Этиология и патогенез анемии при злокачественных
реклама

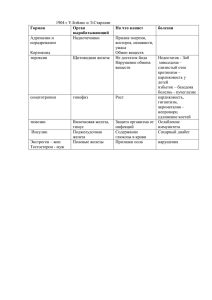
Болезни эритрона Этиология и патогенез анемии при злокачественных новообразованиях А.Д.Павлов, Е.Ф.Морщакова Рязанский филиал НИИ детской гематологии Министерства здравоохранения РФ Ключевые слова: анемия, злокачественные новообразования, патогенетические факторы, эритропоэтин Etiology and pathogenesis of anemia in malignant neoplasms A.D.Pavlov, E.F.Morshchakova Research Institute of Pediatric Haematology, Ministry of Public Health of the Russian Federation, Ryazan Branch Key words: anemia, malignant neoplasms, pathogenetic factors, erythropoietin А немия является обычным проявлением злокачественных новообразований (ЗН). Около 60% пациентов онкологических и онкогематологических клиник страдают анемией, чаще ее выявляют у больных с более выраженными стадиями прогрессивного роста опухолевых клеток и особенно у получавших химио- и лучевую терапию [1]. Анемия при ЗН (АЗН) входит в группу анемий хронических болезней (АХБ) и является частью гематологического стресс-синдрома [2]. АЗН может быть результатом хронической болезни (АХБ) со всеми характерными патофизиологическими чертами, но она может быть также вызвана химиотерапией или лучевой терапией. АЗН относится, как и АХБ, к гипопролиферативным анемиям с относительно уменьшенным числом ретикулоцитов, не соответствующим степени анемии. По данным исследования, включавшего 401 пациента с различными типами ЗН [3], средняя величина гемоглобина (Hb) у всех обследованных всегда была меньше 100 г/л (с колебаниями от 87 до 96 г/л). Среди обследованных данной группы анемия у пациентов с солидными опухолями (СО) была значительно менее выраженной, чем у пациентов с гематологическими ЗН (хронические миелопролиферативные заболевания – ХМЗ, хронический лимфоцитарный лейкоз – ХЛЛ, злокачественные лимфомы – ЗЛ, множественная миелома – ММ, исключая миелодиспластический синдром – МДС. У всех пациентов средние величины среднего объема эритроцитов и концентрация Hb в клетке были в пределах нормы, но имелись различия между группами относительно среднего объема эритДля корреспонденции: Павлов Анатолий Дмитриевич, член-корреспондент РАЕН, доктор медицинских наук, профессор, заместитель директора по науке Рязанского филиала НИИ детской гематологии Министерства здравоохранения РФ Адрес: 390029, Рязань, ул. Строителей, 5В Телефон: (0912) 98-6964 E-mail: [email protected] Статья поступила 14.12.2003 г., принята к печати 26.01.2004 г. 50 роцитов в каждой группе. У значительной части пациентов с СО имелась микроцитарная анемия (по сравнению с другими типами ЗН) и у значительно большей части пациентов с МДС, ХЛЛ, ММ и ЗЛ выявлены макроцитарная анемия (по сравнению с пациентами с СО и хроническими миелопролиферативными заболеваниями, p < 0,0002). Эти результаты указывают, что разные типы АЗН различаются по тяжести и процентному содержанию микро- и макроцитарных форм. В большинстве случаев, однако, АЗН – это гипорегенераторная анемия с низкими числами ретикулоцитов (ретикулоцитопения), с величинами Hb 80–100 г/л и средним объемом эритроцита и средней концентрацией клеточного гемоглобина в пределах нормы [3]. Для анемии характерны такие проявления как снижение физической и умственной активности, быстрая утомляемость и подавленное настроение, что приводит к снижению качества жизни (КЖ). Хотя АЗН может развиваться как результат самого патологического процесса, наиболее часто она возникает ятрогенным путем как следствие химио- или лучевой терапии. Анализ опубликованных клинических наблюдений [5] показал, что частота анемии после химиотерапии составляет 100% (анемия легкой и умеренной степени) и 80% (анемия средней и тяжелой степени). Частота АЗН варьирует в зависимости от типа опухоли и режима химиотерапии. Так, комбинации цисплатина и этопозида наиболее широко используются для лечения мелкоклеточного рака легких, анемия средней и тяжелой степени развивается у 16–55% пациентов. Однако при комбинации 5-флуоурацила и лейковорина, используемой для лечения прогрессирующего колоректального рака, анемия средней и тяжелой степени развивалась только у 2–5% пациентов. Частота развития анемии повышается по мере прохождения всего курса химиотерапии. При анализе данных наблюдения 1064 пациентов с различными ЗН, которые получали химиотерапию, установлено [6], что к третьему циклу терапии примерно у 2/3 пациентов выявлена анемия: у 28% – уме- ùÚËÓÎÓ„Ëfl Ë Ô‡ÚÓ„ÂÌÂÁ ‡ÌÂÏËË ÔË ÁÎÓ͇˜ÂÒÚ‚ÂÌÌ˚ı ÌÓ‚ÓÓ·‡ÁÓ‚‡ÌËflı ренной