поликлинику по месту жительства в декабре 2001 г. с жалобами на одышку при незначительной физической нагрузке и слабость. В анализе крови при обследовании в поликлинике: гемоглобин 92 г/л, эритроциты 3,7 × 1012/л, цветной показатель 0,75, ретикулоциты 16 0/00, лейкоциты 3,8 × 10 9/л, палочкоядерные нейтрофилы 2 %, сегментоядерные нейтрофилы 52 %, эозинофилы 2 %, лимфоциты 35 %, моноциты 9 %, СОЭ 23 мм/ч. Диагностирована железодефицитная анемия. Проводилось лечение препаратами железа. Несмотря на проводимое в течение 4 мес лечение, состояние больного ухудшалось, в повторном анализе крови: гемоглобин 51 г/л, лейкоциты 3,4 × 109/л, тромбоциты 55 × 10 9/л. Госпитализирован в гематологическое отделение с диагнозом панцитопения неясного генеза, выявлена сплено- мегалия 190 × 83 мм, сниженная клеточность костного мозга с остеомиелосклерозом. Установлен диагноз ПМФ и сопутствующее заболевание – желчекаменная болезнь. Проводилась терапия трансфузиями эритроцитарной массы, преднизолоном 40 мг в сутки. Анемический синдром частично купирован, но больной оставался зависим от гемотрансфузий, развился гемолитический синдром, резистентный к кортикостероидной терапии, и глубокая тромбоцитопения до 25 × 10 9/л. В ГНЦ 25.07.2002 проведены спленэктомия и холецистэктомия, биопсия печени. Операционный и послеоперационный периоды протекали без осложнений. Микроскопически в селезенке определялись немногочисленные фолликулы небольших размеров, сформированные малыми лимфоцитами. Красная пульпа очагово полнокровна и неравномерно клеточна. Наряду с малыми лимфоцитами выявлялись очаговые скопления эритроидных клеток с выраженными признаками дисплазии, промежуточных и незрелых клеток гранулоцитопоэза и отдельных мегакариоцитов. Указанные клетки локализовались преимущественно в пульпарных тяжах и местами в синусах (см. рис. 6). В печени имелась дискомплексация печеночных балок, участки фиброза, дистрофические изменения гепатоцитов. В синусоидных капиллярах были скопления клеток эритро- и гранулоцитопоэза, единичных дистрофически измененных мегакариоцитов. В желчном пузыре картина хронического холецистита с очагами обострения. В течение последующих 3 мес после операции у больного сохранялась тяжелая анемия (гемоглобин 69–75 г/л), он получал трансфузии эритроцитарной массы (3–4 раза в мес) и рекормон в дозе 10 000–20 000 ЕД в нед. Учитывая низкую эффективность лечения, больному проведен курс иммуносупрессивной терапии циклоспорином А в дозе 300 мг в сутки, суммарно – 4200 мг. В результате отмечено стойкое повышение гемоглобина до 92 г/л, эритроциты 3,2 × 10 12/л, ретикулоциты 110/00, тромбоциты 259 × 10 9/л, лейкоциты 4,7 × 10 9/л. Затем, в связи с побочными явлениями, терапия циклоспорином А была прервана, а доза рекормона увеличена до 30 000 ЕД в нед. На этой терапии больной находился в течение 24 мес, трансфузии не проводили. При обследовании в сентябре 2004 г.: в анализе крови – гемоглобин 95 г/л, эритроциты 3,35 × 10 12/л, тромбоциты 141 × 10 9/л, ретикулоциты 15 0/00, лейкоциты 7,2 × 10 9/л, бласты 1 %, миелоциты 8 %, метамиелоциты 2 %, палочкоядерные нейтрофилы 16 %, сегментоядерные нейтрофилы 30 %, лимфоциты 32 %, моноциты 11 %, нормобласты 190:100, СОЭ 32 мм/ч, тельца Жолли. В костном мозге эритроидный росток составлял 70,8 %, в трепанобиоптате картина диффузного миелофиброза. С 2004 г. на 3 года терапии доза реаферона уменьшена до 9 млн МЕ в неделю для поддержания субнормального уровня гемоглобина. С октября 2007 г. отмечено ухудшение состояния, снижение гемоглобина до 80 г/л, в трепанобиоптате нарастание явлений остеосклероза и угнетение миелопоэза. Возобновлена терапия рекормоном в дозе 30 000 ЕД в неделю, которая гепарин 10 000 ЕД в сутки и тромбо асс 100 мг в сутки. Больная была выписана на 15-е сутки после операции в удовлетворительном состоянии. Наблюдалась гематологом по месту жительства, сохранялась умеренная анемия (гемоглобин 103 г/л, эритроциты 3,9 × 10 12/л, ретикулоциты 15 0/00, лейкоциты 13,5 × 10 9/л, тромбоцитоз 670–850 × 10 9 /л, поэтому было отменено лечение гидреа и назначен продимин по 100 мг в день, на курс – 8000 мг. Под влиянием проведенной терапии количество тромбоцитов снизилось до 300 × 10 9/л, лейкоцитов до 6,0 × 10 9/л. В связи со снижением гемоглобина до 85 г/л с апреля 2008 г. больной проводится терапия рекормоном по 10 000 ЕД 3 раза в нед, уровень гемоглобина сохраняется в пределах 105–110 г/л. В настоящее время самочувствие и качество жизни удовлетворительные. Таким образом, своевременно проведенная спленэктомия позволила избежать компрессионных осложнений со стороны органов брюшной полости и избавить больную от 2-го злокачественного новообразования. Назначенная цитостатическая терапия позволит сдерживать миелопролиферативный процесс в печени и костном мозге. Наш небольшой опыт подтверждает возможность лечения больных ПМФ препаратами, воздействующими на ангиогенез (леналидомид, талидомид). В результате их применения сокращается миелопролиферация и фиброз в костном мозге, печени и селезенке. В связи с этим предполагается лечение этими препаратами больных с выраженной спленомегалией, у которых невозможна спленэктомия из-за наличия стадии акселерации, а также пациентов с развившейся гепатомегалией после спленэктомии при возникшей резистентности к цитостатическим препаратам. Использование этих лекарственных средств может ограничить применение спленэктомии. Л И Т Е Р А Т У Р А 1. Бутояну Е. Миелоидная метаплазия с миелосклерозом. В кн.: Клиническая гематология (под ред. Ш.Т. Берчану). Изд-во «Бухарест», 1985. С. 672–693. 2. Демидова А.В., Хорошко Н.Д. Сублейкемический миелоз. В кн.: Руководство по гематологии (под ред. А.И.Воробьева). М.: Ньюдиамед, 2007. С. 276–280. 3. Демидова А.В. Хронический идиопатический миелофиброз. В кн.: Клиническая онкогематология (под ред. М.А. Волковой). М.: Медицина, 2007. С. 616–630. 4. Ковалева Л.Г., Карагюлян С.Р., Колосова Л.Ю. и др. Спленэктомия при сублейкемическом миелозе. Гематол и трансфузиол 2004;49(5):14–21. 5. Ковалева Л.Г., Мещерякова Л.М., Колосова Л.Ю. Профилактика и лечение тромбозов при хронических миелопролиферативных заболеваниях. V съезд гематологов и трансфузиологов Украины с международным участием «Итоги и перспективы развития гематологии и трансфузиологии в Украине», Украина, Винница, 20–22 мая 2008 г. В сб.: Гематология и переливание крови 2008;1:197–200. 6. Мещерякова Л.М., Ковалева Л.Г., Колосова Л.Ю. Сублейкемический миелоз – течение и лечение пожилых больных. Научно-практическая конференция «Заболевания крови у пожилых людей: диагностика, лечение, особенности иммуносупрессии», Москва, 25 марта 2004 г. В сб.: Заболевания крови у пожилых людей: диагностика, лечение, особенности иммуносупрессии, 2004. С. 42–45. 7. Колосова Л.Ю., Ковалева Л.Г., Мещерякова Л.М. Лечение анемической формы сублейкемического миелоза. Научно-практическая конференция «Заболевания крови у пожилых людей: диагностика, лечение, особенности иммуносупрессии», Москва, 25 марта 2004 г. В сб.: Заболевания крови у пожилых людей: диагностика, лечение, особенности иммуносупрессии, 2004. С. 45–47. 8. Мещерякова Л.М. Система гемостаза у больных миелопролиферативными заболеваниями с тромбоцитозом при различных режимах терапии. Конференция «Атеротромбоз – проблема современности», Москва, 24–26 марта 1999 г. В сб.: Атеротромбоз – проблема современности, 1999. С. 81–82. 9. Мещерякова Л.М., Ковалева Л.Г. Патофизиологические основы лечения сублейкемического миелоза. В кн.: Патофизиология крови, экстремальные состояния (под ред. акад. А.И. Воробьева и проф. Н.А. Горбуновой). М.: Бивитэк, 2004. С. 122–135. ’2013 продолжается в сочетании с препаратами α-интерферона до настоящего времени. Таким образом, в результате проводимой терапии, включающей спленэктомию с последующей терапией рекомбинантным эритропоэтином в сочетании с препаратами α-интерферона, получена стойкая клиникогематологическая ремиссия. Больная Г., 55 лет. Диагноз ПМФ установлен в 2004 г. на основании спленомегалии (размер селезенки по данным ультразвукового исследования 23 × 10 см); изменений в гемограмме в виде умеренной нормохромной анемии: гемоглобин 105 г/л, эритроциты 3,5 × 1012/л, ретикулоциты 12 0/00, лейкоциты 15,9 × 10 9/л, сдвиг в лейкоформуле до миелоцитов; гистологического анализа костного мозга. Костная ткань с явлениями новообразования в виде напластования остеоида на старую кость. Костномозговые полости сужены, заполнены фиброзной тканью. На этом фоне видны в увеличенном числе деформированные мегакариоциты и группы клеток эритро- и гранулопоэза. Проводили терапию гидреа в дозе 1000 мг в сутки. Несмотря на проводимую терапию, наблюдалось прогрессивное увеличение размеров селезенки. В феврале 2006 г. в связи с немотивированным похуданием (на 5 кг за 2 мес), углублением анемии до 90 г/л, изменением характера анемии с нормохромного на гипохромный произведена гастроскопия, при которой выявлен неопластический процесс в желудке. 18.07.2006 в ГНЦ проведена операция: расширенная гастрэктомия и спленэктомия – удалена селезенка массой 4500 г. Гистологическое заключение: в желудке картина перстневидноклеточного рака. В селезенке и лимфатических узлах изменения, характерные для ПМФ. Постоперационный период осложнился тромбоцитозом до 1200 × 10 9/л, что потребовало увеличения дозы гидреа до 2000 мг в сутки. Был назначен 45 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Современные подходы к лечению цитомегаловирусной инфекции у онкогематологических больных 1 ’2013 46 Д.Н. Балашов ФГБУ ФНКЦ ДГОИ им. Дмитрия Рогачева Минздрава России, Москва Контакты: Дмитрий Николаевич Балашов [email protected] Проблема цитомегаловирусной (ЦМВ) инфекции является чрезвычайно серьезной и актуальной у иммунокомпрометированных пациентов. Препаратом, рутинно используемым для контроля ЦМВ-инфекции в данной когорте пациентов, является ганцикловир. К сожалению, такие проблемы, как миелотоксичность и развитие лекарственной резистентности, нередко ограничивают возможность его применения. Хорошей альтернативой является валганцикловир, эффективность которого подтверждена в клинических исследованиях. Проблема контроля ЦМВ-инфекции не является решенной, даже несмотря на внедрение в практику высокоэффективных методов ранней диагностики и упреждающей терапии. Использование иммуноаблативных препаратов (таких как алемтузумаб или антитимоцитарный глобулин), а также трансплантация гемопоэтических стволовых клеток от альтернативных доноров являются факторами высокого риска развития жизнеугрожающих висцеральных ЦМВ-ассоциированных осложнений. Новые методы контроля ЦМВ активно исследуются в настоящее время. В частности, одним из перспективных направлений является культивирование и клиническое использование ЦМВ-специфических Т-лимфоцитов. Ключевые слова: цитомегаловирусная инфекция, упреждающая терапия, токсичность, лекарственная резистентность, иммунокомпрометированные пациенты Trends in the treatment of cytomegalovirus infection in oncohematological patients D.N. Balashov Dmitriy Rogachev Federal Research Center of Pediatric Hematology, Oncology and Immunology, Ministry of Health of Russia, Moscow Cytomegalovirus (CMV)-related complications remain an extremely serious and urgent problem in immunocompromised patients. Ganciclovir (GCV) is efficient for the treatment of CMV-infection, but myelotoxicity limits the possibilities of their application. In addition, prolonged or intermittent courses of antiviral drugs predispose to the development of CMV drug-resistant strains. Valganciclovir is a safe and effective alternative to intravenous GCV. Despite the well-spread application of effective methods of early detection and pre-emptive treatment, the issue of the control of CMV-infection is not resolved. High intensive immunoablative therapy (alemtuzumab, ATG, еtс.) and hematopoietic stem cell transplantation (HSCT) from alternative donors greatly increase the risk of life-threatening visceral CMV-infections in patients. Thereby, studies of new therapeutic approaches (for example, transfusion of CMV-specific T-cells) are actually in process. Key words: cytomegalovirus (CMV)-infection, pre-emptive treatment, toxicity, drug-resistant strains, immunocompromised patients Цитомегаловирус (ЦМВ) относится к семейству β-герпесвирусов и известен также как вирус герпеса-5 (HHV-5). ЦМВ является самым крупным из известных вирусов, имеет диаметр вирионов, равный 120–150 нм, геном представлен двуспиральной ДНК. ЦМВ относительно термолабилен, инактивируется при температуре +56 °С, его оптимальный рН 7,2–8,0 [1]. Вирусная матрица представляет собой главный протеин pp65, который и является основной мишенью для генерации иммунного ответа. ЦМВ чрезвычайно широко распространен в человеческой популяции, по различным данным к первому году жизни инфицируется каждый 5-й житель планеты, а в зрелом возрасте серопозитивность населения варьирует в пределах от 40 до 100 % [2]. У иммунокомпетентного хозяина после первичного инфицирования и формирования иммунного ответа ЦМВ пожизненно персистирует в виде латентной инфекции. Естественным резервуаром вируса является строма солидных органов, однако есть данные, что гемопоэтические предшественники также могут выполнять эту функцию и передавать вирусный геном периферическим клеткам [3–5]. ЦМВ чаще других известных герпесвирусов является причиной тяжелых и даже жизнеугрожающих инфекций, однако последние встречаются только у пациентов с выраженными дефектами иммунной системы. К группам риска по развитию клинически значимых ЦМВ-инфекций относятся: недоношенные дети в периоде новорожденности, а также пациенты с первичными и вторичными иммунодефицитными состояниями, в том числе онкогематологические больные и пациенты после трансплантации гемопоэтических стволовых клеток (ТГСК). В контексте ТГСК проблема клинически значимой ЦМВ-инфекции стоит наиболее остро, а в некоторых случаях даже Иммуносупрессивная терапия также представляет собой угрозу в отношении развития ЦМВ-инфекции и, безусловно, имеет значение не только у больных после ТГСК, но и в других когортах пациентов. Большое значение играют дозы иммуносупрессивных препаратов и продолжительность их использования [15]. Применение высоких доз кортикостероидов (> 1 мг/кг/сут), микофенолата мофетила, препаратов, направленных на эрадикацию Т-клеток (например, антитимоцитарного глобулина или алемтузумаба), расцениваются как факторы высокого риска развития ЦМВ-инфекции [16]. Интересными свойствами обладает микофенолата мофетил, который не только нарушает иммунологический ответ, но и оказывает непосредственный эффект собственно на ЦМВ-вирион, способствуя ускорению процесса репликации ЦМВ (процесс up-регуляции) [17]. Клинические проявления ЦМВ-инфекции достаточно разнообразны. Наиболее часто встречаются интерстициальная пневмония (рисунок) и поражение гастроинтестинального тракта [16]. По данным Meyers et al. [17], частота развития поражения гастроинтестинального тракта после ТГСК составляет 8–30 %. Несмотря на то, что ЦМВ-пневмония регистрируется несколько реже, смертность от этого осложнения чрезвычайно высока и достигает 30–52 % [18], а по данным отдельных клинических центров – 100 % [19]. К более редким осложнениям относят ЦМВ-ассоциированные гепатиты, ретиниты, энцефалиты, геморрагические циститы, эндотелиальное поражение, тромботическую микроангиопатию [20, 21]. ЦМВ-инфекция у пациентов после ТГСК может стать также причиной развития недостаточности трансплантата и, как следствие, причиной бактериальных и грибковых суперинфекций. По данным Nguyen et al. [22], у 54 % пациентов с ЦМВ-пневмонией, возникшей на поздних сроках после ТГСК, имели место те или иные конкурентные инфекции, причем летальность в этой группе составляла 100 %. ЦМВ-ассоциированная недостаточность трансплантата протекает в виде гипоплазии или тяжелой панцитопении, что нередко связывают с поражением