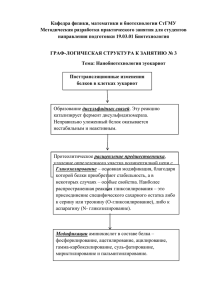
Рубина Ксения Андреевна Т-КАДГЕРИН В ПРОЦЕССАХ РОСТА
реклама

Московский государственный университет имени М.В. Ломоносова Факультет фундаментальной медицины На правах рукописи Рубина Ксения Андреевна Т-КАДГЕРИН В ПРОЦЕССАХ РОСТА, РЕМОДЕЛИРОВАНИЯ КРОВЕНОСНЫХ СОСУДОВ И ОПУХОЛЕВОЙ ПРОГРЕССИИ 03.03.04 – клеточная биология, цитология, гистология Диссертация на соискание учѐной степени доктора биологических наук Научный консультант: доктор биологических наук, профессор, академик РАН В.А. Ткачук Москва – 2015 2 ВВЕДЕНИЕ .................................................................................................................................. 7 СПИСОК СОКРАЩЕНИЙ ....................................................................................................... 12 ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ .......................................................................................... 15 1.1. Регуляция роста и ремоделирования кровеносных сосудов ................................. 15 1.1.1 Клеточные компоненты сосудистой стенки .......................................................... 15 1.1.2 Механизмы формирования сосудов ....................................................................... 17 1.1.3 Ветвление сосудов при ангиогенезе ....................................................................... 20 1.1.4 Специализация клеток при ангиогенезе: ―tip‖ и ―stalk‖ фенотип ........................ 21 1.1.5 Филоподии и ламеллоподии определяют направление роста сосудов ............... 25 1.1.6 Ангиогенные факторы роста ................................................................................... 29 1.1.7 Хемокины и цитокины, их роль в процессах ангиогенеза ................................... 35 1.1.8 Роль интегринов в процессах ангиогенеза ............................................................ 47 1.1.9 Роль протеаз в процессах ангиогенеза ................................................................... 49 1.1.10 Эндогенные ингибиторы ангиогенеза .................................................................. 49 1.2 Навигационные рецепторы в нервной и сердечно-сосудистой системе ................... 51 1.2.1 Эфрины и их рецепторы .......................................................................................... 54 1.2.2 Эфрины и их рецепторы в сосудистой системе .................................................... 60 1.2.3 Семафорины и их рецепторы .................................................................................. 63 1.2.4 Роль Семафоринов и их рецепторы в сосудистой системе .................................. 65 1.2.5 Нетрины и их рецепторы ......................................................................................... 69 1.2.6 Нетрины и их рецепторы в сосудистой системе ................................................... 70 1.2.7 Cлит лиганды и Robo рецепторы ............................................................................ 74 1.2.8 Слит лиганды и Robo рецепторы в сосудистой системе ...................................... 75 1.2.9 Урокиназная система ............................................................................................... 78 1.3 Кадгерины в процессах морфогенеза ............................................................................ 87 1.3.1 «Классические» кадгерины ..................................................................................... 87 1.3.2 Экспрессия кадгеринов в нервной системе в эмбриогенезе ................................ 93 1.3.3 Роль кадгеринов в развитии сердца и сосудов ...................................................... 95 1.3.4 Т-кадгерин................................................................................................................. 97 1.3.5 Навигационная роль Т-кадгерина в нервной системе ........................................ 100 1.3.6 «Гормоноподобные» эффекты липопротеидов низкой плотности (ЛНП). Ткадгерин и ЛНП ............................................................................................................... 104 ГЛАВА 2. МАТЕРИАЛЫ И МЕТОДЫ ................................................................................ 110 2.1 Антитела и реагенты ..................................................................................................... 110 2.2 Культивирование эукариотических клеток ................................................................ 112 2.3 Трансфекция эукариотических клеток ........................................................................ 112 2.4 Создание лентивирусных конструкций для трансдукции эндотелиальных клеток HUVEC ................................................................................................................................. 115 2.5 Создание аденовирусных конструкций для трансдукции эндотелиальных клеток HUVEC ................................................................................................................................. 116 2.6 Вестерн блоттинг (метод иммуноблоттинга) ............................................................. 117 2.6.1 Приготовление лизатов клеток ............................................................................. 117 2.6.2 Определение концентрации белка в лизатах клеток .......................................... 117 2.6.3 Вестерн блоттинг ................................................................................................... 117 2.7 Иммунофлуоресцентное окрашивание эндотелиальных клеток и конфокальная микроскопия ........................................................................................................................ 118 2.8 Измерение SRE активности в NIH3T3 клетках .......................................................... 119 2.9 Выделение активных Rho ГТФаз (GST pull-down assay) .......................................... 120 2.10 Измерение проницаемости эндотелиального монослоя in vitro ............................. 120 2.11 Трипсинизация эндотелиальных клеток ................................................................... 121 2.12 Разделение лизатов клеток на субклеточные фракции ........................................... 121 3 2.13 Оценка пролиферация эндотелиальных клеток ....................................................... 122 2.14 Метод измерения адгезии эндотелиальных клеток ................................................. 122 2.15 Метод измерения апоптоза в эндотелиальных клетках........................................... 123 2.16 Измерение миграции эндотелиальных клеток в камере Бойдена .......................... 123 2.17 Метод изучения формирования капилляро-подобных структур эндотелиальными клетками ............................................................................................................................... 124 2.18 Экспериментальные животные .................................................................................. 124 2.19 Анализ экспрессии Т-кадгерина в раннем эмбриональном развитии у мыши ..... 125 2.19.1 Получение датированной беременности мыши ................................................ 125 2.19.2 Получение эмбрионов мыши на постимплантационной стадии развития ..... 125 2.19.3 Иммунофлуоресцентное окрашивание мышиных эмбрионов антителами против Т-кадгерина ......................................................................................................... 125 2.19.4 Трансформация E.coli, амплификация и очистка плазмид для in situ гибридизации ................................................................................................................... 126 2.19.5 Линеаризация плазмид для in situ гибридизации .............................................. 126 2.19.6 Синтез РНК-содержащей пробы для in situ гибридизации .............................. 127 2.19.7. Гибридизация мышиных эмбрионов in situ ...................................................... 128 2.20 Эксплантная модель ex vivo аорты крысы ................................................................ 129 2.21 Подкожная имплантация Матригеля мышам ........................................................... 129 2.21.1 Оценка содержания гемоглобина ....................................................................... 130 2.21.2 Выявление сосудов в Матригеле с помощью иммунофлуоресцентного окрашивания .................................................................................................................... 130 2.21.3 Подсчѐт сосудов и статистический анализ ........................................................ 131 2.22 Приготовление криосрезов тканей человека и животных ...................................... 131 2.23 Иммунофлуоресцентное окрашивание образцов биопсийного материала кожи человека ................................................................................................................................ 131 2.24 Модель гематогенно метастазирующей в лѐгкие меланомы у мышей in vivo ...... 132 2.25 Анализ кинетики роста и метастазирования клеток меланомы B16F10 ............... 133 2.26 Иммунофлуоресцентное окрашивание криосрезов первичных узлов меланомы 134 2.27 Модель хориоаллантоиcной мембраны куриного эмбриона in vivo ...................... 135 2.28 Оценка пролиферации клеток B16F10 in vitro ......................................................... 136 2.28.1 Метод подсчѐта абсолютного числа клеток ...................................................... 136 2.28.2 Метод измерения клеточного индекса ............................................................... 136 2.28.3 Оценка распределения клеток B16F10 по фазам клеточного цикла in vitro .. 137 2.29 Оценка уровня некроза и апоптоза клеток B16F10 in vitro ..................................... 138 2.30 Анализ экспрессии генов в клетках клонов B16F10 in vitro ................................... 138 2.30.1 Выделение РНК .................................................................................................... 138 2.30.2 Синтез кДНК и полимеразная цепная реакция с детекцией в режиме реального времени (RT-PCR) ......................................................................................... 138 2.30.3 Синтез кДНК и анализ профиля экспрессии генов с помощью наборов RT2 Profiler PCR Array ............................................................................................................ 141 2.31 Концентрирование кондиционированной среды клеток B16F10 ........................... 141 2.32 Оценка инвазивного потенциала клеток B16F10 in vitro ........................................ 141 2.33 Получение мезенхимных стромальных клеток жировой ткани (МСК-ЖТ) мыши ............................................................................................................................................... 143 2.34 Влияние кондиционированной среды клеток B16F10 на пролиферацию и миграцию МСК-ЖТ in vitro................................................................................................ 143 2.34.1 Влияние кондиционированной среды клеток B16F10 на пролиферацию МСКЖТ ..................................................................................................................................... 143 2.34.2 Влияние среды от клеток клонов B16F10 на миграцию МСК-ЖТ ................. 144 2.35 Статистическая обработка результатов .................................................................... 145 2.36 Иммуногистохимическое окрашивание криосрезов аорты человека .................... 145 4 2.37 Выделение и йодирование липопротеидов низкой плотности ............................... 146 2.38 Определение поверхностного связывания 125I-ЛНП с клетками ............................ 147 2.39 Определение концентрации внутриклеточного Ca2+ ............................................... 147 2.40 Оценка влияния липопротеидов низкой плотности на миграцию L929 клеток ... 150 ГЛАВА 3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ ...................................................................... 151 3.1 Выявление экспрессии Т-кадгерина в эмбриогенезе у мыши .................................. 152 3.1.1 Синтез РНК пробы ................................................................................................. 152 3.1.2 Экспрессия Т-кадгерина в эмбриональном головном мозге у мыши ............... 156 3.1.3 Экспрессия Т-кадгерина в эмбриональном сердце ............................................. 161 3.2 Т-кадгерин и опухолевая прогрессия .......................................................................... 163 3.2.1 Т-кадгерин и опухолевый рост ............................................................................. 164 3.2.2 Строма играет важную роль в регуляции роста и прогрессии опухолей ......... 165 3.2.3 Т-кадгерин и васкуляризация опухолей............................................................... 166 3.2.4 Потеря экспрессии Т-кадгерина в кератиноцитах и нарушение его экспрессии в сосудах коррелирует с процессами озлокачествления немеланомных новообразований кожи человека ................................................................................... 168 3.2.4.1 Нормальная кожа ............................................................................................... 169 3.2.4.2 Псориаз................................................................................................................. 171 3.2.4.3 Актинический (солнечный) кератоз (АК) ......................................................... 173 3.2.4.4 Кератоакантома (КА) ....................................................................................... 175 3.2.4.5 Базальноклеточный рак (базалиома) ................................................................ 178 3.2.4.6 Плоскоклеточный рак или плоскоклеточная карцинома (ПР) ....................... 183 3.2.4.7 Метатипический рак (МР) ................................................................................ 186 3.2.4.8 Потеря экспрессии Т-кадгерина в опухолевых клетках и нарушение его экспрессии в сосудах коррелирует с опухолевой прогрессией меланомы .................. 190 3.2.4.9 Экспрессия Т-кадгерина в меланоме человека .................................................. 192 3.3 Т-кадгерин и меланома ................................................................................................. 198 3.3.1 Анализ экспрессии Т-кадгерина в клетках мышиной меланомы В16F10 ........ 198 3.3.2 Влияние экспрессии Т-кадгерина на пролиферацию клеток мышиной меланомы B16F10 in vitro ............................................................................................... 202 3.3.3 Влияние экспрессии Т-кадгерина на распределение клеток клонов B16F10 по фазам клеточного цикла in vitro ..................................................................................... 204 3.3.4 Влияние экспрессии Т-кадгерина на апоптоз и некроз клеток мышиной меланомы B16F10 in vitro ............................................................................................... 204 3.3.5 Влияние экспрессии Т-кадгерина на рост, метастазирование и васкуляризацию первичного узла меланомы B16F10 на модели гематогенно метастазирующей в лѐгкие меланомы у мышей ............................................................................................. 206 3.3.5.1 Влияние экспрессии Т-кадгерина на рост первичного узла мышиной меланомы B16F10 in vivo ................................................................................................ 206 3.3.5.2 Влияние экспрессии Т-кадгерина на частоту очагов некроза на срезах первичных узлов меланомы В16F10 ............................................................................... 208 3.3.5.3 Влияние экспрессии Т-кадгерина на васкуляризацию первичных узлов мышиной меланомы ........................................................................................................ 208 3.3.5.4 Экспрессия Т-кадгерина в клетках меланомы вызывает активацию стромы и увеличивает вклад стромального компонента в формирование первичных узлов мышиной меланомы B16F10 .......................................................................................... 211 3.3.5.5 Изучение влияния экспрессии Т-кадгерина на метастазирование клеток меланомы in vivo .............................................................................................................. 212 3.3.6 Влияние среды роста от клеток клонов B16F10 на пролиферацию и миграцию МСК-ЖТ in vitro .............................................................................................................. 214 3.3.6.1 Влияние среды роста от клеток клонов B16F10 на миграцию МСК-ЖТ ..... 214 5 3.3.6.2 Исследование влияния среды роста от клеток клонов B16F10 на пролиферацию МСК-ЖТ ................................................................................................. 216 3.3.7 Исследование влияния экспрессии Т-кадгерина на инвазивный потенциал клеток клонов B16F10 in vitro ........................................................................................ 217 3.3.8 Исследование влияния Т-кадгерина на экспрессию цитокинов, факторов роста и матриксных металлопротеиназ в клетках B16F10 in vitro ....................................... 219 3.3.9 Влияние экспрессии Т-кадгерина на неоангиогенез в меланоме В16F10 на модели хориоаллантоисной мембраны куриного эмбриона ....................................... 232 3.3.10 Обсуждение результатов ..................................................................................... 234 3.4 Выявление роли Т-кадгерина в процессах ангиогенеза ............................................ 242 3.4.1 Введение клеток, экспрессирующих Т-кадгерин, локально подавляет ангиогенез в бляшках Матригеля .................................................................................. 242 3.4.2 Экспрессия Т-кадгерина подавляет образование капилляров, но не влияет на формирование зрелых сосудов ...................................................................................... 244 3.4.3 Рекомбинантный аминотерминальный ЕС1 домен Т-кадгерина подавляет ангиогенез in vitro............................................................................................................ 248 3.4.4 Т-кадгерин подавляет миграцию эндотелиальных клеток, но не влияет на их адгезию, пролиферацию и апоптоз ................................................................................ 251 3.4.5 Обсуждение результатов ....................................................................................... 256 3.5 Т-кадгерин и проницаемость сосудов. Сигнальные эффекты Т-кадгерина ............ 260 3.5.1 Распределение экспрессии Т-кадгерина во фракциях эндотелиальных клеток после лентивирусной трансдукции ............................................................................... 261 3.5.2 Экспрессия Т-кадгерина регулирует проницаемость монослоя эндотелия ..... 265 3.5.3 Гиперэкспрессия Т-кадгерина вызывает образование межклеточных щелей и исчезновение VE-кадгерина из межклеточных контактов в эндотелиальных клетках ........................................................................................................................................... 267 3.5.4 Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает клатринзависимую интернализацию VE-кадгерина и деградацию в лизосомах ................... 272 3.5.5 Гиперэкспрессия Т-кадгерина увеличивает уровень фосфорилирования VEкадгерина по тирозину Y731 в эндотелиальных клетках ............................................ 279 3.5.6 Т-кадгерин активирует SRE в фибробластах NIH3T3 ........................................ 283 3.5.7 Т-кадгерин активирует Rac1 и Cdc42, но не влияет на активность RhoA в фибробластах NIH3T3 .................................................................................................... 286 3.5.8 Т-кадгерин активирует малые Rho ГТФазы RhoA, Rac1 и, Cdc42 и усиливает сборку стресс-фибрилл актина в эндотелиальных клетках ........................................ 288 3.5.9 Т-кадгерин фосфорилирует LIMK1 и влияет на полимеризацию микротрубочек в эндотелиальных клетках .............................................................................................. 289 3.5.10 Т-кадгерин влияет на полимеризацию стресс-фибрилл актина в эндотелиальных клетках ................................................................................................. 293 3.5.11 Обсуждение результатов ..................................................................................... 299 3.6 Т-кадгерин при атеросклерозе ..................................................................................... 306 3.6.1 Экспрессия Т-кадгерина в здоровых сосудах...................................................... 308 3.6.2 Экспрессия Т-кадгерина в сосудах при атеросклерозе ...................................... 312 3.6.2.1 Липидная полоска ................................................................................................ 312 3.6.2.2 Нестабильная фиброатерома ........................................................................... 315 3.6.2.3 Мягкий кальциноз ................................................................................................ 316 3.6.3 Обсуждение результатов ....................................................................................... 318 3.7 Т-кадгерин-рецептор липопротеидов низкой плотности .......................................... 322 3.7.1 Получение клонов клеток, гиперэкспрессирующих Т-кадгерин. Анализ экспрессии Т-кадгерина в клонах .................................................................................. 322 3.7.2 Связывание [125I]-ЛНП с мембранами клеток L929 и HEK293 ....................... 323 6 3.7.3 Введение ЛНП в среду культивирования клеток, гиперэкспрессирующих Ткадгерин, повышает концентрацию внутриклеточного кальция ............................... 327 3.7.4 ЛНП стимулируют миграцию клеток, гиперэкспрессирующих Т-кадгерин ... 338 3.7.5 Обсуждение результатов ....................................................................................... 340 ГЛАВА 4. ЗАКЛЮЧЕНИЕ ..................................................................................................... 