степени (уровни Hb от 105 до 120 г/л), у 34% – средней степени (Hb от 80 до 105 г/л) и у 5% – тяжелой степени (Hb менее 80 г/л). В отдельном исследовании среди пациентов, получавших цитотоксическую химиотерапию (n = 2821), в среднем у 17% пациентов имелась анемия (Hb менее 80 г/л) перед первым циклом, к шестому циклу эта цифра увеличивалась до 35% [7]. Из этих пациентов 33% нуждались по крайней мере в одной трансфузии эритроцитов вследствие анемии. Анемия после радиотерапии также развивается часто. Так, у 48% пациентов, направленных на лучевую терапию по поводу СО, была анемия (Hb менее 120 г/л) перед лечением и у 57% – после окончания лечения [8]. Многие исследователи обнаружили, что неадекватная оксигенация места локализации опухоли и/или низкие величины Hb являются причиной плохих результатов радиотерпии [9–12]. Оптимальная ответная реакция на радиотерапию требует адекватной оксигенации опухоли, и, таким образом, теоретически возможно, что уменьшение концентрации Hb (что может содействовать снижению оксигенации опухоли) также оказывает влияние на успешное проведение лучевой терапии. В исследовании, проведенном у больных раком шейки матки (n = 74), выживаемость была статистически достоверно связанной с оксигенацией опухоли (p = 0,02) [13]. В других исследованиях показана статистически достоверная связь между анемией и уменьшением локального контроля роста или выживаемости при цервикальном раке или раке головы и шеи [9, 10, 12, 13]. Даже умеренная анемия достоверно коррелировала с плохими результатами лечения при сквамозно-клеточном раке головы и шеи [9]. В нормальных тканях оксигенация обычно регулируется в соответствии с функциональным состоянием или метаболическими потребностями клеток. Напротив, в большинстве СО оксигенация, как и кровоток, не однообразна и нарушена в очень сильной степени. По этой причине гипоксия является характерной чертой локально запущенных СО, она возникает вследствие нарушения баланса между снабжением и потреблением кислорода. Главными патогенетическими механизмами развития гипоксии являются следующие факторы: 1) структурные и функциональные аномалии микроваскуляризации опухоли; 2) повышенные дистанции диффузии и 3) анемия, связанная с опухолевым процессом или индуцированная терапией [14]. Состояние оксигенации перед лучевой и химиотерапией не зависит от клинического размера опухоли, стадии, гистологического типа и биологических особенностей опухоли, но оно несомненно подвержено влиянию уровня гемоглобина. Гипоксия может привести к терапевтическим проблемам, поскольку она вызывает резистентность к действию ионизирующей радиации и к некоторым формам химиотерапии. Резистентность к терапии у пациентов с анемией может быть, по крайней мере, частично, предотвращена или преодолена коррекцией анемии, что приведет к улучшению локально-регионального контроля роста опухоли и выживаемости пациентов. АЗН обычно приводит к гипоксии и может оказывать влияние на жизненно важные органы. Главными проявлениями гипоксии у пациентов с АЗН являются: экспираторная одышка, сердцебиение и другие сердечно-сосудистые нарушения, утомляемость, головокружение, депрессивное состояние, нарушения познавательной функции, анорексия, тошнота, диспепсия, расстройства сна, нарушения менструального цикла и потеря либидо. Температура кожи у пациентов с АЗН понижена, кожа, слизистые оболочки полости рта и конъюнктивы бледные. Тяжесть симптомов зависит не только от степени анемии, но также от быстроты ее развития, основного заболевания и функции легких и сердца у пациента. У пожилых пациентов с АЗН клинические симптомы часто проявляются при более высоких уровнях гемоглобина, чем у пациентов с анемией без злокачественного новообразования. Наиболее важным звеном в патогенезе АЗН является низкая продукция эритроцитов, которая не компенсирует их укороченный период жизни, или, другими словами, относительная недостаточность эритропоэза [15]. До недавнего времени причину этой недостаточности усматривали в дефекте мобилизации железа из клеток макрофагальной системы [16]. Концентрация сывороточного железа при АЗН является характерно низкой, несмотря на достаточные запасы железа в тканях, и перенос железа из клеточного ферритина или гемосидерина к циркулирующему трансферрину нарушен. Отсутствие мобилизации железа и должно, согласно этому представлению, привести к низкому уровню сывороточного железа, к железодефицитному эритропоэзу и анемии. Тот факт, что анемия не всегда гипохромная и микроцитарная, можно было объяснить тем, что