ГСК и с повреждением клеток микроокружения [23]. Если пациентов после ТГСК относят к когорте больных сверхвысокого риска развития ЦМВ-инфекции, то аналогичные проблемы у нетрансплантационных больных нередко уходят на 2-й план и данной проблеме часто не уделяется достаточного внимания. Однако, в соответствии с опубликованными результатами исследования, проведенного в MD Anderson Cancer Center (Хьюстон, США), тяжелые висцеральные ЦМВ-инфекции, в первую очередь, пневмонии, были зарегистрированы у 2,9 % из 2136 онкогематологических пациентов, которым проводилась терапия в рамках программного лечения без использования ТГСК [24]. Причем наибольшая частота развития ЦМВ-заболевания (16 %) зарегистрирована в группе ’2013 выходит на ведущие позиции по частоте развития тяжелых поражений и смертности пациентов. Факторами, влияющими на риск развития и тяжесть течения вирусных инфекций у реципиентов ТГСК, являются особенности и интенсивность подготовительной миело/иммуноаблативной терапии, степень HLA-совместимости донора и реципиента, тип донора (неродственный, родственный, гаплоидентичный и т. д.), а также целый ряд проблем, отдаляющих сроки восстановления специфического иммунологического ответа, в числе которых особое место занимает реакция «трансплантат против хозяина» (РТПХ) и специфическая терапия, направленная на профилактику и лечение этого осложнения. Актуальность ЦМВ-инфекции у пациентов после аутологичной ТГСК не является высокой [6–8]. Основным фактором риска при таком виде трансплантации является ЦМВ-серопозитивность пациента. Вероятность ЦМВ-виремии без клинических проявлений в этом случае может достигать 25 % [9], однако частота развития тяжелых висцеральных осложнений невелика и составляет не более 1,6 % [10]. Вероятность развития тяжелых ЦМВ-ассоциированных осложнений после аллогенной ТГСК существенно выше. К факторам, имеющим одно из ключевых значений при определении риска развития ЦМВ-инфекции, относят ЦМВ-серопозитивность пациента при ЦМВ-серонегативности донора гемопоэтических стволовых клеток (ГСК). По данным Ljungman et al. [11], риск развития ЦМВ при такой комбинации «донор – реципиент» достигает 16 %. Другим фактором риска, влияющим на вероятность развития или реактивации ЦМВ-инфекции после ТГСК, является тип донора ГСК. Трансплантация от неродственного или парциально совместимого донора, как правило, увеличивает риск развития ЦМВинфекции [11, 12]. Естественно, что риск определяет не сам по себе тип донора, а ассоциированные с данным фактором проблемы, такие как тяжесть РТПХ, длительность иммуносупрессивной терапии, сроки иммунореконституции и т. д. [13]. Одним из методов профилактики тяжелой РТПХ у пациентов после трансплантации от парциально совместимого донора является удаление Т-клеток из трансплантата, что может достигаться либо Т-клеточной деплецией, либо CD34+-позитивной селекцией продукта, достигаемыми с помощью иммуномагнитных методик. Естественно, что удаление Т-клеток, в числе которых содержатся ЦМВ-специфические цитотоксические клетки, может стать триггером развития или реактивации ЦМВ-инфекции. Кроме того, такой вариант ТГСК сопровождается чрезвычайно длительным периодом иммунологической реконституции, в том числе отсроченным восстановлением CD3+, CD4+ и CD8+-клеток, что также компрометирует противовирусный потенциал реципиента ГСК [14]. 47 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 48 Компьютерная томография органов грудной клетки. ЦМВ-пневмония у пациента с синдромом Краббе–Бенеке после алло-ТГСК (собственное наблюдение) пациентов с хроническим лимфолейкозом (ХЛЛ), компрометирующим фактором развития ЦМВ у которых являлось применение алемтузумаба. Частота реактивации инфекции после терапии данным препаратом достигает по некоторым данным 30 % [25]. Среди пациентов, которым не проводилась ТГСК, реактивация ЦМВ чаще встречается при миеломной болезни, ХЛЛ, остром лимфобластном лейкозе и различных лимфомах (13,6 %), в то время как при миелоидных лейкозах данный показатель не превышает 3,9 % [26]. Таким образом, проблема ЦМВ-инфекций актуальна и у пациентов, не получающих ТГСК, но имеющих такие факторы риска, как серопозитивность пациента и проведение иммуноаблативной терапии, в том числе с применением алемтузумаба [27]. Современные алгоритмы контроля ЦМВ неоднократно претерпевали изменения. Важной мерой профилактики первичного инфицирования ЦМВ-серонегативного пациента является использование препаратов крови, полученных от ЦМВ-серонегативных доноров. При отсутствии возможности подбора донора по ЦМВ-серологическому статусу важным требованием является применение для компонентов крови современных лейкоцитарных фильтров IV поколения. Риск инфицирования как в первом, так и во втором случае не превышает 3 % [28]. Применение препаратов гипериммунных ЦМВиммуноглобулинов по данным большинства исследований, изучавших эффективность ЦМВ-специфических внутривенных иммуноглобулинов для профилактики инфекции, не приводит к уменьшению частоты развития ЦМВ-инфекции и улучшению выживаемости пациентов [29–32]. Существует целый ряд исследований, посвященных оценке эффективности медикаментозной профи- лактики ЦМВ-инфекции у пациентов после ТГСК. В частности, в исследованиях Ljungman et al. [21] было продемонстрировано, что использование ацикловира и валацикловира может быть эффективным, однако при применении 2-го препарата были зарегистрированы некоторые преимущества. Активная профилактика с использованием ганцикловира у пациентов после ТГСК обычно не проводится в связи с миелотоксичностью препарата в отношении трансплантата и более поздней специфической противовирусной реконституции в посттрансплантационный период. Наиболее адекватным и хорошо зарекомендовавшим себя подходом к контролю ЦМВ-инфекции у пациентов после ТГСК является упреждающая терапия, основанная на рутинном использовании полимеразной цепной реакции в режиме реального времени для раннего выявления ЦМВ-виремии c ее количественной оценкой и начала специфической вирусостатической терапии до появления признаков висцеральной инфекции. Точно так же, как и у пациентов трансплантационных центров, многие клинические центры рекомендуют использовать принципы упреждающей терапии ЦМВ у больных, получавших алемтузумаб [33–35]. Однако есть данные и об эффективности медикаментозной профилактики, в частности валганцикловиром, у пациентов с ХЛЛ после курса алемтузумаба [25]. Не так давно единственным вирусостатиком, рутинно используемым в контексте упреждающей терапии и лечения ЦМВ-инфекции, являлся ганцикловир. К сожалению, одним из ведущих побочных эффектов данного препарата является его миелотоксичность и, как следствие, нейтропения и увеличение частоты развития тяжелых бактериальных и грибковых инфек- доступности валганцикловира в зависимости от наличия или отсутствия признаков острой РТПХ с поражением желудочно-кишечного тракта. Полученные результаты свидетельствовали, что биодоступность валганцикловира снижалась на 15 %, однако различий в токсичности или частоте развития ЦМВ-инфекций при сравнении с группой пациентов без РТПХ получено не было. У больных, которым не проводилась ТГСК, применение валганцикловира также может быть обоснованным и эффективным. По результатам исследования S. O’Brien et al. [25], изучавших возможности различных режимов профилактики ЦМВ у больных ХЛЛ после терапии алемтузумабом, частота реактивации инфекции в группе, получавшей валганцикловир, составила 0 % (0 из 20 пациентов), а в группе валацикловира – 35 % (7 из 20 пациентов). Одной из весьма интересных и прогрессивных стратегий контроля ЦМВ-инфекции в группах риска является разработка и внедрение в практику методов искусственного моделирования ЦМВ-специфического иммунитета. Не так давно начаты исследования эффективности вакцинации от ЦМВ донора ГСК, когда в качестве вакцин используют препараты, содержащие ослабленные вирусы, рекомбинантные вирусы и наиболее часто встречающиеся ЦМВ-протеины. Ведутся активные работы в отношении развития технологии культивирования и последующих трансфузий ЦМВ-специфических Т-лимфоцитов [46, 47]. До появления и внедрения в практику активного мониторинга реактивации ЦМВ и актуальных сегодня принципов профилактики и упреждающей терапии данная инфекция довольно часто являлась причиной тяжелых органных дисфункций и высокой смертности в группах пациентов высокого риска. Исследования, направленные на оптимизацию алгоритмов контроля ЦМВ-инфекции, позволили некоторое время назад приблизить проблему ЦМВ к решению. Тем не менее появление новых высокоинтенсивных протоколов лечения онкогематологических заболеваний, подразумевающих активное применение препаратов с выраженными иммуноаблативными свойствами, в том числе в контексте ТГСК, а также широкое использование трансплантаций от «альтернативных» доноров, вновь способствовало увеличению серьезности проблемы контроля ЦМВ. Использование сегодня современных методов диагностики и активных в отношении ЦМВ вирусостатических препаратов, таких как ганцикловир, фоскарнет и валганцикловир, является достаточно эффективным, однако по-прежнему не снимает актуальность данного осложнения, что является веским аргументом для продолжения исследований, направленных на отработку новых подходов и альтернативных принципов контроля ЦМВ-инфекции в группах риска. ’2013 ций [36]. Снижение уровня нейтрофилов ниже 1000/мкл на фоне применения ганцикловира, по данным некоторых публикаций, регистрируется у 41–58 % больных [37, 38]. Альтернативой ганцикловиру является фоскарнет, имеющий другой токсический профиль (нефротоксичность и электролитные нарушения), часто лимитирующий возможность его использования [39]. Одной из серьезных проблем, связанных с длительным и частым использованием ганцикловира, является развитие лекарственной резистентности, которая связана с появлением у пациента либо мутации в гене вирусной киназы (UL97), отвечающей за фосфорилирование и активацию ганцикловира, либо мутации в гене вирусной полимеразы (UL54), отвечающем за ингибирование ганцикловиром ДНК-полимеразы ЦМВ, либо мутаций в обоих генах одновременно. Есть данные, что частота развития такой ситуации достигает 10 % [40]. Кроме того, известно, что появление мутации в гене UL54 может вызывать резистентность не только к ганцикловиру, но и к фоскарнету [40]. В настоящее время достаточно активную позицию в решении вопроса профилактики и лечения ЦМВ занимает препарат валганцикловир, который представляет собой пероральную форму L-валилового эфира (про-препарат) ганцикловира. Его биодоступность чрезвычайно высока и составляет приблизительно 60 % при сравнении с внутривенной формой ганцикловира, и более чем в 10 раз выше, чем биодоступность у пероральной формы ганцикловира [40]. Токсический профиль валганцикловира схож с ганцикловиром, однако уровень тромбоцитов, как правило, не снижается, а частота тяжелых валганцикловир-индуцированных нейтропений при упреждающей терапии после ТГСК по некоторым данным ниже, чем при использовании ганцикловира [41, 42]. Несмотря на то, что валганцикловир является пропрепаратом ганцикловира, есть интересные сведения об отличительных особенностях этих 2 препаратов. В частности, T. Allice et al. [40] провели сравнительное исследование ганцикловира и валганцикловира в отношении потенциала развития мутаций в генах UL97 и UL54, приводящих к резистентности ЦМВ к проводимой терапии, в результате чего доказательств о развитии резистентности к валганцикловиру получено не было, в отличие от пациентов, получавших ганцикловир, где частота появления мутаций составила 7,7 %. Результаты исследований валганцикловира у пациентов после ТГСК выглядят многообещающе [42–45]. По данным P.L. van der Heiden et al. [41], частота элиминации вируса при упреждающей терапии ганцикловиром в дозе 10 мг/кг/сут составила 76 %, а при применении валганцикловира в дозе 1800 мг/сут – 80 %, при схожем профиле побочных эффектов. В исследовании E. Ayala et al. [42] был проведен анализ био- 49 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 50 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Л И Т Е Р А Т У Р А 1. Avery R.K., Bolwell B.J., Yen-Lieberman B. et al. Use of leflunomide in an allogeneic bone marrow transplant recipient with refractory cytomegalovirus infection. Bone Marrow Transplant 2004;34:1071–5. 2. Stocchi R.,Ward K.N., Fanin R., Baccarani M., Apperley J.F. Management of human cytomegalovirus infection and disease after allogeneic bone marrow transplantation. Haematol 1998;84:71–9. 3. Taylor-Wiedeman J., Sissons J.G., Borysiewicz L.K., Sinclair J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 1991;72:2059–64. 4. Soderberg-Naucler C., Fish K.N., Nelson J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997;91:119–26. 5. Michelson S. Interaction of human CMV with monocytes/macrophages: a love-hate relationship. Pathol Biol Paris 1997; 45:146–58. 6. Meyers J.D., Flournoy N., Thomas E.D. Risk factors for cytomegalovirus infection after human marrow transplantation. J Infect Dis 1986;153:478–88. 7. Reusser P., Fisher L.D., Buckner C.D. et al. Cytomegalovirus infection after autologous bone marrow transplantation: occurrence of cytomegalovirus disease and effect on engraftment. Blood 1990;75:1888–94. 8. Ljungman P., Biron P., Bosi A. et al. Cytomegalovirus interstitial pneumonia in autologous bone marrow transplant recipients. Infectious Disease Working Party of the European Group for Bone Marrow Transplantation. Bone Marrow Transplant 1994;13(2):209–12. 9. Konoplev S., Champlin R.E., Giralt S. et al. Cytomegalovirus pneumonia in adult autologous blood and marrow transplant recipients. Bone Marrow Transplant 2001;27:877–81. 10. Osarogiagbon R.U., Defor T.E., Weisdorf M.A. et al. CMV antigenemia following bone marrow transplantation: risk factors and outcomes. Biol Blood Marrow Transplant 2000;6:280–8. 11. Ljungman P., Aschan J., LewensohnFuchs I. et al. Results of different strategies for reducing cytomegalovirus-associated mortality in allogeneic stem cell transplant recipients. Transplantation 1998;66:1330–4. 12. Takenaka R., Gongo H., Tanimoto K. et al. Increased incidence of cytomegalovirus (CMV) infections and CMV-associated disease after allogeneic bone marrow transplantation from unrelated donors. Bone Marrow Transplant 1997;19:241–8. 13. Reusser P., Riddell S., Meyers J., Greenberg P.D. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: pattern of recovery and correlation with cytomegalovirus infection and disease. Blood 1991;78:1373–80. 14. Small T.N., Papadopoulos E.B., Boulad F. et al. Comparison of immune reconstitution after unrelated and related T-celldepleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood 1999;93:467–80. 15. Rubin R.H. Clinical approach to infection in the compromised host. In: Infection in the Organ Transplant Recipient; Rubin R.H., Young L.S. (eds.). New York, NY, Kluwer Academic Press, 2002. Pp. 573–679. 16. Boeckh M., Nichols W.G., Papanicolaou G. et al. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant 2003;9:543–58. 17. Meyers J.D., Ljungman P., Fisher L.D. Cytomegalovirus excretion as a predictor of cytomegalovirus disease after marrow transplantation: importance of cytomegalovirus viremia. J Infect Dis 1990;162:373–80. 18. Ljungman P., Engelhard D., Link H. et al. Treatment of interstitial pneumonitis due to cytomegalovirus with ganciclovir and intravenous immune globulin: experience of European Bone Marrow Transplant Group. Clin Infect Dis 1992;14:831–5. 19. Yanada M., Yamamoto K., Emi N. et al. Cytomegalovirus antigenemia and outcome of patients treated with preemptive ganciclovir: retrospective analysis of 241 consecutive patients undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2003;32:801–7. 20. Takatsuka H., Wakae T., Mori A. et al. Endothelial damage caused by cytomegalovirus and human herpesvirus-6. Bone Marrow Transplant 2003;31:475–9. 