343 ВЫВОДЫ ................................................................................................................................. 351 СПИСОК ЛИТЕРАТУРЫ....................................................................................................... 352 7 ВВЕДЕНИЕ В современной биологической науке достаточно хорошо изучены морфогенетические процессы, происходящие в эмбриогенезе, однако, отсутствуют серьезные обоснованные представления о генетических и эпигенетических механизмах морфогенеза во взрослом организме, делении и дифференцировке клеток, направленной клеточной миграции, построении архитектуры и поддержания целостности органов и тканей, обеспечении их кровоснабжением и иннервацией. Известно, что в норме существует динамическое равновесие между клеточной гибелью и восполнением утраченных клеточных элементов, то есть поддерживается баланс качественного и количественного содержания клеток в органах и тканях, однако, основные молекулярные механизмы этих процессов требуют более детального изучения. Формирование, развитие и поддержание структуры и функции большинства органов и тканей напрямую зависит от их кровоснабжения. Кровеносные сосуды обеспечивают ткани кислородом, питательными веществами, обеспечивают эндокринную регуляцию, транспорт клеток иммунной системы и стволовых клеток, и поддержание гомеостаза. Кровеносные сосуды являются одними из первых функциональных структур, которые формируются во время эмбрионального развития. Дефекты формирования кровеносных сосудов часто приводят к врожденным порокам развития, которые в дальнейшем необходимо корректировать путем хирургического вмешательства или с помощью активно развивающихся в последнее время новых подходов регенеративной медицины. После рождения сформировавшиеся кровеносные сосуды активно растут и подвергаются ремоделированию. В отличие от эмбрионального развития, во взрослом организме в норме кровеносные сосуды образуются существенно реже: формирование кровеносных сосудов de novo происходит при заживлении ран, при циклических изменениях в женской половой системе или при различных патологических состояниях, таких как ревматоидный артрит, ретинопатия, ишемические состояния, опухоли и метастазы. Сердечно-сосудистые и онкологические заболевания являются ведущими причинами заболеваемости и смертности во всем мире. В основе этих патологий лежит недостаточный или избыточный рост сосудов (ангиогенез), соответственно. Известно, что рост солидных опухолей и метастазирование зависит напрямую от интенсивности их васкуляризации, в этой связи одним из перспективных направлений в современной онкологии является разработка лекарственных препаратов, подавляющих опухолевый 8 неоангиогенез [Folkman, 2007; Carmeliet, 2003]. Однако в клинике общая эффективность нескольких одобренных методов лечения, которые направлены на подавление проангиогенных сигнальных путей, контролируемых фактором роста эндотелия (VEGF), была кратковременной и, в целом, довольно невысокой. Это связано с тем, что опухоли могут активировать альтернативые пути адаптации к недостаточному росту сосудов, например, сами формировать сосудистые структуры или использовать другие механизмы индукции опухолевого неоангиогенеза [Rivera and Bergers, 2014]. При старении организма состояние и функционирование кровеносных сосудов ухудшается в результате процессов окисления липидов и отложение кальция в сосудистой стенке, воспаления, аутоиммунных реакций и различных инфекций, что потенциально ухудшает функционирование всех основных органов и систем организма вследствие нарушения кровотока. При инфаркте происходит гибель кардиомиоцитов как непосредственно в момент самого инфаркта, так и в последующие дни и неделе в результате нарушения кровоснабжения в зоне самого инфаркта и в периинфарктной зоне, аналогично происходит гибель нейронов при инсульте, гепатоцитов при гепатите или циррозе печени. В этой связи актуальна проблема индукции направленного регулируемого роста новых кровеносных сосудов. В этой связи помимо решения проблемы понимания фундаментальных основ организации и функционирования органов и тканей, существуют практические задачи регенеративной медицины, такие как восстановление структуры и функции при различных ишемических и дегенеративных заболеваниях (инфаркт, инсульт, ишемия конечностей, цирроз), при лечении трофических язв, после травм и ранений, приживлении при трансплантации органов и тканей, при лечении заболеваний суставов, предотвращении фиброза тканей и др. За последние годы стало очевидно, что в основе многих регенеративных процессов также лежит обеспечение кровоснабжением поврежденных органов и тканей. Детальное изучение молекулярно-биологических, биохимических и клеточных механизмов этих процессов является, безусловно, актуальной задачей современной биологии и медицины. Понимание регуляции основных процессов ангиогенеза, таких как активация и специализация сосудистых клеток, их пролиферация, миграция эндотелиальных клеток по градиенту хемоаттрактанта, выбор траектории роста и стабилизация вновь сформированных сосудов за счет взаимодействия с муральными клетками необходимо для разработки современных подходов в регенеративной медицине. Эти же знания необходимы при разработке новых лекарств, направленных на блокирование процессов роста сосудов при избыточном или 9 аберрантном ангиогенезе, которые выходят из-под физиологического контроля при различных заболеваниях. В последнее время помимо основных молекул, участвующих в процессах ангиогенеза, таких как факторы роста, цитокины и хемокины, большое внимание уделяется изучению навигационных рецепторов и их лигандов, которые определяют направление роста вновь формирующихся сосудов (Эфрины и их рецепторы, Нейропилины и Плексины - рецепторы Семафоринов, Robo - рецепторы Слит белков, и UNC5B рецепторы, связывающие Нетрины). Помимо контролирования траектории роста сосудов в эмбриогенезе и регенерации навигационные молекулы играют важную роль при патологическом ангиогенезе. Т-кадгерин является навигационной молекулой, для которой ранее было показано участие в регуляции направленного роста аксонов. Однако известно, что сосуды и нервы растут параллельно, а одни и те же молекулы часто контролируют как рост аксонов нейронов, так и кровеносных сосудов [Weinstein, 2005]. В этой связи целью настоящего исследования было изучение роли Т-кадгерина в процессах роста и функционирования кровеносных сосудов Для достижения указанной цели были поставлены следующие задачи: 1. Выявить экспрессию мРНК и белка Т-кадгерина в головном мозге и сердце в раннем эмбриональном развитии у мыши. 2. Определить характер экспрессии Т-кадгерина в кератиноцитах и кровеносных сосудах новообразований кожи различной степени злокачественности, а также в кровеносных сосудах и опухолевых клетках меланомы человека. 3. Изучить влияние экспрессии Т-кадгерина на пролиферативную активность и уровень апоптоза в клетках мышиной меланомы B16F10 in vitro. Оценить влияние экспрессии Т-кадгерина на скорость роста первичных узлов мышиной меланомы и характер их васкуляризации in vivo. Сравнить клетки мышиной меланомы с разным уровнем экспрессии Т-кадгерина по их способности к метастазированию in vivo и инвазии in vitro. 4. Оценить влияние экспрессии Т-кадгерина на уровень экспрессии ингибиторов и активаторов ангиогенеза, хемокинов и их рецепторов, факторов роста, молекул адгезии и протеаз в клетках мышиной меланомы in vitro. 5. Изучить влияние экспрессии Т-кадгерина в клетках мышиной меланомы на их способность стимулировать неоангиогенез на модели хориоаллантоисной мембраны куриного эмбриона. 10 6. Оценить влияние Т-кадгерина на васкуляризацию бляшек Матригеля у мышей при подкожном введении клеток с разным уровнем экспрессии Т-кадгерина. 7. Изучить влияние Т-кадгерина на пролиферацию, адгезию, апоптоз, миграцию эндотелиальных клеток и формирование ими капилляро-подобных трубочек in vitro. 8. Изучить влияние гиперэкспрессии Т-кадгерина в эндотелиальных клетках на барьерную функцию эндотелия in vitro. 9. Выявить экспрессию Т-кадгерина в различных слоях нормальной аорты и в сосудах человека при атеросклерозе. 10. Изучить влияние гиперэкспрессии Т-кадгерина на характер связывания ЛНП с мембранами клеток, внутриклеточную сигнализацию и миграцию клеток по градиенту ЛНП. Работа выполнялась на базе факультета фундаментальной медицины Московского государственного университета имени М.В. Ломоносова и Федерального государственного бюджетного учреждения «Российский кардиологический научнопроизводственный комплекс» Министерства здравоохранения Российской Федерации. Исследования проводили in vitro и in vivo на экспериментальных моделях, наиболее близких к заболеваниям человека и позволяющих изучать ангиогенез во взрослом организме. Глава «Обзор литературы» посвящена описанию основных хемотактических и ростовых факторов роста, навигационным рецепторам и их лигандам, а также основным клеточным и молекулярным механизмам ангиогенеза и нейрогенеза. Проведенные нами исследования показали, что Т-кадгерин действительно является навигационной молекулой, которая регулирует направленный рост не только аксонов нейронов, но и сосудов. Впервые обнаружено, что Т-кадгерин экспрессируется в раннем эмбриональном развитии у мыши в головном мозге и в области развивающегося сердца в зонах наиболее активного ангиогенеза. На экспериментальных моделях показано, что Т-кадгерин ингибирует начальные этапы ангиогенеза, в основе этих эффектов лежит гомофильное взаимодействие между молекулами Т-кадгерина, следствием которого является подавление миграции эндотелиальных клеток, формирования, роста и ветвления капилляро-подобных трубочек. Далее обнаружено, что Т-кадгерин участвует в регуляции проницаемости эндотелия, вызывает перестройки цитоскелета и эндоцитоз VE-кадгерина, основного белка адгезивных контактов эндотелия, ответственного за его барьерную функцию. Раскрыты биохимические механизмы сигнализации с участием Т-кадгерина в этих процессах. Экспрессия Ткадгерина изменяется при различных патологических состояниях. Снижение экспрессии 11 Т-кадгерина коррелирует с малигнизацией новообразований кожи и меланомы человека, в опухолевых клетках и в сосудах выявляется частичная или полная потеря экспрессии Т-кадгерина. На экспериментальных опухолевых моделях обнаружено, что, несмотря на антиангиогенные свойства, Т-кадгерин не может считаться опухолевым супрессором, поскольку экспрессия Т-кадгерина в опухолевых клетках вызывает активацию компенсаторных механизмов в виде экспрессии генов, способствующих пролиферации, выживанию, миграции и инвазии опухолевых клеток. Кроме того, экспрессия Ткадгерина стимулирует продукцию хемокинов опухолевыми клетками, что способствует активации мезенхимальных клеток стромы ближайшего микроокружения и опухолевой прогрессии в целом. Экспрессия Т-кадгерина повышается при атеросклерозе. Показано, что Т-кадгерин является низкоаффинным рецептором ЛНП, опосредующим их сигнальные эффекты. Этот материал изложен в главе «Результаты и обсуждение», которой предшествует раздел «Материалы и методы». Завершается диссертация разделами «Заключение» и «Выводы», в котором мы излагаем свою точку зрения на участие Т-кадгерина и сопряженных с ним сигнальных молекул на рост и функционирование кровеносных сосудов животных и человека. 12 СПИСОК СОКРАЩЕНИЙ AGP - Angiopoietin 1, ангиопоэтин-1 AGPopt – optimized Angiopoietin 1, оптимизированный ген ангиопоэтина-1 Ang-1 - ангиопоэтин bFGF - основной фактор роста фибробластов CD31 - маркѐр эндотелиальных клеток CHO – Chinese hamster ovary, опухоль яичника китайского хомячка c-Met – MET, hepatocyte growth factor receptor (HGFR), рецептор фактора роста гепатоцитов DAPI – 4,6-diamidino-2-phenylindole, 4',6-диамидино-2-фенилиндол, маркѐр ядерной ДНК DEPC – Diethylpyrocarbonate, диэтилпирокарбонат DMEM- Dulbecco's Modified Eagle Medium, среда Игла, модифицированная Дульбекко EEA1- маркер для выявления мембранной фракции и фракции белков ранних эндосом EGF - Epidermal growth factor, эпидермальный фактор роста EGM-2 - полноценная среда роста ELISA – Enzyme-Linked ImmunoSorbent Assay, иммуноферментный анализ EST - expressed sequence tag FGF - фактор роста фибробластов FITC –декстран- флуоресцеинизотиоцианат-декстран GDNF - Glial cell-derived neurotrophic factor, глиальный фактор роста нервов GFP- green fluorescent protein, зелѐный флуоресцирующий белок GFR Матригель - Матригель без факторов роста GFR- общий коэффициент фертильности GPI – Glycophosphatidylinositol, гликозилфосфатидилинозитол HBSS буфер - Hank's Balanced Salt Solution, буфер Хэнкса HGF- hepatocyte growth factor, фактор роста гепатоцитов HGFopt – optimized Hepatocyte growth factor, оптимизированный ген фактора роста гепатоцитов HUVEC – human vein umbilical endothelial cell IL-8- интерлейкин-8 LAMP1- маркер лизосом для выявления цитоплазматических белков LB - Lysogeny broth, богатая среда для роста культур бактерий LIMK1- Серин-треониновая киназа 1 типа LIM MAP киназы- митоген-активируемые белковые киназы 13 MCP-1 -белок, вызывающий хемотаксис моноцитов M-CSF- гранулоцитарно-макрофагальный колониестимулирующий фактор MDCK - Madin-Darby canine kidney, клетки почки собаки MMP2 – Matrix metalloproteinase 2, матриксная металлопротеиназа 2 типа MMP9 - Matrix metalloproteinase 9, матриксная металлопротеиназа 9 типа MOI – multiplicity of infection, число вирусных частиц в клетке NTC - non-templatecontrol, отрицательный контроль без добавления матрицы NUDE - линия бестимусных мышей OD – optical density, оптическая плотность PAK1- Rho-ассоциированная киназа, является мишенью для активированных Rac1 и Rho ГТФазы - семейство клеточных сигнальных белков, «малых» (около 21 кДа) G-белков. относящихся к суперсемейству Ras PCMV- промотор цитомегаловируса PCNA- ядерный антиген пролиферирующих клеток PCR - polymerase chain reaction, полимеразная цепная реакция PDGF - тромбоцитарный фактор роста PDGF-BB – platelet derived growth factor-BB PHD2 - пролил гидроксилаза 2 PKC – протеинкиназа С PLC - фосфолипаза С PVDF - Polyvinylidene fluoride, мембрана, специально разработана для вестерн блоттинга ROCK-II- Rho-ассоциированная киназа, активируется RhoA RT-PCR – real time polymerase chain reaction, полимеразная цепная реакция в режиме реальногот времени SDF1 – Stromal cell-derived factor-1, хемокин подсемейства CXC shRNA- короткие шпилечные РНК олигонуклеотиды si-T-cad – подавление экспрессии Т-кадгерина РНК интерференцией SRE – serum response element, транскрипционный фактор, активируемый сывороткой T-cad – Т-кадгерин, гиперэкспрессия Т-кадгерина TCF- Ternary Complex Factor TGF-beta 1- трансформирующий фактор роста-бета 1 TNFα- фактор некроза опухолей tPA- тканевой активатор плазминогена uPA – urokinase, активатор плазминогена урокиназного типа, урокиназа uPAR –Urokinase receptor, рецептор урокиназы 14 VEGF – vascular endothelial growth factor, фактор роста эндотелия сосудов VSVG - псевдотипичные лентивирусные частицы vWF - Von Willebrand factor, маркѐр эндотелиальных клеток Y27632 – ингибитор Rho киназы ROCK-II АК – актинический кератоз БСА – бычий сывороточный альбумин ГМК – гладкомышечные клетки ДНК – дезоксирибонуклеиновая кислота ДНКаза – дезоксирибонуклеаза КА - кератоакантома кДНК – копийная ДНК ЛК - легочный коэффициент ЛНП - липопротеиды низкой плотности МР – метатипический рак мРНК – матриксная РНК МСК - мезенхимные стволовые клетки МСК-ЖТ - мезенхимные стромальные клетки жировой ткани МТ - микротрубочки ПААГ - полиакриламидный гель ПР – плоскоклеточный рак ПЦР – полимеразная цепная реакция РНК – рибонуклеиновая кислота РНКаза - рибонуклеаза СК – солнечный кератоз ФБС – фетальная бычья сыворотка ФСБ – фосфатно-солевой буфер ЭДТА - этилендиаминтетраацетат ЭК – эндотелиальные клетки ЭПР - эндоплазматический ретикулум 15 ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ 1.1. Регуляция роста и ремоделирования кровеносных сосудов 1.1.1 Клеточные компоненты сосудистой стенки Во взрослом организме в норме эндотелиальные клетки пребывают в покое и живут продолжительное время, их жизнеспособность и функциональная активность поддерживается аутокринно за счѐт секреции таких факторов как VEGF (фактор роста эндотелия сосудов), NOTCH, ангиопоэтин-1 (Ang-1) и фактор роста фибробластов (FGF) [DeSmet et al., 2009; Carmeliet and Jain, 2011]. Эндотелиальные клетки чувствительны к изменениям концентрации кислорода в крови за счет кислородных сенсоров и способны продуцировать гипоксия-индуцируемые факторы такие, как пролил гидроксилазы (PHD2) и гипоксия-индуцируемый фактор-2α (HIF-2α), что позволяет им регулировать диаметр сосудов и скорость кровотока. Покоящиеся эндотелиальные клетки образуют монослой фаланговых клеток («phalanx cells») с гладкой поверхностью, взаимосвязанных между собой с помощью адгезивных VE-кадгерин содержащих контактов и плотных контактов, содержащих окклюдин и клаудин. Эндотелиальные клетки покрыты муральными клетками (перицитами и гладомышечными клетками - ГМК), c которыми они взаимодействуют за счет N-кадгерин содержащих адгезивных контактов. Муральные клетки подавляют пролиферацию эндотелиальных клеток и секретируют факторы, такие как VEGF и Ang1, обеспечивающие выживание эндотелия (Рисунок 1.1) [Erez et al., 2004; Cavallaro et al., 2006; Koutsouki et al., 2005; George and Beeching, 2006]. Эндотелиальные клетки и перициты в состоянии покоя формируют общую базальную мембрану. 16 Рисунок 1.1. Кадгерины в сосудистых клетках. Эндотелиальные клетки формируют гомофильные VE-кадгерин-содержащие адгезивные контакты друг с другом, обеспечивающие барьерную функцию эндотелия. Стабильность сосудов обеспечивается взаимодействием эндотелиальных клеток с перицитами и ГМК (муральные клетки) за счет формирования N-кадгерин-содержащих адгезивных контактов. R-кадгерин также обнаруживаются в адгезивных контактах между эндотелиальными и муральными клетками и участвует в стабилизации сосудов. Эндотелиальные клетки и перициты в состоянии покоя формируют общую базальную мембрану. T-кадгерин не локализуется в зоне межклеточных контактов. 17 1.1.2 Механизмы формирования сосудов В литературе существует несколько моделей, описывающих формирования сосудов в норме и при патологии [Carmeliet and Jain, 2011]. В развивающихся эмбрионах млекопитающих, ангиобласты, клетки-предшественники сосудов, дифференцируются в эндотелиальные клетки, которые в дальнейшем формируют сосудистый лабиринт (первичная сосудистая сеть), и этот процесс формирования сосудов de novo называется васкулогенез (Рисунок 1.2). Для первичной сосудистой сети характерен одинаковый размер всех сосудистых структур и отсутствие клеточной иерархии. Неспособность сформировать первичный лабиринт приводит к гибели эмбрионов в промежутке между E9.5 и E12.5 стадиями развития у мыши. Первичное сосудистое сплетение является временной структурой и в дальнейшем подвергается ремоделированию. Активация NOTCH сигнального пути регулирует артериальную или венозную дифференцировку клеток: NOTCH контролирует взаимодействие эфринов со своими рецепторами (Eph-Ephrin), которые в свою очередь определяют формирование артериовенозных границ [Adams an dAlitalo, 2007; Swift and Weinstein, 2009]. Дальнейшее ветвление и разрастание первичной сосудистой сети обеспечивается в процессе ангиогенеза за счет формирования новых сосудов от предсуществующих. В процессе артериогенеза вновь образованные капилляры, состоящие из эндотелиальных клеток, покрываютcя гладкомышечными клетками (ГМК) и перицитами, что обеспечивает их стабильность и регулирует проницаемость. При этом происходят перестройки уже сформированного сосуда, связанные с ремоделированием его стенки и увеличением просвета. Васкуляризация тканей может происходить также с использованием других механизмов, однако, их распространенность и физиологическое значение до конца не ясны. Например, уже существующие сосуды могут ветвиться за счет инвагинации и и бифуркации (intussusception), что приводит к образованию дочерних сосудов или разветвления исходных (Рисунок 1.2). В других случаях клетки опухолей, располагаясь в тканях с хорошо развитой сосудистой сетью вокруг этих сосудов, могут использовать сосуды для обеспечения своей жизнедеятельности (co-option) (Рисунок 1.2). Кроме того, в случае меланомы, опухолевые клетки сами способны формировать характерную сеть сосудистых каналов и обеспечивать опухоль питательными веществами и кислородом, это явление было названо «васкулогенной (сосудистой) мимикрией» (Рисунок 1.2) [Carmeliet and Jain, 2011]. Однако лакуны, в собственном смысле слова, не являются васкулогенными или ангиогенными, поскольку образованы опухолевыми клетками 18 самой меланомы [Folberg et al., 2000; Maniotis et al., 1999]. Кроме того, т.н. «раковые стволовые клетки» могут дифференцироваться в эндотелиальные клетки и формировать сосуды внутри опухоли (Рисунок 1.2) [Wang et al., 2010]. В настоящее время в литературе обсуждается вопрос о степени участия стволовых/прогениторных клеток в процессах регенерации в сосудистой стенке и их вклада в васкуляризацию тканей и органов при различных патологиях. Эндотелиальные прогениторные клетки периферической крови и/или стволовые/прогениторные клеток из костного мозга могут быть рекрутированы в кровеносное русло и мигрировать по градиенту хемоаттрактанта в зону повреждения. Прогениторные клетки могут включаться в состав эндотелия, выстилающего сосуд, в процессе известном как постнатальный васкулогенез (Рисунок 1.2). При реваскуляризации коллатеральные сосуды, снабжающие ишемизированные ткани, увеличиваются в диаметре в ответ на соответствующие факторы роста, которые продуцируют клетки в зоне ишемии, а также за счет привлечения и активации миелоидных клеток [Schaper, 2008]. 19 Рисунок 1.2. Модели формирования сосудов в норме и при опухолевом росте. Новые сосуды могут образовываться из предсуществующих сосудов в процессе ангиогенеза (А), рекрутирования эндотелиальных клеток из костного мозга при васкулогенезе (Б), за счет бифуркации сосудов (intussusception) (В), за счѐт использования опухолевыми клетками предсуществующих сосудов (co-option) (Г), в процессе «васкулогенной мимикрии» (Д), дифференцировки «раковых стволовых клеток» в эндотелиальные клетки (Е). В отличие от нормальных тканей, в которых реализуется ангиогенез и ветвление сосудов, васкулогенез и ветвление с бифуркацией(intussusceptions), в опухолях реализуются все типы формирования новых сосудов (Адаптировано из Carmeliet and Jain, 2011). 