концентрация трансферрина у пациентов с АЗН ниже нормального уровня и не повышается как у пациентов с истинной железодефицитной анемией. Такая ситуация позволила бы железу из относительно более насыщенного трансферрина переходить с большей легкостью к рецепторам эритроидных клеток, снижая до минимума проявления дефицита железа в клетках эритрона. Патофизиология хронической анемии при ЗН включает интенсивное взаимодействие между популяцией опухолевых клеток и иммунной системой, что приводит к активации макрофагов и к повышенной экспрессии различных цитокинов (см. рисунок). Все характерные патофизиологические черты АЗН (уменьшение периода жизни эритроцитов, уменьшенная реутилизация железа костным мозгом, неадекватная продукция эритропоэтина – ЭПО и супрессия эрит- Опухолевые клетки Активация иммунной системы AIS Эритроциты Эритрофагоцитоз Дисэритропоэз Макрофаги TNF Укорочение выживаемости IL-1α, IL-1β TNF АНЕМИЯ Снижение продукции эритропоэтина IFN-γ IL-1 TNF α1-antitrypsin Супрессия БОЕ-Э КОЕ-Э IFN-γ IL-1 TNF Нарушение утилизации железа Рисунок. Патогенетические механизмы, связанные с развитием АЗН [3]. 51 Ä.Ñ.臂ÎÓ‚, Ö.î.åÓ˘‡ÍÓ‚‡ // ÇÓÔÓÒ˚ „ÂχÚÓÎÓ„ËË/ÓÌÍÓÎÓ„ËË Ë ËÏÏÛÌÓÔ‡ÚÓÎÓ„ËË ‚ Ô‰ˇÚËË, 2004, Ú. 3, ‹1, Ò. 50–55 роидных предшественников) являются результатом активации иммунной и воспалительной реакции злокачественными клетками, и определенные иммунные и воспалительные цитокины могут потенциально способствовать развитию АЗН. Рассмотрим патофизиологические черты, характерные для АЗН (напомним, что они свойственны и АХБ). Этими чертами являются укорочение периода жизни эритроцитов; нарушенная утилизация железа костным мозгом; ингибирование эритроидных предшественников костного мозга и неадекватная продукция ЭПО. Для решения вопроса о целесообразности использования рекомбинантного человеческого ЭПО (рчЭПО) в качестве альтернативного способа лечения АЗН понимание патогенеза этого типа анемии имеет решающее значение. Таблица. Параметры метаболизма железа при злокачественных новообразованиях Болезнь Ферритин, Железо, Трансферрин, Насыщение мкг/л мкг/дл мг/дл трансферрина, % МДС 1000 192 199 71 (155–10090) (56–1153) (117–9000) (5–102) ХМЗ 556 111 200 39 (30–6000) (30–299) (130–325) (10–106) ХЛЛ 580 24 230 7 (34–7320) (6–1160) (97–18000) (2–71) ММ 535 21 204 7 (45–2643) (2–1253) (135–23000) (1–75) ЗЛ 340 22 240 7 (21–2500) (5–212) (115–448) (2–74) СО 208 14 235 4 (11–3916) (1–126) (84–408) (0–42) Указаны средние значения (М), в скобках – минимальные и максимальные значения (разброс). Укороченный период жизни эритроцитов та с различными типами ЗН у пациентов с ХЛЛ, ММ, миелоидной лейкемией (МЛ) или СО имелись значительно уменьшенные уровни сывороточного железа и насыщения трансферрина, несмотря на нормальные или даже повышенные уровни сывороточного ферритина, подобно другим типам хронических болезней и АХБ [3]. Напротив, у пациентов с МДС или ХМЗ имелись нормальные уровни сывороточного железа и насыщения трансферрина и повышенные уровни сывороточного ферритина. Уровни сывороточного железа и насыщения трансферрина в этих 2 группах были значительно выше (p < 0,05), чем в других 4 группах. Эти данные свидетельствуют, что у пациентов с МДС или ХМЗ отсутствуют такие же нарушения метаболизма железа, как у пациентов с АХБ (см. таблицу). У пациентов с АХБ и АЗН существует обратное соотношение между уровнем гемоглобина и ферритина, с одной стороны, и маркерами клеточной иммунной активации (интерферон-гамма – IFN-γ, неоптерин) – с другой, это свидетельствует, что активированные макрофаги могут быть вовлечены в изменение метаболизма железа и развитие АХБ [20–22]. Недавно проведенные исследования показали, что человеческие мононуклеарные фагоциты ухудшают экспрессию своих трансферриновых рецепторов (ТФР) и содержание ферритина, когда они подвергаются воздействию IFN-γ [23]. Уменьшение включения железа, однако, количественно ниже, чем уменьшение экспрессии ТФР и содержания ферритина. В результате ферритин остается в этих клетках приблизительно в 3 раза более насыщенным включенным железом, чем ферритин в неактивированных клетках [23]. Более того, ферритин в IFN-γ-активированных моноцитах является, по-видимому, типом «высокого железа», т.