21. Ljungman P., Griffiths P., Paya C. Definitions of CMV infection and disease in transplant recipients. Clin Infect Dis 2002;34:1094–7. 22. Nguyen Q., Champlin R., Giralt S. et al. Late cytomegalovirus pneumonia in adult allogeneic blood and marrow transplant recipients. Clin Infect Dis 1999;28:618–23. 23. Randolph-Habecker J., Iwata M, Torok-Storb B. Cytomegalovirus mediated myelosuppression. J Clin Virol 2002;25 (Suppl 2):S51–6. 24. Nguyen Q., Estey E., Raad I. et al. Cytomegalovirus pneumonia in adults with leukemia: an emerging problem. Clin Infect Dis 2001;32:539–45. 25. O’Brien S., Ravandi F., Riehl T. et al. Valganciclovir prevents cytomegalovirus reactivation in patients receiving. Blood 2008;111(4):1816–9. 26. Han X.Y. Epidemiologic analysis of reactivated cytomegalovirus antigenemia in patients with cancer. J Clin Microbiol 2007;45:1126–32. 27. Ljungman P., de la Camara R., Cordonnier C. et al. Management of CMV, HHV-6, HHV-7 and Kaposi-sarcoma herpesvirus (HHV-8) infections in patients with hematological malignancies and after SCT. Bone Marrow Transplant 2008;42: 227–40. 28. Bowden R.A., Slichter S.J., Sayers M. A comparison of filtered leukocyte-reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfusion-associated CMV infection after marrow transplantation. Blood 1995;86:3598–603. 29. Bass E.B., Powe N.R., Goodman S.N. et al. Efficacy of immune globulin in preventing complications of bone marrow transplantation: a meta-analysis. Bone Marrow Transplant 1993;12:273–82. 30. Guglielmo B.J., Wong-Beringer A., Linker C.A. Immune globulin therapy in allogeneic bone marrow transplant: a critical review. Bone Marrow Transplant 1994;13:499–510. 31. Messori A., Rampazzo R., Scroccaro G., Martini N. Efficacy of hyperimmune anticytomegalovirus immunoglobulins for the prevention of cytomegalovirus infection in recipients of allogeneic bone marrow transplantation: a meta-analysis. Bone Marrow Transplant 1994;13:163–7. 32. Zikos P., Van Lint M.T., Lamparelli T. et al. A randomized trial of high dose polyvalent intravenous immunoglobulin (HDIgG) vs. cytomegalovirus (CMV) hyperimmune IgG in allogeneic hemopoietic stem cell transplants (HSCT). Haematologica 1998;83:132–7. 33. Keating M.J., Flinn I., Jain V. et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61. 34. Rai K.R., Freter C.E., Mercier R.J. et al. Alemtuzumab in previously treated chronic lymphocytic leucemia patients who also had received fludarabine. J Clin Oncol 2002;20:3891–7. 35. Osterborg A., Dyer M.J., Bunjes D. et al. Phase II multicenter study of human CD52 antibody in previously treated chronic lymphocytic leukemia: European study group of CAMPATH-1H Treatment in chronic lymphocytic leukemia. J Clin Oncol 1997;15:1567–74. 36. Boeckh M., Zaia J.A., Jung D. et al. A study of the pharmacokinetics, antiviral activity, and tolerability of oral ganciclovir for CMV prophylaxis in marrow transplantation. Biol Blood Marrow Transplant 1998;4:13–9. stem cell transplantation: implications for the emergence of drug-resistant cytomegalovirus. J Antimicrob Chemother 2009;63(3):600–8. 41. van der Heiden P.L., Kalpoe J.S., Barge R.M. et al. Oral valganciclovir as pre-emptive therapy has similar efficacy on cytomegalovirus DNA load reduction as intravenous ganciclovir in allogeneic stem cell transplantation recipients. Bone Marrow Transplant 2006;37:693–8. 42. Ayala E., Greene J., Sandin R. et al. Valganciclovir is safe and effective as preemptive therapy for CMV infection in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2006;37:851–6. 43. Busca A., de Fabritiis P., Ghisetti V. et al. Oral valganciclovir as preemptive therapy for cytomegalovirus infection post allogeneic stem cell transplantation. Transpl Infect Dis 2007;9:102–7. 44. Candoni A., Simeone E., Tiribelli M. et al. What is the optimal dosage of valganciclovir as preemptive therapy for CMV infection in allogeneic hematopoietic SCT. Bone Marrow Transplant 2008;42:207–8. 45. Takenaka K., Eto T., Nagafuji K. et al. Fukuoka Blood and Marrow Transplant Group (FBMTG). Oral valganciclovir as preemptive therapy is effective for cytomegalovirus infection in allogeneic hematopoietic stem cell transplant recipients. Int J Hematol 2009;89:231–7. 46. Peggs K.S., Verfuerth S., Mackinnon S. Induction of cytomegalovirus (CMV)specific T-cell responses using dendritic cells pulsed with CMV antigen: a novel culture system free of live CMV virions. Blood 2001;97:994–1000. 47. Zaia J.A., Sissons J.G., Riddell S. et al. Status of cytomegalovirus prevention and treatment in 2000. Hematology (Am Soc Hematol EducProgram) 2000;2000:339–55. ’2013 37. Goodrich J.M., Bowden R.A., Fisher L. et al. Ganciclovir prophylaxis to prevent cytomegalovirus disease after marrow transplant. Ann Intern Med 1993;118:173–8. 38. Winston D.J., Ho W.G., Bartoni K. et al. Ganciclovir prophylaxis of cytomegalovirus infection and disease in allogeneic bone marrow transplant recipients. Results of placebo-controlled, double-blind trial. Ann Intern Med 1993;118:179–84. 39. Reusser P., Einsele H., Lee J. et al. Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Randomized multicenter trial of foscarnet versus ganciclovir for preemptive therapy of cytomegalovirus infection after allogeneic stem cell transplantation. Blood 2002;99: 1159–64. 40. Allice T., Busca A., Locatelli F. et al. Valganciclovir as pre-emptive therapy for cytomegalovirus infection post-allogenic 51 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Случай успешного лечения мукормикоза легких у ребенка с апластической анемией 1 ’2013 52 Е.А. Никитина1, Н.Н. Климко2, А.С. Колбин3, Э.Г. Бойченко1, И.А. Гарбузова1, М.В. Голубева1, Т.С. Богомолова2 1 Детская городская больница № 1, Санкт-Петербург; ГБОУ ВПО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России, Санкт-Петербург; 3 медицинский факультет ФГБОУ ВПО «Санкт-Петербургский государственный университет» 2 Контакты: Екатерина Андреевна Никитина [email protected] Мукормикоз (зигомикоз) характеризуется высокой летальностью у иммунокомпрометированных больных. Представлен клинический случай успешного лечения мукормикоза у 11-летнего ребенка со сверхтяжелой формой идиопатической апластической анемии. Диагноз инвазивного микоза был установлен на основании критериев EORTC 2008. При компьютерной томографии определена субтотальная инфильтрация правого легкого. При исследовании бронхоальвеолярного лаважа выделили Lichtheimia corymbifera. В результате длительной комбинированной антифунгальной терапии липидным комплексом амфотерицина B и позаконазолом удалось добиться полного купирования клинико-лабораторных и инструментальных проявлений мукормикоза и продолжить успешное лечение апластической анемии. Ключевые слова: мукормикоз, апластическая анемия, амфотерицин B, позаконазол, дети Successful treatment of pulmonary mucormycosis in a child with aplastic anemia Ye.A. Nikitina1, N.N. Klimko2, A.S. Kolbin3, E.G. Boychenko1, I.A. Garbuzova1, M.V. Golubeva1, T.S. Bogomolova2 1 Children Municipal Hospital № 1, Saint Petersburg; I.I. Mechnikov North-Western State Medical University, Ministry of Health of Russia, Saint Petersburg; 3 Saint Petersburg State University 2 Mucormycosis (zygomycosis) is a frequently fatal fungal infection in immunocompromized patients. We describe a case of a successfull treatment of pulmonary mucormycosis in 11-year-child with a very severe aplastic anemia. The diagnosis of invasive mycosis has been proved according to EORTC 2008 criteria. Computed tomography showed bilobar right-sided pulmonary infiltration. Lichtheimia corymbifera cultured from BAL. The child achieved complete clinical, laboratory and instrumental response on long-term amphotericin B lipid-formulated therapy combined with posaconazole. Key words: mucormycosis, aplastic anemia, amphotericin B, posaconazole, children Актуальность У больных с онкогематологической патологией проблема инвазивных микозов (ИМ) приобрела особую актуальность после внедрения в клиническую практику программ интенсивной полихимиотерапии. Частота грибковых инфекций в отделениях онкологии, гематологии и трансплантации достигает 15–20 % [1]. До недавнего времени наиболее частыми этиологическими агентами были Candida spp. и Aspergillus spp. Однако в последние годы отмечено нарастание частоты выделения возбудителей, вызывающих такие редкие инфекции, как мукормикоз. Количество сообщений о результатах лечения мукормикоза у детей в отечественной и зарубежной литературе крайне ограниченно. В связи с этим целью настоящей публикации явилось описание собственного клинического наблюдения успешного лечения доказанного легочного мукормикоза у ребенка со сверхтяжелой формой апластической анемии. Пациенты и методы Представлено описание клинического случая успешного лечения мукормикоза у ребенка, получившего курс иммуносупрессивной терапии (ИСТ) по поводу апластической анемии в отделении гематологии, онкологии и химиотерапии Детской городской больницы № 1 г. Санкт-Петербурга. Для постановки диагноза ИМ использовали критерии Европейской организации по изучению и лечению рака (EORTC) и Национального института здоровья США (NIH) [2]. Общим клиническим критерием мукормикоза являлась персистирующая более 96 ч лихорадка, рефрактерная к назначению антибиотиков широкого спектра действия. Клиническими критериями мукормикоза нижних дыхательных путей считали признаки инфекции нижних дыхательных путей, такие как кашель, хрипы в легких и обнаружение инфильтратов при компьютерной томографии (КТ). Мукормикоз считали доказанным, если при гистопатологическом исследо- ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Описание клинического случая Пациент 11 лет поступил в клинику 01.09.2011 с жалобами на бледность кожных покровов, кожный геморрагический синдром, рецидивирующие лакунарные ангины (2 эпизода за предшествующие 3 мес). В анализе крови при поступлении – панцитопения, агранулоцитоз (таблица). При цитологическом и гистологическом исследовании костного мозга – аплазия кроветворения. На основании клинико-гематологических данных был установлен диагноз сверхтяжелой формы апластической анемии. Через неделю от момента поступления у ребенка на фоне агранулоцитоза развилась фебрильная лихорадка и тяжелая некротическая ангина, потребовавшая массивной антибактериальной терапии (меропенем, амикацин, ванкомицин). Лабораторные показатели пациента при поступлении и через 8 мес от начала лечения Лабораторный параметр При поступлении Через 8 мес Гемоглобин (г/л) 57 100 Лейкоциты (× 109/л) 1,5 5,2 < 200 > 1500 СОЭ (мм/ч) 75 16 Тромбоциты (× 109/л) 10 60 Мочевина (мг/дл) 36 57 Креатинин (мг/дл) 0,5 1,1 Абсолютное число нейтрофилов (кл/мкл) В связи с отсутствием родственного совместимого донора после купирования инфекционного процесса был инициирован курс ИСТ антитимоцитарным иммуноглобулином (ATGAM), циклоспорином и глюкокортикостероидами. Перед началом ИСТ было проведено обследование для исключения ИМ: мультиспиральная КТ (МСКТ) грудной клетки и исследование крови на галактоманнан. Признаков очагового или инфильтративного поражения легких выявлено не было. Галактоманнан в сыворотке – отрицательный. С 16-х суток пребывания в стационаре ребенку была инициирована первичная антифунгальная профилактика вориконазолом с учетом высокого риска развития инвазивного аспергиллеза. Через 2 нед от начала ИСТ развился сухой малопродуктивный кашель. При аускультативном обследовании определялось ослабление дыхания и влажные мелкопузырчатые хрипы в нижних отделах правого легкого. При рентгенографии легких выявлена инфильтрация в средней доле (S4, S5) правого легкого (рис. 1). На МСКТ грудной клетки – тотальная инфильтрация правого легкого смешанного характера 1 ’2013 вании была установлена инфекция глубоких тканей: присутствие в тканях широких (10–15 мкм), нечасто септированных тонкостенных гиф и выделение из стерильного в норме биосубстрата грибов рода Mucor культуральным методом. 53 Рис. 1. Рентгенограмма грудной клетки через двое суток от появления дыхательной симптоматики (интерстициальная с элементами альвеолярной), симптом «булыжной мостовой», участок уплотнения легочной ткани по типу «матового стекла» на фоне выраженного усиления и отека междолькового интерстиция, а также выраженная прикорневая инфильтрация с элементами аденопатии бронхопульмональных лимфатических узлов (рис. 2). При бронхоскопии визуализированы грибковые разрастания в нижнедолевом бронхе справа, обтурирующие просвет. При микроскопии биоптата был обнаружен обильный несептированный мицелий, ветвящийся под прямым углом, а при посеве бронхоальвеолярного лаважа (БАЛ) выделена Lichtheimia corymbifera. В связи с развившимся угрожающим жизни инфекционным осложнением терапия циклоспорином и глюкокортикостероидами была приостановлена. Начата терапия липидным комплексом амфотерицина B (ЛКАB, 5 мг/кг/сут), которая сопровождалась персистирующими признаками нефротоксичности: гипокалиемия (K = 2,7–3,2 μm/l), нарушение азотовыделительной функции (мочевина max = 88,8 mg/dl, креатинин = 1,1 mg/dl). Проводилась постоянная стимуляция миелопоэза колониестимулирующим фактором (G-CSF), на фоне которой через 3 нед от начала курса ИСТ уровень гранулоцитов достиг 500 кл/мкл. Состояние пациента характеризовалось высокой лихорадкой, периодическими болями в правой половине грудной клетки и частым непродуктивным кашлем. Признаков дыхательной недостаточности и кровохарканья не отмечалось. По данным ультразвукового исследования (УЗИ) выявлялось умеренное количество плеврального выпота в правом синусе, не требующее дренирования. На 15-й день терапии ЛКАB, в связи с сохранением стойкой фебрильной лихорадки с тенденцией к нарастанию количества фебрильных подъемов, антифунгальная терапия была усилена позаконазолом в дозе 800 мг/сут. При повторной фибробронхоскопии через месяц от начала противогрибковой терапии была выявлена язва в устье промежуточного бронха справа, без признаков ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 54 Рис. 2. МСКТ грудной клетки на момент установления диагноза ИМ кровотечения, диаметром 2 × 1 см. При посеве и микроскопии БАЛ грибов выявлено не было. УЗИ в динамике установило нарастание количества жидкости в правой плевральной полости. Под УЗИ-контролем произведена плевральная пункция, получено 1100 мл мутного геморрагического экссудата, клеточный состав которого составляли: эритроциты – 5 %, нейтрофилы – 56 %, эозинофилы – 1 %, гистиоциты – 24 %. Грибы не обнаружены. Через 2 мес от момента диагностики мукормикоза МСКТ в режиме ангиографии с контрастированием (рис. 3) продемонстрировала субтотальные деструктивные изменения в нижней и средней долях правого легкого с максимальными изменениями в S6 по типу «абсцедирования». Умеренное количество плеврального выпота справа с признаками организации. В левом легком очаговых и инфильтративных изменений не было. Клинически состояние пациента оставалось стабильным, фебрильная лихорадка регрессировала, кашель не нарастал, кровохарканья не отмечалось. С учетом полученной динамики изменений с формированием деструкции правого легкого в непосредствен- ной близости к крупным сосудам, у ребенка имелся высокий риск развития угрожающего жизни кровотечения. В связи с этим интенсивно обсуждался вопрос о проведении срочного оперативного вмешательства при условии обеспечения адекватной трансфузионной поддержки компонентами донорской крови. На этот момент ребенок находился в постоянной зависимости от переливания компонентов крови, базальный уровень тромбоцитов на тот момент составлял 10 × 10 9/л. Родители приняли решение о переводе ребенка в зарубежную клинику для проведения хирургического лечения. С 20.12.2011 по 30.12.2011 ребенка наблюдали в отделении торакальной хирургии университетской клиники Гейдельберга (Германия), где было принято решение