20 1.1.3 Ветвление сосудов при ангиогенезе При гипоксии, воспалении, растяжении или опухолевом росте клетки различного происхождения продуцируют ангиогенные факторы (VEGF, Ang-2, FGF) и хемокины (SDF-1 и др.). Факторы роста и хемокины стимулируют миграцию, инвазию эндотелиальных клеток в окружающие ткани и их пролиферацию. На такой стимул покоящийся сосуд в первую очередь отвечает откреплением перицитов от базальной мембраны (в ответ на Ang-2) и локальной протеолитической деградацией внеклеточного матрикса. Параллельно происходит повышение локальной концентрации NO за счет активации синтазы оксида азота (NO), что приводит к вазодилатации [Distler et al., 2003]. Под действием ангиогенных факторов на первом этапе происходит активация и формирование ―tip‖ эндотелиальных клеток. При этом межклеточные адгезивные контакты, содержащие VE-кадгерин, частично разбираются, что позволяет эндотелиальным клеткам мигрировать по градиенту хемоаттрактанта. VEGF увеличивает проницаемость эндотелиального монослоя, в результате чего белки плазмы (фибронектин и фибриноген) крови вытекают из сосудов в ткань и служат подложкойматриксом для мигрирующих эндотелиальных клеток [Carmeliet and Jain, 2011]. Важную роль при миграции играют интегрины. Компоненты внеклеточного матрикса, такие как фибронектин, ламинин, гиалуроновая кислота и тромбоспондин содержат участки связывания для интегринов, экспрессирующихся на эндотелиальных и муральных клетках [Distler et al., 2003]. Активированные эндотелиальные клетки и перициты продуцируют протеазы, способствующие разрушению базальной мембраны и повышению подвижности этих клеток. Протеазы, такие как активаторы плазминогена, химазы, гепариназы, триптазы, катепсины и матриксные металлопротеиназы, вызывают протеолитическую деградацию и разрыхление компонентов внеклеточного матрикса, высвобождение и протеолитическую активацию ангиогенных факторов роста, депонированных в матриксе, таких как bFGF, VEGF, HGF (фактор роста гепатоцитов), TGFβ (трансформирующий фактор роста бета) и других, что, в целом, создает ангиогенную среду и способствует эффективному функциональных сосудов [Carmeliet and Jain, 2011]. формированию новых 21 1.1.4 Специализация клеток при ангиогенезе: “tip” и “stalk” фенотип Для формирования в строго определенном месте функционально зрелых сосудов и предотвращения массовой хаотичной миграции эндотелиальных клеток при ангиогенезе лишь одна из многих эндотелиальных клеток становится ―tip‖ клеткой («концевой» клеткой), в соседних клетках формируется ―stalk‖ фенотип. Характерной чертой ―tip‖ клетки является ее локализация на конце растущего сосуда, большое количество филоподий, с помощью которых ―tip‖ клеткой «тестирует» микроокружение. Именно ―tip‖ клетка преобразует комбинации различных навигационных сигналов микроокружения в направленную миграцию следующих за ней эндотелиальных клеток и рост всего сосуда. В ответ на навигационные сигналы, поступающие от Эфринов и Семафоринов, а также и других навигационных молекул, ―tip‖ клетка мигрирует по градиенту хемоаттрактанта и направляет рост всего сосуда (Рисунок 1.3). ―Tip‖ клетка не участвует в формировании просвета сосуда и практически не пролиферирует. У ―tip‖ клетки есть характерный молекулярный профиль, она экспрессирует VEGFR2, VEGFR3, тромбоцитарный фактор роста PDGF-BB, Unc5B, NOTCH лиганды (Dll4и JAGGED1), Нейропилины (NRP) и ряд других белков [DeSmet et al., 2009]. Главную роль в специализации ―tip‖ и ―stalk‖ клеток играет градиент VEGF. В условиях высокого уровня VEGF происходит активация и формирование клеток ―tip‖ клеточного фенотипа с высокой миграционной способностью, в то время как средний уровень VEGF способствует пролиферации и поддержанию в соседних с концевыми клетками ―stalk‖ фенотипа. VEGF, связываясь с VEGFR2, инициирует сигнальный каскад, приводящий к тому, что одна клетка становится ―tip‖ клеткой, а другие – ―stalk‖ клетками. В условиях гипоксии происходит конкуренция за положение ―tip‖ клетки: клетка, которая связывает максимальное количество VEGF, становится ―tip‖ клеткой, при этом в соседних с «концевой» клетки подавляется ―tip‖ фенотип, и они становятся ведомыми (―stalk‖ клетки) [Quaegebeur et al., 2011]. Активация внутриклеточной сигнализации при связывании VEGF с VEGFR2 в ―tip‖ клетке приводит к экспрессии лиганда Dll4 и экспонированию его на поверхности (Рисунок 1.3 и Рисунок 1.4). Начальная специализация и дальнейшее поддержание эндотелиальных клеток в состоянии ―tip‖ или ―stalk‖ фенотипа регулируется на уровне Dll4/NOTCH сигнализации. Dll4, являясь лигандом NOTCH, связывается с NOTCH рецептором на ―stalk‖ клетках, что приводит к подавлению в них ―tip‖ клеточного фенотипа. NOTCH внутри ―stalk‖ клеток расщепляется с образованием NOTCH внутриклеточного домена (NICD), который является фактором, регулирующим экспрессию генов. 22 По принципу обратной связи активация сигнализации от NOTCH блокирует экспрессию генов VEGFR2 и NRPs в ―stalk‖ клетках. Помимо VEGFR2 ―tip‖ клетки экспрессируют рецептор VEGFR3, который участвует в формировании филоподий и ламеллоподий в ―tip‖ клетке. Соседние ―stalk‖ клетки делятся и формируют стенки растущего сосуда, в этом процессе важную роль играют такие факторы как, Notch-регулируемый белок, содержащий анкириновые повторы (NRARP), белки Wnt сигнального пути, плацентарный фактор роста (PlGF) и FGF [Carmeliet and Jain, 2011]. В отличие от Dll4 другой лиганд NOTCH- JAGGED1 экспрессируется только в ―stalk‖ клетках и поддерживает фенотип этих клеток. Некоторые другие сигнальные пути, в том числе и Wnt/-катенин путь также участвуют в регуляции экспрессии Dll4 в ―tip‖ клетке на уровне транскрипции генов (Рисунок 1.3 и Рисунок 1.4) [Quaegebeur et al., 2011]. В процессах более тонкой регуляции и поддержания специализированного состояния также участвует VEGFR1: VEGFR1 присутствует в ―tip‖ клетках в небольшом количестве в трансмембранной форме, а в ―stalk‖ клетках в основном представлен растворимой формой и служит ловушкой для VEGF, что блокирует специализацию последних в ―tip‖ клетках [DeSmet et al., 2009]. Нейропилины (NRP1 и NRP2) также играют важную роль при ангиогенезе. NRPs в эндотелиальных клетках выступают либо как ко-рецепторы VEGFR (VEGFR1, 2, 3) и участвуют в морфогенезе и ветвлении сосудов, либо как рецепторы Семафоринов (Sema3), регулируя траекторию роста сосудов за счет отталкивания и подавления миграции эндотелиальных клеток в область с высокой концентрацией Sema3 [DeSmet et al., 2009]. Добавление антител против NRP1, которые связываются только с VEGF, но не связываются с Sema3, не влияет существенно на морфогенез сосудов в эмбриогенезе [Geretti et al., 2008]. Однако Sema3 играет важную роль при опухолевом росте и является перспективной мишенью для подавления опухолевого неоангиогенеза и метастазирования. Генетические исследования показали, что в отсутствие NRP1 формирующиеся в заднем мозге сосуды не ветвятся, а сливаются с формированием дефектного сосудистого сплетения; блокирование процесса связывания VEGF с NRP1 препятствует дальнейшему ремоделированию этого сплетения и формированию функционально зрелой сосудистой сети [Geretti et al., 2008]. Дефекты ветвления сосудов и образование аберрантной сосудистой сети в отсутствии NRP1 вызваны нарушением формирования латеральных филоподий, которые важны для изменения направления миграции ―tip‖ клетки в момент поворота и слияния растущих по направлению друг к другу сосудов (Рисунок 1.3 и Рисунок 1.4) [Geretti et al., 2008]. 23 Рисунок 1.3. Сигнальные пути, определяющие специализацию эндотелиальных клеток в растущем сосуде: “tip” клетки, осуществляющие навигацию, и “stalk”клетки, ответственные за пролиферацию клеток растущего сосуда. Связывание VEGF и активация сигнализации в “tip”клетках вызывает увеличение экспрессии Dll4, который активирует NOTCH сигнализацию в соседней “stalk” клетках. Wnt сигнализация через опосредованную-катенином активацию транскрипции определенных генов стимулирует пролиферацию “stalk” клеток. Сигнализация от VEGFR2 в “tip”клетках стимулирует экспрессию VEGFR3, который участвует в формировании филоподий и ламеллоподий в “tip”клетке. (Адаптировано из Quaegebeur et al., 2011). 24 Рисунок 1.4. Начальные этапы ангиогенеза. Формирование “tip” клеток («концевых» клеток), навигация и выбор направления миграции по градиенту хемоаттрактанта (VEGF), пролиферация “stalk” клетке, ветвление сосудов, дальнейший рост и элонгация, формирование просвета сосуда (А). VEGF/NOTCH сигнализация определяет “tip” или “stalk” специализацию клеток и поддержание выбранного фенотипа. VEGF/VEGFR2 взаимодействие приводит к увеличению экспрессии Dll4 в “tip” клетках, в соседних “stalk” клетках NOTCH подавляет“tip” фенотип за счѐт снижения экспрессии VEGFR2, VEGFR3 и NRP1, и увеличения экспрессии VEGFR1 (Б) (Адаптировано из Potente et al., 2011). 25 1.1.5 Филоподии и ламеллоподии определяют направление роста сосудов Основной функцией ―tip‖ клеток является навигация, которая выражается в тестировании микроокружения и преобразовании его сигналов в динамические процессы адгезии (на переднем крае клетки) и открепления (на заднем конце клетки), что, в конечном счете, приводит к направленной миграции эндотелиальных клеток растущего сосуда. Для того чтобы осуществлять навигацию, клетка должна поляризоваться, то есть у нее должен появиться передний и задний край и направление движение. Ламеллоподия (ведущая ламелла, лидирующий край) - это тонкий слой цитоплазмы, который выступает на переднем конце распластывающейся и мигрирующей клетки. Ламеллоподии содержат сильно разветвленную сеть актина (Рисунок 1.5). Филоподии, в свою очередь, содержат длинные, параллельные, плотно упакованные пучки филаментов актина (F-актин), которые, как правило, простираются от ламеллоподий вперед по направлению движения клетки (Рисунок 1.5). Полимеризация актиновых филаментов на переднем крае мигрирующей клетки является движущей силой образования филоподий и клеточной протрузии. Ламеллоподии и филоподии являются очень динамичными образованиями, именно они осуществляют зондирование микроокружения мигрирующей ―tip‖ клетки и распознавание навигационных сигналов (attraction and repulsion). Аттрактивные сигналы вызывают полимеризацию F-актина и удлинение филоподий по градиенту аттрактивного сигнала, а сигналы отталкивания вызывают разборку актина, его ретроградный транспорт и ретракцию лидирующего края клетки [DeSmet et al., 2009]. Филоподии и ламеллоподии участвуют в адгезии клетки на субстрате за счет формирования интегринсодержащих фокальных контактов, которые опосредуют взаимодействие компонентов актинового цитоскелета с внеклеточным матриксом (ламинин, фибронктин, коллаген и др.). Это, в свою очередь, делает возможным работу акто-миозинового комплекса, который обеспечивает подтягивание тела клетки вдоль стресс фибрилл актина и тем самым движение клетки вперед. Полимеризация субъединиц мономерного G-актина, формирование фибрилл Fактина и их удлинение происходит с участием белков Arp2/3 (Actin-related protein-2/3) комплекса, профилина, Ena/Vasp и белков семейства форминов. Ветвление актиновых филаментов происходит с участием белков Arp2/3 и WASP (Wiscott-Aldrich Syndrome protein), деполимеризация актиновых филаментов происходит с участием кофилина (cofilin) [DeSmet et al., 2009]. Важную роль в упаковке растущих филаментов актина в параллельные пучки играет миозин Х и белки семейства фасцинов (Fascin) [DeSmet et al., 2009]. 26 Рисунок 1.5. Схематичное представление организации актина и активности Rho ГТФаз на лидирующем крае мигрирующей “tip” клетки. Стресс-фибриллы обозначены зеленым цветом, филаменты актина красным, разветвленная сеть актина в зоне ламеллоподии - фиолетовым. Филоподии формируют фокальные контакты (выделено оранжевым цветом) с внеклеточным матриксом (черный) (Адаптировано из De Smet et al., 2009). 27 Основными регуляторами формирования филоподий и ламеллоподий являются Rho ГТФазы (Cdc42, Rac1и RhoA) (Рисунок 1.5), относящиеся к семейству малых Gбелков [Pollard and Borisy, 2003; Huber et al., 2003]. Активация Rho ГТФаз происходит в ответ на связывание многих мембранных рецепторов со своими лигандами, включая GPCRs рецепторы [Huber et al., 2003]. Rho ГТФазы активируются путем связывания с ГТФ и инактивируются за счет своей собственной ГТФазной активности, приводящей к гидролизу ГТФ до ГДФ. GEFs (Guanine nucleotide Exchange Factors) ускоряют обмен ГДФ на ГТФ и тем самым активируют Rho ГТФазы, GAPs (GTFase Activating proteins) стимулируют гидролиз и инактивацию Rho ГТФаз. Активированные Cdc42 и Rac1 взаимодействуют с Arp2/3 и WASP белками, которые обеспечивает нуклеацию, рост и ветвление филаментов актина на переднем края мигрирующей клетки, а также важны для функционирования и динамики актиновых филаментов в ламеллоподии [Pollard and Borisy, 2003]. Cdc42 иRac1 регулируют формирование ламеллоподий и филоподий через активацию РАК (p21-activated kinase). RhoA участвует в адгезии и направленном движении клеток за счет активации ROCK (Rho-associated serine-threonine protein kinase) и полимеризации актиновых стресс фибрилл [Pollard and Borisy, 2003; Huber et al., 2003; Defilippi et al., 1999]. РАК и ROCK активируют LIM-киназу, которая ингибирует кофилин, вызывающий деполимеризацию актина, и тем самым участвует в сборке актиновых филаментов [Huber et al., 2003]. Формирующиеся филоподии образуют адгезивные контакты с внеклеточным матриксом (фокальные контакты), важным регулятором этого процесса является киназа фокальных контактов (FAK). В свою очередь интегрины и рецепторы факторов роста в филоподиях передают сигналы извне внутрь клетки (outside-inside signaling) на компоненты цитоскелета, опосредуя петлю обратной связи и информацию от микроокружения [DeSmet et al., 2009]. Как уже упоминалось выше, основную роль в специализации ―tip‖ клеток играет VEGF, он же определяет поляризацию клетки и выбор направление движения. Рецептор VEGFR2 активирует Rho ГТФазы, которые регулируют процессы поляризации и миграции ―tip‖ клеток. В ответ на связывание VEGF с VEGFR2 активируется Cdc42, который индуцирует формирование филоподий и поляризацию клеток через организацию актинового цитоскелета и микротрубочек, в то время как Rac1 вместе с РАК регулирует динамичное поведение актина и формирование ламеллоподий. В эндотелиальных клетках Cdc42 также активирует Rac1. Потеря Rac1 приводит к гибели эмбрионов из-за отсутствия нормального процесса ветвления сосудов. RhoA в ответ на 28 VEGF индуцирует формирование стресс фибрилл, увеличивает проницаемость сосуда, клеточную миграцию, а позднее стабилизирует формирующиеся капилляры. На активность RhoA влияет также напряжение сдвига [DeSmet et al., 2009]. Помимо названных факторов, есть еще ряд молекул, влияющих на формирование филоподий. Известно, что воспаление активирует эндотелиальные клетки, ангиогенез и ветвление сосудов. TNF-(фактор некроза опухоли альфа) стимулирует формирование ―tip‖ фенотипа и ветвление вновь формирующихся сосудов. В патологических условиях (ангиогенез при воспалении) TNF- через активацию транскрипционного фактора NF-B стимулирует экспрессию молекул JAGGED1/NOTCH сигнального пути в ―tip‖ клетках, при этом JAGGED1 выполняет функцию, аналогичную той, что выполняет Dll4 «концевых» клетках в физиологических условиях [Sainson et al., 2008]. Даже кратковременное инкубирование эндотелиальных клеток в присутствии TNF-приводит к экcпрессии маркеров ―tip‖ клеток, таких как VEGFR2 и PDGF-BB. Другой медиатор воспаления, брадикинин, вызывает формирование филоподий через активацию Cdc42 сигнализации. S1P, сфингозин-1-фосфат (sphingosine-1-phosphate) – противовоспалительный фосфолипид, продуцируемый активированными тромбоцитами, также стимулирует формирование ламеллоподий [DeSmet et al., 2009]. Сигнализация с участием S1P вызывает перемещение Arp2/3 комплекса из цитоплазмы на лидирующий край, что важно для обеспечения локальной полимеризации актина и направленной миграции клеток [DeSmet et al., 2009]. Для формирования и роста нового сосуда ―stalk‖ клетки должны обладать рядом свойств: они должны активно делиться, образовывать и поддерживать адгезивные контакты с «концевой» клеткой и формировать просвет сосуда. В отличие от ―tip‖ клеток, миграционная активность ―stalk‖ клеток в растущем сосуде подавлена, ингибирование формирования филоподий происходит за счет NOTCH сигналинга и подавления экспрессии VEGFR2, VEGFR3, NRP1 и CXCR4. Важную роль в формировании межклеточных VE-кадгерин содержащих адгезивных контактах играет FGF и NRARP-индуцируемый Wnt сигналинг [Carmeliet and Jain, 2011]. Формирование просвета сосудов зависит от образования вакуолей и их слияния как внутри эндотелиальных клеток, так и между клетками. Для формирования вакуолей важно взаимодействие интегринов с внеклеточным матриксом, а для их слияния необходима реорганизация актинового и микротрубочкового цитоскелета. Вакуоли формируются и накапливаются в районе центросомы/аппарата Гольджи, и важную роль при этом играют локализующиеся на мембране вакуолей Rac1 и Сdc42 [DeSmet et al., 2009]. 29 Как только формирование нового сосуда завершается, эндотелиальные клетки переходят в состояние покоя (phalanx cells), формируют прочные межклеточные VEкадгериновые контакты и обеспечивают барьерную функцию эндотелия. Покоящиеся клетки слабо отвечают на стимуляцию небольшими концентрациями VEGF, практически не формируют филоподий и обладают низкой миграционной и пролиферативной активностью. В этом состоянии присутствие VEGF важно для поддержания функции покоящихся эндотелиальных клеток и защиты их от апоптоза. Высокие концентрации VEGF активируют специализацию эндотелиальных клеток и процессы ангиогенеза [DeSmet et al., 2009]. По мере формирования нового сосуда процессы миграции и пролиферации постепенно замедляются, эндотелиальные клетки формируют межклеточные контакты и образуют капиллярные трубочки. VE-кадгерин, CD34, сиаломуцины, VEGF и hedgehog участвуют в формировании просвета растущего сосуда; если просвет сосуда не формируется, то такой сосуд подвергается регрессии. Финальным этапом ангиогенеза является стабилизация вновь образованных сосудов за счет взаимодействия с перицитами и ГМК и формирование новой базальной мембраны. В этих процессах важную роль играют ингибиторы матриксных металлопротеиназ (TIMPs) и ингибиторы активатора плазминогена (PAI-1) [Semenza, 2007; Carmeliet and Jain, 2011; Potente et al., 2011]. 1.1.6 Ангиогенные факторы роста Источником ангиогенных факторов и ингибиторов ангиогенеза могут быть различные типы клеток: моноциты и макрофаги, эндотелиальные клетки, тучные, муральные, а также мезенхимные стволовые клетки. Важнейшими проангиогенными факторами является семейство VEGF. VEGF действует как специфический митоген для эндотелиальных клеток, он также стимулирует их выживание и миграцию, увеличивает проницаемость сосудов, а также продукцию факторов, способствующих ремоделированию внеклеточного матрикса [Distler et al., 2003; Carmeliet and Jain, 2011]. VEGF необходимым для развития сосудистой сети в норме и при патологии. Семейство VEGF представляет собой гетеродимерные гликопротеиды и включают VEGF-A, VEGF-B, VEGF-C, VEGF-D (Рисунок 1.6) [Ferrara, 2009; Nagy et al., 2007]. На сегодняшний день известно 5 изоформ VEGF-А, образующихся в результате альтернативного сплайсинга: VEGF121, VEGF165, VEGF183, VEGF189, VEGF206. VEGF165 экспрессируется в большинстве органов и тканей и является основной изоформой. VEGF-С, лиганд VEGFR-2 и VEGFR-3 30 рецепторов, участвует в активации и специализации эндотелиальных клеток в ―tip‖ клетки [Tvorogov et al., 2010]. Дефицит или мутации по VEGF-В не являются летальными и не влияют на нормальное развитие сосудистой системы в эмбриогенезе у мышей. VEGF-В обладает ангиогенным эффектом и стимулирует рост сосудов лишь в некоторых тканях, например, в миокарде, кроме того VEGF-В важен для выживания нейронов [Fischer et al., 2008]. VEGF может существовать в растворимой форме или быть связан с внеклеточным матриксом, и в зависимости от формы VEGF оказывает разные биологические эффекты: в растворимой форме VEGF способствует увеличению просвета сосудов, в связанной стимулирует ангиогенез и ветвление сосудов [Carmeliet and Jain, 2011]. VEGF, секретируемый паракринно клетками опухоли, миелоидными или стромальными клетками, стимулирует формирование аберрантных сосудов и усиливает их ветвление, в то время как VEGF, аутокринно секретируемый эндотелиальными клетками, стимулирует поддержание клеточного гомеостаза в сосудах [Lee et al., 2007]. Блокирование VEGF сигнализации лежит в основе антиангиогенных подходов в противоопухолевой терапии и ряда офтальмологических заболеваний. Работы по введению рекомбинантного белка VEGF или по генной терапии с использованием конструкций для экспрессии VEGF легли в основу терапевтического ангиогенеза, подхода для стимуляции ангиогенеза в ишемизированных органах и тканях, разрабатываемого с конца 20-го века [Парфенова и Ткачук, 2007; Deveza et al., 2012]. Существуют три типа рецепторов VEGF-A в эндотелиальных клетках: VEGFR1 (Flt-1), VEGFR2 (KDR) и VEGFR3 (Flt-4) (Рисунок 1.6). Все VEGF рецепторы являются трансмембранными белками и обладают собственной тирозинкиназной активностью за исключение sVEGFR1, который представляет собой растворимую форму рецептора, участвует в регуляции процессов ангиогенеза за счѐт секвестрирования VEGF и ингибирования его сигнализации. VEGFR2 экспрессирован практически во всех органах и тканях и обнаруживается во всех типах эндотелиальных клеток, VEGFR1 менее распространен. VEGFR1 связывается с VEGF-A, VEGF-C и VEGF-D. VEGFR3 является специфичным рецептором VEGF эндотелиальных клеток лимфатических сосудов. Данные последних лет свидетельствуют о том, что биологические эффекты VEGF и соответствующие сигнальные пути зависят от локализации VEGFR-2. Так, сигнальные пути, приводящие к морфогенезу артерий, активируются при сигнализации от VEGFR-2, находящегося во внутриклеточном компартменте после его эндоцитоза с поверхности мембраны [Lanahan et al., 2010]. 31 Рисунок 1.6. Семейство VEGF и VEGF рецепторов (VEGFR). 