е. типом ферритина, который быстро захватывает железо и освобождает его относительно медленно по сравнению с типом ферритина «низкого железа» [23, 24]. Такие количественные и качественные изменения метаболизма железа в активированных макрофагах могут уменьшать доступность железа для клеток эритрона и способствовать развитию АЗН. Метаболизм железа может быть изменен не только IFN-γ, но также TNF. В экспериментах на крысах и мышах с введением TNF обнаружена анемия и гипоферремия, последняя связана со значительным уменьшением освобождения железа из макрофагов и включения железа в эритроциты [17, 25]. Другим цитокином, возможно, также вовлеченным в изменения метаболизма железа, является IL-1. Этот цитокин способен У пациентов с хроническими болезнями средняя величина периода жизни эритроцитов обычно колеблется от 60 до 90 сут (120 сут у здоровых лиц). Когда проводили трансфузии эритроцитов от здоровых доноров пациентам со злокачественными новообразованиями, период жизни перелитых эритроцитов укорачивался, что свидетельствует о внеэритроцитарных факторах гемолиза [15]. Клинические и экспериментальные данные указывают, что такой гемолиз и укорочение периода жизни эритроцитов опосредуются интерлейкином-1 (IL-1) и фактором некроза опухолей (TNF-α). У пациентов с ревматоидным артритом укороченный период жизни эритроцитов коррелировал с уровнями IL-1 [17] и повторные инъекции крысам TNF-α вызывали дисэритропоэз и анемию, обусловленную уменьшенным образованием эритроцитов и уменьшением периода их жизни в циркуляции [16]. TNF, кроме того, способен вызывать дисэритропоэз и эритрофагоцитоз у мышей с экспериментальной малярией [18]. Уменьшение эритропоэза и укорочение периода жизни эритроцитов у пациентов с АЗН также могут быть обусловлены действием TNF. Недавно был обнаружен фактор, индуцирующий развитие анемии (AIS) в плазме пациентов с раковыми опухолями [3]. AIS уменьшает осмотическую резистентность эритроцитов и обнаруживается не только в плазме, но также в цитозоле и ядерной фракции неопластических клеток [19]. Механизм, лежащий в основе пониженной осмотической резистентности эритроцитов, связанный с AIS, зависит от ингибирования метаболизма (поступление глюкозы, активность пируваткиназы и концентрация АТФ) в этих клетках. AIS, повидимому, является специфическим для злокачественных новообразований и не связан с хроническими воспалительными заболеваниями [19]. Нарушенная утилизация железа Отличительными чертами АЗН, как и других АХБ, являются: низкие уровни сывороточного железа (СЖ), общей железосвязывающей способности сыворотки (ОЖСС), низкое насыщение трансферрина и присутствие адекватных запасов железа, о которых судили по уровню сывороточного ферритина или по результатам исследования костного мозга [20]. При оценке и сравнении метаболизма железа у 401 пациен- 52 ùÚËÓÎÓ„Ëfl Ë Ô‡ÚÓ„ÂÌÂÁ ‡ÌÂÏËË ÔË ÁÎÓ͇˜ÂÒÚ‚ÂÌÌ˚ı ÌÓ‚ÓÓ·‡ÁÓ‚‡ÌËflı увеличивать продукцию ферритина, который может действовать в качестве «ловушки» для железа, иначе оно могло бы быть доступно для эритропоэза [25, 26]. Другим механизмом, который может вызывать нарушение метаболизма железа при АХБ, является изменение ТФР на эритроидных клетках. В эритробластах у пациентов с АХБ и АЗН снижено число ТФР, кроме того, эритробласты имеют более низкое сродство к трансферрину по сравнению с эритробластами от здоровых индивидуумов [27]. В течение инфекции, злокачественных новообразований и иммунологических нарушений IL-1, IL-6 и TNF инициируют повышение концентрации белка острой фазы α1-антитрипсина, способного ингибировать эритропоэз путем нарушения связывания трансферрина с ТФР и последующее образование (интернализацию) ТФР-трансферринового комплекса [28]. При АЗН или других хронических болезнях вначале нормальные или повышенные уровни ферритина уменьшаются, когда пациентов лечат успешно рчЭПО [29–32]. Уровень сывороточного ферритина во время ранней фазы лечения, кроме того, является прогностическим признаком ответной реакции на рчЭПО [32]. Эти наблюдения показывают, что фармакологически высокие дозировки рчЭПО способны преодолеть нарушенную мобилизацию железа у некоторых пациентов с АХБ или АЗН. Супрессия ранних эритроидных предшественников Другим механизмом, который может способствовать развитию АЗН, является супрессивное действие цитокинов на эритроидные предшественники. IFN-γ, IL-1 и TNF ингибируют эритропоэз in vitro и in vivo и все эти три цитокина подавляют эритропоэз синергически или повышая экспрессию друг друга [33-37]. IL-1 и TNF-α обычно повышены у пациентов с хроническими воспалительными заболеваниями и косвенно ответственны за отсутствие эффекта ЭПО на костный мозг. IL-1 стимулирует Т-лимфоциты к выработке IFN-γ, тогда как TNF стимулирует костно-мозговые стромальные клетки к выработке IFN-β. Оба IFN-γ и IFN-β являются потенциальными супрессорами