отказаться от проведения хирургического вмешательства и рекомендовано продолжить антимикотическую терапию по месту жительства. С 31.12.2011 ребенок наблюдался в ДГБ № 1 г. Санкт-Петербурга, продолжая антимикотическую терапию (ЛКАB и позаконазол), прием циклоспорина А не возобновляли. От проведения хирургического лечения родители ребенка отказались. Состояние ребен- Рис. 3. МСКТ легких в режиме ангиографии через 2 мес от постановки диагноза ИМ воспалительного процесса в легких и высокого риска потенцирования нефротоксических осложнений на фоне планируемого возобновления ИСТ циклоспорином, было принято решение о снижении дозы ЛКАB с 5 до 2,5 мг/ кг/сут. Пациент получал ЛКАB в дозе 5 мг/кг/сут в течение 3 мес, еще 1,5 мес – в 50 % дозе. Всего терапию ЛКАB проводили почти 5 мес (суммарная доза составила 23 г), и прервали в связи с прогрессирующей нефротоксичностью: нарушением азотовыделительной функции и снижением концентрационной способности почек. Терапию позаконазолом в суточной дозе 800 мг проводили непрерывно в течение 6 мес, нежелательных явлений отмечено не было. Позднее, через 5 мес от момента его отмены, по улучшении скорости клубочковой фильтрации с 32 до 56 мл/мин × 1,73 м2, был возобновлен прием циклоспорина А в дозе 2,5 мг/кг (50 % от расчетной). По данным легочных проб функция внешнего дыхания восстановилась полностью. Клинически, по данным аускультативного исследования, наблюдалась дальнейшая положительная динамика. МСКТ через 8 мес от момента диагностирования мукормикоза продемонстрировала уменьшение размеров зоны инфильтративных изменений в виде полной регрессии фокуса уплотнения легочной ткани по типу «матового стекла» в средней доле правого легкого, сохранялась картина тотального ателектаза нижней доли на фоне перераздутой верхней доли правого легкого (рис. 5). При бронхоскопии выявлено уменьшение в объеме грануляций в промежуточном бронхе. При микроскопии и посеве БАЛ мукормицеты не обнаружены. Рис. 4. МСКТ грудной клетки через 4 мес от начала антифунгальной терапии Рис. 5. МСКТ грудной клетки через 8 мес от начала заболевания ’2013 ка оставалось достаточно стабильным, кровохарканья не наблюдали. Отмечено достижение частичной ремиссии апластической анемии: с конца декабря не получал G-CSF, уровень гранулоцитов стабильно > 2500 кл/мкл, больной перестал нуждаться в трансфузиях эритроцитарной массы. Однако сохранялась глубокая тромбоцитопения и зависимость от трансфузий тромбоконцентрата. По данным миелограммы, через 4 мес от проведения курса ИСТ получено восстановление всех ростков кроветворения, увеличение клеточности костного мозга. В середине января при аускультации отмечено увеличение зоны проведения дыхания над средней долей правого легкого. МСКТ (рис. 4) продемонстрировала восстановление пневматизации верхней и средней долей правого легкого, с сохранением тотальной инфильтрации нижней доли. По данным фибробронхоскопии: язва в устье промежуточного бронха справа в стадии рубцевания, просвет промежуточного бронха частично обтурирован грануляциями слизистой, но проходимость его восстановилась. При микроскопии БАЛ были выявлены гифы мукормицета. Клинически получена отчетливая положительная динамика: самочувствие хорошее, состояние пациента ближе к удовлетворительному, кровохарканье и признаки дыхательной недостаточности отсутствовали. По данным функциональных дыхательных проб пневматизация правого легкого улучшилась, что было обусловлено восстановлением проходимости промежуточного и среднедолевого бронхов. С учетом полученной регрессии 55 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 56 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ Таким образом, на основании данных комплексного обследования через 8 мес от постановки диагноза мукормикоза противогрибковая терапия позаконазолом была отменена. В настоящее время, спустя 15 мес от развития апластической анемии и мукормикоза легких, у ребенка сохраняется ремиссия заболевания, независимость от трансфузий гемокомпонентов, самочувствие ребенка не страдает, дыхательная симптоматика отсутствует, одышки нет. Обсуждение Мукормикоз является наиболее агрессивной, быстро прогрессирующей инвазивной грибковой инфекцией, вызываемой мицелиальными возбудителями [3, 4]. Заболевание характеризуется высокой атрибутивной летальностью. Описано около 867 разновидностей мукормицетов, однако более 80 % культурально доказанных случаев мукормикоза вызывают Rhizopus oryzae (arrhizus), R. microsporus var., Rhizopodiformis, Lichtheimia corymbifera и Rhizomucor pusillus. Основными факторами риска развития мукормикоза являются длительный агранулоцитоз, ИСТ и высокодозная цитостатическая терапия, длительное применение глюкокортикостероидов, хелаторная терапия дефероксамином, а также диабетический кетоацидоз [5]. Таким образом, основной группой риска развития мукормикоза являются пациенты с гемобластозами, апластической анемией и реципиенты трансплантатов гемопоэтических стволовых клеток. Поражение нижних дыхательных путей – наиболее частый вариант локализации инвазивного мукормикоза у пациентов с длительной нейтропенией и иммуносупрессией [6]. Клинические проявления неспецифичны и сходны с любой грибковой инфекцией нижних дыхательных путей: гипертермия, не отвечающая на массивную антибактериальную терапию, хрипы и кашель, возможны боли в грудной клетке и кровохарканье. При КТ в легких определяются инфильтраты. Ангиоинвазивный рост мукормицетов приводит к некрозу паренхимы легких, появлению полостей и кровотечения, которое может быть фатальным при поражении крупного сосуда [7]. Общая летальность при легочном мукормикозе составляет около 65 % [8], а при диссеминированном процессе, который чаще является осложнением инфекции нижних дыхательных путей, почти 100 % [9]. Успех в лечении мукормикоза зависит от нескольких факторов: это, прежде всего, – ранняя диагностика, активная антифунгальная терапия, устранение факторов риска и своевременная хирургическая санация очагов. К сожалению, не существует серологических тестов или методов полимеразной цепной реакции, позволяющих быстро установить предполагаемый диагноз мукормикоза. Для постановки диагноза доказанного мукормикоза необходимым является биопсия и гистологическое исследование тканей. Большое зна- чение имеет устранение или коррекция предрасполагающих факторов, таких как нейтропения, а также отмена или снижение дозы глюкокортикостероидов и иммуносупрессивных препаратов. Третье и четвертое звено успеха – это немедленное начало интенсивной противогрибковой терапии и хирургическое лечение. В одном из исследований летальность при мукормикозе в случае применения только противогрибковой терапии составила 68 %, в то время как в группе, где противогрибковую терапию сочетали с адекватным хирургическим удалением очагов, летальность снизилась до 11 % [10]. К сожалению, применение агрессивных хирургических методов может быть затруднено у онкогематологических пациентов и реципиентов трансплантатов гемопоэтических стволовых клеток в связи с сопутствующей тромбоцитопенией и коагулопатией. Возбудители мукормикоза полирезистентны, поэтому перечень противогрибковых препаратов для лечения этого заболевания крайне ограничен. Лекарственными средствами (ЛС) выбора для стартовой терапии являются липидные формы амфотерицина В. Для ЛКАB стандартная дозировка при мукормикозе составляет 5 мг/кг/сут как у детей, так и у взрослых. Для обычной формы амфотерицина В, который является альтернативным ЛС, максимальная дозировка составляет 1,5 мг/кг/сут [11]. Позаконазол – пероральное ЛС из группы азолов, показал эффективность против мукормицетов in vitro при экспериментальном мукормикозе, а также в клинических исследованиях у пациентов, которые были рефрактерны или не переносили лечение различными формами амфотерицина B. Позаконазол применяют как в комбинации с липидными формами амфотерицина B при мукормикозе, рефрактерном к стартовой терапии, так и в качестве монотерапии при непереносимой токсичности амфотерицина B [12, 13]. Немногочисленные обзоры по мукормикозу у детей основываются на единичных или небольших серийных описаниях случаев. Тем не менее они суммируют имеющийся опыт лечения и способствуют пониманию течения такого редкого заболевания, как мукормикоз. В одном из опубликованных обзоров, посвященных мукормикозу у детей, общее число включенных пациентов составило 157 (медиана возраста – 5 лет) [14]. В группу анализа летальности вошли 33 новорожденных; смертность новорожденных с мукормикозом составила 64 %. В группе детей в возрасте от 1 месяца до 18 лет имела место летальность 56 %, сравнимая с показателями у взрослых, – 53 % (408/772) [15]. У детей и новорожденных, которым был рано установлен диагноз и начата антифунгальная терапия (большинство выживших пациентов получали липидные формы амфотерицина B), смертность снизилась до 36 % по сравнению с 88 % среди пациентов, которые не получали антифунгальной терапии (р < 0,0001) [16]. Еди- менная отмена глюкокортикостероидов и циклоспорина. Безусловно, при подобном массивном легочном поражении целесообразным представлялось добавление к консервативному лечению хирургического удаления очагов, однако в нашем случае это было невозможным из-за отказа родителей пациента от операции. Таким образом, мукормикоз является жизнеугрожающей инфекцией у детей и новорожденных, при которой наиболее целесообразной стратегией лечения является раннее начало комбинированной антифунгальной терапии липидными формами амфотерицина B и позаконазолом, с добавлением хирургического удаления очагов вовлечения. Выводы и рекомендации 1. У онкогематологических пациентов и реципиентов трансплантатов гемопоэтических стволовых клеток с длительной нейтропенией и выраженной иммуносупрессией существует вероятность развития мукормикоза. 2. Мукормикоз – это агрессивная, быстро (в течение 2–3 нед) прогрессирующая и нередко фатальная инфекция. 3. Для ранней диагностики мукормикоза необходимо проведение прямой микроскопии и посева материала (БАЛ, мокрота, выделения из носа, аспират из пазух носа) из очага поражения. 4. Для эффективного лечения мукормикоза необходимы стабилизация основного заболевания, уменьшение выраженности нейтропении и иммуносупрессии, адекватная антифунгальная терапия, а также хирургическое удаление пораженных тканей. Л И Т Е Р А Т У Р А 1. Климко Н.Н. Микозы: диагностика и лечение. Руководство для врачей. 2-е изд. перераб. и доп. М.: Премьер МТ, 2008. 336 с. 2. De Pauw B., Walsh T.J., Donnelly J.P. et al. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008;46:1813–21. 3. Климко Н.Н., Васильева Н.В., Елинов Н.П. и др. Перечень основных методов и критериев диагностики микозов. СПб., 2001. 24 с. 4. Gonzalez C.E., Rinaldi M.G., Sugar A.M. Zygomycosis. Infect Dis Clin North Am 2002;16(4):895–914. 5. Kontoyiannis D.P., Lionakis M.S., Lewis R.E. et al. Zygomycosis in a tertiary- care cancer center in the era of Aspergillusactive antifungal therapy: A case-control observational study of 27 recent cases. J Infect Dis 2005;191:1350–60. 6. Marr K.A., Carter R.A., Crippa F. et al. Epidemiology and outcome of mould infections in hematopoietic stem cell transplant recipients. Clin Infect Dis 2002;34:909–17. 7. Harada M., Manabe T., Yamashita K. et al. Pulmonary mucormycosis with fatal massive hemoptysis. Acta Pathol Jpn 1992;42:49–55. 8. Tedder M., Spratt J.A., Anstadt M.P. et al. Pulmonary mucormycosis: Results of medical and surgical therapy. Ann Thorac Surg 1994;57:1044–50. 9. Gleissner B., Schilling A., Anagnostopolous I. et al. Improved outcome of zygomycosis in patients with hematological diseases? Leukemia Lymphoma 2004;45:1351–60. 10. Tedder M., Spratt J.A., Anstadt M.P. et al. Pulmonary mucormycosis: results of medical and surgical therapy. Ann Thorac Surg 1994;57:1044–50. 11. Spellberg B., Walsh T.J., Kontoyiannis D.P. et al. Recent advances in the management of mucormycosis: from bench to bedside. Clin Infect Dis 2009;48(12):1743–51. 12. Van Burik J-A., Hare R.S., Solomon H.F. et al. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006;42:e61–5. 13. Spellberg B., Ibrahim A., Roilides E. et al. Combination therapy for mucormycosis: why, what, and how? Clin Infect Dis 2012;54 Suppl 1:S73. 14. Zaoutis T.E., Roilides E., Chiou C.C. et al. Zygomycosis in children: a review and analysis of reported cases. Pediatr Infect Dis J 2007;26:723–7. ’2013 ничные опубликованные случаи применения терапии позаконазолом у детей с диагностированным мукормикозом показали хорошую его эффективность, как для лечения, так и для профилактики при высоком риске внутригоспитальной инфекции, и отсутствие побочных явлений даже у пациентов младшего возраста [17]. У представленного в нашей статье пациента возбудителем заболевания был Lichtheimia corymbifera (другие названия – Absidia corymbifera, Mycocladus corymbifer), микромицет, ассоциированный с более высоким риском диссеминации легочного мукормикоза у реципиентов после органной трансплантации [18]. Успех лечения данного больного, как нам представляется, обусловлен многими факторами, как со стороны макроорганизма и характеристик самого возбудителя, так и со стороны активной тактики ведения пациента. Безусловно, большую роль сыграло и то, что в ходе ИСТ по поводу апластической анемии был достигнут быстрый ответ: в ранние сроки после проведения курса ATGAM восстановились гранулоциты, а с ними и напряженность иммунитета в борьбе с возникшим жизнеугрожающим инфекционным осложнением. Наряду с эффективностью курса лечения по поводу основного заболевания, быстрая специфическая диагностика и активное начало терапии мукормикоза адекватными дозами ЛКАB, усиленной затем позаконазолом, а также длительная суммарная продолжительность терапии (5 мес терапии ЛКАB в суммарной дозе 23 г и почти 8 мес непрерывной терапии позаконазолом) позволили в данном случае справиться с легочным мукормикозом. Этому также способствовала своевре- 57 1 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 1 ’2013 58 ГЕМОБЛАСТОЗЫ: ДИАГНОСТИКА, ЛЕЧЕНИЕ, СОПРОВОДИТЕЛЬНАЯ ТЕРАПИЯ 15. Roden M.M., Zaoutis T.E., Buchanan W.L. et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis 2005;41:634–53. 16. Roilides E., Zaoutis T.E. and Walsh T.J. Invasive zygomycosis in neonates and children. Clin Microbiol Infect 2009;15:50–4. doi: 10.1111/j.1469-0691.2009.02981.x 17. Garner D., Machin K. Investigation and management of an outbreak of mucormycosis in a paediatric oncology unit. J Hosp Infect 2008;70(1):53–9. 18. Sun H.Y., Aguado J.M., Bonatti H. et al. Zygomycosis Transplant Study Group. Pulmonary zygomycosis in solid organ transplant recipients in the currentera. Am J Transplant 2009;9(9): 2166–71. 1 Современное представление о биоаналогах в гематологии и онкологии 59 ’2013 ФАРМАКОТЕРАПИЯ В.