32 Активация внутриклеточной сигнализации при связывании с VEGF зависит также от ко-рецепторов, с которыми взаимодействуют VEGFR на поверхности эндотелиальных клеток. К ним относится протеогликан гепаран сульфат, Нейропилины и VE-кадгерин. У этих белков нет собственного тирозинкиназного домена, однако, они в значительной степени модулируют эффекты VEGF, например, активация пролиферации эндотелиальных клеток реализуется через взаимодействие VEGFR с VE-кадгерином [Neufeld and Kessler, 2008; Distler et al., 2003]. Плацентарный фактор роста (PlGF) также принадлежит к семейству VEGF. В отличие от VEGF отсутствие PlGF не оказывает никакого влияния на развитие сосудистой системы в эмбриогенезе; для PlGF было показано участие в заживлении ран и обнаружены ангиогенные эффекты при патологии [Carmeliet andJain, 2011]. PlGF является цитокином, участвующим в процессах ангиогенеза, напрямую или опосредованно, за счѐт активации стволовых и прогениторных клеток. Было показано, что PlGF стимулирует выход эндотелиальных предшественников из костного мозга, активирует миелоидные клетки и клетки стромы, что может способствовать опухолевой прогрессии [Fischer et al., 2008]. Блокирование PlGF стимулирует созревание сосудов опухоли и улучшает перфузию ткани, что повышает эффективность лечения при химиотерапии [Rolny et al., 2011]. Помимо белков семейства VEGF существует целый ряд факторов роста и цитокинов, которые вызывают рекрутирование перицитов, ГМК и мезенхимальных стволовых клеток. Среди этих факторов важнейшими являются PDGF (фактор роста из тромбоцитов), ангиопоэтин (Ang-1) и TGFβ (фактор роста опухоли) [Jain, 2003]. Ангиогенные эндотелиальные клетки продуцируют PDGF-BВ, который связывается с рецептором PDGFR-β на поверхности перицитов и служит для них хемоаттрактантом [Hellberg et al.; 2010, Gaengel, 2009]. Рекрутированные перициты формируют межклеточные пролиферацию, контакты с обеспечивает эндотелиальными выживание и клетками, способствует что ограничивает стабилизации их вновь сформированных сосудов. Кроме локального происхождения, перициты могут также дифференцироваться из периваскулярных PDGFR-β+ клеток-предшественников или мигрировать из костного мозга [Song et al., 2005]. В первичных опухолях перициты служат физическим барьером и лимитируют инвазию и метастазирование опухолевых клеток за счет формирования более плотной сосудистой стенки [Gerhardt and Semb, 2008]. При метастазировании PDGFR-β+ клетки играют двоякую роль. В некоторых опухолях блокирование PDGFR-β ингибирует опухолевый рост, поскольку вызывает открепление перицитов, регрессию незрелых и 33 нестабильных сосудов опухоли [Bergers et al., 2003]. По других данным, блокирование PDGF-BВ в клинических исследованиях вызывало слишком сильное повышение проницаемости сосудов и коррелировало с частотой метастазирования у пациентов [Carmeliet and Jain, 2011]. Важную роль в регуляции процессов ангиогенеза играет семейство ангиопоэтинов (Ang) и их рецепторов - Tie. У человека семейство Ang состоит из трех лигандов (Ang-1, Ang-2 и Ang-4) и двух рецепторов (Tie-1 и Tie-2). Все ангиопоэтины связываются с Tie-2 рецептором. На начальном этапе ангиогенеза секреция Ang-2 «концевой» клеткой стимулирует открепление перицитов, что облегчает миграцию и пролиферацию эндотелиальных клеток [Carmeliet and Jain, 2011]. Ang-1 необходим для ветвления, ремоделирования и стабилизации формирующихся сосудов [Distler et al., 2003]. Ang-1 конкурирует с Ang-2 за связывание с Tie-2 и подавляет его дестабилизирующий эффект. В конфлюентном эндотелии Ang-1 стимулирует кластеризацию Tie-2 in trans в зоне межклеточных контактов, что обеспечивает поддержание эндотелиальных клеток в состоянии покоя. Действие Ang-2 может быть как про-, так и антиангиогенным, в зависимости от присутствия других факторов и цитокинов. Так, при высокой концентрации VEGF-A Ang-2 усиливает ангиогенное действие VEGF, увеличивает ветвление сосудов; однако, при низкой концентрации VEGF-A Ang-2 подавляет ангиогенез, способствует регрессии существующих сосудов и гибели эндотелиальных клеток за счѐт апоптоза [Shibuya, 2008]. До сих пор неизвестен лиганд для Tie-1, возможно, что это орфанный рецептор, и функционирует он как негативный регулятор Tie-2, однако, точных данных о роли Tie-1 в литературе нет [Augustin et al., 2009]. Ang-1 секретируется муральными и опухолевыми клетками, в то время как Ang-2 является продуктом именно «концевой» клетки и в нормальных сосудах стимулирует формирование базальной мембраны, привлечению муральных клеток и обеспечения барьерной функции эндотелия. Роль Ang-Tie системы в опухолевом росте неоднозначна. С одной стороны, Ang-1 стимулирует рост опухоли за счет влияния на эндотелиальные клетки: повышения их выживаемости и стабилизации сосудов, снабжающих опухоль. С другой стороны, это же свойство Ang-1 стабилизировать сосуды препятствует инвазии опухолевых клеток [Carmeliet and Jain, 2011]. Ang-2 стимулирует опухолевый ангиогенез и привлекает проангиогенные TIE-экспрессирующие моноциты (TEMs), а ингибирование Ang-2 способствует регрессии опухолевых сосудов [Falcon et al., 2009]. В этой связи Ang-2 считается перспективной мишенью в противоопухолевой терапии. Различные препараты, блокирующие Tie-2 или Ang-2, в настоящее время проходят первые стадии клинических исследований [Carmeliet and Jain, 2011]. 34 Еще одним ангиогенным фактором является TGFβ. TGFβ стимулирует дифференцировку гладкомышечных клеток сосудов и ремоделирование внеклеточного матрикса [Distler, 2003; Semenza, 2007; Carmeliet, 2011]. Мутации по рецепторам TGFβ ALK-1, TGFR-1, TGFR-2 или ENG (эндоглин) приводят к нарушениям формирования артерио-венозной системы наблюдающиеся у у мышей пациентов с и напоминают наследственной сосудистые формой патологии, геморрагической телеангиэктазии [Pardali et al., 2010]. Суперсемейство FGFs и их рецепторов контролирует многие биологические процессы в органах и тканях [Beenken and Mohammadi, 2009]. bFGF (основный FGF), как FGF1, обладает ангиогенными и артериогенными свойствами. FGFs способны либо напрямую активировать ангиогенез за счет связывания с рецепторами FGF (FGFRs) на эндотелиальных клетках, либо действовать опосредованно, за счет стимуляции секреции ангиогенных факторов другими типами клеток [Beenken and Mohammadi, 2009]. Подавление сигнализации от FGF рецептора в покоящихся эндотелиальных клетках приводит к нарушению целостности сосудов, в то время как присутствие FGF в небольшой концентрации оказывается достаточно для ее поддержания [Murakami et al., 2008]. В некоторых случаях устойчивость опухолей к антиангиогенной терапии с использованием ингибиторов рецепторов VEGF или EGF (эпидермальный фактор роста) обусловлена именно нарушением FGF сигнализации, что, в конечном счете, способствует опухолевому неоангиогенезу и опосредует опухолевый рост [Bergers and Hanahan, 2008]. Рекомбинантный белок FGF и генетическая конструкция для экспрессии FGF в тканях человека были протестированы в пилотных клинических исследованиях с целью стимуляции терапевтического ангиогенеза в ишемизированных тканях, однако, существенного эффекта зарегистрировано не было [Carmeliet and Jain, 2011]. Важную роль в активации процессов ангиогенеза и их регуляции, играет ишемия или гипоксия. Понижение содержания кислорода в поврежденных тканях активирует продукцию и секрецию факторов роста, стимулирующих ангиогенез. Одним из наиболее хорошо изученных механизмов клеточного ответа на гипоксию является стабилизация гипоксия-индуцибельного фактора (hypoxia inducible factor, HIF) HIF-1α, активирующего транскрипцию генов факторов роста, стимулирующих ангиогенез, а именно VEGF и его рецептора VEGFR2, ангиопоэтинов, NO-синтазы, PDGF-BB, TGFβ3, эндотелина-1, металлопротеиназ и рецептора урокиназы является одним из хорошо изученных механизмов влияния гипоксии на клетки [Semenza, 2010]. HIF-1 и HIF-1 (aryl hydrocarbon receptor nuclear translocator; ARNT) образуют транскрипционный комплекс HIF-1, обеспечивающий клеточный ответ на уровне экспрессии генов. В то время как 35 содержание HIF-1 в клетках поддерживается на постоянном уровне, HIF-1при нормальном содержании кислорода гидроксилируется с помощью пролилгидроксилаз (PHD) и связывается с продуктом гена VHL, что вызывает убиквитин-зависимую деградацию комплекса в протеосомах. Гипоксия приводит к стабилизации HIF-1 и формированию комплекса HIF-1, который при участии белка р300 взаимодействует с участком HRE (hypoxia responsive element) промотерной части регулируемых генов и стимулирует их транскрипцию [Semenza, 2010]. В условиях ишемии и гипоксии не только повышается HIF-1-опосредованная транскрипция генов ангиогенных факторов, но и увеличивается стабильность мРНК, кодирующих эти белки, в частности, VEGF [Stein, 1998]. Кроме того, снижение уровня кислорода в тканях вызывает активацию систем вторичных посредников (например, PI3К-Akt сигнального пути), что повышает выживаемость эндотелиальных клеток, активирует их пролиферацию и защищает от апоптоза [Hung, 2007; Semenza, 2010]. В последнее время накапливаются данные о посттранскрипционной регуляции ангиогенеза, а именно, с участием некодирующих микроРНК, которые активируют деградацию своих мишеней-мРНК или блокируют их трансляцию. В условиях гипоксии или гиперэкспрессии VEGF в сосудистых клетках экспрессируется целый спектр различных микроРНК, которые участвуют в регуляции процессов ангиогенеза [Carmeliet, 2011]. Повышение активности HIF-1 комплекса отмечается во многих типах опухолей, что связано с их активным ростом и высокой потребностью в кровоснабжении [Semenza, 2010]. 1.1.7 Хемокины и цитокины, их роль в процессах ангиогенеза Цитокины. К цитокинам относят интерфероны, колониестимулирующие факторы, хемокины, трансформирующие ростовые факторы (TGF); фактор некроза опухолей (TNF; интерлейкины и некоторые другие эндогенные медиаторы. Цитокины по своему биологическому действию делятся на провоспалительные (интерлейкины 1, 2, 6, 8 и др., TNF, INFγ), противовоспалительные (интерлейкины 4, 10 и др., TGFβ) и регуляторы клеточного и гуморального иммунитета. Цитокины влияют на межклеточные взаимодействия, регулируют выживаемость клеток, стимулируют или подавляют их пролиферацию, дифференцировку, функциональную активность и апоптоз, а также координируют и обеспечивают согласованность действий иммунной, эндокринной и нервной систем в нормальных условиях и в ответ на патологические воздействия [Strieter 36 et al., 1992; Koch et al. 1992; Gupta et al., 1998; Kasama et al., 2002; Losordo and Dimmeler, 2004; Kohgo et al., 2005; Li et al., 2003]. Основными продуцентами цитокинов являются лимфоциты, их также секретируют макрофаги, гранулоциты, ретикулярные фибробласты, эндотелиальные клетки и другие типы клеток. Провоспалительные интерлейкины IL-1, IL-6, IL-15, IL-18 и IL-17 и ряд других цитокинов обладают способностью стимулировать ангиогенез. В модельных экспериментах in vivo и in vitro было показано, что IL-1, IL-8, IL-15, IL-18 стимулируют выживание и пролиферацию эндотелиальных клеток, формирования ими капилляро-подобных трубочек, роста сосудов на Матригеле [Li et al., 2003]. В зависимости от экспериментальных условий IL6 может, как стимулировать, так и ингибировать ангиогенез. Экспрессия ряда провоспалительных цитокинов оказывается повышена при аутоиммунных заболеваний и вносит свой вклад в развитие заболевания. Повышенное содержание IL-1, IL-6, IL-15, IL18 и IL-17, а также G-CSF (гранулоцит колониестимулирующий фактор), GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор) и oncostatinM обнаруживается при ревматоидном артрите [Koch et al. 1992]. Некоторые цитокины за счет своих ангиогененых свойств способствуют опухолевому росту, так, IL-17 стимулирует CXCR2-зависимую васкуляризацию немелкоклеточного рака легкого у SCID мышей [Numasaki et al., 2005]. Другие цитокины, такие как IFN-, IFN-, IL-4, IL12 и LIF (фактор ингибирования лейкемии) опосредованно способны ингибировать ангиогенез, за счет подавления секреции клетками ангиогенных факторов роста, хемокинов и цитокинов [Koch et al., 1996]. Например, IL-4 блокирует ангиогенные эффекты TNFи IL-1 [Koch et al., 1996], а IL-12 блокирует ангиогенез за счет индукции экспрессии IFN-и IP-10/CXCL10, который обладает ингибирующими ангиогенез свойствами [Strieter et al., 2005]. Хемокины. Хемокины являются уникальным семейством цитокинов и представляют собой низкомолекулярные (8-10 кДа) гепарин-связывающие белки, о функции которых долгое время было известно, что они являются факторами, привлекающими лейкоциты в зону воспаления [Mehrad et al., 2007]. В настоящее время считается, что хемокины играют важную роль в регуляции ангиогенеза как в норме, так и при патологии. Семейство хемокинов включает как проангиогенные, так и антиангиогенные молекулы. В основе стимуляции ангиогенеза с участием хемокинов лежит рекрутирование проангиогенных гематопоэтических и иммунных клеток, стромальных клетоки эндотелиальных клеток-предшественников, индукции миграции эндотелиальных клеток и формирования капилляров; хемокины также модулируют сигнализацию от рецепторов факторов роста, таких как VEGF и FGF [Singh et al., 2007]. 37 Хемокины, ингибиторы ангиогенеза, связываясь с рецепторами на эндотелиальных клетках, вызывают их апоптоз или регрессию сосудов. Эти хемокины также могут формировать комплексы с факторами роста и тем самым препятствовать их связыванию со своими рецепторами, ингибировать тирозин-киназнную сигализацию от рецепторов факторов роста и вызывать хемотаксис Т-клеток, которые продуцируют ингибиторы ангиогенеза, в том числе и секрецию самих антиангиогенных хемокинов [Mehrad et al., 2007]. На сегодняшний день у человека известно 20 рецепторов хемокинов и 50 лигандов, которые условно разделены на 4 подсемейства в зависимости от структуры (C, CC, CXC, and CX3C): так, CXC хемокины между первым и вторым цистеинами на Nконце содержат неконсервативную аминокислоту (Х); CC хемокины, самое большое подсемейство, содержат два последовательных цистеина; С хемокины содержат один цистеин и представлены единственным хемокином lymphotactin (ХCL1 и ХCL2), CX3C хемокины между цистеинами содержат 3 неконсервативные аминокислоты, имеют единственного представителя Fractalkine, который является уникальным мембранносвязанным хемокином [Taub 2004; Singh et al., 2007]. Далее, CXC хемокины разделяют на 2 подгруппы: ELR-позитивные хемокины (ELR+) и ELR-негативные хемокины (ELR), в зависимости от наличия консервативной аминокислотной последовательности (GluLeu-Arg) перед первым цистеином на N-конце. Наличие ELR определяет специфичность связывания с рецептором и биологическую функцию: ELR+хемокины проангиогенны, ELR-хемокины – антиангиогенны [Singh et al., 2007]. Связывание хемокинов с GPCR рецепторами приводит к замене ГДФ на ГТФ в αсубъединице, диссоциации гетеротримерного белкового комплекса (G) на и cубъединицы, которые в свою очередь активируют фосфолипазу С (Cβ1 иCβ2), вызывают гидролиз фосфатидилинозитол 4,5-биcфосфата (PIP2), что приводит к образованию инозитол трифосфата (IP3) и диацилглицерола (DAG) с последующим увеличением содержания внутриклеточного кальция за счет его выхода из эндоплазматического ретикулума в цитоплазму [Thelen, 2001; Salanga et al., 2009]. Сами хемокиновые рецепторы не обладают тирозин киназной активностью, однако, в результате кросс-тока с другими рецепторами могут стимулировать фосфорилирование белков цитоскелета p130 Cas и paxillin [Dutt et al., 1998] и активировать киназу фокальных адгезий (FAK), а также сигнальные пути Ras/Raf/MEK/JNK/p38/ERK1/ERK2 и сигнализацию с участием фосфоинозитид-3-киназы (PI3Kinase)/AKT/mTOR с последующей активацией транскрипционных факторов и экспрессией генов [Ganju et al., 1998; Mellado et al., 1998; Thelen, 2001; Takami et al., 2002; Strieter et al., 2005]. Кроме 38 того, имеются данные о том, что внутриклеточная сигнализация с участием рецепторов, сопряжѐнных с G-белками, приводит к активации Rho и Rac сигнальных путей, полимеризации актина, сборке фокальных контактов и клеточной миграции [Schraufstatter et al., 2001]. Хемокины ингибиторы ангиогенеза. Для четкой регуляции процессов ангиогенеза необходим баланс проангиогенных и антиангиогенных факторов, такие факторы как гипоксия сдвигают этот баланс в сторону стимуляторов ангиогенеза. В физиологических условиях при формировании необходимого количества сосудов ангиогенез прекращается за счет индукции соответствующих ингибиторов. При патологии, например, при хроническом воспалении, опухолевом росте и других заболеваниях этот баланс нарушается, что приводит к неконтролируемому ангиогенезу с формированием аберрантных и нефункциональных сосудов. В норме в ответ на стимуляцию ангиогенеза факторами роста и ангиогенными хемокинами параллельно начинает продуцироваться определенный спектр антиангиогенных хемокинов [Balestrieri et al., 2008]. Среди них интерферон (IFN)-индуцируемые ELR-хемокины CXC подсемейства, которые являются важными ингибиторами ангиогенеза, включая CXCL4/фактор тромбоцитов 4 (PF4), CXCL10/интерферон индуцируемый белок 10 (IP-10), CXCL9/-индуцируемый монокин (MIG), CXCL11/интерферон индуцируемый -хемоаттрактант Т-клеток (ITAC), и CXCL4L1 аналог CXCL4 [Strieter et al., 1995; Maione et al., 1990; Angiolillo et al., 1995; Sato et al., 1990; Romagnani et al., 2001; Struyf et al., 2004]. В основном, все эти антиангиогенные хемокины связываются рецепторами с CXCR3a и CXCR3b на эндотелиальных клетках, которые представляют собой сплайсинг варианты одного и того же гена [Lasagni et al., 2003]. У мышей, у которых отсутствует CXCR3, отмечается избыточное формирование сосудов при заживлении ран, что указывает на важную функцию этих хемокинов в регуляции сосудистого гомеостаза. Такие хемокины как CXCL4, CXCL9, CXCL10 и CXCL11 напрямую ингибируют индуцированный факторами роста хемотаксис эндотелиальных клеток и формирование капилляро-подобных трубочек in vitro, а также ангиогенез на моделях хориоаллантоисной мембраны и бляшек Матригеля при подкожном введении мышам in vivo, что указывает на значительную автономность хемокиновой системы в регуляции процессов ангиогенеза [Strieter et al., 1995; Maione et al., 1990; Angiolillo et al., 1995; Sato et al., 1990; Romagnani et al., 2001; Struyf et al., 2004]. CXCL10 может вызывать регрессию уже сформированных сосудов при заживлении ран и апоптоз эндотелиальных клеток даже в присутствии факторов роста [Bodnar et al. 2009]. CXCL4 тормозит FGFиндуцированный ангиогенез за счет прямого формирования комплекса CXCL4-bFGF, 39 что физически препятствует связыванию FGF с рецептором, димеризации и активации FGFR2 [Perollet et al., 1998]. Аналогичный механизм реализуется при блокировании VEGF/VEGFR сигнального пути в присутствии CXCL4 [Gengrinovitch et al., 1995]. CXCL4 также может взаимодействовать с интегринами и блокировать интегринопосредованную адгезию эндотелиальных клеток, что приводит к подавлению ангиогенеза и апоптозу [Aidoudi et al., 2008]. По-видимому, хемокиновая система особенно важна для регуляции процессов индукции и ингибирования ангиогенеза, поскольку только активированные пролиферирующие эндотелиальные клетки реагируют на лиганды CXCR3 рецепторов, а сам CXCR3рецептор обнаруживается в эндотелиальных клетках только в S-фазе клеточного цикла [Romagnani et al., 2001]. Данные in vivo о специфическом связывании CXCL4 с рецепторами на эндотелиальных клетках только в зоне активного ангиогенеза свидетельствуют о том же [Hansell et al., 1995]. Известно, что хемокиновые рецепторы экспрессируются на нескольких типах иммунных клеток, которые мигрируют по градиенту лигандов-хемокинов, сопрягая процессы регуляции иммунного ответа и ангиогенза. Существует взаимная регуляция активности IFN-индуцируемых ELR-хемокинов и индукции иммунного ответа Th1 типа [Balestrieri et al., 2008]. Хемокины связываются с CXCR3 рецептором на поверхности Tклеток Th1 типа, натуральных (NK) киллеров и моноцитов, что стимулирует продукцию этими клетками IFN-. IFN-в свою очередь вызывает в клетках экспрессию CXCL9, CXCL10 и CXCL11 хемокинов, которые стимулируют еще большее рекрутирование CXCR3-экспрессирующих клеток и увеличивает их секреторную активность. Непосредственное взаимодействие между моноцитами и эндотелиальными клетками также повышает экспрессию CXCL10 в эндотелии [Kasama et al., 2002]. Это петля положительной обратной связи может приводить к феномену «иммуноангиостаза», при котором иммунный ответ по 1 типу и подавление ангиогенеза происходят одновременно. Аналогичным образом, интерлейкин 12 (IL-12), который является ключевым цитокином, активирующим Т-клетки, индуцирует секрецию ингибирующих ангиогенез хемокинов CXCL10 и CXCL9 лейкоцитами [Strasly et al., 2001]. Следует отметить, что IL-12 вызывает подавление FGF-индуцированного ангиогенеза, и это ингибирование непосредственно зависит от CXCL10 и не реализуется в его отсутствии [Sgadari et al., 1996]. Концепция хемокин-зависимого «иммуноангиостаза» предполагает, что IFNиндуцирует экспрессию ELR-хемокинов, что в сочетании с рекрутированием иммунных клеток Th1-типа может вызывать ингибирование роста опухоли. На 40 экспериментальных животных моделях у SCID мышей было показано, что подавление экспрессии CXCL10 или нейтрализация антителами увеличивает рост, метастазирование, и васкуляризацию немелкоклеточного рака легкого (NSCLC) [Arenberg et al. 1996]. Геннотерапевтические подходы с использованием вирусных конструкций для экспрессии CXCL4 продемонстрировали блокирование роста и васкуляризации глиом у мышей и увеличение выживаемости животных [Tanaka et al., 1997]. Аналогично, CXCL4L1, аналог CXCL4, оказался достаточно мощным ингибитором роста и метастазирования меланомы за счет подавления ее васкуляризации [Struyf et al., 2007]. В целом, эти данные позволяют рассматривать терапию с использование антиангиогенных хемокинов в качестве перспективного подхода к лечению онкологических заболеваний. Ангиогенные хемокины. В условиях in vivo хемокины подгруппы ELR+ СХС стимулируют ангиогенез за счет привлечения полиморфноядерных нейтрофилов в зону повреждения, а также за счет активации, пролиферации и миграции эндотелиальных клеток [Strieter et al., 1992; Koch et al., 1992]. Первым среди этих хемокинов был выявлен CXCL8/IL-8, затем были обнаружены CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, и CXCL7 [Strieter et al., 1995]. Общим рецептором для всех ELR+ хемокинов, опосредующим их ангиогенные эффекты, является CXCR2; исключение составляют CXCL8 и CXCL6, которые также связываются с CXCR1 [Keeley et al., 