эритропоэтинчувствительных предшественников (например, КОЕ-Э). Хотя IFN-γ ингибирует как миелоидные, так и эритроидные предшественники, в действительности наблюдается только эритроидная супрессия. Это, вероятно, объясняется тем, что IL-1 стимулирует продукцию Г-КСФ и ГМ-КСФ, которые компенсируют миелоидную супрессию, вызванную IFN-γ. In vitro эритроидное ингибирование, вызванное IFN-β или IFN-γ, может быть устранено добавлением рчЭПО к среде. Эти результаты указывают на то, что терапевтический эффект рчЭПО, наблюдаемый у пациентов с АЗН, может быть частично обусловлен преодолением супрессорного эффекта этих цитокинов в отношении эритроидных предшественников. Неадекватная продукция эритропоэтина У пациентов с АЗН эритроидные предшественники реагируют нормально на ЭПО в условиях in vitro, но реакция ЭПО на анемию, по-видимому, нарушена, т. е. она не соответст- вует падению уровня гемоглобина. Адекватный анализ ответной реакции ЭПО на данную степень анемии может быть выполнен только в тех случаях, когда измеренные уровни сывороточного эритропоэтина (сЭПО) и величины гематокрита или гемоглобина коррелируют индивидуально у каждого пациента и результат сравнивается с реакцией ЭПО на данную степень «стандартной» анемии (железодефицитной или после кровопотери). У таких пациентов с анемией, обусловленной кровопотерей или дефицитом железа (т.е. с нормальной эритропоэтической системой), уровень сЭПО является экспоненциальной функцией степени анемии, посредством чего продукция ЭПО повышается с увеличением дефицита гемоглобина. Это положение не соответствует результатам измерений продукции ЭПО у многих пациентов с хронической АЗН. Например, продукция ЭПО была неадекватной приблизительно у 50% пациентов с выраженной множественной миеломой и практически у всех больных раком с тяжелыми повреждениями почек [38]. Повреждение почек может быть вызвано основной болезнью (например, множественной миеломой), а также химиотерапией, при которой у пациентов также нарушается реакция ЭПО [39]. У многих пациентов с ММ уровень сЭПО ниже стандартного, в то время как у большинства пациентов с гематологическими нарушениями стволовых клеток, такими как МДС или миелофиброз с миелоидной метаплазией (МММ), величины сЭПО находятся в пределах стандартного диапазона [41–43]. Низкие уровни сЭПО постоянно обнаруживают у пациентов с СО, и потеря взаимоотношения между уровнями сЭПО и гемоглобина свидетельствует, что нормальный механизм обратной связи, характерный для нефрогенной анемии, здесь отсутствует [44, 45]. Часть противоречивых результатов может быть объяснена множественными патогенетическими механизмами, действующими при АЗН. Три главных фактора могут нарушать продукцию ЭПО в этих условиях: 1) воспалительный компонент, присущий опухоли; 2) повреждение почек, обусловленное развитием опухоли и 3) химиопрепараты, используемые в лечении опухоли. Повреждение почек как причину заторможенной продукции ЭПО гипотетически связывают с ММ [46] и лимфомами [47]. Однако постулирована и другая причина для неадекватной продукции ЭПО при ММ. Установлено, что образование ЭПО находится в обратном соотношении с вязкостью плазмы пациентов с ММ или болезнью Вальденстрема, и уменьшение уровней сЭПО при более высоких величинах вязкости плазмы происходит параллельно с уменьшением уровней почечной ЭПО мРНК [48]. Эти данные указывают, что вязкость плазмы может быть значительным ингибиторным модулятором образования ЭПО, индуцированного анемией. Заторможенная реакция ЭПО у пациентов с АЗН ухудшается во время курса химиотерапии [49, 50, 56]. Этот эффект не зависит от вызванной терапией нефротоксичности и, повидимому, является специфичным для химиотерапевтических агентов, которые ингибируют синтез РНК [51]. Опубликовано интересное наблюдение, согласно которому злокачественные эритролейкемические клетки способны синтезировать ЭПО [52]. В другой работе допускается возможность, что аутокринная продукция ЭПО и его освобожде- 53 Ä.Ñ.臂ÎÓ‚, Ö.î.