В. Птушкин ФГБУ ФНКЦ ДГОИ им. Дмитрия Рогачева Минздрава России, Москва Контакты: Вадим Вадимович Птушкин [email protected] Биопрепараты – крупные белковые или полипептидные молекулы, продуцируемые живыми организмами – во многом определяют эффективность современной противоопухолевой терапии. Биопрепараты, разрушающие злокачественные клетки и биопрепараты, защищающие нормальные ткани пациента, позволили добиться максимального прогресса в лечении целого ряда нозологий: рака молочной железы, колоректального рака, рака почки, злокачественных лимфом. К негативным сторонам применения биопрепаратов относятся их высокая стоимость и сложности производства. Истечение срока патентной защиты ряда биопрепаратов создает предпосылки снижения их стоимости при выпуске альтернативным производителем. В то же время биоаналоги, как и оригинальные белковые молекулы, являются продуктами производства живых клеток, что создает серьезные трудности в достижении их идентичности. Для того чтобы исключить риск недостатка эффективности или повышения токсичности лечения этими препаратами, в Европейском союзе были разработаны специальные правила для регистрации данной категории лекарственных средств. Разработанные Европейским медицинским агентством, они включают регламенты по применяемым методикам для определения качества биопродукта, подробное описание требований к доклиническим и клиническим исследованиям, в зависимости от его специфических свойств, а также требования по фармаконадзору. Реализация подобной стратегии позволила зарегистрировать в странах Евросоюза несколько биоаналогов гранулоцитарного колониестимулирующего фактора, один из которых – Зарсио – в нескольких клинических исследованиях показал полностью сопоставимую с оригинальным биопрепаратом эффективность и переносимость, что позволило говорить о значимом снижении стоимости лечения при равной эффективности и токсичности. Ключевые слова: онкология, биологические препараты, биоаналоги, нейтропения, гранулоцитарный колониестимулирующий фактор, Зарсио Modern concepts of biosimilars in hematology and oncology V.V. Ptushkin Dmitry Rogachev Federal Research Center of Pediatric Hematology, Oncology and Immunology, Ministry of Health of Russia, Moscow Biologics are large protein or polypeptide molecules produced by living organisms, which largely determine the efficiency of modern anticancer therapy. Biological products that destroy cancer cells and protect normal patient tissue led to progress in the treatment of breast cancer, colon cancer, kidney cancer, malignant lymphomas and other diseases. But the high cost and complexity of production limit their use. The expiration of patent protection for a number of biological products resulting to the possibility of reduces their costs when issuing an alternative manufacturer. At the same time, biosimilars are produced by living cells (as the original protein molecules), which led to serious difficulties in reaching their identity. The European Union has developed special registration rules for these preparations in order to avoid lack of efficacy or increased toxicity. They include regulations to determine the quality of biological products, requirements for pre-clinical and clinical studies, according to its specific characteristics, as well as the requirements for pharmacovigilance. Implementation of such a strategy has to register in the EU several biosimilars of granulocyte colony-stimulating factor. For one of them – Zarzio – in several clinical studies fully comparable efficacy and tolerability with the original preparation was shown, thus providing a significant reduction of treatment cost with equal efficacy and toxicity. Key words: oncology, biological agents, biosimilars, neutropenia, granulocyte colony-stimulating factor, Zarzio По определению Управления по контролю качества пищевых продуктов и лекарственных препаратов США (Food and Drug Administration, FDA, USFDA) биологические лекарственные препараты, также называемые биологические продукты, представляют собой лекарственные средства, такие как вакцины, производные крови или соматических клеток, продукты генной терапии, ткани, рекомбинантные терапевтические белки, или живые клетки, применяемые для лечения заболеваний [1]. Общим в этой крайне разнородной группе является то, что они продуцируются живыми организмами или клетками, а не синтезируются химически. Ассоциация международных фармацевтических производителей (AIPM) предлагает свое определение. Биологическое лекарственное средство – лекарственное средство, действующим началом которого является вещество белковой структуры, полученное или выделенное из биологического источника, в том числе при помощи одного или нескольких перечисленных биотехнологических методов: технология рекомбинантной ДНК (рДНК); контролируемая экспрессия 1 ’2013 60 ФАРМАКОТЕРАПИЯ генов, кодирующих выработку биологически активных белков; методы гибридизации и моноклональных антител [2]. Это определение несколько сужает круг препаратов, относящихся к биологическим, но фокусирует внимание на самой быстроразвивающейся и технологичной подгруппе этого гетерогенного семейства. За последние 20 лет биологические препараты завоевали прочные позиции в медицине и терапевтические достижения в таких областях, как онкология, аутоиммунные заболевания, нефрология, эндокринология, во многом связаны именно с этой лекарственной группой [3]. В онкологии биопрепараты, разрушающие злокачественные клетки, и биопрепараты, защищающие нормальные ткани пациента, позволили добиться максимального прогресса в лечении целого ряда наиболее значимых нозологий: рака молочной железы, колоректального рака, рака почки, злокачественных лимфом [4]. К сожалению, сложности разработки, производства и тестирования лекарств данной группы определяют их высокую стоимость. Сегодня по приблизительным оценкам доля биопрепаратов в лекарственном бюджете современных онкологических центров составляет до 70 %. Это приводит к его существенному обременению и, как следствие, не все больные, выигрывающие от назначения биопрепаратов, получают их в полном объеме. Особенно остро эта проблема стоит в развивающихся странах в связи с ограниченностью средств на лекарственное обеспечение. В то же время первое поколение биофармацевтических препаратов, являющихся копиями человеческих эндогенных белков, таких как эритропоэтин, гранулоцитарный колониестимулирующий фактор (Г-КСФ), инсулин, гормон роста, которые были разработаны с использованием рДНК или гибридомной техники, исчерпало сроки патентной защиты. Это открыло рынок для воспроизведенных версий этих продуктов, так называемых «биоаналогов», во всем мире. Создание копий биологических препаратов позволяет рассчитывать на существенное снижение их стоимости за счет сокращения затрат на разработку и проверку клинической эффективности. Однако в отличие от традиционных лекарственных малых молекул, включающих от 20 до 100 атомов и легко воспроизводящихся на современном химическом производстве, биопрепараты существенно больше и сложнее. Даже такие относительно простые биопрепараты, как гормоны, включают от 200 до 3000 атомов, а большие (антитела) включают до 50 000 атомов. Можно представить соотношение структурной комбинационности аспирина (малая молекула), соматотропина (гормон роста человека) и трастузумаба (антитело к рецептору эпидермального фактора роста, применяемое при лечении рака молочной железы) на примере велосипеда, автомобиля и реактивного самолета. Число деталей в этих устройствах приблизительно соответствует количеству атомов в сравниваемых молекулах. Этот пример пока- зывает, насколько сложны молекулы биопрепаратов и потенциальные проблемы при их воспроизводстве. Получение биопрепаратов значительно отличается от производства малых лекарственных молекул. Малые молекулы синтезируются с использованием химических реакций. Биопрепараты, как правило, производят искусственно измененные клетки. Малые молекулы легко охарактеризовать, очистить и проверить с помощью обычных лабораторных тестов. Биопрепараты – особенно большие – производятся в составе смесей молекул, которые отличаются очень незначительно, что затрудняет их выделение, очистку и контроль качества [5]. Отсюда следует, что свойства биопрепаратов часто напрямую зависят от характера производственного процесса, который может длиться очень долго (до года) и зависеть от небольших изменений условий в реакторе. Кроме того, белки имеют уникальную третичную структуру, которая влияет на их работу в организме, и молекулы, полностью идентичные химически, могут иметь различные биологические эффекты. Примером этого может служить различие между сырым яйцом и сваренным: химически они идентичны, но физически и биологически очень разные. В связи с вышеизложенным во многих странах, включая ЕЭС и США, требования к разработке и лицензированию биоаналогов значительно серьезнее, чем к генерическим препаратам малых молекул. В последнем случае, как правило, достаточно физикохимической идентичности и сходности фармакокинетического профиля (биоэквивалентности). Европейский союз принял законы, а Европейское медицинское агентство (ЕМЕА) разработало нормативные принципы для утверждения биоаналогов [6, 7]. Из-за существенных различий между биофармацевтическими продуктами процесс утверждения варьирует в зависимости от препарата. В частности, характер и объем клинических исследований могут зависеть от изменчивости молекулы действующего вещества, критериев эффективности и наличия подтвержденных суррогатных маркеров терапевтической действенности. В общем, утверждение биоаналогов основано на демонстрации сопоставимой эффективности и безопасности нового продукта в соответствующей популяции пациентов (т. е. «сопоставимости»). Руководство ЕМЕА по биоаналогам позволяет, при наличии надлежащего обоснования, экстраполировать данные по эффективности и безопасности применения нового препарата по одному терапевтическому показанию на другие, принятые для референтного биопрепарата, показания, позволяя использовать биоаналоги в показаниях, для которых они не были официально изучены [8]. Применение биофармацевтических препаратов может быть связано с серьезными побочными эффектами [9], поэтому в руководстве ЕМЕА требуется проведение исследования иммуногенности и последующего фармаконадзора после вывода препарата ние содержания нейтрофилов в периферической крови после интенсивной химиотерапии (ХТ) значительно повышает риск присоединения инфекции [13]. Как следствие, существенно возрастают затраты на лечение и снижается качество жизни пациентов. Коррекция нейтропении возможна благодаря применению Г-КСФ – человеческого белка, производимого по рекомбинантной технологии и способного поддержать выживание и пролиферацию предшественников нейтрофилов в костном мозге. С 1990-х годов Г-КСФ широко применяется во всем мире, включая Россию. Значение этих препаратов как непременной составляющей сопроводительной терапии в современных интенсивных протоколах лечения опухолей трудно переоценить. Фармакоэкономические исследования показали, что применение Г-КСФ снижает риск развития инфекций, особенно тяжелых, вследствие своевременной коррекции нейтропении и в итоге уменьшает потребность в повторных госпитализациях, позволяет выиграть в затратах на лечение [14]. При этом применение Г-КСФ позволяет снизить раннюю и инфекционную летальность после ХТ, обеспечивая предпосылки продления жизни, особенно для пожилых (старше 65 лет) и ослабленных пациентов [15]. Высокая стоимость референтного препарата Нейпоген® ограничивала его назначение всем нуждающимся пациентам, что косвенно способствовало снижению дозоинтенсивности ХТ и повышению риска неудачи противоопухолевого лечения [16]. Это послужило основанием для инициирования процедуры регистрации в ЕЭС биоаналогов препарата Нейпоген® после истечения срока его патентной защиты. Эти новые препараты – Зарсио®, Теваграстим® и Рациограстим® – перед получением разрешения на клиническое использование должны были пройти преклинический и затем клинический этап оценки. Конкретные результаты можно проиллюстрировать на примере биоаналога Г-КСФ Зарсио®. На первом этапе в соответствии с рекомендациями ЕМЕА препарат был подвергнут тщательному контролю на молекулярном уровне. Пептидное картирование показало сопоставимость следующих параметров – аминокислотная последовательность и расположение дисульфидных соединений частей молекулы – препарата Зарсио® и референтного образца. В ядерном магнитно-резонансном исследовании препарат Зарсио® демонстрирует подобный референтному продукту спектр ядерного магнитного резонанса, что подтверждает сопоставимость их вторичной и третичной структур [17]. Кроме того, оба препарата показали равно высокую аффинность к рецепторам Г-КСФ на клеточной модели, что открыло возможности для клинического этапа испытаний. В I фазе клинических испытаний в 4 перекрестных исследованиях эффективности и токсичности препарата Зарсио® по сравнению с референтным продуктом было включено 146 здоровых добровольцев (81 муж- ’2013 на рынок – программы для мониторинга эффективности и безопасности биоаналогов после их утверждения. В США первоначально биоаналоги приравнивались к референтным препаратам и должны были проходить все процедуры регистрации, необходимые для нового лекарственного вещества. В настоящее время разрабатываются правовые пути для утверждения биоаналогов по сходным с ЕМЕА принципам [10]. Как и дженерики, биоаналоги предназначены для использования в той же дозе и таком же режиме введения для лечения того же заболевания, как и референтный (оригинальный) биопрепарат. Таким образом, основная цель проверки биоаналога состоит не в установлении для пациента выгоды как таковой, это уже было сделано для оригинального продукта, а в демонстрации высокого сходства с эталонным продуктом, опираясь, в частности, на его эффективность и безопасность. По этим причинам дизайн исследований, направленных на подтверждение сходства биологических продуктов, может отличаться от исследований референтных образцов. Тип и объем клинических данных, требуемых для биоаналогов, варьирует в зависимости от сложности активного вещества и возможности его характеристики, наличия принятого суррогатного критерия эффективности, от типа и серьезности проблем безопасности, а также от возможности экстраполировать эффективность и безопасность данных, полученных при одном показании, на другие, которые не были проверены. Проведение исследований для всех показаний не является обязательным и даже может считаться неэтичным, хотя для ряда клинических ситуаций это положение дискутируется [11]. Следует подчеркнуть, что принципы, лежащие в основе подтверждения сопоставимости для референтных препаратов и биоаналогов, используются и для референтного образца при внесении изменения в процесс его производства. Однако, учитывая тот факт, что новый производитель биоаналога разрабатывает собственную технологию, то требования для демонстрации его соответствия референтному препарату, как правило, более объемные, чем для демонстрации сопоставимости референтного биологического препарата до и после внесения изменений в технологию производства у того же производителя. Выполнение всех тестов, входящих в перечень необходимых исследований для каждого конкретного биопрепарата, позволит производителю биоаналога получить уверенность в том, что его препарат окажется эффективным и не опасным для больного. В короткой истории биоаналогов накопился уже определенный опыт как положительный, так и отрицательный, на основании которого можно проиллюстрировать плюсы и минусы создания этой группы лекарственных средств [12]. Одним из широко востребованных в онкологии рекомбинантных белков является Г-КСФ. Еще в середине прошлого века было установлено, что сниже- 61 1 ФАРМАКОТЕРАПИЯ 1 ’2013 62 ФАРМАКОТЕРАПИЯ чина и 65 женщин). Ответ на сравниваемые препараты оценивали по увеличению абсолютного числа нейтрофилов крови (АЧН) после однократного и многократного введения препаратов в интервале доз 1–10 мкг/кг, а также по уровню миграции CD34+-клеток из костного мозга в периферическую кровь после их многократного введения в интервале доз 2,5–10 мкг/кг [18]. Кроме того, в этих исследованиях анализу подвергались все возможные побочные действия и формирование нейтрализующих антител. Полученные результаты показали, что и кривые увеличения содержания АЧН крови, и концентрации CD34+-клеток после подкожных и внутривенных однократных и многократных введений бионалога и референтного продукта были сопоставимы. Не было также отмечено значимых различий в выраженности, тяжести и спектре побочных действий между препаратами Зарсио® и Нейпоген®. Биоаналог Г-КСФ Зарсио® прошел испытания в контролируемом клиническом исследовании [18] у 170 пациенток, страдающих раком молочной железы II стадии высокого риска (3 %), III стадии (66 %), а также IV стадии при наличии метастазов (30 %). ХТ включала 4 цикла доксорубицина 60 мг/м2 и доцетаксела 75 мг/м2 каждые 3 нед. Препарат вводился со 2-го дня