2008]. На экспериментальных моделях васкуляризации роговицы у крысы было показано, что блокирование CXCR2 с использованием антител подавляет ангиогенез [Addison et al., 2000]. У нокаутных по гену CXCR2 мышей наблюдается нарушение заживления ран за счет отсутствия необходимой васкуляризации, что обусловлено недостаточным рекрутированием нейтрофилов в зону повреждения и нарушением миграции эндотелиальных клеток [Devalaraja et al., 2000]. Стимуляция ангиогенеза с участием CXCR2 рецептора и его лигандов чаще всего происходит при активации транскрипционного фактора NF-B. NF-B является центральным медиатором многих сигнальных путей, которые запускаются при активации эндотелия или при воспалении с участием фактора некроза опухоли альфа (TNF-) и IL-1Нарушения в сигнализации именно NF-B сигнальных путей были обнаружены при разных онкологических заболеваниях. Так, было показано, что экспрессия CXCL2 увеличена в IL-1-экспрессирующих клетках карциномы легкого, и именно связывание CXCL2с рецептором CXCR2 стимулирует неоангиогенез и опухолевый рост [Saijo et al., 2002]. NF-B-зависимая сигнализация запускается также при активации HIF-1в условиях гипоксии. У трансгенных мышей конститутивная 41 экспрессия HIF-1 в кератиноцитах вызывает активацию NF-B и экспрессию генов ангиогенных хемокинов CXCL1, CXCL2 и CXCL3 [Scortegagna et al., 2008]. Глиобластома является одной из наиболее агрессивных опухолей мозга с летальностью 80% в первый год диагностики. Значительную роль в высокой туморогенности глиобластомы играет опухолевый неоангиогенез, обеспечивающий ее быстрый рост и метастазирование. Было обнаружено, что в глиобастомах снижена экспрессия белка ING4 - опухолевого супрессора, который связывает субъединицу NF-B и препятствует его транслокации в ядро и последующей активации генов. Высокую агрессивность и ангиогенную активность в глиобластомах связывают с низким уровнем ING4 и высоким уровнем CXCL8, подавление экспрессии CXCL8 значительно ингибирует васкуляризацию опухолей и снижает опухолевый рост in vivo [Garkavtsev et al., 2004]. Эти данные свидетельствует о том, что активация NF-B и последующая экспрессия ELR+ хемокинов играют важную роль в патогенезе различных опухолей, а ангиогенные хемокины являются возможной перспективной мишенью для противоопухолевой терапии. Исследования с использованием экспериментальных животных моделей роста меланомы in vivo свидетельствуют о том, что экспрессия хемокинов CXCL1, CXCL2 и CXCL3 стимулирует пролиферацию опухолевых клеток и опухолевую васкуляризацию [Strieter et al., 2005]. Экспрессия этих хемокинов также повышена в образцах меланомы человека. Более того, иммортализованные мышиные меланоциты после трансфекции генами CXCL1, CXCL2 и CXCL3 человека приобретали способность расти независимо от прикрепления к субстрату in vitro и формировать опухоли у иммунодефицитных мышей in vivo [Strieter et al., 2005]. В случае подавления экспрессии CXCL1, CXCL2 и CXCL3 в клетках мышиной меланомы В16, которая в стандартных условиях быстро растет и метастазирует в легкие, отмечалось существенное снижение степени васкуляризации и, как следствие, уменьшение размеров первичного узла и способности формировать метастазы [Strieter et al., 2005]. Эти данные позволяют предполагать, что ELR+хемокины стимулируют опухолевую прогрессию меланомы и как аутокринные стимуляторы пролиферации опухолевых клеток, и как паракринные медиаторы неоангиогенеза. Известно, что CXC хемокины играют важную роль и при других заболеваниях, связанных с избыточным ангиогенезом. По данным ряда авторов, основными ангиогенными факторами, присутствующими в синовиальной жидкости у больных ревматоидным артритом, являются CXCL8 и CXCL5 [Koch et al., 2001]. При псориазе, который является заболеванием кожи с избыточным ростом сосудов, кератиноциты 42 секретируют повышенное количество ангиогенных факторов, в том числе и CXCL8 [Nickoloff et al., 1994]. Для формирования атеросклеротических бляшек также характерен избыточный ангиогенез и пролиферация клеток, рекрутирование моноцитов и прогениторных клеток из периферической крови [Fowler et al., 1980; Joris et al., 1983; Munro et al., 1988; Koch et al., 2001; Folkman, 1995; Folkman, 2008; Clark, 1991]. В образцах коронарных артерий с атеросклеротическими повреждениями уровень CXCL8 был существенно повышен по сравнению с контрольными здоровыми сосудами [Simonini et al., 2000]. Эти данные позволяют предполагать, что хемокины вносят важный вклад в патогенез этих заболеваний. Проангиогеннные хемокины влияют на внутриклеточные сигнальные пути, которые активируются от рецепторов факторов роста. Часто это происходит по аутокринному механизму. Так, в некоторых опухолях мозга VEGF, секретируемый эндотелиальными клетками сосудов, прорастающих в опухоль, стимулирует секрецию CXCL8 [Charalambous et al., 2005]. CXCL8 через активацию NF-B стимулирует экспрессию VEGF в этих же клетках, что в свою очередь приводит к связыванию VEGF с VEGFR2 и активации внутриклеточной сигнализации [Martin et al., 2009]. Более того, было обнаружено прямое взаимодействие между CXCL8 и компонентами VEGFсигнального пути: во-первых, CXCL8 может стимулировать фосфорилирование и активацию сигнализации от VEGFR2 независимо от присутствия VEGF, во-вторых, добавление рекомбинантного CXCL8 может приводить к формированию комплекса между CXCR1/CXCR2 фосфорилирования по и VEGFR2, Src-зависимому трансактивации механизму. VEGFR2 за Фосфорилирование счет его VEGFR2 вызывает активацию RhoA, сборку стресс фибрилл актина и увеличение проницаемости эндотелия [Petreaca et al., 2007]. Кроме того, увеличение экспрессии CXCL8 приводит к повышению выживаемости и защите от апоптоза эндотелиальных клеток через регуляцию Bcl-2 и ингибирование Вах, предполагая существование петли обратной связи между экспрессией хемокинов и белков семейства Bcl-2, регулирующих апоптоз [Li et al., 2003]. Единственным проангиогенным хемокином, у которого отсутствует ELR мотив, является CXCL12/SDF-1 (stromal cell-derived factor-1). У мышей, нокаутных по CXCL12 или CXCR4/CXCR7, выявляются серьезные аномалии формирования сердечнососудистой системы в эмбриогенезе, что подтверждает важность этой лигандрецепторной системы для морфогенеза сердца и сосудов [Tachibana et al., 1998; Ara et al., 2005; Sierro et al., 2007]. Связывание SDF-1 с рецепторами CXCR4 и CXCR7 на «концевых» эндотелиальных клетках важно для их миграции при ангиогенезе, что было показано на клеточных культурах in vitro [Duda et al., 2011]. На эксплантной модели 43 колечка аорты крысы было продемонстрировано, что связывание CXCL12с CXCR4 на эндотелиальных клетках активирует их хемотаксис и формирование капилляроподобных структур ex vivo [Gupta et al., 1998; Salcedo et al., 1999; Deshane et al., 2007]. CXCL12/CXCR4 сигнализация важна для реализации ангиогенных программ, которые активируют факторы роста VEGF и FGF. А именно, блокирование CXCL12 или CXCR4 ингибирует VEGF и FGF-индуцированное формирование капилляро-подобных структур эндотелиальными клетками in vitro [Salvucci et al., 2002; Salcedo et al., 2001]. В условиях гипоксии in vivo создается градиент кислорода и его недостаток непосредственно индуцируют экспрессию CXCL12 через активацию HIF- 1транскрипционных факторов. В костном мозге давление кислорода ниже, чем в периферической крови, поэтому CXCL12 постоянно секретируется клетками стромы и эндотелиальными клетками, что обеспечивает хоуминг CXCR4-экспрессирующих гематопоэтических стволовых клеток [Petit et al., 2007; Mendez-Ferrer and Frenette, 2007; Burger and Burkle, 2007]. Помимо костного мозга CXCL12 экспрессируется во многих типах клеток, его экспрессия повышается при повреждении и ишемии, что приводит к мобилизацииCXCR4+ гематопоэтических и эндотелиальных прогениторных клеток из костного мозга и их рекрутирование в ишемизированные ткани [Schioppa et al., 2003; Ceradini et al., 2004]. CXCL12 играет также важную роль в удержании в зоне образования новых сосудов рекрутированных клеток, которые формируют ангиогенные ниши и обеспечивает продолжительную секрецию ангиогенных факторов, что способствуют васкуляризации ишемизированных тканей, в том числе и опухолей [Grunewald et al., 2006; Petit et al., 2007]. Считается, что in vivo ангиогенные эффекты CXCL12 в основном обусловлены именно рекрутированием стволовых и прогениторных клеток в зону ишемии или повреждения. Благодаря своим проангиогенным свойствам, CXCL12 известен как мощный стимулятор опухолевого роста и метастазирования [Guleng et al., 2005; Orimo et al., 2005; Singh et al., 2007; Yoon et al., 2007; Li and Ransohoff, 2009]. Известно, что фибробласты, ассоциированные с опухолью, секретируют значительные количества CXCL12, что способствует ее росту и васкуляризации. Ингибирование CXCL12/CXCR4 сигнализации приводит к подавлению опухолевого роста: у CXCR4-дефицитных мышей значительно снижен рост и метастазирование почечно-клеточной карциномы (RENCA), что связано с блокированием ангиогенеза, увеличением площади некроза и снижением метастатического потенциала [Mestas et al., 2005]. При раке предстательной железы CXCL12/CXCR4 сигнальный путь стимулирует ангиогенез за счет подавления активности фосфоглицераткиназы, что приводит к 44 увеличению секреции VEGF и CXCL8 и снижению экспрессии антиангиогенного ангиостатина [Wang et al., 2007]. CXCL8 в свою очередь является хемоаттрактантом моноцитов и нейтрофилов, которые обеспечивают продукцию ангиогенных факторов и матриксных металлопротеиназ. Введение клеток меланомы человека, экспрессирующих CXCL8, Nude мышам, дефицитным по CXCR2 рецептору, приводит к снижению инфильтрации нейтрофилами и, как следствие, к подавлению роста, васкуляризации и метастазированию опухоли [Singh et al., 2009]. Трансгенная экспрессия CXCR2 в эндотелиальных клетках у таких мышей стимулирует опухолевый неоангиогенез и рост меланомы, клетки которой экспрессируют функциональный гомолог человеческого CXCL8/CXCL1 [Horton et al., 2007], что еще раз напрямую подтверждает роль CXC хемокинов и их рецепторов в васкуляризации и опухолевой прогрессии. Тем не менее, в некоторых типах опухолей стимулирующий эффект CXCL12 не связан с его влиянием на ангиогенез. Так, на модели SCID (severe combined immunodeficiency) мышей при немелкоклеточном раке легкого (NSCLC) было показано, что нейтрализующие CXCL12 антитела блокируют рост NSCLC и метастазирование, но не влияют на неоангиогенез [Phillips et al., 2003]. Подсемейство CCL хемокинов является самой крупной подгруппой хемокинов, участвует в воспалительных реакциях при различных инфекциях и повреждениях органов и тканей, а также при инфильтрации и хоуминге лимфоцитов. На клеточных моделях было показано, что некоторые CCL хемокины, например, CCL1 и CCL2 стимулируют ангиогенез за счет активации миграции, инвазии эндотелиальных клеток и формирования ими капилляро-подобных трубочек [Weber et al., 1999; Salcedo et al., 2000]. На этих же моделях in vitro было показано, что CCL15, CCL16 и CCL23 хемокины стимулируют ангиогенез, а in vivo - васкуляризацию Матригелей при подкожном введении мышам и ангиогенез на модели хориоаллантоисной мембраны [Hwang et al., 2004; Hwang et al., 2005; Strasly et al., 2004]. Принято считать, что ангиогенез in vivo с участием CCL хемокинов, в основном, обусловлен рекрутированием лейкоцитов и секрецией ими различных факторов. CCL1 участвует в рекрутировании T-клеткок Th2 типа и моноцитов, и, как было показано на моделях ангиогенеза на роговице кролика и хориоаллантоисной мембраны, стимулирует ангиогенез in vivo [Bernardini et al., 2000]. CCL11/eotaxin является хемоаттрактантом эозинофилов, которые, приходя в зону формирования сосудов, секретируют, в том числе, и ангиогенные факторы; CCL11 также участвует в ангиогенезе за счет связывания с CCR3 на эндотелиальных клетках и индукции их хемотаксиса in vitro и in vivo [Salcedo et al., 2001]. При воспалении CCL3 стимулирует ангиогенез опосредованно, за счет 45 привлечения макрофагов и секреции ими проангиогенных факторов [Barcelos et al., 2009; Wu et al., 2008] Однако в зоне активного опухолевого ангиогенеза, как это было показано на модели гепатокарциномы у мышей, CCL3 может аутокринно стимулировать пролиферацию эндотелиальных клеток за счет связывания с CCR5 рецептором на поверхности эндотелия [Ryschich et al., 2006]. Продукция CCL хемокинов новообразованными опухолевыми сосудами также способствует рекрутированию циркулирующих CCR2+ и CCR5+ эндотелиальных прогениторных клеток из периферической крови и васкуляризации опухоли [Spring et al., 2005]. Для CCL хемокинов характерен «кросс-ток» (cross-talk) c другими ангиогенными сигнальными путями. Так, например, при связывании CCL16 c CCR1 рецептором, происходит активация экспрессии CXCL8 и CCL2 хемокинов, которые, в свою очередь, повышают чувствительность эндотелиальных клеток к VEGF и другим митогенным сигналам [Strasly et al., 2004]. CCL2 способствует также увеличению проницаемости эндотелильного монослоя в ответ на повышение концентрации VEGF, что важно для начальных этапов ангиогенеза [Yamada et al., 2003; Marumo et al., 1999]. Подсемейство CCL хемокинов регулирует не только ангиогенез, но и артериогенез. Так, CCL2/MCP-1 является хемоаттрактантом для моноцитов. В ответ на связывание CCL с CCR2 на поверхности моноцитов последние мигрируют в сосудистую стенку, где экспрессируют цитокины, факторы роста и мембранно-связанные матриксные металлопротеиназы (MTI-MMP - membrane type-1 matrix metalloproteinase), которые, в свою очередь, стимулируют пролиферацию и миграцию эндотелиальных клеток, ГМК и фибробластов [Galvez et al., 2005; Stamatovic et al., 2006; Niu et al., 2008]. Рекрутирование моноцитов оказывается особенно важно для формирования коллатеральных сосудов при окклюзии или ишемии тканей. В отсутствии CCL2 или CCR2 у мышей, дефицитных по этим генам, миграция моноцитов нарушается, что приводит к патологическим реакциям воспаления и недостатку функционально зрелых сосудов [Charo and Taubman, 2004]. Индукция экспрессии CCL хемокинов происходит с участием факторов роста и цитокинов. Так, TGF- напрямую стимулирует экспрессию CCL2 через SMAD белки и их сигнализацию, что приводит к активации миграции ГМК и стабилизации вновь формирующихся сосудов [Ma et al., 2007]. bFGF увеличивает секрецию CCL2 мезенхимальными клетками и тем самым способствует артериогенезу и восстановлению кровоснабжения в ишемизированных конечностях у экспериментальных мышей [Fuji et al., 2006]. CX3CL/Fractalkine является единственным известным членом подсемейства CX3C-хемокинов и экспрессируется как трансмембранный белок, в определенных 46 условиях он может расщепляться с образованием растворимой формы. CX3CL стимулирует хемотаксис и формирование капилляро-подобных структур эндотелиальными клетками на различных моделях in vivo и in vitro [Volin et al., 2001; Ryu et al., 2008; You et al., 2007]. Ангиогенные эффекты CX3CL обусловлены индукцией HIF транскрипционных факторов, экспрессия/стабилизация которых приводит к увеличению секреции VEGF; блокирование VEGFR2 подавляет ангиогенный эффект CX3CL [Ryu et al., 2008]. Уровень CXC3L повышен в синовиальной жидкости у больных ревматоидным артритом и сахарным диабетом, поэтому CX3CL рассматривают как потенциальную мишень для подавления избыточного ангиогенеза при лечении этих заболеваний [Volin et al., 2001; You et al., 2007]. Биологически активный липид сфингозин-1-фосфат (S1P), который связывается с семейством S1P рецепторов, сопряжѐнных с G-белками (S1PRs), также участвует в регуляции процессов ангиогенеза, барьерной функции эндотелия и стабильности сосудов. S1P cтимулирует процессы ангиогенеза за счет непосредственного взаимодействия с рецепторами PDGF и VEGF и их активации [Visentin et al., 2006]. В последнее время накапливаются данные о том, что агонисты некоторых рецепторов, сопряжѐнных с G-белками, включая гормоны, нейромедиаторы и другие вещества, способны активировать сигнализацию от рецепторов факторов роста (growth factor receptor tyrosine kinases - RTKs), таких как рецепторы Нейропилинов, PDGF и FGF, в отсутствии самих факторов роста. Этот феномен называется трансактивацией и представляет собой дополнительный механизм регуляции пролиферации, роста и миграции клеток с участием GPCR-лигандов. С другой стороны, рецепторы факторов роста, обладающие собственной тирозин-киназной активностью, могут использовать сигнальные молекулы, такие как G-белки и аррестины, которые обычно участвуют в GPCR сигнализации, для передачи сигнала, а сами факторы роста могут трансактивировать GPCRs рецепторы [Delcourt et al., 2007]. Взаимодействия между разными типами рецепторов создают дополнительные «кросс-токи» и уровни регуляции в рамках комплексной сети сигнализации от рецепторов факторов роста, рецепторов гормонов и хемокинов, а также других рецепторов, сопряжѐнных с G-белками. В настоящее время проходят клинические исследования эффективности и безопасности использования ингибиторов CXCL12, CXCR4 и S1P для лечения онкологических заболеваний [Duda et al., 2011]. 47 1.1.8 Роль интегринов в процессах ангиогенеза Внеклеточный матрикс является связующим звеном между сосудистыми клетками и окружающими их тканями. Эндотелиальные клетки взаимодействуют с компонентами внеклеточного матрикса и способны его моделировать в зависимости от сигналов микроокружения. Интегрины представляют собой гетеродимеры и функционируют как рецепторы, опосредующие адгезию клеток к внеклеточному матриксу и молекулам семейства иммуноглобулинов [Desgrosellier and Cheresh, 2010; Hodivala-Dilke, 2008]. Гетеродимерная организация интегринов позволяет им взаимодействовать с несколькими типами внеклеточного матрикса (фибронектином, коллагенами, витронектином, остеопонтином, тромбоспондином, фибриногеном и другими) [Weis and Cheresh, 2011]. Взаимодействие клеток с внеклеточным матриксом с участием интегринов приводит к активации внутриклеточного сигнального каскада, необходимого для выживания клеток. Среди наиболее распространенных интегринов, присутствующих в сосудах, следует назвать αvβ3, αvβ5, α1β1, α2β1, α4β1, α5β1, α6β1, α9β1 и α6β4 [Carmeliet and Jain, 2011]. Интегрины αvβ3 и αvβ5 характерны для ангиогенных эндотелиальных клеток (но не обнаруживаются в покоящихся эндотелиальных клетках), они участвуют в заживлении ран, ремоделировании тканей и в таких патологических процессах как ревматоидный артрит и опухолевая прогрессия [Seed and Walsh, 2008]. Помимо обеспечения клеточной адгезии на внеклеточном матриксе интегрины могут взаимодействовать с факторами роста и цитокинами (VEGF, bFGFs, Ang-1, TNF-, TGF- (трансформирующий ростовой фактор бета) или их рецепторами (VEGFR-2 и FGFRs) и регулировать внутриклеточную сигнализацию, которую те активируют. Так, для индукции ангиогенеза с участием bFGF или TNF- необходима функциональная активность αvβ3, в то время как для стимуляции ангиогенеза с участием VEGF или TGF необходима экспрессия αvβ5. Такое различие обусловлено разными сигнальными путями, которые активируют эти интегрины. Сигнализация от VEGF c участием αvβ5 активирует FAK (киназа фокальных адгезий), Src киназу, и приводит к фосфорилированию Raf-1 по серинам 338/339, что защищает эндотелиальные клетки от активации внутреннего пути апоптоза; в то время как αvβ3 активирует РАК (р21активируемая киназа), фосфорилирует Raf по серинам 340/341 и МЕК1, что обеспечивает защиту клеток от индукции внешнего пути апоптоза [Weis and Cheresh, 2011]. Некоторые интегрины способны напрямую связываться с факторами роста, например, α9β1 и αvβ3 связываются с изоформами VEGF, α5β1 может формировать комплекс с Tie-2, рецептором Ang, и усиливать ангиогенный сигнал этих факторов [Weis 48 and Cheresh, 2011]. Многие сигнальные пути нормальных клеток используются опухолевыми клетками для адгезии, миграции и инвазии. Хотя сами по себе интегрины не являются онкогенными, их экспрессия часто ассоциирована или оказывается необходима клеткам для опухолевого роста и метастазирования. Известно, что экспрессия таких интегринов, как αvβ3, αvβ5, αvβ6, α2β1, α5β1, α6β1 и α6β4, повышается при опухолевом росте [Weis and Cheresh, 2011]. Экспрессия αvβ3 в опухолевых клетках увеличивает их способность к трансэндотелиальной миграции и продукции металлопротеиназ, что повышает их метастатический потенциал. Интегрины важны для выживания, миграции и инвазии в опухоль эндотелиальных клеток, мигрирующих по градиенту хемоаттрактанта вдоль фибрилл внеклеточного матрикса. Увеличение экспрессии αvβ3 и αvβ5 на эндотелиальных клетках, мигрирующих в опухоль, позволяет им адгезировать на компонентах временного матрикса, который продуцируют опухолевые клетки, таких как витронектин, фибриноген и фибронектин внутри растущей опухоли и способствует их выживанию в условиях опухолевого микроокружения [Carmeliet and Jain, 2011]. Другие интегрины участвуют в адгезии эндотелиальных клеток предшественников, мигрирующих из периферической крови и костного мозга и адгезирующих на эндотелиальных клетках опухоли [Carmeliet and Jain, 2011]. Эффективность использования блокаторов интегринов для противоопухолевой терапии в настоящее время тестируется в клинических исследованиях [Weis and Cheresh, 2011]. 49 1.1.9 Роль протеаз в процессах ангиогенеза Важную роль в процессах ангиогенеза и ремоделирования сосудистой стенки играют матриксные металлопротеиназы (ММР). Покоящиеся эндотелиальные и муральные клетки имеют общую базальную мембрану, которая ограничивает их миграцию и пролиферацию. При индукции ангиогенеза ММРs облегчают миграцию эндотелиальных клеток и формирование капилляро-подобных трубочек либо за счет того, что они располагаются на лидирующем края путем направленного протеолиза ремоделируют базальную мембрану, как в случае мембранно-связанной ММР-1 (MT1MMP или ММР14), или за счет частичного протеолиза внеклеточного матрикса, приводящего к экспозиции определенных белковых сайтов. Кроме того, важную роль в ангиогенезе играют сериновые протеазы. Так, например, плазмин высвобождает VEGF и FGF, иммобилизованные во внеклеточном матриксе [Bergers et al., 2000]. Изоформы VEGF, высвобождаемые из внеклеточного матрикса (растворимая форма VEGF), важен для увеличения просвета вновь формирующихся сосудов, в то время как устойчивый к протеолизу и остающийся связанный с матриксом VEGF необходим для ветвления сосудов [Iruela-Arispe and Davis, 2009]. Макрофаги, нейтрофилы и тучные клетки индуцируют ангиогенез за счет ММР-9 опосредованной активации VEGF [De et al., 2008]. Ряд других протеаз, в том числе и ММР-9, участвуют в мобилизации клеток предшественников из костного мозга, миграция которых в опухоль создает преметастатическую нишу и способствует опухолевой прогрессии [Kaplan et al., 2005]. Однако нормальный ангиогенез невозможен без баланса между активностью протеаз и их ингибиторов. Так, потеря PAI-1 (ингибитора активатора плазминогена) блокирует ангиогенез, поскольку избыточная деградация внеклеточного матрикса приводит к нарушению адгезии и миграции клеток [Blasi and Carmeliet, 2002]. Поскольку активность матриксных металлопротеиназ приводит также к формированию антиангиогенных фрагментов, тумстатина и ангиостатина, важную роль при созревании сосуда и формировании базальной мембраны между эндотелиальными и муральными клетками играют ингибиторы протеаз TIMPs (тканевой ингибитор металлопротеиназ) [Folkman, 2007]. 