åÓ˘‡ÍÓ‚‡ // ÇÓÔÓÒ˚ „ÂχÚÓÎÓ„ËË/ÓÌÍÓÎÓ„ËË Ë ËÏÏÛÌÓÔ‡ÚÓÎÓ„ËË ‚ Ô‰ˇÚËË, 2004, Ú. 3, ‹1, Ò. 50–55 ние в окружение костного мозга могут происходить при клональных заболеваниях эритрона и, таким образом, ингибировать почечную продукцию ЭПО и уменьшать уровни сЭПО [53]. Таким образом, АЗН может быть результатом активации иммунной и воспалительной систем злокачественными клетками, и определенные иммунные и воспалительные цитокины, такие как IFN, TNF и IL-1, могут потенциально содействовать развитию АЗН. У пациентов с гематологическими злокачественными заболеваниями и АЗН значительно повышены уровни IL-1, TNF и IFN в плазме, а также неоптерина – маркера активации клеточного иммунитета. Уровни неоптерина находятся в прямой корреляционной зависимости с уровнями IFN-γ и в обратной с уровнями гемоглобина и железа [21]. Эти корреляции указывают на активацию клеточного иммунитета и возможную связь между активированными макрофагами и анемией у этих пациентов. Концентрации IFN-γ также повышены при других хронических болезнях и коррелируют с активностью процесса [54]. Другим цитокином, образованным макрофагами, является TNF. Уровни TNF у пациентов с АЗН повышаются и, по-видимому, зависят от типа и активности лежащего в основе злокачественного заболевания. У пациентов с активным процессом уровни TNF выше, чем у пациентов без видимых проявлений болезни [55]. В соответствии с результатами некоторых клинических и экспериментальных исследований хроническое воздействие TNF может привести к анемии. При клинических испытаниях I фазы у пациентов после введения TNF обнаруживались признаки анемии [21]. Более того, многократное введение TNF крысам и мышам вызывало анемию, во многом сходную с АЗН у человека [16, 34, 56]. Концентрации IL-1 также оказались повышенными при ревматоидном артрите и других хронических болезнях и ЗН [57, 58]. Это повышение, кроме того, оказалось прямо пропорциональным степени анемии. В итоге можно сделать следующее заключение. Анемия является частым осложнением у пациентов со ЗН. Имеется целый ряд факторов, предрасполагающих к развитию АЗН. У значительного числа пациентов, однако, нет причин, которые могли бы объяснить анемию, кроме самой болезни, т.е. ЗН. Такие анемии (АЗН) имеют многие гематологические и биохимические сходства с анемиями при других хронических болезнях. В большинстве случаев АЗН – гипорегенераторная, нормоцитарная и нормохромная, характеризующаяся уменьшением содержания сывороточного железа, уменьшенным насыщением трансферрина, несмотря на нормальные или повышенные уровни сывороточного ферритина. Недавние исследования показали, что АЗН является результатом многофакторного процесса, который запускается активацией иммунной и воспалительной систем с образованием определенных цитокинов, таких как IFN, TNF или IL-1, участвующих в развитии АЗН. Концентрации этих цитокинов повышены у пациентов со ЗН или хроническими болезнями. При АЗН период жизни эритроцитов укорочен, но более важным фактором, способствующим развитию анемии, повидимому, является относительная недостаточность эритропоэза для компенсации укороченного периода жизни эрит- 54 роцитов. Постулировано три патогенетических механизма в развитии АЗН, они опосредованы цитокинами и ответственны за относительную недостаточность эритропоэза: 1) нарушенная утилизация железа костным мозгом; 2) супрессия эритроидных предшественников; 3) неадекватная продукция ЭПО. У большинства пациентов с АЗН снижена реакция ЭПО относительно степени анемии, и введение рчЭПО у ряда пациентов корригирует анемию. Экспериментальные и клинические исследования свидетельствуют, что у пациентов с АЗН фармакологическая дозировка рчЭПО может не только корригировать относительную недостаточность ЭПО, но также преодолеть супрессию эритроидных предшественников и нарушенную утилизацию железа. Литература 1. Ludwig H., Fritz E. Anemia in cancer patients. Semin Oncol 1998, 25: 2–6. 2. Beguin Y. Prediction of response and other improvments on the limitations of recombinant human erythropoietin therapy in anemic cancer patients. Haematologica, 2002, 87, 1209–21. 3. Howrousian M.R., Kasper C., Oberhoff C., et al. Pathophysiology of cancerrelated anemia. In: Smith J.F., et al., eds. rhErythropoietin in cancer supportive treatment. New York: Marcel Dekker Inc. 1996; 13–34. 4. Vogelzang N.J., Breitbart W., Cella D., et al. Patients, caregiver, and oncologist perceptions of cancer-related fatique: results of a tripart assessment survey. Semin Hematol 1997, 34 (suppl 2): 4–12. 5. Groopman J.E., Itri L.M. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst 1999; 91: 1616–34 6. Coiffier B. Retrospective analysis of hematological parameters and transfusion requirements in non-platinum chemotherapy-treated patients. Proc Am Soc Clin Oncol 1998; 17: 90 a (abstr 346). 7. Dalton J.D., Bailey N.P., Barrett-Lee P.J., O’Brien M.F.R. Multicenter UK audit of anemia in patients receiving cytotoxic chemotherapy. Proc Am Soc Clin Oncol 1998; 17: 418 a (abstr 1611). 8. Harrison L.B., Shasha D., White C., Ramdeen B. Radiotherapy-associated anemia: the scope of the problem. Oncologist 2000; 5: 1–7. 