после окончания ХТ подкожно ежедневно до 14-го дня или до достижения АЧН крови 10 × 109/л. Средний период назначения Зарсио® составил 31 день (от 6 до 48 дней) в ежедневной дозе 300 μg (30 MIU) или 480 μg (48 MIU) в зависимости от того, была ли масса больного менее или более 60 кг. Средняя доза препарата составила, таким образом, 6,1 ± 0,9 мкг/кг массы тела в день (от 3,7 до 8,4 мкг/кг). Учитывая колебания доз, был проведен дополнительный анализ эффекта препарата Зарсио® в зависимости от массы тела пациента. Общая частота нейтропении III или IV степени (АЧН < 1,0 × 109/л) при стратификации по массе тела и реально полученной дозе не менялась (p = 0,66). Всего в исследовании было зафиксировано 10 (6 %) случаев развития фебрильной нейтропении (ФН) (95 % ДИ от 2,9 до 10,6 %) – все в течение первого цикла ХТ [18]. Во время последующих циклов случаев ФН не наблюдалось. Госпитализация по причине ФН потребовалась 6 (3,5 %) больным и длительность ее составила 12 ± 8,1 дня. Состояние ни одной из этих пациенток не потребовало перевода в отделение интенсивной терапии. Антибиотики внутривенно получали лишь 9 (5,3 %) из лихорадивших пациентов и только 1 (0,6 %) пациент потребовал трансфузионной заместительной терапии в связи с анемией. Средняя продолжительность тяжелой нейтропении у больных, получавших Зарсио®, составила 1,8 дня в цикле 1 по сравнению с 7 днями в контроле без поддержки факторами роста [18]. Продолжительность тяжелой нейтропении в данном исследовании была сопоставима с данными, полученными в исследованиях препарата Нейпоген® [19, 20], хотя частота тяжелой нейтро- пении на протяжении нескольких циклов ХТ была несколько ниже по сравнению с опубликованными данными для референтного продукта (47 % в цикле 1 для Зарсио® по сравнению с 79 % и 83 % для препарата Нейпоген®). В то же время низкую частоту тяжелой нейтропении можно объяснить гетерогенностью характеристик включенных пациентов: в исследование с препаратом Зарсио® были включены больные, не получавшие ранее ХТ и не имевшие предшествующего миелотоксического фона, в отличие от исследования сравнения, где около 20 % пациентов получали ХТ и ранее [19]. В исследовании у здоровых добровольцев [18] ожидаемые побочные действия препарата (костномышечная боль, лейкоцитоз, тромбоцитопения и головные боли) наблюдали с равной частотой в сравнении с Нейпогеном®. Все осложнения в основном были легкими (89 %) или умеренными (11 %) по степени тяжести. В ходе исследований серьезных нежелательных явлений не было, равно как и случаев смерти. Кроме того, анализ ожидаемых побочных действий, связанных с применением Г-КСФ, не показал различий в частоте их возникновения в зависимости от реально полученной дозы при высокой или низкой массе тела пациента. Результаты лабораторных исследований, мониторирование жизненно важных функций и физикальное обследование подтвердили отсутствие заметных изменений в состоянии добровольцев и пациентов. В ходе клинических исследований с применением препарата Зарсио® для выявления образования антител к препарату были протестированы методом радиоиммунопреципитации в общей сложности 1060 проб сыворотки в период начального скрининга и 29 тестовых проб для подтверждающего анализа [18]. Среди них 3 пробы дали положительный результат на антитела. Эти 3 образца принадлежали здоровым добровольцам в исследовании EP06-102 и были взяты до лечения. Увеличения содержания антител к Г-КСФ в процессе терапии Зарсио® не отмечено. Не было обнаружено образования нейтрализующих антител в этих образцах сыворотки с помощью NAB-анализа. В заключение следует отметить, что ни у одного из участвующих в исследовании пациентов не было образования связывающих RHg-CSF антител. Данные этих исследований позволили ЕМЕА разрешить применение биоаналога Г-КСФ Зарсио® для лечения пациентов в странах Евросоюза по всем показаниям, зарегистрированным для референтного препарата Нейпоген®. С 2009 г. препарат успешно используется у онкологических и гематологических больных, что позволило в рамках расширенного фармаконадзора провести ретроспективный анализ карт пациентов после перехода с референтного препарата Г-КСФ (Нейпоген®) на биоаналог Г-КСФ (Зарсио® / Филграстим Hexal®) в широкой онкологической практике [21]. В общей сложности были оценены медицинские шинства пациентов оказалось успешным. ФН развилась только у 1 пациента. Выраженное снижение АЧН привело к редукции дозы цитостатиков у 5 (6,5 %) пациентов и прекращению ХТ у 2 (2,5 %) пациентов. Не было отмечено каких-либо побочных действий, выходящих за рамки стандартных осложнений при назначении Г-КСФ. Интенсивную ХТ с вероятностью развития ФН более 20 % получали только 24 % пациентов, 36 % больных получали Нейпоген® в качестве первичной профилактики. ФН развилась у 1 пациента. Редукция дозы химиопрепаратов потребовалась у 2 (8 %) пациентов, лечение было преждевременно прекращено также у 2 (8 %) пациентов. Выводы авторов данного исследования свидетельствуют о высокой эффективности биоаналога Г-КСФ Зарсио® и доказанной эквивалентности – химической, биологической и терапевтической – по сравнению с референтным образцом. Л И Т Е Р А Т У Р А 1. Center for Biologics Evaluation and Research (2007-10-29). “What is a biological product?” U.S. Food and Drug Administration. Retrieved 2007-12-17. 2. Avidor Y., Mabjeesh N.J., Matzkin H. Biotechnology and drug discovery: from bench to bedside. South Med J 2003;96:1174–86. 3. Guide to Biological Medicines Guide to Biological Medicines http://www. europabio.org/sites/default/files/report/ guide_to_biological_medicines_a_focus_ on_biosimilar_medicines.pdf. 4. Cheson B.D., Leonard J.P. Monoclonal antibody therapy for B-cell non-Hodgkin's lymphoma. N Engl J Med 2008;359:613–26. 5. Crommelin D.J., Bermejo T., Bissig M. et al. Pharmaceutical evaluation of biosimilars: important differences from generic low-molecular weight pharm. Eur J Hosp Pharm Sci 2005;1:11–7. 6. European Medicines Agency, Committee for Medicinal Products for Human Use. Biosimilar Guidelines. https://www.ema. europa.eu/ema/ index.jsp?curlpages/ regulation/general/general_ content_ 000408.jsp&murlmenus/regulations/ regulations.jsp&midWC0b01ac058002958c. Accessed October 20, 2012. 7. Combe C., Tredree R.L., Schellekens H. Biosimilar epoetins: an analysis based on recently implemented European Medicines Evaluation Agency guidelines on comparability of biopharmaceutical proteins. Pharmacotherapy 2005;25:954–62. 8. European Medicines Agency. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant granulocyte colony stimulating factor (G-CSF) 2006. http://www.emea.europa.eu/ pdfs/human/biosimilar/3132905en.pdf. 9. Deechongkit S., Aoki K.H., Park S.S. et al. Biophysical comparability of the same protein from different manufacturers: a case study using Epoetin alfa from Epogen and Eprex. J Pharm Sci 2006;95:1931–43. 10. U.S. Food and Drug Administration. Draft guidance on biosimilar product development. http://www.fda.gov/Drugs/ DevelopmentApproval Process/HowDrugsare DevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm. Accessed October 20, 2012. 11. Cri-report - Risk – Reward of Developing a Herceptin Biosimilar – A Thorough Assessment. http://www. basearticles.com/Art/1026247/24/Crireport-Risk--Reward-of-Developing-a-HerceptinBiosimilar--A-Thorough-Assessment.html. 12. Boven K., Stryker S., Knight J. et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005 Jun;67(6):2346–5398. 13. Bodey G.P., Buckley M., Sathe Y.S. et al. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med 1966;64:328–40. 14. Lyman G.H., Lyman C.G., Sanderson R.A. et al. Decision analysis of hematopoietic growth factor use in patients receiving cancer chemotherapy. J Nattl Cancer Inst 1993;85:488–93. 15. Kuderer N.M., Dale D.C., Crawford J. et al. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. JCO 2007;25:3158–67. 16. Lyman G.H., Dale D.C., Crawford J. et al. Incidence and predictors of low doseintensity in adjuvant breast cancer chemotherapy: a nationwide study of community practices. JCO 2003;21:4524–31. 17. Sörgel F., Lerch H., Lauber T. Physicochemical and biologic comparability of a biosimilar granulocyte colonystimulating factor with its reference product. BioDrugs 2010;24:347–57. 18. Gascon P., Fuhr U., Sörgel F. et al. Development of a new G-CSF product based on biosimilarity assessment. Ann Oncol 2010;21:1419–29. 19. Green M.D., Koelbl H., Baselga J. et al. A randomized double-blind multicenter phase III study of fixed-dose singleadministration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 2003;14:29–35. 20. Holmes F.A., O’Shaughnessy J.A., Vukelja S. et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J Clin Oncol 2002;20:727–31. 21. Verpoort K., Möhler T.M. A non-interventional study of biosimilar granulocyte colony-stimulating factor as prophylaxis for chemotherapy-induced neutropenia in a community oncology centre. Ther Adv Med Oncol 2012;4(6):289–93. ’2013 документы 77 пациентов с различными новообразованиями, получавших в связи с нейтропенией после ХТ биоаналог Г-КСФ Зарсио®. Полученные данные сравнивали с результатами 25 пациентов, получавших референтный Г-КСФ в том же центре. Средний возраст пациентов в группе биоаналога Зарсио® составил 67 (20–83) лет. В этой группе 48 % пациентов получали режимы ХТ с риском развития ФН более 20 %. У большинства больных (в 52 % случаев) биоаналог Г-КСФ был назначен в качестве первичной профилактики. В остальных случаях имела место вторичная профилактика ФН. В части случаев Г-КСФ назначали при режимах ХТ с риском развития нейтропении и инфекции менее 20 %, в этих случаях причиной назначения являлись пожилой возраст и эпизоды ФН в анамнезе, равно как и другие факторы, повышающие опасность инфекции в период постцитостатической нейтропении. Проведение профилактики нейтропении у боль- 63 1 ФАРМАКОТЕРАПИЯ НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ ОТ РЕДАКЦИИ / FROM EDITION 1 ’2013 64 Уважаемые коллеги! Продолжаем публиковать краткие лекции, которые можно назвать введением в проблему науки о наночастицах. Нет сомнений, что достижения наномедицины – которых пока реально нет – придут, прежде всего, в теорию и практику онкологии и гематологии, что становится очевидным уже при беглом просмотре списка литературных источников. Возможности использования наночастиц изучают или на моделях опухолевого роста, или на гемопоэтических культуральных системах. Поэтому нам кажется важным, чтобы первичное знакомство с тайнами нанонауки произошло на страницах журнала «Онкогематология». Надеемся, что эта информация найдет своих читателей. Начинаем, естественно, с участия наночастиц в процессах апоптоза – удивительного процесса жизни клеток, КОТОРЫЙ ЗАВЕРШАЕТ ИХ СУЩЕСТВОВАНИЕ, являясь феноменом запрограммированной клеточной смерти. Это тот процесс, который мечтают задержать, направить в нужное русло, ускорить или вообще отменить, и без понимания которого нельзя решать практические задачи лечения и профилактики всех болезней. Итак, продолжаем! Научный совет журнала Фуллерены и апоптоз ’2013 65 М.А. Орлова1, Т.П. Трофимова1, А.П. Орлов2, О.А. Шаталов3, Ю.К. Наполов3, А.А. Свистунов3, В.П. Чехонин2 1 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ 1 Химический факультет, кафедра радиохимии ФГБОУ ВПО «Московский государственный университет им. М.В. Ломоносова»; 2 медико-биологический факультет, кафедра медицинских нанобиотехнологий ГБОУ ВПО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, Москва; 3 фармацевтический факультет, кафедра фармакологии ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России Контакты: Марина Алексеевна Орлова [email protected] Многие представители обширного семейства водорастворимых аддуктов фуллеренов и наночастиц на их основе привлекают серьезное внимание как противовирусные агенты, противоопухолевые агенты и средства адресной доставки лекарств. Сегодня получено огромное количество таких производных фуллерена С60. Однако для внедрения фуллереновых производных в медицинскую практику необходимо понимание причин и механизмов прямых и отдаленных последствий их эффектов in vivo. В первую очередь это касается их влияния на регуляцию процессов пролиферации, апоптоза и некроза. Огромное значение имеют способ получения, функционализации и морфология фуллереновых наночастиц (их размеры, форма, рельеф поверхности, аффинность к клеточным структурам), т. е. параметры, в зависимости от которых биологические эффекты наночастиц могут меняться от цитопротекторного до цитотоксического. В данной лекции содержится анализ современных представлений о влиянии фуллеренов и их производных на сигнальные пути апоптоза нормальных и опухолевых клеток. Ключевые слова: фуллерены, апоптоз, сигнальные пути Fullerene and apoptosis M.A. Orlova1, T.P. Trofimova1, A.P. Orlov2, O.A. Shatalov3, Yu.K. Napolov3, A.A. Svistunov3, V.P. Chekhonin2 1 M.V. Lomonosov Moscow State University; N.I. Pirogov Russian National Research Medical University, Ministry of Health of Russia, Moscow; 3 I.M. Sechenov Moscow State Medical University, Ministry of Health of Russia 2 Fullerene derivatives superfamily attracts a serious attention as antiviral and anticancer agents and drug delivery carriers as well. A large number of such fullerene С60 derivatives obtained to date. However, there is an obvious deficit of information about causes and mechanisms of immediately and long-term consequences of their effects in vivo which is a true obstacle on the way leading to practical medical use of them. First, this concerns their impact on the proliferation, apoptosis and necrosis regulation. Fullerene nanoparticle functionalization type, their sizes and surface nanopathology are of great importance to further promoting of either cytoprotective or cytotoxic effects. This lecture provides modern concept analysis regarding fullerenes effects on apoptosis pathway in normal and tumor cells. Key words: fullerenes, apoptosis, signaling pathway Апоптоз (греч. απόπτωσις – опадание листьев) – программируемая клеточная смерть, регулируемый процесс самоликвидации на клеточном уровне, в результате которого клетка фрагментируется на отдельные апоптотические тельца, ограниченные плазматической мембраной. Фрагменты погибшей клетки обычно очень быстро (в среднем за 90 мин) фагоцитируются макрофагами либо соседними клетками, минуя развитие воспалительной реакции. Морфологически регистрируемый процесс апоптоза продолжается 1–3 ч. Одной из биологических «задач» апоптоза является уничтожение дефектных (поврежденных, мутантных, инфицированных) клеток. В многоклеточных организмах апоптоз задействован в процессах дифференцировки и морфогенеза, в поддержании клеточного гомеостаза, в обеспечении развития и функционирования иммунной системы. Апоптоз присущ всем эукариотам, начиная от одноклеточных простейших вплоть до высших организмов. Апоптоз регулируется каскадом внутриклеточных молекул, взаимодействие которых приводит к активации или торможению процесса. Этот каскад последовательных взаимодействий и превращений называется сигнальным путем. Основная задача системы, регулирующей апоптоз, – держать эффекторные ферменты – каспазы, демонтирующие клетки, в неактивном состоянии, но быстро переводить их в активную форму в ответ на минимальное действие соответствующих индукторов. Тенденция исследований в области фуллереновых производных в основном направлена на расширение их классов, в то время как механизм и интенсивность воздействия на пролиферативные процессы, а также взаимосвязь с действием других про- или антиапоптотических факторов изучена недостаточно. Нет четкой прогностической модели того, какие производные фуллерена и в каких условиях будут проявлять антиапоптотические свойства, а какие можно сразу причислять к возможным антиопухолевым агентам. Приведенная ниже градация условна, однако она помогает выделить некоторые взаимосвязи. 