1.1.10 Эндогенные ингибиторы ангиогенеза Для регуляции нормального физиологического ангиогенеза важен баланс проангиогенных и антингиогенных факторов, обычно этот баланс нарушается при патологическом ангиогенезе. Связывание интегринов с компонентами внеклеточного 50 матрикса приводит к адгезии и выживанию клеток на субстрате, связывание интегринов с растворимыми фрагментами белков внеклеточного матрикса блокирует адгезию, выживание и пролиферацию клеток. Так, например, Ангиостатин представляет собой фрагмент плазмина, связывающийся с αvβ3, Канстатин – фрагмент коллагена IV, связывающийся с αvβ5 и αvβ3, Эндостатин – фрагмент коллагена XVIII, связывающий αvβ5, αvβ3 и α5β1, Тумстатин – фрагмент коллагена IV, связывающий αvβ3 и α5β1 [Weis and Cheresh, 2011]. Протеолитическая деградация компонентов внеклеточного матрикса с образованием целого спектра белковых фрагментов представляет собой эндогенный механизм регуляции ангиогенеза за счет блокирования функции определенных интегринов. В последнее время также появились данные о том, что помимо растворимых белковых фрагментов активное участие в регуляции функциональной активности интегринов принимают микроРНК [Weis and Cheresh, 2011]. В настоящее время эта область активно развивается и формируются новые методы и подходы для управления процессами ангиогенеза с использованием микроРНК. 51 1.2 Навигационные рецепторы в нервной и сердечно-сосудистой системе Несмотря на имеющиеся функциональные различия, кровеносная и нервная системы имеют большое структурное сходство. Обе системы представляют собой упорядоченные разветвленные сети, пронизывающие весь организм. Кровеносные сосуды и нервы ко-локализуются во взрослом организме, а в эмбриогенезе растут параллельно друг другу. В литературе описана роль одних и тех же факторов роста в стимуляции роста как нервов, так и сосудов (VEGF, BDNF, bFGF, IGF-I и TGF-α). В отличие от нервной системы, механизмы, определяющие направление роста и морфогенез сосудов, изучены существенно меньше. Однако имеются данные том, что непосредственная анатомическая ко-локализация сосудов и нервов отражает взаимное влияние этих двух систем друг на друга и их согласованное функционирование. Так, например, активность клеток нервной системы регулирует кровоток в мозге, а уровень перфузии после инсульта коррелирует со степенью сохранности нервной ткани [Melani and Weinstein, 2010]. Важную роль при этом играют нейротрофины, которые регулируют выживание или гибель клеток нервной системы, дифференцировку, высвобождение нейротрансмиттеров, функционирование синапсов и др. Семейство нейротрофинов включает фактор роста нервов (NGF – nerve growth factor), нейроторфический фактор роста мозга (BDNF – brain-derived neurotrophic factor) и Нейропилины (NRP 3 и NRP 4). Нейротрофины связываются с Trk рецепторами (neurotrophin-selective tropomyosinreceptor-kinase receptors) совместно с p75NTR рецепторами (pan-neurotrophin-binding receptor) и вызывают активацию внутриклеточной сигнализации. В последнее время появились данные о том, что эти хорошо изученные нейротрофические факторы помимо собственно своего основного действия в нервной системе оказывают выраженные ангиогенные эффекты. Например, эндотелиальные клетки в культуре экспрессирют оба рецептора NGF, Trk и p75NTR, а добавление NGF в среду культивирования эндотелиальных клеток стимулирует их пролиферацию и миграцию. In vivo введение экзогенного NGF стимулирует ангиогенез, что было показано на моделях роста сосудов хориоаллантоисной мембраны эмбрионов куропатки, куриных эмбрионов, а также на экспериментальной модели васкуляризации роговицы крысы [Melani and Weinstein, 2010]. Не вполне ясно, являются ли ангиогенные эффекты нейротрофических факторов прямыми (прямое действие NGF на эндотелиальные клетки) или опосредованными через NGF-опосредованное рекрутирование проангиогенных клеток или NGF- 52 стимулированную индукцию секреции других ангиогенных факторов. Ангиогенная роль нейротрофина BDNF и его TrkВ рецептора также была продемонстрирована на моделях in vivo и in vitro. Эндотелиальные клетки артерий, скелетной мускулатуры и миокарда экспрессируют и BDNF, и TrkВ. У мышей нокаут по BDNF вызывает снижение выживаемости эндотелиальных клеток внутримиокардиальных артерий и капилляров, а у мышей, нокаутных по TrkВ, наблюдается уменьшение количества внутримиокардиальных сосудов по сравнению с контролем [Melani and Weinstein, 2010]. На сегодняшний день очевидно, что регуляция процессов ангиогенеза и направленного роста и ветвления нейритов осуществляется с использованием сходных молекулярных механизмов [Carmeliet, 2003]. «Концевые» эндотелиальные клетки растущего сосуда ведут себя подобно конусу роста аксона, тестируя микроокружение и выбирая направление миграции в зависимости от навигационных сигналов. Одни и те же молекулы, такие как VEGF, стимулируют выживание и рост как сосудов, так и нервов. «Концевые» клетки используют те же навигационные молекулы, что и аксоны, включая Эфрины (Ephrin) и их рецепторы (Eph), Нейропилины (NRPs) и Плексины (Plexin-D1), которые связывают Семафорины (Semaphorins), Robo 4, которые взаимодействуют со Слит белками (Slit), и UNC5B рецепторы, связывающие Нетрины (Netrins) (Рисунок 1.7). Для всех этих белков продемонстрировано участие в прорастании аксонов в развивающейся нервной системе позвоночных и беспозвоночных, а для некоторых показано роль в формировании и ремоделировании сосудистой системы. Кроме того, многие из этих белков экспрессируются и в других органах и тканях за пределами нервной системы и участвуют в развитии некоторых патологий, в частности в опухолевом росте и метастазировании [Carmeliet, 2003; Eichmann et al., 2005; Weinstein, 2005]. 53 Рисунок 1.7. Доменная организация навигационных рецепторов, участвующих в регуляции направленного роста аксонов и сосудов. Мембранно-связанные лигандыСемафорины и Эфрины В, растворимые лигандыЭфрины А, VEGF, Нетрины и Слит. Мембранно-связанные рецепторы Плексины, Нейропилины, рецептор VEGF (VEGFR2), рецепторы, UNC-5 (Uncoordinated-5), рецептор DCC (deletedincolorectalcancer) и рецепторы Roundabout (Robo1 и Robo 4). Консервативныедомены: CC, Robo-conserved domain; CUB, complement-binding domain; DB, DСС-binding domain; DD, death domain; Eph LBD, Ephrin ligand-binding domain; FN3, fibronectin type 3-like; Ig, immunoglobulin domain; IPT, Ig-like fold/plexin; Lam, laminin domain; LRR, leucine-rich repeat; MAM, meprin/A5 protein/phosphatase-μ-related; P1/2/3, DСС conserved domains; PSI, Plexin-Semaphorin-Integrin; SP1/2, serine/threonine protein kinase catalytic domain; TSP1, thrombospondin-1-like; TyK, tyrosine kinase; ZU5, zona occludens-1-like. (Адаптированоиз Melani and Weinstein, 2010). 54 1.2.1 Эфрины и их рецепторы Система Эфринов и Eph рецепторов играет важную роль в эмбриогенезе при гаструляции, формировании сомитов, морфогенезе скелетной, нервной и сердечнососудистой систем, при опухолевом росте, неоангиогенезе и метастазировании [Lackmann, 2010]. Наиболее хорошо роль Эфринов и их рецепторов изучена в эмбриогенезе, получены данные об их участии в регуляции траектории роста аксонов, формировании границ между органами и тканями, сегментации, топографическом картировании и формировании связей между нейронами в нервной системе [Poliakov et al., 2004]. Семейство Эфриновых рецепторов представляет собой рецепторные тирозин киназы, которые активируются за счет связывания с лигандами - Эфинами. Существует два подкласса Эфринов: Эфрины В (Эфрины В1-3) представляют собой трансмембранные белки, а Эфрины А (Эфрины А1-5) крепятся на цитоплазматической мембране при помощи гликозилфосфатидилинозитольного (GPI) якоря [Pasquale, 2008]. Рецепторы Эфринов также делятся на подклассы Eph А и Eph В типа. Эфрины разных классов достаточно широко взаимодействуют с рецепторами без соблюдения строгой специфичности между подклассами. Для Ephrin-Eph взаимодействия характерна активация внутриклеточной сигнализации как в клетке, экспрессирующей рецептор (forward signaling), так и в клетке экспрессирующий лиганд (reverse signaling) (Рисунок 1.8). При активации сигнализации в клетках, экспрессирующих Эфрины В (reverse signaling), происходит характерное фосфорилирование Эфринов по нескольким высоко консервативным тирозиновым остаткам, расположенным в цитоплазматическом домене, а наличие С-терминального PDZ домена в Эфринах (Postsynaptic density protein/Disclarge/Zona occludens) служит основой для сборки сигнальных белковых комплексов и их взаимодействия с другими PDZ- содержащими белками внутри клетки [Pitulescu and Adams, 2010; Lackmann, 2010]. Активация сигнализации от Эфринов В внутри клетки может также приводить к дефосфорилированию ряда белков, активации киназ семейства Src и Rho ГТФаз, а также влиять на сигнальные пути, идущие от GPCR белков [Poliakov et al., 2004; Pasquale, 2008; Lackmann, 2010]. Несмотря на отсутствие трансмембранной и цитоплазматической части у Эфринов А, они способны активировать внутриклеточную сигнализацию с участием белков семейства Src киназ и фосфатидилинозитол-3-киназы (PI3K), которые, в свою очередь, взаимодействуют с адапторными белками или ко-рецепторами. Такими ко-рецепторами являются, например, рецепторы нейротрофина Trk/B и p75, сигнализация которых усиливается при колокализации с Эфринами А [Pitulescu and Adams, 2010]. 55 Рисунок 1.8. Доменная организации Эфринов и их рецепторов. Лиганд связывающий и рецептор связывающий домены, Cis-домен (домен, богатый цистеинами),FNtypeIII (фибронектиновый домен IIIтипа), трансмембранный домен, домен, обладающий киназной активностью, SAMдомен (sterilemotif домен), СтериминальныйPDZ-связывающий домен, Y-сайты фосфорилирования по тирозину (АдаптированоизPitulescu and Adams., 2010). 56 Активация внутриклеточной сигнализации от рецепторов Эфринов (forward signaling) представляет собой еще более сложную цепь взаимодействий. Она приводит к фосфорилированию нескольких консервативных тирозиновых остатков в цитоплазматическом домене (Рисунок 1.8), что создает сайты для взаимодействия с различными SH2-домен (Src Homology 2), содержащими адапторными и scaffoldбелками, а также Abelson (Abl) киназой, белками семейства Src киназ, белкамирегуляторами активности Rho ГТФаз [Kuijper et al., 2007; Lackmann, 2010]. Основной мишенью внутриклеточной сигнализации, которая активируется в результате Ephrin-Eph взаимодействия (forward signaling), являются компоненты цитоскелета. Ключевыми при этом являются белки, регулирующие динамичное поведение актиновых филаментов и микротрубочек (paxillin), компоненты адгезивных контактов (FAK), белки, участвующие в клеточной подвижности (PI3K, Crk, Nck, Src, Fyn, Rho ГТФазы - Rho, Rac, Cdc42) и регулирующие их факторы, такие как RasGAP, Grb и др. [Lackmann, 2010]. Помимо того, что Эфрины влияют на морфологию и миграцию клеток (а именно, регулируют сигнальные пути, идущие от интегринов и FAK), Eph рецепторы участвуют в кросс-токе сигнальных путей, регулирующих выживаемость клеток, пролиферацию и дифференцировку, таких как Ras/Erk/MAPK и Wnt сигнальный каскад. В отличие от многих рецепторных тирозин киназ, которые активируют MAPK сигнальный каскад, активация Eph рецепторов при связывании с соответствующими Эфринами подавляет MAPK сигнализацию, что было продемонстрировано, например, для Эфрина А2 и ряда других лигандов. Еще одной характерной особенностью ЭФриновой системы является кросс-ток между Eph и Wnt сигналингом, который определяет положение и миграцию клеток в криптах кишечного эпителия во взрослом организме и важен при формировании кишечной трубки в эмбриогенезе [Lackmann, 2010]. Известно, что взаимодействие Эфринов с рецепторами может вызывать как адгезию (attraction), так и отталкивание (repulsion). Взаимодействие Эфринов с рецепторами приводит к отталкиванию клеток при высоком уровне экспрессии/кластеризации Eph/Ephrin комплексов или в случае протеолитического расщепления Эфринов с последующим эндоцитозом Eph/Ephrin комплекса внутрь клетки, экспрессирующей рецептор [Palmer and Klein, 2003; Poliakov et al., 2004]. Если процесс расщепления Eph/Ephrin комплекса или его последующего эндоцитоза в клетках оказывается заблокирован, результатом взаимодействия клеток будет адгезия. Так, существует сплайс-вариант Эфрина А1b, рецептор-связывающие характеристики которого отличаются от исходного варианта. Экспрессия Эфрина А1b в клетках 57 блокирует протеолитическое расщепление Эфрина А1а, что подавляет активацию внутриклеточной сигнализации от Эфринового рецептора и приводит к адгезии вместо отталкивания [Finne et al., 2004]. Адгезия клеток также происходит при низком уровне экспрессии лиганда и/или рецептора, или при низком уровне кластеризации лигандрецепторных комплексов. Как правило, взаимодействие Эфринов с Eph рецепторами происходит на поверхности клеток (in trans), однако, в ряде последних исследований было обнаружено, что латеральное взаимодействие Эфринов с рецепторами (in cis) также возможно. Так, Эфрин А5 может латерально взаимодействовать с фибронектиновым доменом III типа Эфринового рецептора Eph А3, что блокирует in trans взаимодействие Эфринов с рецепторами между соседними клетками, ингибирует активацию внутриклеточной сигнализации (forward signaling) и приводит к адгезии [Melani and Weinstein, 2010]. В последние годы стало ясно, что клетки могут переключаться от одного на другой тип ответа; клеточный ответ - адгезия или отталкивание - лежит в основе организации клеток в органы и формирования границ между тканями. Так, было показано, что именно такое переключение ответа важно для пространственно-временной регуляции миграции клеток нервного гребня из нервной трубки [Santiago and Erickson, 2002]. На ранней стадии развития куриных эмбрионов клетки нервного гребня экспрессируют нейральные маркеры и мигрируют вентрально через ростральную часть сомитов (Рисунок 1.11), в то время как на более поздней стадии клетки нервного гребня имеют фенотип меланобластов и мигрируют по дорзо-латеральному направлению. Миграцию в первом случае регулируют рецепторы Eph В2/Eph В3: клетки нервного гребня, экспрессирующие эти рецепторы, избегают каудальной части сомитов и мезенхимы в дорзальной части эмбриона, где клетки экспрессируют Эфрины В типа [Poliakov et al., 2004]. Однако на более поздней стадии эти же Эфрины В по-прежнему экспрессируются дорзо-латерально, но теперь служат аттрактивным сигналом для мигрирующих клеток нервного гребня [Santiago and Erickson, 2002]. Аналогичным образом активация Ephrin-Eph системы в эндотелиальных клетках может приводить как адгезии/инвазии, так и де-адгезии/отталкиванию. Например, активация Eph В4 (forward signaling) или Ephrin B2 (reverse signaling) в эндотелии может как стимулировать, так и подавлять адгезию и миграцию клеток in vitro [Poliakov et al., 2004]. В основе такого различного клеточного ответа лежат разные механизмы активации внутриклеточной сигнализации и последующий биохимический каскад реакций. Более того, для топографического картирования аксонов в головном мозге и установления соответствующих связей между нейронами оказывается важен градиент экспрессии 58 Эфринов и их рецепторов. Так, в ряде in vivo и in vitro исследований было показано, что мигрирующие аксоны с высоким уровнем экспрессии Eph более чувствительны к градиенту экспрессии Эфринов А, чем аксоны с низким уровнем экспрессии Eph. Для прорастающих аксонов сетчатки было обнаружено, что существует некоторая пороговая концентрация Эфринов А в окружающих тканях, при которой мигрирующие аксоны воспринимают этот сигнал как аттрактивный, при превышении пороговой концентрации Эфринов происходит отталкивание [Poliakov et al., 2004]. Эфриновая система играет важную роль в позиционировании клеток за счет регуляции клеточной адгезии и/или ограничения миграции и инвазии путем формирования границ между тканями [Palmer and Klein, 2003; Poliakov et al., 2004]. При формировании границ между тканями Эфриновая система может контролировать функцию кадгеринов. Так, у позвоночных при формировании границы между нотохордом и мезодермой, из которой в дальнейшем будут формироваться сомиты, формирование адгезивных кадгерин-содержащих комплексов блокируется в результате взаимодействия Эфринов и их рецепторов на двух соседних типах клеток. Сигнализация от Эфринов и Eph рецепторов вдоль формирующейся границы вызывает локальное увеличение активности актомиозинового комплекса и сокращение клеток, что препятствует формированию стабильных кадгериновых контактов и прочной межклеточной адгезии [Fagotto et al., 2013]. Кроме того, Эфриновая системы участвует в кросс-токе различных сигнальных путей, регулирующих пролиферацию и дифференцировку. Нарушение экспрессии или сигнализации, идущей от Эфринов или их рецепторов, определяющих клеточную дифференцировку также могут способствовать канцерогенезу. Так, в криптах кишечника в норме взаимодействие Эфрина В1 с Eph B3 вызывает отталкивание, что определяет позицию стволовых и дифференцированных клеток, ограничивает их избыточную миграцию и пролиферацию. При аденоме кишечника и на начальной стадии рака конститутивная активация Wnt/catenin/Tcf сигнального пути приводит к увеличению экспрессии Eph B рецепторов (Eph B2 и Eph B3), которые при этом играют роль опухолевых супрессоров и сдерживают опухолевый рост, опосредуя отталкивание. На более поздних стадиях рака клетки теряют экспрессию белков Ephrin-Eph системы, что способствует росту и инвазии клеток без ограничений, что было продемонстировано на экспериментальных моделях у мышей, у которых отсутствует экспрессия Eph B3 или Эфрина В1. По-видимому, в основе механизма, сдерживающего пролиферацию и миграцию Eph B позитивных клеток опухоли, лежит их взаимодействие с окружающими 59 эпителиальными клетками, которые экспрессируют Эфрины В и поддерживают прочные межклеточные Е-кадгерин содержащие контакты [Pasquale, 2008]. Еще одним примером кросс-тока между Эфриновыми рецепторами и другими сигнальными путями является опухолевая прогрессия при раке молочной железы. Гиперэкспрессия Eph A2 в клетках нормального эпителия молочной железы приводит к их опухолевой трансформации и потери чувствительности к эстрогенной регуляции, что, по всей видимости, свидетельствует о кросс-токе между Eph рецепторами и рецепторами гормонов. В то же время, подавление экспрессии Eph A2 и Eph B4 с помощью siRNA или антисенс олигонуклеотидов в клетках опухоли молочной железы человека ингибирует миграцию клеток, их инвазию и рост первичной опухоли на ксенографтной модели у мыши. На этой же модели было показано, что определяющую роль в том, будет ли сигнал от Eph рецепторов проонкогенным (низкий уровень) или антионкогенным (высокий уровень) играет уровень экспрессии Eph рецепторов, степень их кластеризации и уровень фосфорилирования [Pasquale, 2008]. Возможно, что для определения того или иного типа клеточного ответа также имеет значение происхождение опухолевых клеток и стадия заболевания. Помимо экспрессии непосредственно в опухолевых клетках Эфрины и Eph рецепторы экспрессируюся в клетках кровеносных сосудов, прорастающих в опухоль, что важно для ее питания и диссеминации. Основную роль в опухолевом неоангиогенезе приписывают Eph А2 (forward signaling) и Эфрину В2 (reverse signaling). Следует отметить, что Eph А2, в отличие от Эфрина В2, появляется только в опухолевых сосудах, он не экспрессирутся ни в формирующейся кровеносной системе в эмбриогенезе, ни в нормальных стабильных сосудах во взрослом организме [Palmer and Klein, 2003; Pasquale, 2008]. Использование антител, блокирующих Eph А2-Эфрин А или Eph ВЭфрин В2 взаимодействия, подавляет опухолевый рост за счет блокирования опухолевого неоангиогенеза, что было показано с использованием нескольких опухолевых моделей на мышах [Pasquale, 2008]. В тоже время в некоторых опухолях уровень Эфрина А1 повышается в ответ на использование блокаторов VEGF в качестве противоопухолевой терапии, что позволяет предполагать, что Эфрина А1 вносит определенный вклад в развитие данного типа резистентности [Carmeliet and Jain, 2011]. Некоторые препараты на основе антител, блокирующих Eph-Эфрин взаимодействия, в настоящее время исследований, проходят однако, этот доклинические подход вряд и ли начальные является этапы клинических перспективным для противоопухолевой терапии, что следует из приведенных выше данных, поскольку блокирование Eph-Эфрин взаимодействий влияет также на сами опухолевые клетки, 60 способность которых к миграции и инвазии по данным литературы увеличивается при исчезновении ограничивающего фактора. 