9. Dubray B., Mosser V., Brunin F., et al. Anemia is associated with lower localregional control and survival after radiation therapy for head and neck cancer: a prospectiva study. Radiology 1996; 201: 553–8. 10. Fein D.A., Lee W.R., Hanlon A.L., et al. Pretreatment hemoglobin level influences local control and survival of T1–T2 squamous cell carcinomas of the glottic larynx. J Clin Oncol 1995; 13: 2077–83. 11. Fyles A.W., Milosevic M., Wong R., et al. Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol 1998; 48: 149–56. 12. Grogan M., Thomas G.M., Melamed J., et al. The importance of hemoglobin levels during radiotherapy for carcinoma of the cervix. Cancer 1999; 86: 1528–36. 13. Glaspy J., Cavill J. Role of iron in optimizing responses of anemic cancer patients to erythropoietin. Oncology 1995; 13: 461–73 14. Vaupel P., Thews O., Hoekel M. Tumor oxigenation: characterization and clinical implications. In: Smith J.F., eds. rhErythropoietin in cancer supportive treatment New York: Marcel Dekker Inc.; 1996; 205–39. 15. Means R.T., Krantz S.B. Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 1992; 80: 1639–47. 16. Moldawer L.L., Marano M.A., Wei H., et al. Cachectin / tumor necrosis factor-α alters red blood cell kinetics and induces anemia in vivo. FASEB J 1989; 3: 1637–43. 17. Salvazini C., Casali B., Salvo D., et al. The role of interleukin 1, erythropoietin and red cell bound immunoglobulins in the anemia of rheumatoid artritis. Clin Exp Rheumatol 1991; 9: 241–6. ùÚËÓÎÓ„Ëfl Ë Ô‡ÚÓ„ÂÌÂÁ ‡ÌÂÏËË ÔË ÁÎÓ͇˜ÂÒÚ‚ÂÌÌ˚ı ÌÓ‚ÓÓ·‡ÁÓ‚‡ÌËflı 18. Clark J.A., Chaudhri G. Tumor necrosis factor may contribute to the anemia of malaria by causing dyserythropoiesis and erythrophagocytosis. Br J Haematol 1988; 70: 99–103. 19. Honda K., Ishiko O., Tatsuta J., et al. Anemia-inducing substance from plasma of patients with advanced malignant neoplasms. Cancer Res 1995; 55: 3623–8. 20. Zucker S. Anemia in cancer. Cancer Invest., 1985, 3, 249–260 21. Denz H., Fuch D., Huber H., et al. Correlation between neopterin, interferongamma and haemoglobin in patients with haematological disorders. Eur J Haematol 1990; 44: 186–9. 22. Fuchs D., Zangerle R., Artner-Dworzak E., et al. Association between immune activation, changes of iron metabolism and anemia in patients with HIV infection. Eur J Haematol 1993; 50: 90–4. 23. Byrd T.F., Horwitz M.A. Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes: Coordinate upregulation by iron transferrin and downregulation by interferon-gamma. J Clin Invest 1993; 91: 969–76. 24. Mertz J.R., Theil E.C. Subunit dimer in sheep spleen apoferritin: The effect on iron storage. J Biol Chem 1983; 258: 11719–26. 25. Rogers J., Durmowicz G., Kasschau K., et al. A motif within the 5’noncoding region of hepatic acute phase mRNAs mediates ferritin translation by interleukin1β and may contribute to the anemia of chronic disease. Blood 1991; 78(Suppl 1): 367. 26. Alvarez-Hernandez X., Liceaga J., McKay J.C., Brock J.H. Induction of hypoferremia and modulation of macrophage iron metabolism by tumor necrosis factor. Lab Invest 1989; 61: 319–22. 27. Feelders R.A., Vreugdenhil G., van Dijk J.P., et al. Decreased affinity and number of transferrin receptors on erythroblasts in the anemia of rheumatoid arthritis. Am J Hematol 1993; 43: 200–4. 28. Graziadei J., Gaggl. S., Kaserbacher R., et al. The acute-phase protein α1-antitrypsin inhibits growth and proliferation of human early erythroid progenitor cells (burst-forming units – erythroid) and of human erythroleukemic cells (K562) in vitro by interfering with transferrin iron uptake. Blood 1994; 83: 260–8. 29. Oster W., Herrmann F., Gamm H., et al. Erythropoietin for the treatment of anemia of malignancy associated with neoplastic bone marrow infiltration. J Clin Oncol 1990; 8: 956–62. 30. Ludwig H., Fritz E., Kotzmann H., et al. Erythropoietin treatment of anemia associated with multiple myeloma. N Engl J Med 1990; 322: 1693–9. 31. Vreugdenhil G., Manger B., Nieuwenhuizen C., et al. Iron stores and serum transferrin receptor levels during combination human erythropoietin treatment of anemia in rheumatoid arthritis. Ann Hematol 1992; 65: 265. 32. Ludwig H., Fritz E., Laitgeb C., et al. Prediction of response to erythropoietin treatment in chronic anemia of cancer. Blood 1994; 84: 1056–63. 33. Blick M., Sherwin S.A., Rosenblum M., Gutterman J. Phase I study of recombinant tumor necrosis factor in cancer patients. Cancer Res 1987; 47: 2986–9. 