1 ’2013 66 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ Показано, что дендритные однозамещенные фуллерены с большим количеством гидроксильных групп (-ОН) в заместителе, а также С3-карбоксифуллерены являются ингибиторами апоптоза [1]. Однако для всех ли клеток это справедливо, и насколько универсально это действие, остается под вопросом. Водорастворимые фуллерены влияют на ионный гомеостаз, тогда как некоторые производные С60 проявляют свойства блокаторов ионных каналов биомембран [2, 3], за исключением ClC-3 каналов. Ингибирование носит обратимый характер, и, как предполагают, взаимодействие происходит во внеклеточных участках канала. Чтобы оценить возможность проникновения наночастиц сквозь мембрану, вычислили величину энергии Гиббса, необходимой для переноса С60 вдоль оси, перпендикулярной к поверхности двойного слоя мембраны [4], и была показана принципиальная возможность проникновения С60. В целом, водорастворимые производные фуллерена легко проникают через биологические мембраны различных типов клеток [5]. Митоген-активируемые протеинкиназы (MAPK) осуществляют контроль клеточного деления и являются критическими регуляторами процессов транскрипции [6], участвуя в нескольких сигнальных путях апоптоза. Токсическое действие n-C60 аналогично влиянию на апоптоз одностенных углеродных нанотрубок [7] и может быть связано с активацией MAPK, что, в свою очередь, взаимосвязано с активацией транс- H2O2 крипционного фактора NF-κB. Процесс зависит от дозы препарата [8]. Водорастворимые фуллерены in vitro и in vivo [9] выступают в роли защитных агентов от индукции остеокластогенеза по RANK (receptor activator NF-κB)сигнальному пути и деструкции костных остеокластов при артритах. В экспериментах показано, что у крыс они способны понижать RANK-индуцированную дифференцировку остеокластов и подавлять их разрушение при внутриартикулярном введении. Наночастицы С60(С(СООН)2)2 подавляют апоптоз, связанный с JNK (с-Jun amino-terminal kinases)-сигнальными путями, уменьшая JNK-фосфорилирование, активацию АР-1 (активатора протеина 1) и каспазы-3 (рис. 1). При этом не наблюдалось влияния на активацию классической митоген-активируемой киназы (ERK). Имеются данные, что протекторная роль n-C60 может быть опосредована активацией p38 [10, 11]. Каспазный путь апоптоза является наиболее распространенным, к активации каспазного каскада могут приводить изменения многочисленных компонентов биосистемы. Однако эти пути прослежены в очень немногих работах по фуллеренам, чаще рассматриваются отдельные факторы, способные привести к активации каспаз, или констатируется активация каспазы-3. Вопрос о влиянии заместителя рассматривали на примере апоптоза человеческих моноцитов (линия THP1) под действием разных производных фуллерена [12]. С60 (C(COOH)2)2 nanoparticles ROS Cyt c Рис. 1. Схема событий в CMECs, индуцированных пероксидом водорода и обработкой наночастицами C60 (C(COOH)2 )2. Изменения опосредованы уменьшением фосфорилирования JNK и ингибированием ROS-продукции. Наблюдается супрессия данного сигнального пути с уменьшением активации c-Jun и каспазы-3 и с ингибированием гидролиза PARP, высвобождающего цитохром С митохондрий [57] тирующих факторов. Происходит снижение выпуска катепсина из лизосом и ингибирование апоптоза, индуцированного TNF-α (рис. 2) [15]. Исследования, проведенные на гепатоцитах крыс, показали, что фуллеролы С60(ОН)24 проявляют зависимую от концентрации препарата (0–0,25 ммоль) и времени экспозиции (0–3 ч) цитотоксичность вследствие индукции процесса перекисного окисления липидов, потери клеточного аденозинтрифосфата (ATФ) и глутатиона, увеличения уровней малонового диальдегида и окисленного глутатиона (GSSG) [16]. Считается, что основной мишенью фуллеролов являются митохондрии, поэтому на ранней стадии наблюдается митохондриальная недостаточность, деполяризация и ингибирование синтеза АТФ. Лишь на более поздней стадии наблюдается перекисное окисление липидов в результате оксидативного стресса. Для производных С60-аминокислоты показана концентрационная зависимость при воздействии на линию человеческих эпидермальных кератиноцитов в культуре. В концентрации 0,4 и 0,04 мг/мл они снижали жизнеспособность клеток и развитие воспалительной реакции, а в концентрации ниже 0,04 мг/мл инициировали потерю активности цитокинов и поддерживали жизнеспособность клеток [17]. Перспективными можно признать исследования с целью оценки возможности использовать фуллерены в качестве модулятора действия лекарств. Например, гибрид С60-дексаметазон сохраняет противовоспалительную активность дексаметазона, но оказывает гораздо более низкое иммуносупрессивное действие [18]. Фуллерен [Gd2C82(OH)22]n может вызвать фенотипическое созревание дендритных клеток. Эти наночастицы являются мощным активатором как для Aggregate [С60 (C(COOH)2)2]n pH 7.0 Lysosomal lipid membrane Lysosomal membrane pore С60 (C(COOH)2)2 nanoparticles Рис. 2. Влияние С60 (С(СООН)2 )2 на лизосомально-митохондриальный апоптотический путь [35] ’2013 Оказалось, что роль заместителя особенно важна для регуляции апоптоза. Некоторые фуллерены способны, по крайней мере, частично защищать клетки от апоптоза, вызванного оксидативным стрессом, путем повышения экспрессии антиоксидантных белков. При действии n-С60 на рост опухолевых клеток меланомы мышей В16 in vitro наблюдали оксидативный стресс, митохондриальную деполяризацию и, как результат, активацию каспазного каскада [13]. Наоборот, наночастицы С60(С(СООН)2)2 ингибировали расщепление поли(АДФ-рибоза)-полимеразы (PARP) и выход митохондриального цитохрома С, что приводило к эффективному подавлению каспаззависимого апоптоза, вызванного окислительным стрессом. Производные С60-гексакарбоновой кислоты описаны как блокаторы апоптозного сигнала тромбоцитарного фактора роста β (TGF-β) в клетках человеческой гепатомы [14]. С60(С(СООН)2)2 может ингибировать апоптоз, инициированный TNF-α в клетках HeLa, с помощью стабилизации лизосом. При интернализации С60(С(СООН)2)2 повышается экспрессия молекулярного шаперона Hsp70, что способствует выживаемости клеток путем ингибирования пермеабилизации лизосомальных мембран. Важным фактором является кислая среда внутри лизосом, которая оказывает заметное, хотя и временное влияние на агрегированность наночастиц. Распределение по размерам изменяется в среднем со 100 до 10 нм и даже до отдельных молекул. Дезагрегированные частицы могут встраиваться в лизосомальные мембраны, образуя фуллерен-содержащие смешанные бислойные мембраны. Это стабилизирует лизосомы, уменьшая проницаемость их мембран, в частности для деструк- 67 1 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ 1 ’2013 68 дендритных клеток, так и для Th1-опосредованного иммунного ответа. Препарат вызывает увеличение продукции IFN-γ, IL-1β, IL-12, p70 и IL-2 и ко-стимуляторов СD80, CD83, CD86 [19]. Механохимически солюбилизированный С60 способен выступать цитопротектором против NO-индуцированной цитотоксичности, но не в результате прямого взаимодействия [20]. Предполагают, что происходит нейтрализация митохондриально-продуцированных супероксид радикалов, и это пресекает цепочку образования пероксинитрита и активации каспаз. Показано, что именно индукция пероксинитрита может быть критическим сигнальным событием для генотоксичности нано-С60 [21]. Растворимые фуллерены, модифицированные β-аланином, валином и фолиевой кислотой, проявляют непосредственную акцепторную активность по отношению к NO-радикалам [22]. NO-индуцированные события при контакте клеток феохромоцитомы крыс с 1 ммоль/л нитропруссида натрия (SNP), который вызывал заметное снижение митохондриального мембранного потенциала, активности супероксиддисмутазы (SOD), каталазы (CAT) и глутатионпероксидазы (Gpx), уменьшение жизнеспособности клеток, увеличение уровня накопления внутриклеточного NO и малонового диальдегида и повышение активности каспазы-3, заметно ингибировались предобработкой клеток до контакта с SNP аминокислота-С60-производными. Степень ингибирования зависела от морфологии агрегации фуллереновых наночастиц, которая влияла и на их защитный эффект против Н2О2-индуцированного апоптоза. Сульфонат-производные С60 после внутривенного введения повышали содержание NO в плазме крови [23]. Фуллеролы С60(ОН)24 и С60(ОН)х вызывали прямое NO-тушение [24], результатом чего явилось уменьшение NO-индуцированного снижения активностей антиоксидантов – CAT, глутатион-S-трансферазы (GST) и Gpx в денуклеированной фракции интерстиа б циальных тестикулярных клеток взрослых крыс после инъекции SNP. Предполагается, что in vivo может происходить конкуренция между аргиназой и индуцибельной NO-синтазой (iNOS) за аргинин как субстрат. В подтверждение этого для С60(ОН)х действительно наблюдалось увеличение активности аргиназы и подавление продукции NO. Полигидроксилированные C60-производные [25], С60-(глюкозамин)6 [26], карбоксифуллерены [20, 24] и С60-(цистин)5 показали способность включаться во взаимосвязи и реакции с белками и биоструктурами, что приводит к модуляции апоптоза в различных типах клеток, включая нейроны. Известно воздействие наночастиц модифицированных фуллеренов на клеточный ионный гомеостаз [27] и репликацию вирусов [2], а также их участие в расщеплении ДНК [28], ингибировании амилоидных образований [29]. Пытаются моделировать взаимодействие С60 и ряда белков, представляющих возможные мишени для лекарств [30], с целью создания в дальнейшем биоконъюгатных материалов. На основании компьютерного моделирования получены структуры комплексов С60 с NOS, GST (рис. 3), β-секретазой-1, гипоксантин фосфорибозил трансферазой [31], с некоторыми другими белками. Имеются экспериментальные подтверждения участия производных фуллеренов в ингибировании NOS [32], глутатион-редуктазы [33], а также цистеиновых и сериновых протеаз [34]. Известна анти-ВИЧ (ингибиторная) активность фуллеренпроизводных [2, 35, 36], а также образование фуллерен-специфичных антител [37]. Таким образом, С60-производные способны вмешиваться в биохимические процессы организма за счет связывания с белками и ферментами. С одной стороны, важно научиться использовать эти возможности в терапевтических целях, предсказывая докингом наиболее благоприятные мишени для целенаправленного действия с использованием производных фуллерена. Однако эти же процессы могут представлять в Рис. 3. Кристаллографическая структура глутатион-S-трансферазы с интеркалированной молекулой глутатиона (а) и докинг-комплексы С60-глутатион-S-трансфераза (б) и С60-NO-синтаза (в) [58] представление о модели транспорта наночастиц в биологической системе. При прямом измерении митохондриальной Mg2+АТФазной активности оказалось, что фермент ингибируется фуллеролом с IC50 = 7,1 ± 0,3 μмоль/л [46] и процесс является концентрационно-зависимым. Применение водорастворимых гексасульфонатных производных фуллерена С60 у крыс оказывало нейропротективное действие, снижая концентрацию лактатдегидрогеназы в крови [23]. Фуллеропирролидины являются неконкурентными ингибиторами по отношению к ацетилхолинэстеразе, IC50 варьируют в пределах 15,6–31,4 μмоль/л [47], а смешанный карбоксифуллерен-С60 проявлял ингибиторную активность против цистеиновых и сериновых протеиназ. Ингибирование тромбина составляло 24 % при концентрации препарата 10 μмоль/л [44, 48]. Как ингибиторы, такие фуллерены могут использоваться при ожогах и в качестве радиопротекторов, т. е. в тех случаях, когда происходит сильнейшая активация протеиназ. Водорастворимые фуллерены, имеющие в качестве боковой группы спиртовую, аминную или аминокислотную, являются ингибиторами Zn-содержащего фермента карбоангидразы (CAs) [49]. Механизм связан с закупоркой входа в активный центр сеткой фуллерена (размер и того и другого ~ 1 нм), при этом боковые фрагменты осуществляют взаимодействия с аминокислотными остатками активного центра, среди которых гистидин в положении 64, 94 и 96, валин в положении 121 и тирозин в положении 200. Получен урациловый аддукт С60 [50], способный к комплементарному связыванию с аденином, аденозином и АТФ. Разнообразные свойства могут проявлять и порфирин-фуллерены. Образуя металлоком- His440 Trp214 Trp214 His288 His440 His288 Рис. 4. Докинг-прогнозируемый сайт связывания карбоксифуллерена с HSA [59] ’2013 опасность для организма, непредсказуемо нарушая пролиферацию и вмешиваясь в сигнальные пути апоптоза, которые до сих пор изучены недостаточно. Особую важность представляет способность фуллеренов взаимодействовать с BSA (бычий сывороточный альбумин) [38, 39], HSА (человеческий сывороточный альбумин, коэффициент связывания Ксвяз. = 1,2 × 107 моль-1) [40] и лизоцимом [41], так как BSA и HSА выполняют в организме функцию универсального транспортера белков и биологически важных микроэлементов (металлов) [42]. Высокое сродство к HSA показали органофосфат-содержащие фуллеролы [43]. При этом происходило изменение третичной структуры HSА в сторону большей компактности с увеличением процента α-спиралей и β-складок и увеличением полярности окружения триптофанового остатка. HSА может обеспечивать доставку наночастиц к органам и тканям. Однако одновременно создается конкуренция другим физиологически важным компонентам за счет изменения конформационного состояния молекулы и простой конкурентности. О реальном влиянии фуллеренов на структуру и функции белков пока известно мало. Более того, хотя некоторые белки и взаимодействуют с фуллеренами, до настоящего времени кристаллической структуры таких комплексов не было получено. Однако докингом было показано [44] высокое сходство между физико-химическими свойствами и геометрией поверхности сайтов связывания фуллерена с ВИЧпротеазой, BSA и HSA (рис. 4). Известно образование комплекса карбоксифуллерена с β-лактоглобулином (типичным представителем семейства белков барьерной жидкости) [45], и показана возможность переноса карбоксифуллерена из образованного комплекса в альбумин, что расширяет 69 1 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ 1 ’2013 70 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ плексы, они способны встраиваться в белки, например, в апо-миоглобин, с получением С60-миоглобина [51]. Один из наиболее известных путей апоптоза реализуется через повреждения ДНК, что активирует как минимум р53-сигнальный путь [52]. Показано, что водорастворимые производные C60 могут резко усиливать эффективность полимеразной реакции при относительно низкой концентрации ДНК [53], что способно помешать нормальной репликации ДНК in vivo и приводит в действие различные сигнальные пути апоптоза. Моно- и бис-метанофосфонаты С60 выступают в ряде случаев ингибиторами ДНК-рестриктивных эндонуклеаз. Ингибирование ДНК-эндонуклеазы Exo III бис-метанофосфонатом-С60 (BMPF) прямо зависит от дозы фуллерена, а ингибирование полимеразной цепной реакции (ПЦР) с Taq ДНК-полимеразой (IC50 = 2,7 μмоль/л) с помощью BMPF уменьшалось при увеличении концентрации Taq полимеразы в системе ПЦР [54]. Предполагается, что этот процесс ингибирования не коррелирует с АФК и зависит от связывания фуллеренового аддукта с полимеразой, а не с ДНК. При рН < 9 размер фуллерола удобен для связывания с бактериальной ДНК [55]. В этом случае происходит увеличение термической и ферментативной (по отношению к ДНКазе I и Hind III рестриктазе) стабильности ДНК дозозависимым образом. Защитная концентрация фуллерола составляла 50–500 нг/μл. В то же время при связывании этой же ДНК с нано-С60 для защиты от ферментативного расщепления требовалась концентрация не менее 500 нг/μл. Наблюдалась также зависимость влияния размеров фуллереновых наночастиц на результат воздействия на ДНК и на токсичность. Например, нано-С60 с размером 100 нм являются неблагоприятными для связывания с ДНК. ДНК-фуллерен связывание способно изменять третичную структуру молекул ДНК и усложнять их узнавание белками, которое базируется на специфической и строго определенной форме молекулы ДНК [56]. Конкретные изменения были продемонстрированы на примере комплекса С60(ОН)24-ДНК, где фуллерол прочно связывался с фосфатным остовом за пределами цепи нативной ДНК и с парами оснований в пределах главного канала между спиралями [57]. Благодаря способности вызывать в определенных условиях гибель клеток фуллерены являются потенциальными противоопухолевыми агентами. Однако однозначное прогнозирование про- и антиапоптотических свойств различных производных фуллерена, тем более для различных типов клеток, пока еще невозможно. Л И Т Е Р А Т У Р А 1. Lebovits R., Rosenblum M. Substuted fullerenes and their use as inhibitors of cell death, US Patent, № 0197950 A1, 2009. 