1.2.2 Эфрины и их рецепторы в сосудистой системе Первые данные о роли Эфринов и Eph рецепторов в организации сосудистой системы были получены при изучении артерио-венозной дифференцировки в эмбриогенезе. Именно Эфриновая система определяет эту дифференцировку, и эта специализация является генетически детерминированной. В то же время последние исследования свидетельствуют о том, что эндотелиальные клетки обладают определенной пластичностью, хотя и ограниченной, и могут быть репрограммированы в зависимости от микроокружения или гемодинамических условий. Об определенной пластичности свидетельствуют экспериментальные работы на животных по пересадке небольших участков артерий или вен от куропаток цыплятам на разных этапах развития с последующим анализом состава и экспрессии маркеров на эндотелиальных клетках формирующихся вен и артерий [Melani and Weinstein, 2010]. Наличие пластичности эндотелиальных клеток важно для современной сосудистой хирургии, например, при проведении операций шунтирования успешно используются участки вен для замены участков артерий, пораженных атеросклерозом. Сегодня ясно, что эндотелиальные клетки вен и артерий различаются как функционально, так и по экспрессии молекулярных маркеров. Эфрин В2 специфически экспрессируется в артериях, но не встречается в венах, а Eph В4 экспрессирован только в венах. Существует несколько механизмов, за счет которых Эфрин В2 и его рецептор Eph В4 участвуют в морфогенезе кровеносной системы. Во-первых, в эмбриогенезе в процессе образования первичного сосудистого сплетения путем васкулогенеза происходит зонирование на территории, где клетки экспрессируют Эфрин В2 и в дальнейшем будут дифференцироваться в клетки артерий, и клетки с венозной судьбой, экспрессирующие Eph В4. Взаимодействие Эфрина В2 с рецептором Eph В4 приводит к отталкиванию и сегрегации эндотелиальных клеток венозной и артериальной системы, в результате чего эти две популяции клеток оказываются функционально и морфогенетически разделены и не перемешиваются [Carmeliet and Jain, 2011]. Вовторых, Эфрин В2 играет важную роль при формировании филоподий в ―tip‖ клетках в процессе ангиогенеза: сигнализация от Эфрин В2 (reverse signalling) необходима для интернализации лиганд-рецепторного комплекса VEGF с VEGFR-2, и последующей активации внутриклеточной сигнализации от VEGFR-2. В-третьих, Эфрин В2 участвует в процессах рекрутирования муральных клеток и циркулирующих в крови 61 эндотелиальных прогениторных клеток костномозгового происхождения, которые участвуют в росте сосудов за счет васкулогенеза [Carmeliet and Jain, 2011]. Экспрессия генов Эфрина В2 и Eph В4 в эндотелиальных клетках начинается в эмбриогенезе у мыши задолго до начала собственно циркуляции крови; на дифференциальную экспрессию этих генов в эндотелиальных клетках влияют гемодинамические параметры и микроокружение. Таргетная инактивация генов, кодирующих Эфрин В2 и Eph В4 у мышей, приводит к патологическому развитию сосудов и свидетельствует о важной роли этих молекул в формировании нормальной артерио-венозной сети [Adams and Eichmann, 2010]. У нокаутных мышей, гомозиготных по гену Eph В4, наблюдаются дефекты артерио-венозной сети в головном мозге и желточном мешке, а также патологии формирования трабекул сердца [Melani and Weinstein, 2010]. У мышей, нокаутных по Эфрину В2, обнаруживается сходный фенотип, что позволяет предполагать, что эти белки функционируют как лиганд-рецепторная пара [Gerety et al., 1999]. Эфрин В2 и и Eph В4 также важны для формирования лимфатической системы в эмбриогенезе [Adams and Eichmann, 2010]. Данные литературы о том, какая сигнализация является определяющей для правильного формирования сосудов в эмбриогенезе, в клетках экспрессирующих лиганд (reverse signaling), или в клетках, экспрессирующих рецептор (forward signaling), достаточно противоречивы. У личинки шпорцевой лягушки (Xenopus laevis larva) гиперэкспрессия полноразмерной или мутантной формы Эфрина В2, лишенной цитоплазматического домена, как и экспрессия доминантно-негативной формы Eph В4 приводят к сходных дефектам формирования межсомитных сосудов, что по мнению авторов свидетельствует о важной роли forward сигнализации [Helbling et al., 2000]. В другом исследовании мыши, у которых Эфрин В2 лишен способности активировать внутриклеточную сигнализацию в силу делеции цитоплазматического домена, имеют сходные дефекты формирования сосудистой системы, что и мыши, у которых полностью отсутствует Эфрин В2 или Eph В4, что свидетельствует о важности reverse signalling [Adams et al., 2001]. Данные in vitro также не позволяют сделать однозначного вывода. В эндотелиальных клетках активация сигнализации через Eph В рецепторы может стимулировать пролиферацию и миграцию через сигнализацию с участием PI3-киназы и серин/треонин специфической киназы Akt/PKB [Maekawa et al., 2003]. Однако другими авторами было обнаружено, что активация сигнализации через Eph В4, наоборот, подавляет адгезию, миграцию и пролиферацию эндотелиальных клеток in vitro за счет ингибирования малой Rho ГТФазы Ras и МАР (mitogen-activated protein) киназы [Fuller et al., 2003; Hamada et al., 2003]. Возможно, что разница между полученными данными in 62 vitro обусловлена условиями культивирования и проведения эксперимента, поскольку известно, что Эфрин-Eph сигнализация может также влиять на адгезию клеток за счет модулирования функции интегринов [Adams and Eichmann, 2010], и, таким образом, результаты при культивировании клеток на различных типах матриксов могут различаться вплоть до противоположных. Эфрин В2 помимо эндотелиальных клеток экспрессируется перицитами и ГМК сосудов, его экспрессия важна для взаимодействия эндотелиальных клеток с муральными и стабилизации вновь формирующихся сосудов. Мутации, специфически инактивирующие экспрессию Эфрина В2 в муральных клетках, летальны и характеризуются дефектами формирования сосудов в коже, легких, желудочнокишечном тракте, почках и дефектами лимфатических сосудов [Melani and Weinstein, 2010]. В культуре ГМК клетки, лишенные Эфрина В2, не способны нормально распластываться и формировать фокальные контакты, для них характерно ускоренное хаотичное движение без определенного направления [Adams and Eichmann, 2010]. Помимо артерио-венозной дифференцировки эндотелиальных клеток эфриновая система определяет направление роста и навигацию кровеносных сосудов, подобно навигации аксонов нейронов, прорастающих к своим мишеням в эмбриогенезе. Так, в эмбриогенезе личинок мезенхимальными шпорцевой клетками сомитов, лягушки Эфрины расположенными по В экспрессируются направлению роста межсомитных сосудов. Инъекции РНК, кодирующей доминантно-негативные формы Eph В4 или формы Эфринов В, блокирующие сигнализацию от Эфриновых рецепторов, вызывают потерю правильного направления роста сосудов и различные дефекты их формирования в развивающихся эмбрионах. Авторы предположили, что Eph В4 и Эфринов В система вызывает негативное регулирование миграции эндотелиальных клеток и обеспечивает навигационные сигналы для правильного формировании сосудов в эмбриогенезе. Схожие результаты о роли Эфриновой системы в регуляции направленного роста сосудов и формирования сомитов были получены в работах на рыбках Данио (Zebrafish) [Melani and Weinstein, 2010]. Специфическая для артерий экспрессия Эфрина В2 сохраняется и во взрослом организме и обнаруживается даже в самых мелких капиллярах между терминальными артериолами и посткапиллярными венулами, что позволяет сделать вывод об их артериальном происхождении [Melani and Weinstein, 2010]. Во взрослом организме высокая экспрессия Эфрина В2 наблюдается в зонах ангиогенеза при заживлении ран, созревании фолликулов и формировании желтого тела в яичнике, а также при опухолевом ангиогенезе. В целом, в эмбриогенезе уровень экспрессии Эфринов и их 63 рецепторов существенно выше, чем во взрослом организме, однако, при различных патологиях, в том числе, при онкологических заболеваниях, таких как рак молочной железы, рак легкого, почечная карцинома, меланома, саркома, нейробластома, рак яичников и простаты человека их экспрессия может значительно увеличиваться [Pasquale, 2008; Lackmann, 2010]. Так, гиперэкспрессия Эфрина А1 и его рецепторов Eph А2 и/или Eph A3 наблюдается в клетках меланомы, при раке молочной железы, раке легкого и раке простаты [Kuijper et al., 2007]. В образцах меланомы человека опухолевый рост и метастазирование коррелирует с увеличением экспрессии EphA2 и Eph A3, увеличение эксрессии Eph А2 коррелирует со способностью клеток меланомы к «васкулогенной мимикрии», что, в целом, свидетельствует об опухолевой прогрессии и негативном прогнозе у пациентов. Связывание Эфрина А1 с Eph А2 на клетках меланомы человека вызывает увеличение их пролиферативной активности, кроме того, Eph А2 на клетках меланомы может латерально взаимодействовать с VE-кадгерином, что стимулирует опухолевую неоваскуляризацию [Lackmann, 2010]. In vitro одновременная экспрессия Eph В4 и Эфрина В2 в клетках агрессивной мышиной меланомы SW1 активирует RhoA сигнализацию и способствует миграции клеток, в то же время in vivo активация Eph В4 в клетках меланомы человека рекомбинантным химерным Эфрином В2/Fc на ксенографтной модели у мышей блокирует опухолевый рост [Pasquale, 2008]. Чем обусловлены столь разные эффекты на разных моделях роста меланомы до конца не ясно. 1.2.3 Семафорины и их рецепторы Семафорины (Sema) представляют собой большое семейство трансмембранных и секретируемых молекул, которые обладают одним общим свойством – в своей структуре они имеют Sema домен (Рисунок 1.7). Мембранно-связанные Семафорины связываются с Плексинами (Plexin), в то время как секретируемые Семафорины III типа (Sema 3A-Sema 3G) связываются с Нейропилинами (NRP), которые не сигнализируют сами по себе, а функционирует как ко-рецепторы Плексинов в нервной системе или как ко-рецепторы VEGFR в сосудах [Larrivée et al., 2009]. Исключение составляет лишь Sema 3Е, который напрямую связывается с Плексином D1, одним из девяти Плексинов, экспрессированных в эндотелиальных клетках сосудов млекопитающих в эмбриогенезе. Роль Семафоринов при формировании нервной системы в эмбриогенезе как навигационных молекул, негативных регуляторов роста аксонов, достаточно хорошо изучена [Melani and Weinstein, 2010]. Первый Семафорин, трансмембранный Sema 1А, был обнаружен и охарактеризован в нервной системе кузнечиков [Kolodkin et al., 1992]. 64 Следующим из эмбрионального мозга цыпленка был выделен Sema 3А и назван Коллапсином в связи с обнаруженной функцией подавления роста аксонов [Luo et al., 1993]. В основном, связывание Семафоринов с рецепторами вызывает отталкивание, однако, в ряде случаев было обнаружено, что Семафорины могут функционировать как аттрактивные молекулы. Связывание трансмембранных Семафоринов вызывает активацию внутриклеточной сигнализации в клетках, экспрессирующих рецептор, однако, подобно трансмембранным Эфринам (reverse связывание signaling), Семафоринов может также активировать сигнализацию в клетках экспрессирующих лиганд [Melani and Weinstein, 2010]. В нервной системе в эмбриогенезе, когда рост аксонов идет по направлению к своим мишеням, новые аксоны часто растут вдоль уже имеющихся аксонов, с которыми они объединяются в пучки (fasciculation). Nrp рецепторы важны для правильной организации аксонов в пучки и навигации, например, черепных нервов: у мутантных Nrp1-/- мышей наблюдаются дефекты формирования VII, IX и X черепных нервов, которые не обнаруживаются у Nrp 2-/- мышей. А у Nrp 2-/- мышей наоборот, II и IV черепные нервы, которые у Nrp 1-/- мышей развиваются нормально, формируют проекции с нарушениями. Аналогичным образом аксоны нейронов симпатических ганглиев у Nrp 2-/- мышей не отвечают на навигационный стимул Sema 3F, который является основным лигандом Nrp 2 при формировании целого ряда проекций, включая черепные нервы, лимбические структуры, аксоны спинальных мотонейронов и обонятельных нейронов, что проявляется в дефектах развития этих структур. Направление роста аксонов чувствительных нейронов спинальных ганглиев регулируется с участием Sema 3A. Аксоны чувствительных нейронов, выделенные из спинальных ганглиев Nrp1-/- мышей, не чувствительны к Sema 3A и не способны отвечать изменением направления на добавление Sema 3A в среду культивирования in vitro. Кроме того, дефекты развития черепных нервов, которые наблюдаются у Nrp1-/мышей полностью совпадают с дефектами Sema3а мутантных мышей, что свидетельствует о том, что Sema 3A является лигандом, необходимым для отталкивания и правильной навигации растущих аксонов черепных нервов, экспрессирующих Nrp. В некоторых работах было показано участие Nrp-Sema системы в регуляции направленной миграции клеток нервного гребня, что свидетельствует о том, что Нейропилины и их лиганды играют сложную и многоплановую роль в развитии периферической нервной системы в целом [Larrivée et al., 2009]. 65 1.2.4 Роль Семафоринов и их рецепторы в сосудистой системе Помимо нервной системы, Семафорины играют навигационную роль при формировании других органов и тканей. В эмбриогенезе цыпленка растущие кровеносные сосуды избегают сомитов, клетки которых экспрессируют Sema 3, что указывает на навигационную функцию Sema 3 для мигрирующих эндотелиальных клеток [Adams and Eichmann, 2010]. Эти данные были подтверждены in vitro, Sema 3 подавляет выживание и миграцию NRP 1-экспрессирующих эндотелиальных клеток в культуре и формирование капилляро-подобных трубочек на модели сосудистого колечка ex vivo. В то же время, было обнаружено, что у мышей, нокаутных по Sema 3A или экспрессирующих мутантный вариант NRP 1, который неспособен связывать Sema 3A, формируется нормальная кровеносная система. Авторы высказали предположение о том, что Семафорины не являются необходимыми для нормального формирования сосудов в эмбриогенезе у мыши, где основной контроль осуществляет VEGF164, или, что функция Семафоринов дублируется другими белками [Larrivée et al., 2009]. В отличие от сосудистой системы, в нервной системе именно Sema 3A, а не VEGF164, необходим для выбора правильной траектории аксонов мотонейронов, растущих к своим мишеням в конечностях у мыши [Larrivée et al., 2009]. Помимо NRP Sema 3 способны связываться с Плексином D1 на поверхности эндотелиальных клеток, что вызывает активацию внутриклеточной сигнализации, результатом которой является отталкивание. Мигрирующие эндотелиальные клетки используют навигационный Sema 3 сигнал микроокружения для выбора направления роста сосуда [Adams and Eichmann, 2010; Carmeliet and Jain, 2011]. Так, в эмбриогенезе у мыши связывание Sema 3Е с Плексином D1 определяет выбор правильной траектории роста межсомитных сосудов. У рыбок Данио (Zebrafish) мутации в гене Plxnd1, вызывающие потерю функции этого белка, при нормальной пролиферации эндотелиальных клеток и артерио-венозной дифференцировке, приводят к избыточному ангиогенезу и формированию аберрантных межсомитных сосудов [Adams and Eichmann, 2010]. Мутации по Плексину D1 у мышей летальны в силу дефектов формирования сердечно-сосудистой системы и скелета, мыши погибают сразу после рождения. В эмбрионах у мыши при кондиционной инактивации гена Plxnd1 в эндотелиальных клетках наблюдается тот же фенотип, что и у полных нокаутов по гену Plxnd1, что свидетельствует о важной роли Плексина D1 в формировании сосудистой системы [Adams and Eichmann, 2010]. У нокаутных мышей по гену Sema3e выявляется похожий на Plxnd1 дефектный паттерн формирования сосудов туловища, однако, у Plxnd1-/мышей наблюдается более тяжелый фенотип по сравнению с Sema3е-/- мышами, что 66 может свидетельствовать о том, что помимо Sema 3Е Плексин D1 взаимодействует с другими лигандами или формирует более сложный комплекс, включающий NRP 1 и NRP 2 рецепторы и несколько Семафоринов (Sema 3А, Sema 3С, Sema 4А) [Adams and Eichmann, 2010]. Однако у мышей, мутантных по сайту связывания NRP1 с Sema 3 и при одновременном нокауте по гену NRP 2, формируется нормальные сосуды в туловище, что позволяет предполагать, что Plxnd1 и Sema3e фенотип формируется независимо от Нейропилинов. Нейропилины (NRP 1 и NRP 2) представляют собой трансмембранные белки с тремя внеклеточными доменами (два CUB домена, ответственных за связывание с Семафоринами, и домены, ответственные за димеризацию и взаимодействие с корецепторами) и цитоплазматическим доменом, имеющий сайт связывания с PDZ белками [Adams and Eichmann, 2010; Ellis, 2006]. Основной функцией NRP является регуляция миграции клеток как в нервной, так и в сосудистой системе. Свою навигационную функцию NRP реализуют при связывании с Семафоринами и VEGF. В нервной системе это взаимодействие приводит в основном к отталкиванию и опосредует коллапс конуса роста аксонов с изменением направление роста, в то время как в сосудах взаимодействие NRP с различными представителями семейства VEGF вызывает аттракцию, миграцию «концевых» клеток и рост сосудов по градиенту VEGF. В эндотелиальных клетках NRP формируют комплекс с VEGF рецепторами, причем NRP 1 является ко-рецептором VEGFR-2, а NRP 2 ко-иммунопреципитируется с VEGFR-2 и VEGFR-3 [Larrivée et al., 2009]. NRP важен для правильного морфогенеза сосудистой сети в эмбриогенезе, поскольку полный нокаут по NRP 1 приводит к дефектам артериальной дифференцировки; в отличие от NRP 1, у NRP 2 мутантов артериовенозная дифференцировка происходит нормально, однако, у этих животных наблюдается дефекты ветвления лимфатических сосудов [Larrivée et al., 2009]. NRP взаимодействуют со структурно не похожими друг на друга Семафоринами III типа и белками семейства VEGF [Bagri et al., 2009; Ellis, 2006]. В самых первых исследованиях было сделано предположение о том, что Семафорины и VEGF конкурируют за связывание с NRP. Однако рентгено-структурный анализ фрагментов внеклеточных доменов NRP 1 и NRP 2 показал, что Семафорины и VEGF не являются прямыми конкурентами за связывание с NRP [Appleton et al., 2007], что объясняет отсутствие выраженного фенотипа у нокаутных по Sema 3 мышей, а также другие противоречивые данные, имеющиеся в литературе [Adams and Eichmann, 2010; Melani and Weinstein, 2010]. Более того, Семафорины и VEGF вызывают эндоцитоз рецепторов NRP и последующую сигнализацию внутри клетки по-разному: в случае связывания 67 VEGF с NRP эндоцитоз идет по клатриновому пути, а в случае Sema 3C с NRP - по кавеолиновому пути с участием липидных плотов [Larrivée et al., 2009]. В этой связи и связывание NRP с разными лигандами обеспечивает избирательность активации эффекторных белков в клетке и специфичность клеточного ответа в зависимости от конкретного лиганда. Недавно были созданы антитела, специфически блокирующие связывание либо Семафоринов с NRP, либо белков семейства VEGF с NRP. Анти-NRP 1А антитела блокируют связывание Sema3A, а анти-NRP 1В антитела ориентированы на сайт связывания NRP 1 с VEGF, хотя оба антитела подавляют формирование комплекса NRP 1 с VEGFR-2. Анти-NRP 2В антитела ингибируют VEGF165 и VEGF-C связывание с NRP 2 и подавляют формирование комплекса NRP 2 с VEGFR-2 и VEGFR-3. In vitro добавление всех антител в среду культивирования подавляет VEGF-индуцированный ангиогенез, причем анти-NRP 1 антитела специфичны для эндотелиальных клеток из пупочной вены (HUVEC), а анти-NRP 1В антитела – для эндотелиальных клеток лимфатических сосудов, предварительно стимулированных VEGF-C. Использование этих антител эффективно подавляет опухолевый неоангиогенез (NRP 1) и лимфангиогенез (NRP 2) на животных моделях in vivo, что позволяет рассматривать стратегию использования антител к NRP перспективным подходом для лечения онкологических заболеваний в клинике [Larrivée et al., 2009]. Данные биохимических экспериментов показали, что добавление анти-NRP 1В антител или анти-NRP 2В антител в среду культивирования эндотелиальных клеток, независимо от присутствия в ней VEGF, не оказывает прямого влияния на уровень фосфорилирования VEGFR или известных нисходящих сигнальных белков-посредников VEGF рецепторов, таких как ERK-1/2 (extracellular signal-regulated kinase) или AKT киназу [Larrivée et al., 2009]. Возможно, что NRP активирует какие-то дополнительные сигнальные пути или рекрутирует неизвестные сигнальные молекулы в VEGFR комплекс. Данные о том, что NRP могут активировать внутриклеточную сигнализацию независимо от рецепторов VEGF, были получены с использованием «фьюжн белка» (fusion protein), содержащего внеклеточный домен рецептора эпидермального фактора роста EGF (epidermal growth factor receptor) и внутриклеточный и трансмембранный домен NRP. Такой химерный белок стимулировал клеточную миграцию в ответ на добавление EGF, что означает, что внутриклеточный домен NRP может самостоятельно активировать внутриклеточную сигнализацию и миграцию без ко-рецепторов [Larrivée et al., 2009]. Единственный известный на сегодняшний день белок, способный связываться с внутриклеточным доменом NRP 1, - это Синектин (Synectin). Synectin является PDZ 68 адапторным белком, который связывает неокаймленные везикулы (эндоцитических пузырьки) с моторным белком Миозином VI, а последний необходим для транспорта и рециркулирования (recycling) эндоцитированных мембранных рецепторов. У мышей, нокаутных по Синектину, и у рыбок Данио (Zebrafish) при подавлении экспрессии Синектина с использованием олигонуклеотидов (нокдаун гена) наблюдаются дефекты формирования и ветвления артерий и подавление NRP 1-зависимой миграции эндотелиальных клеток. В эндотелиальных клетках, у которых отсутствует экспрессия Синектина, формирование NRP 1/VEGFR-2 комплекса подавлено, что позволяет предположить, что Синектин играет роль мостика, связывающего NRP 1 и VEGFR-2 в общий сигнальный рецепторный комплекс [Prahst et al., 2008]. В ряде работ было показано, что экспрессия NRP 1 может усиливать ангиогенный сигнал, возникающий при связывании VEGF рецепторов с изоформами VEGF А (VEGF164/165), способными также связывать NRP 1 [Parker et al., 2012]. В этих работах было обнаружено, что эксрессия NRP 1 необходима для VEGF-индуцированной активации p38 МАРК (mitogen-activated protein kinase), а также что этот сигнальный путь важен для взаимодействия эндотелиальных клеток с перицитами, необходимого для стабилизации сосудов. В ряде работ было обнаружено, что одни и те же эндотелиальные клетки могут одновременно экспрессировать NRP, рецепторы VEGF и Плексин D1. Эти данные позволяют предполагать, что эндотелиальные клетки могут по-разному отвечать на связывание с лигандами (Семафоринами и/или VEGF), в зависимости от того, с какими рецепторами они взаимодействуют (NRP и VEGFR) и какие сигнальные пути активируются. Более того, недавно было обнаружено, что в эндотелиальных клетках NRP 1 и NRP 2 могут функционировать как ко-рецепторы других рецепторов и стимулировать ангиогенез. Так, NRP могут быть ко-рецепторами c-met (рецептор фактора роста гепатоцитов, HGF), рецептора фактора роста фибробластов (FGF) и рецептора плацентарного фактора роста PlGF (VEGFR1) [Larrivée et al., 2009; Melani and Weinstein, 2010]. Нейропилины экспрессируются в большом количестве на опухолевых клетках различного происхождения и в сосудах, прорастающих в опухоль. У человека высокий уровень экспрессии NRP 1 и NRP 2 коррелирует с опухолевым ростом и метастазированием рака простаты, толстого кишечника, рака легкого и астроцитомы [Ellis, 2006]. В экспериментах in vitro и in vivo было обнаружено, что связывание VEGF с Нейропилинами защищает опухолевые клетки от апоптоза. Эти данные были получены на культуре клеток MDA-MB-231 рака молочной железы, раке простаты in vitro и экспериментальных моделях in vivo. Добавление Sema 3 в среду культивирования 69 блокирует этот эффект VEGF, подавляет миграцию и пролиферацию опухолевых клеток. Проведенный с помощью ДНК-микрочип технологии анализ экспрессии генов при раке поджелудочной железы показал, что гиперэкспрессия NRP 1 активирует гены, экспрессия которых способствует выживанию этих клеток. Известно, что некоторые Семафорины, Sema 3C и Sema 4D, обладают способностью стимулировать опухолевый неоангиогенез, в то время как другие, Sema 3A, Sema 3B, Sema 3D, Sema 3F, Sema 4A, наоборот, его подавляют [Carmeliet and Jain, 2011; Ellis, 2006]. 