34. Tracey K.J., Wei H., Manogue K.R., et al. Cachectin/tumor necrosis factor induces cachexia, anemia, and inflammation. J Exp Med 1988; 167: 1211–27. 35. Broxmeyer H.E., Williams D.E., Lu L., et al. The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: synergism of tumor necrosis factor and interferon-γ. J Immunol 1986; 136: 4487–95. 36. Roodman G.D., Bird A., Hutzler D., Montgomery W. Tumor necrosis factor-alpha and hematopoietic progenitors: effect of tumor necrosis factor on the growth of erythroid progenitors CFU-E and BFU-E and the hematopoietic cell lines K562, HL60 and HEL cells. Exp Hematol 1987; 15: 928–35. 37. Furmanski P., Johnson C.S. Macrophage control of normal and leukemic erythro- poiesis: identification of the macrophage-derived erythroid suppressing activity as interleukin-1 and the mediator of its in vivo action as tumor necrosis factor. Blood 1990; 75: 2328–34. 38. Henry D.H. Clinical application of recombinant erythropoietin in anemic cancer patients. Hematol/Oncol Clin N Am 1994; 8: 961–74. 39. Miller C.B., Jones R., Piantadosi S., et al. Decreased erythropoietin response in patients with anaemia of cancer. N Engl J Med 1990; 322: 1689–92. 40. Begnin Y., Yerna M., Loo M., et al. Erythropoiesis in multiple myeloma: defective red cell production due to inappropriate erythropoietin production. Br J Haematol 1992; 82: 648–53. 41. Barosi G., Liberato N.L., Guarnone R. Serum erythropoietin in patients with myelofibrosis with myeloid metaplasia. Br J Haematol 1993; 83: 365–9. 42. Bosi A., Vannucchi A.M., Grossi A., et al. Inadequate erythropoietin production in allogeneic bone marrow transplant patients. Haematologica 1991; 76: 280–4. 43. Cazzola M., Ponchio L., Beguin Y., et al. Subcutaneous erythropoietin for treatment a refractory anemia in hematologic disorders. Results of a phase – I/II clinical trial. Blood 1992; 79: 29–37. 44. Cox R., Musial T., Gyde O.H.B. Reduced erythropoietin levels as a cause of anaemia in patients with lung cancer. Eur J Cancer 1986; 22: 511–4. 45. Kettelhack C., Sch(ter D., Matthias D., Schlag P.M. Serum erythropoietin levels in patients with solid tumors. Eur J Cancer 1994; 30A: 1289–91. 46. Takagi M., Miyamoto Y., Kosaka M., Saito S. Clinical significance of serum erythropoietin levels in patients with multiple myeloma. Rinsho Ketsucki 1992; 33: 1151–7. 47. Ariad S., Clifford D., Penfold G., et al. Erythropoietin responce in anaemic patients with multiple myeloma and other lymphoid malignancies infiltrating the bone marrow. Eur J Haematol 1992; 49: 59–62. 48. Singh A., Eckardt K.U., Zimmermann A., et al. Increased plasma viscosity as a reason for inappropriate erythropoietin formation. J Clin Invest 1993; 91: 251–6. 49. Schapira L., Antin J.H., Ransil B., et al. Serum erythropoietin levels in patients receiving intensive chemotherapy and radiotherapy. Blood 1990; 76: 2354–9. 50. Smith D.H., Guarnieri C.M., Whaling S.M., Vokes E.E. Erythropoietin response in cancer patients receving cisplatin. Proc. Am. Assoc. Cancer Res 1988; 29: 52–6. 51. Wolff M., Jelkmann W. Effects of chemotherapeutic and immunosuppressive drugs on the production of erythropoietin in human hepatoma cultures. Ann Hematol 1993, 66, 27–31 52. Mitjavilla M., Le Couedic J.P., Casadevall N., et al. Autocrine stimulation by erythropoietin and autonomous growth of human erythroid leukemia cells in vitro. Clin Invest 1991; 8: 789. 53. Rafanelli D., Grossi A., Longo G., et al. Recombinant human erythropoietin for treatment of myelodysplastic syndromes. Leukemia 1992; 6: 323–7. 54. Hooks J.J., Moutsopoulos H.M., Geis S.A., et al. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med 1979; 301: 5–8. 55. Balkwill F., Osborne R., Burke F., et al. Evidence for tumour necrosis factor / cachectin production in cancer. Lancet 1987; 2: 1229–32. 56. Johnson R.Y., Waddelow T.A., Caro J., et al. Chronic exposure to tumour necrosis factor in vivo preferentially inhibits erythropoiesis in nude mice. Blood 1989; 74: 130–8. 57. Eastgate J.A., Symons J.A., Wood N.C., et al. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet 1988; 2: 706–9. 58. Maury C.P.J., Andersson L.C., Teppo A.M., et al. Mechanism of anaemia in rheumatoid artritis: demonstration of raised interleukin 1α concentration in anaemic patients and of interleukin 1 mediated suppression of normal erythropoiesis and proliferation of human erythroleukemia (HEL) cells in vitro. Ann Rheum Dis 1988; 47: 972–8. 55