2. Friedman S.H., De Camp D.L., Sijbesma R.P. et al. Inhibition of the HIV-1 protease by fullerene derivatives: model building studies and experimental verification. J Am Chem Soc 1993;115:6506–9. 3. Park K.H., Chhowalla M., Iqbal Z., Sesti F. Single-walled carbon nanotubes are a new class of ion channel blockers. J Biol Chem 2003;278:50212–6. 4. Redmill P.S., McCabe C. Molecular dynamics study of the behavior of selected nanoscale building blocks in a gel-phase lipid bilayer. J Phys Chem B 2010;114:9165–72. 5. Andreev I., Petrukhina A., Garmanova A. et al. Penetration of fullerene C60 derivatives through biological membranes. Fullerenes Nanotubes & Carbon Nanostructures 2008;16:89–102. 6. Johnson G.L., Lapadat R.L. Mitogenactivated protein kinase pathways mediated by ERK, JNK and p38 protein kinases. Science 2002;298:1911–2. 7. Фатхутдинова Л.М., Халиулин Т.О., Зальялов Р.Р. Токсичность инженерных наночастиц. Казанский мед журн 2009;90:578–84. 8. Manna S.K., Sarkar S., Barr J. et al. Single-walled carbon nanotube induces oxidative stress and activates nuclear transcription factor-κB in human keratinocytes. Nano Lett 2005;5:1676–84. 9. Yudoh K., Karasawa R., Masuko K., Kato T. Water-soluble fullerene (C60) inhibits the osteoclast differentiation and bone destruction in arthritis. Int J Nanomed 2009;4:233–9. 10. Okada T., Otani H., Wu Y. et al. Role of F-actin organization in p38 MAPkinasemediated apoptosis and necrosis in neonatal rat cardiomyocytes subjected to simulated ischemia and reoxygenation. Am J Physiol Heart Circ Physiol 2005;289:2310–8. 11. Luschen S., Scherer G., Ussat S. et al. Inhibition of p38 mitogen-activated protein kinase reduces TNF-induced activation of NF-kB, elicits caspase activity, and enhances cytotoxicity. Exp Cell Res 2004;293:196–206. 12. Rebecca M., Hsing-Lin W., Jun G. et al. Impact of physicochemical properties of engineered fullerenes on key biological responses. Toxicol Appl Pharmacol 2009;234:58–67. 13. Zogovic N.S., Nikolic N.S., Vranjes-Djuric S.D. et al. Opposite effects of nanocrystalline fullerene (C60) on tumour cell growth in vitro and in vivo and a possible role of immunosupression in the cancerpromoting activity of C60. Biomaterials 2009;30:6940–6. 14. Huang Y.L., Shen C.K.F., Luh T.Y. et al. Blockage of apoptotic signaling of transforming growth factor-beta in human hepatoma cells by carboxy-fullerene. Eur J Biochem 1998;254:38–43. 15. Li W., Zhao L., Wei T. et al. The inhibition of death receptor mediated apoptosis through lysosome stabilization following internalization of carboxyfullerene nanoparticles. Biomaterials 2011;32: 4030–41. 16. Nakagawa Y., Suzuki T., Ishii H. et al. Cytotoxic effects of hydroxylated fullerenes on isolated rat hepatocytes via mitochondrial dysfunction. Arch Toxicol 2011;85:1429–40. 17. Rouse J.G., Yang J.Z., Barron A.R., Monteiro-Riviere N.A. Fullerene-based amino acid nanoparticle interactions with human epidermal keratinocytes. Toxicol In Vitro 2006;20(8):1313–20. 18. Liu R.L., Cai X.Q., Wang J.D. et al. Research on the bioactivities of C60dexamethasone. J Nanosci Nanotechnol 2009;9:3171–6. 19. Yang D., Zhao Y., Guo H. et al. [Gd@C82(OH)22]n nanoparticles induce dendritic cell maturation and activate Th1 synthase by acting as highly potent calmodulin antagonists. Arch Biochem Biophys 2002;399:130–41. 33. Mashino T., Okuda K., Hirota T. et al. Inhibitory effect of fullerene derivatives on glutathione reductase. Fullerene Sci Technol 2001;9:191–6. 34. Tokuyama H., Yamago S., Nakamura E. et al. Photoinduced biochemical-activity of fullerene carboxylic-acid. J Am Chem Soc 1993;115:7918–9. 35. Marcorin G.L., Da Ros T., Castellano S. et al. Design and synthesis of novel [60] fullerene derivatives as potential HIV aspartic protease inhibitors. Org Lett 2000;2:3955–8. 36. Schuster D.I., Wilson S.R., Schinazi R.F. Anti-human immunodeficiency virus activity and cytotoxicity of derivatized buckminsterfullerenes. Bioorg Med Chem Lett 1996;6(11):1253–6. 37. Braden B.C., Goldbaum F.A., Chen B.X. et al. X-Ray crystal structure of an anti-buckminsterfullerene antibody fab fragment: biomolecular recognition of C60. Proc Natl Acad Sci U.S.A. 2000;97:12193–7. 38. Rozhkov S.P., Goryunov A.S., Sukhanova G.A. et al. Protein interaction with hydrated C60 fullerene in aqueous solutions. Biochem Biophys Res Commun 2003;303:562–6. 39. Belgorodsky B., Fadeev L., Kolsenik J., Gozin M. Formation of a soluble stable complex between pristine C60-fullerene and a native blood protein. Chembiochem 2006;7:1783–9. 40. Belgorodsky B., Fadeev L., Ittah V. et al. Formation and characterization of stable human serum albumin-tris-malonic acid [C60] fullerene complex. Bioconjug Chem 2005;16:1058–62. 41. Yang S.T., Wang H., Guo L. et al. Interaction of fullerenol with lysozyme investigated by experimental and computational approaches. Nanotechnology 2008;19:395–401. 42. Zhang X.F., Shu C.Y., Xie L. et al. Protein conformation changes induced by a novel organophosphate-containing watersoluble derivative of a C60 fullerene nanoparticle. J Phys Chem C 2007;111:14327–33. 43. Benyamini H., Shulman-Peleg A., Wolfson H.J. et al. Interaction of C60-fullerene and carboxyfullerene with proteins: docking and binding site alignment. Bioconjug Chem 2006;17:378–86. 44. Belgorodsky B., Fadeev L., Kolsenik J., Gozin M. Biodelivery of a fullerene derivative. Bioconjug Chem 2007;18:1095–100. 45. Ueng T.H., Kang J.J., Wang H.W. et al. Suppression of microsomal cytochrome P450-Dependent monooxygenases and mitochondrial oxidative phosphorylation by fullerenol, a polyhydroxylated fullerene C60. Toxicol Lett 1997;93:29–37. 46. Pastorin G., Marchesan S., Hoebeke J. et al. Design and activity of cationic fullerene derivatives as inhibitors of acetylcholinesteras. Org Biomol Chem 2006;4:2556–62. 47. Iwata N., Mukai T., Yamakoshi Y.N. et al. Effect of C60, a fullerene, on the activities of glutathione S-transferase and glutathion-related enzymes. Fullerenes, Nanotubes, Carbon Nanostruct 1998; 6:213–26. 48. Innocenti A., Durdagi S., Doostdar N. et al. Nanoscale enzyme inhibitors: fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg Med Chem 2010;18:2822–8. 49. Marczak R., Hoang V.T., Noworyta K. et al. Molecular recognition of adenine, adenosine and ATP at the air–water interface by a uracil appended fullerene. J Mater Chem 2002;12:2123–9. 50. Ito M., Nakashima N. Design, synthesis and photophysical properties of C60-modified proteins. J Mater Chem 2002;12:2026–33. 51. Желтухин А.О., Чумаков П.М. Повседневные и индуцированные функции гена p53. Усп биол наук 2010;50:447–516. 52. Liang Y., Luo F., Lin Y. et al. C60 affects DNA replication in vitro by decreasing the melting temperature of DNA templates. Carbon 2009;47:1457–65. 53. Kang F., Song G.G. Inhibition of Taq DNA polymerase and DNA exonuclease ExoIII by an aqueous nanoparticle suspension of a bis-methanophosphonate fullerene. Mater Sci Forum 2011;685:345–51. 54. An H., Jin B. DNA exposure to buckminsterfullerene (C60): toward DNA stability, reactivity, and replication. Environ Sci Technol 2011;45:6608–16. 55. Rohs R., West S.M., Sosinsky A. et al. The role of DNA shape in protein-DNA recognition. Nature 2009;461:1248–53. 56. Pinteala M., Dascalu A., Ungurenasu C. Binding fullerenol C60(OH)24 to dsDNA. Int J Nanomed 2009;4:193–9. 57. Shinohara N., Matsumoto K., Endoh S. et al. In vitro and in vivo genotoxicity tests on fullerene C60 nanoparticles. Toxicol Lett 2009;191:289–96. 58. Baker G.L., Gupta A., Clark M.L. et al. Inhalation toxicity and lung toxicokinetics of C60 fullerene nanoparticles and microparticles. Toxicol Sci 2008;101:122–31. 59. Seki M., Fujishima S., Gondo Y. et al. Acute toxicity of fullerene C60 in aquatic organism. Environ Sci 2008;21:53–62. ’2013 immune responses. ACS Nano 2010;4(2):1178–86. 20. Misirkic M.S., Todorovic-Markovic B.M., Vucicevic L.M. et al. The protection of cells from nitric oxide-mediated apoptotic death by mechanochemically synthesized fullerene (C60) nanoparticles. Biomaterials 2009;30:2319–28. 21. Xu A., Chai Y., Nohmi T., Hei T.K. Genotoxic responses to titanium dioxide nanoparticles and fullerene in gpt delta transgenic MEF cells. Part Fibre Toxicol 2009;6:3–16. 22. Hu Z., Huang Y., Guan W. et al. The protective activities of water-soluble C60 derivatives against nitric oxide-induced cytotoxicity in rat pheochromocytoma cells. Biomaterials 2010;31:8872–81. 23. Huang S.S., Tsai S.K., Chin C.L. et al. Neuroprotective effect of hexasulfobutylated C60 on rats subjected to focal cerebral ischemia. Free Rad Biol Med 2001;30(6):643–9. 24. Bisaglia M., Natalini B.L., Pellicciari R. et al. Carboxyfullerenes as neuroprotective agents C3-fullerotris-methanodicarboxylic acid protects cerebellar granule cells from apoptosis. J Neurochem 2000;74:1197–204. 25. Chen Y.W., Hwang K.C.H., Yen C.C., Lai Y.L. Fullerene derivatives protect against oxidative stress in RAW 264.7 cells and ischemia-reperfused lungs. Am J Physiol 2004;287:21–6. 26. Chien C.T., Chen C.F., Hsu S.M. et al. Forced expression of bcl-2 and bcl-xL by novel water-soluble fullerene, C60(glucosamine)6, reduces renal ischemia/ reperfusion-induced oxidative stress. Fuller Nanotub Car 2001;9:77–88. 27. Kotelnikova R.A., Kotelnikov A.I., Bogdanov G.N. et al. Membranotropic properties of the water soluble amino acid and peptide derivatives of fullerene C60. FEBS Lett 1996;389:111–4. 28. Boutorine A.S., Tokuyama H., Takasugi M. et al. Fullerene-oligonucleotide conjugates: photoinduced sequence-specific DNA cleavage. Angew Chem Int Ed 1994;33:2426–65. 29. Kim J.E., Lee M. Fullerene inhibitsamyloid peptide aggregation. Biochem Biophys Res Commun 2003;303:576–9. 30. Calvaresi M., Zerbetto F. Baiting proteins with C60. ACS Nano 2010;4:2283–99. 31. Gupta S., Dhawan A., Shanker R. In silico approaches: prediction of biological targets for fullerene derivatives. J Biomed Nanotechnol 2011;7:91–2. 32. Wolff D.J., Barbieri C.M., Richardson C.F. et al. Trisamine C60-fullerene adducts inhibit neuronal nitric oxide 71 1 НОВЫЕ НАПРАВЛЕНИЯ МЕДИЦИНСКОЙ НАУКИ КОНФЕРЕНЦИИ, СИМПОЗИУМЫ, СОВЕЩАНИЯ Научно-практическая конференция «Актуальные вопросы детской онкологии и гематологии» 1 ’2013 72 К.И. Киргизов1, С.Р. Варфоломеева1, Г.И. Бишарова2 1 ФНКЦ ДГОИ им. Дмитрия Рогачева Минздрава России, Москва; Читинский филиал Научного центра проблем здоровья семьи и репродукции человека Сибирского отделения РАМН 2 15–16 января 2013 г. в Чите состоялось открытие Клиники детской онкогематологии в новом корпусе Забайкальского краевого онкологического диспансера, а также была проведена научно-практическая конференция «Актуальные вопросы детской онкологии и гематологии». Клиника детской онкогематологии Читинского филиала Научного центра проблем здоровья семьи и репродукции человека Сибирского отделения РАМН является одним из ведущих центров в области детской онкологии/гематологии Сибири. 15 января 2013 г. во вновь построенном корпусе Забайкальского краевого онкологического диспансера состоялось открытие отделения детской онкогематологии, где предусмотрены специальные асептические палаты для проведения трансплантации гемопоэтических стволовых клеток. Отделение оснащено современным оборудованием, созданы условия для проведения афереза стволовых клеток. Открытие отделения привлекло большое внимание общественности, на торжественной церемонии присутствовали губернатор края Р.Ф. Гениатулин и министр здравоохранения краевого правительства Б.П. Сормолотов. Руководитель московской делегации, директор Федерального научно-клинического центра детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева, академик РАМН А.Г. Румянцев отметил, что открытие такого отделения в Забайкальском крае позволит вывести на новый уровень краевую науку и практику. Семинар по программе «Дальние регионы» традиционно открыли заместитель министра здравоохранения Забайкальского края, начальник отдела материнства и детства Н.Г. Игнатьева и главный врач Забайкальского краевого онкологического диспансера С.В. Лесков. С докладом о приоритетных направлениях развития детской онкогематологии выступил акад. А.Г. Румянцев. Особое внимание он уделил изучению молекулярно-генетических механизмов развития заболеваний и современных способов их диагностики и лечения. Профессор Читинского филиала Научного центра проблем здоровья семьи и репродукции человека Сибирского отделения РАМН Г.И. Бишарова рассказала о развитии детской онкогематологии в Забайкальском крае, отметив значение нового отделения в развитии данного направления. Представители забайкальской школы детской гематологии-онкологии О.И. Кряжева и Е.П. Мацеха рассказали об особенностях эпидемиологии злокачественных новообразований в Забайкальском крае и организации их мониторинга с помощью канцер-регистра. В рамках программы «Дальние регионы» состоялся научнопрактический семинар и консультация пациентов, организованные ФНКЦ ДГОИ им. Дмитрия Рогачева, благотворительным фондом «Подари Жизнь» и Национальным обществом детских гематологов и онкологов. ТЕРАПИЯ ВЫСОКИХ ДОСТИЖЕНИЙ Показания. Неходжкинская лимфома Рецидивирующая или химиоустойчивая В-клеточная, СD20-положительная неходжкинская лимфома низкой степени злокачественности или фолликулярная. Фолликулярная лимфома III-IV стадии в комбинации с химиотерапией у ранее нелеченных пациентов. Фолликулярная лимфома в качестве поддерживающей терапии после ответа на индукционную терапию. СD20-положительная диффузная В-крупноклеточная неходжкинская лимфома в комбинации с химиотерапией по схеме CHOP. Хронический лимфолейкоз в комбинации с химиотерапией у пациентов, ранее не получавших стандартную терапию. Рецидивирующий или химиоустойчивый хронический лимфолейкоз в комбинации с химиотерапией. Ревматоидный артрит (активная форма) у взрослых в комбинации с метотрексатом при непереносимости или неадекватном ответе на текущие режимы терапии, включающие один или более ингибиторов фактора некроза опухолей (ФНО-а). Безопасность и эффективность препарата у детей не установлены. Противопоказания. Гиперчувствительность к ритуксимабу, любому компоненту препарата или к белкам мыши, острые инфекционные заболевания, выраженный первичный или вторичный иммунодефицит. Правила приготовления и хранения раствора. Необходимое количество препарата набирают в асептических условиях и разводят до расчетной концентрации (1-4 мг/мл) в инфузионном флаконе (пакете) с 0.9% раствором натрия хлорида для инфузий или 5% раствором декстрозы (растворы должны быть стерильными и апирогенными). Приготовленный инфузионный раствор Мабтеры физически и химически стабилен в течение 12 ч при комнатной температуре или в течение не более 24 ч при температуре от 2 до 8 °С. Мабтеру вводят внутривенно, инфузионно (медленно), через отдельный катетер. Нельзя вводить в/в болюсно или в виде в/в инъекций. Дополнительная информация в инструкции по применению. ЗАО «Рош-Москва» Официальный дистрибьютор «Ф. Хоффманн - Ля Рош Лтд.» (Швейцария) Россия, 107031 Москва Трубная площадь, дом 2 Бизнес-центр «Неглинная Плаза» Тел.: + 7 (495) 229-29-99 Факс: + 7 (495) 229-79-99 www.roche.ru