1.2.5 Нетрины и их рецепторы Нетрины (Netrins) представляют собой еще одно семейство белков, выполняющих навигационную функцию для растущих аксонов и мигрирующих клеток. Нетрины содержат VI ламининовый домен, EGF-подобные повторы, домен, аналогичный V домену ламинина, и гепарин-связывающий С-терминальный домен (Рисунок 1.7) [Larrivée et al., 2009; Melani and Weinstein, 2010]. Впервые Нетрины были описаны у мутантов C.elegans при скрининиге генов, ответственных за подвижность нематод [Hedgecock et al., 1990]. Далее было обнаружено, что Нетрины, выделенные из экстракта мозга позвоночных, стимулируют рост аксонов из эксплантов спинного мозга ex vivo. У млекопитающих известны Нетрин 1, Нетрин 3, Нетрин 4 [Melani and Weinstein, 2010]. Основные рецепторы Нетринов у млекопитающих представлены семействами белков DCC/Neogenin и UNC-5 (UNC-5A, UNC-B, UNC-C и UNC-D). Нетрины являются бифункциональными навигационными молекулами, которые в нервной системе взаимодействуют с обоими типами рецепторов и могут опосредовать как отталивание растущих аксонов, так и рост аксонов по градиенту хемоаттрактанта. DCC рецепторы состоят из внеклеточного домена, включающего шесть фибронектино-подобных повторов третьего типа (FNIII3) и четыре иммуноглобулиновых повтора (Ig), трансмембранный домен и цитоплазматический домен, состоящий из трех coined доменов (P1, P2 и P3) (Рисунок 1.7). Внутриклеточный домен DCC рецептора содержит несколько сайтов фосфорилирования и сайты связывания с адапторными белками. Сайт связывания с Нетринами содержится в одном из фибронектино-подобных трансмембранные повторов рецепторы, иммуноглобулиновых доменов 3 внеклеточная и двух типа. часть UNC-5 которых представляют собой состоит двух тромбоспондиноподобных из доменов; цитоплазматическая часть включает zonula occludens 5 домен, DCC связывающий и death 70 домены. Нетрин 1 связывающий сайт расположен в области иммуноглобулиновых повторов у UNC-5 рецептора [Larrivée et al., 2009]. Исследования, проведенные на мышах, нематодах и дрозофиле продемонстрировали, что связывание Нетрина 1 с гомодимерами UNC-5 или с UNC5/DCC гетеродимерами вызывает отталкивание, а взаимодействие Нетрина 1 с DCC и DCC/Neogenin – наоборот, аттракцию. В эмбриогенезе в центральной нервной системе у мышей клетки в вентрикулярной части средней линии секретируют Нетрин 1, что привлекает комиссуральные аксоны, которые растут в этом направлении. Результаты генетических исследований показывают, что у мышей, нокаутных по генам netrin-1 и dcc, рост комиссуральных аксонов останавливается, и аксоны не достигают средней линии. Для других аксонов Нетрины служат негативным навигационным сигналом, например, для аксонов мотонейронов блокового нерва (IV пара черепномозговых нервов), иннервирующего верхнюю косую мышцу глаза [Larrivée et al., 2009]. Эктопическая экспрессия UNC-5 рецепторов в спинальных нейронах Xenopus вызывает трансформацию ответа и отталкивание вместо аттракции. Это происходит в силу того, что UNC-5 и DCC рецепторы после связывания с Нетринами могут взаимодействовать между собой своими цитоплазматическими доменами, что вызывает трансформацию аттрактивного сигнала от DCC рецептора в реакцию отталкивания [Melani and Weinstein, 2010]. Помимо описанных DCC и UNC-5 рецепторов недавно было обнаружено, что Нетрины могут связываться с DSCAM белками, которые относятся к семейству иммуноглобулинов. DSCAM участвуют в регуляции поворота растущих аксонов по градиенту Нетрина 1. Так, известно, что связывание Нетрина 1 с DSCAM белками и дальнейшее взаимодействие этого комплекса с DCC рецепторами важно для своевременного поворота растущих комиссуральных аксонов, что было показано в модельных экспериментах на нейронах млекопитающих и Xenopus [Larrivée et al., 2009]. 1.2.6 Нетрины и их рецепторы в сосудистой системе Нетрины и их рецепторы участвуют в регуляции морфогенетических процессов не только в нервной системе, но и в других органах и тканях и способны модулировать клеточную адгезию, подвижность, дифференцировку и выживание клеток. В сосудах Нетрины могут играть двоякую роль и быть как проангиогенными, так и антиангиогенными. Исследования, проведенные группой под руководством профессора Эйчман (Professor Anna Eichmann), на моделях подкожного введения Матригеля и опухолевого роста in vivo, на эксплантной модели аорты крысы ex vivo и трехмерной культуре эндотелиальных клеток продемонстировали, что Нетрин 1 в определенных 71 условиях подавляет ангиогенез [Larrivée et al., 2009]. Свои антиангиогенные эффекты Нетрин 1 опосредует через связывание с UNC-5B рецептором. Нетрин 4 также способен ингибировать ангиогенез, скорее всего за счет связывания с UNC-5B, хотя в одном из исследований было показано, что Нетрин 4 связывается с UNC-5B не напрямую, а опосредованно через Неогенин [Leong et al., 2005]. Добавление Нетрина 1 в среду культивирования эндотелиальных клеток, экспрессирующих UNC-5B, вызывает ретракцию филоподий in vitro [Larrivée et al., 2009]. Высокий уровень UNC-5B характерен как для эндотелиальных клеток формирующихся артерий в эмбриогенезе, так и для «концевых» клеток в зоне активного ангиогенеза во взрослом организме. Экспрессия UNC-5B на покоящихся эндотелиальных клетках (phalanx cells) во взрослом организме минимальна, однако, она повышается в местах активного физиологического ангиогенеза или опухолевого роста, что позволяет рассматривать UNC-5B как маркер ангиогенеза или потенциальную мишень для блокирования избыточного или аберрантного роста сосудов при патологии [Larrivée et al., 2009]. Инактивация гена unc5 приводит к нарушениям формирования филоподий «концевыми» клетками, избыточному ветвлению формирующихся сосудов и ошибке при выборе траектории роста. Гиперэкспрессия Нетрина 1 с использованием ретровирусной конструкции в линейных опухолевых клетках человека с последующей ксенотрансплантацией этих клеток мышам приводит к снижению неоваскуляризации первичных опухолевых узлов по сравнению с контрольным вирусом. Аналогичные результаты in vivo были получены при трансплантации гиперэкспрессирующих Нетрин 1 опухолевых клеток на модели хориоаллантоисной мембраны куриных эмбрионах. На обеих моделях гиперэкспрессия Нетрина 1 блокирует начальные этапы ангиогенеза (миграцию «концевых» эндотелиальных клеток) и подавляет прорастание UNC-5Bэкспрессирующих сосудов в опухолевые узлы, но не влияет на дальнейшую пролиферацию эндотелиальных клеток и удлинение сосудистых отростков. При со-культивировании опухолевых клеток, экспрессирующих Нетрин 1, и эндотелиальных клеток из аорты, экспрессирующих UNC-5B, происходит подавление формирования капилляро-подобных трубочек, причем для этого необходима сигнализация с участием UNC-5B, поскольку, если в эндотелиальных клетках экспрессировать UNC-5B рецептор без цитоплазматического домена, то ингибирующие эффекты Нетрина 1 исчезают. Нетрин 4 может также оказывать антиангиогенное действие. При подавлении экспрессии Нетрина 4 способность эндотелиальных клеток к миграции и формированию капилляро-подобных трубочек на Матригеле in vitro увеличивается. А подавление экспрессии одного из рецепторов, Неогенина или UNC-5B, 72 приводит к исчезновению in vitro ингибирующих ангиогенез эффектов Нетрина 4 [Melani and Weinstein, 2010; Larrivée et al., 2009]. Суммируя эти данные, следует отметить, что активация внутриклеточной сигнализации от Нетринов с участием UNC-5B приводит к подавлению формирования филоподий эндотелиальными клетками и, как следствие, к подавлению процессов ветвления сосудов, что и опосредует ингибирующие ангиогенез эффекты Нетринов in vitro. Не ясно, оказывают ли эндогенные Нетрины подобное действие in vivo, поскольку дефектов формирования сосудистой сети у мышей, дефицитных по Нетрину 1, обнаружено не было, а данных по мышам, нокаутным по генам netrin-3 и netrin-4 в литературе пока нет [Larrivée et al., 2009]. Возможно, Нетрины по принципу обратной связи блокируют избыточный ангиогенез при длительной стимуляции ангиогенными факторами и тем самым участвуют в регуляции процессов ангиогенеза. Об этом свидетельствуют некоторые данные in vivo и in vitro: при долгом экспонировании эндотелиальных клеток в присутствии VEGF экспрессия ими Нетрина 4 повышается. Добавление экзогенного Нетрина 1 или Нетрина 4 в среду культивирования блокирует VEGF-индуцированную миграцию эндотелиальных клеток. Кроме того, введение рекомбинантного офтальмологической Нетрина модели 4 ингибирует хороидальной патологический неоваскуляризации ангиогенез при на лазерном повреждении, а рекомбинантный Нетрин 1 подавляет ангиогенез на моделях васкуляризации бляшек Матригеля, несмотря на введение в бляшки FGF [Leong et al., 2005]. В то же время в литературе имеется достаточное количество данных, убедительно свидетельствующих о том, что Нетрины и их рецепторы, а именно DCC и Неогенин, могут играть проангиогенную роль [Nguyen et al., 2006; Park et al., 2004; Yang et al., 2007]. В этих работах было показано, что Нетрины могут стимулировать пролиферацию, адгезию и миграцию эндотелиальных и гладкомышечных сосудистых клеток in vitro, а также ангиогенез in vivo на модели хориаллантоисной мембраны и экспериментальной модели васкуляризации роговицы. В первых двух работах не удалось идентифицировать рецептор, ответственный за проангиогенные эффекты Нетринов [Nguyen et al., 2006], в то время как в двух других работах было показано, что именно DCC рецептор опосредует промиграторные эффекты Нетринов [Park et al., 2004; Yang et al., 2007]. Кроме этого было обнаружено, что Неогенин экспрессируется в гладкомышечных клетках сосудов, а антитела к Неогенину блокируют миграцию ГМК по градиенту Нетрина 1 [Melani and Weinstein, 2010]. Еще в одном исследовании было обнаружено, что Нетрин 1 стимулирует ангиогенез за счет увеличения активности эндотелиальной 73 NO синтазы (eNOS) и продукции NO эндотелиальными клетками. Эти эффекты Нетрина 1 исчезали при добавлении антител, блокирующих DCC, или siRNA для подавления экспрессии мРНК DCC, что свидетельствует о том, что стимулирующие ангиогенез эффекты Нетрина 1 напрямую зависят от DCC рецептора [Melani and Weinstein, 2010]. Имеются данные об участии MEK/ERK сигнализации в проангиогенных эффектах Нетринов [Melani and Weinstein, 2010]. In vivo проангиогенные эффекты Нетринов были обнаружены при экзогенном введении этих белков во взрослый мозг экспериментальных животных. Увеличение экспрессии эндогенного Нетрина 4 также было отмечено в зоне инсульта, в то время как экспрессия DCC, но не UNC-5A и не UNC-5B обнаруживалась на нейритах нейронов в периинсультной зоне. Кроме того, интрацеребровентрикулярное введение Нетрина 4 на модели экспериментального инсульта у мышей (при дистальный окклюзии среднемозговой артерии у мышей) приводило к увеличению плотности кровеносных сосудов, стимулировало пролиферацию эндотелиальных клеток и восстановление нормального поведенческого статуса уже спустя 1 неделю после введения [Hoang et al., 2008]. Инъекции плазмид, кодирующих гены Нетрина 1 или Нетрина 4, ускоряли реваскуляризацию ишемизированных мышц in vivo на модели ишемии задней конечности у мыши, происходило это за счет рекрутирование ГМК в зону ишемии [Wilson et al., 2006; Larrivée et al., 2009]. В этой работе авторам, однако, не удалось выявить рецептор, который опосредовал эффекты Нетринов в ишемизированных тканях in vivo, экспрессии ни одного из известных рецепторов Нетринов в клетках в зоне ишемии обнаружено не было. В том, будет ли ответ на Нетрины проангиогенным или антиангиогенным, важную роль может играть их концентрация [Yang et al., 2007]. Так, было обнаружено, что Нетрин 1 в низкой концентрации может стимулировать миграцию и пролиферацию эндотелиальных клеток (HUVECs) in vitro, а в высокой -ингибировать. Аналогичные проангиогенные и антиангиогенные эффекты различных концентраций Нетрина 1 были получены на модели васкуляризации роговицы in vivo, причем эти эффекты были опосредованы связыванием Нетрина 1 с UNC-5B, поскольку при подавлении экспрессии UNC-5B ингибирующие и частично стимулирующие эффекты Нетрина 1 исчезали. Аналогичные механизмы концентрационной зависимости типа клеточного ответа уже были описаны для Эфринов и их рецепторов: для растущих аксонов в развивающейся нервной системе градиент Эфринов или их рецепторов в низкой концентрации служит аттрактивным сигналом, превышение некоторой пороговой концентрации вызывает отталкивание [Poliakov et al., 2004]. Возможно, это явление отражает некий общий 74 механизм, присущий навигационным рецепторам, а тип клеточного ответа зависит от адапторных белков, взаимодействующих с рецепторами, и конкретных путей внутриклеточной сигнализации, которые при этом активируются. Однако это предположение нуждается в дальнейшей проверке. Еще в одной группе работ Нетрины и их рецепторы рассматриваются как молекулы, регулирующие выживание эндотелиальных клеток и апоптоз [Larrivée et al., 2009]. Нетрины при этом играют роль факторов, обеспечивающих выживание эндотелиальных клеток, за счет того, что, связываясь с UNC-5B, они блокируют апоптотический сигнал, идущий от этого рецептора и его адапторного белка серин/треониновой киназы DAPK. Эта модель объясняет, почему подавление экспрессии UNC-5B (подавление проапоптотического сигнала) приводит к выживанию эндотелиальных клеток и стимулирует ангиогенез, в то время как потеря экспрессии Нетрина 1 (потеря антиапоптотического сигнала или фактора, способствующего выживанию) вызывает гибель эндотелиальных клеток и подавляет ангиогенез [Wilson et al., 2006]. Очевидно, что Нетрины и их рецепторы играют двоякую роль как в нервной системе, где они могут оказывать как стимулирующий рост аксонов эффект, так и ингибирующий, так и в сосудистой системе, где связывание Нетринов с рецепторами может опосредовать как их ангиогенные эффекты, так и антиангиогенные. 1.2.7 Cлит лиганды и Robo рецепторы Слит лиганды (Slits) представляют собой секретируемые гликопротеиды, которые исходно были описаны как молекулы, вызывающие отталкивание в нервной системе в эмбриогенезе. В дальнейшем была продемонстрирована их роль в определении места закладки и формирования почек в эмбриогенезе, формировании кровеносных сосудов и регуляции миграции активированных хемокинами CXCR4+ лейкоцитов [Melani and Weinstein, 2010]. Структурно Слит белки состоят из четырех повторов с высоким содержанием лейцина, от семи до девяти EGF-подобных повторов и LamG домена (Рисунок 1.7). Слит белки взаимодействуют с рецепторами из семейства Robo (Roundabout), кроме того они могут связываться с протеогликанами внеклеточного матрикса (гепарином и гепаран сульфатом), что, возможно, стабилизирует лиганд-рецепторный Slit/Robo комплекс или может опосредовать его взаимодействие с другими рецепторами [Larrivée et al., 2009; Adams and Eichmann, 2010]. У млекопитающих известно три Слит белка (Slit 1, Slit 2, Slit 3) и четыре Robo рецептора (Robo 1, Robo 2, Robo 3/Rig-1, Robo 4). Robo 1, Robo 2, Robo 3 в основном экспрессированы в нервной системе и содержат пять иммуноглобулиновых 75 повторов (Ig), три фибронектино-подобных повтора 3 типа (FNIII3), трансмембранный домен и цитоплазматический домен, состоящий из четырех консервативных Roboспецифичных мотивов (СС0, СС1, СС2, СС3) (Рисунок 1.7). Roundabout (Robo) рецепторы были так названы, потому что у Drosophila при их делеции был обнаружен фенотип, при котором в центральной нервной системе ипсилатеральные аксоны, в норме избегающие пересечения средней линии, ее пересекают, а комиссуральные аксоны пересекают среднюю линию туда и обратно несколько раз. Слит выступают в роли молекул отталкивания, связывание Слит лигандов с Robo рецепторами важно для выбора траектории роста аксонов в зоне средней линии развивающегося эмбриона. Была высказана гипотеза о существовании «Robo кода», согласно которой финальная латеральная позиция комиссуральных аксонов после пересечения средней линии определяется набором Robo рецепторов на каждом конкретном аксоне. Robo рецепторы, в свою очередь, взаимодействуют со Слит белками, градиент экспрессии последних создают клетки средней линии, что является определенной картой роста. Помимо хорошо изученной роли в регуляции траектории роста комиссуральных и ипсилатеральных аксонов, Slit/Robo белки участвуют в формировании продольных трактов аксонов в ЦНС, проекций вомероназальных аксонов в добавочную обонятельную луковицу в переднем мозге, а также ветвлении центральных аксонов тройничного нерва [Larrivée et al., 2009]. Кроме описанных механизмов известно, что в нервной системе Robo рецепторы, активированные Слит, могут взаимодействовать своими внутриклеточными доменами с DCC рецепторами Нетринов и подавлять аттрактивный сигнал Нетринов, что предполагает дополнительные уровни взаимодействия между навигационными молекулами разных групп и регуляции клеточного ответа [Melani and Weinstein, 2010]. 1.2.8 Слит лиганды и Robo рецепторы в сосудистой системе Как и белки других групп навигационных рецепторов, исходно обнаруженных в нервной системе, Slit/Robo также участвуют в формировании сердца и сосудов в эмбриогенезе. У млекопитающих данные об участии Slit/Robo белков в мофрогенезе сердечно-сосудистой системы немногочисленны и достаточно противоречивы. А у Drosophila с использованием методов in situ гибридизации и генетического анализа было обнаружено, что «Robo код» определяет миграцию и позиционирование отдельных популяций клеток формирующегося сердца (кардиобластов и перикардиоцитов) относительно друг друга и средней линии. Кардиобласты (Slit) и перикардиоциты (Robo) экспрессируют разный набор лигандов и рецепторов, что на ранних этапах обеспечивает 76 сегрегацию этих двух популяций и важно для их миграции и взаимного расположения. На более поздних этапах при слиянии кардиобластов и формировании полости сердца Slit/Robo сигнализация регулирует адгезию клеток, что важно для мофрогенеза сердца, формирования его правильной формы и размера [Melani and Weinstein, 2010]. Основным представителем Robo рецепторов в сосудистой системе является Robo 4, он экспрессирован у млекопитающих только в эндотелиальных клетках. В отличие от остальных Robo рецепторов, Robo 4 содержит только два иммуноглобулиновых повтора, два фибронектино-подобных повтора третьего типа и цитоплазматический домен, состоящий из двух консервативных Robo-специфичных мотивов (СС1 и СС2) [Melani and Weinstein, 2010]. У млекопитающих использование метода in situ гибридизации и рекомбинантных технологий позволило выявить экспрессию Robo 4 в эндотелиальных клетках в развивающихся эмбрионах мыши, во взрослом организме на сосудах мелкого диаметра и капиллярах, а также в сосудах, прорастающих в опухоль, на ксенографтной модели опухолевого роста [Larrivée et al., 2009]. У Zebrafish гомолог Robo 4 экспрессируется и в развивающейся нервной системе, и формирующихся сосудах, включая дорзальную аорту и кардинальные вены, а также в «концевых» эндотелиальных клетках ветвящихся межсомитных сосудов. Подавление экспрессии Robo 4 с использованием Морфолино нуклеотидов или гиперэкспрессия Robo 4 вызывает пространственно-временные дефекты формирования межсомитных сосудов, что свидетельствует о важной роли Robo 4 в регуляции направленного роста этого типа сосудов. Выделенные из таких эмбрионов ангиобласты в культуре мигрируют более активно и беспорядочно по сравнению с ангиобластами из контрольных рыбок. В ангиобластах из мутантных рыбок содержание активных форм Cdc42 и Rac снижено, из чего авторы сделали вывод о том, что Robo 4 участвует в передаче аттрактивного навигационного сигнала [Melani and Weinstein, 2010]. В отличие от результатов исследований на Zebrafish у млекопитающих при таргетном нарушении экспрессии гена Robo4 явной роли этого белка в эмбриональном развитии сосудистой системы выявить не удалось. Robo4-/- мутантные мыши являются жизнеспособными и не обладают выраженным фенотипом, однако, при локальном введении VEGF165 у них наблюдается избыточная проницаемость сосудов сетчатки и ее гиперваскуляризация при O2–индуцированной ретинопатии. Повышенная проницаемость эндотелия у Robo-/- мышей может быть снижена с использованием PP2, ингибитора Src киназы, что свидетельствует о том, что Robo 4, напрямую или косвенно, блокирует Src-опосредованные эффекты VEGF. Дальнейший анализ фенотипа Robo4-/мышей и эксперименты на культуре эндотелиальных клеток in vitro позволили 77 предположить, что Robo 4 действует по принципу обратной связи и нейтрализует ангиогенные эффекты VEGF, а именно миграцию эндотелиальнх клеток, формирование капилляро-подобных трубочек и увеличение проницаемости эндотелия. Авторы высказали предположение, что тем самым Robo 4 участвует в поддержании целостности сосудистой стенки [Adams and Eichmann, 2010; Melani and Weinstein, 2010]. Кроме того, недавно было показано, что при связывании Слит с Robo 4 цитоплазматический домен рецептора взаимодействует с адапторным белком паксиллином (paxillin). Взаимодействие Robo 4/paxillin блокирует активацию малых ГТФаз Rаc и Arf-6 (компоненты сигнального пути, активируемого VEGF). In vivo ингибирование активности Arf-6 имитирует эффекты Robo 4 и также, как и Robo 4, подавляет гиперпроницаемость сосудов сетчатки, наблюдаемую при введении VEGF165 [Jones et al., 2009]. Эти данные свидетельствуют о том, Robo 4 может рассматриваться как потенциальная мишень при разработке лекарств для лечения заболеваний, связанных с избыточным ангиогенезом или гиперпроницаемостью сосудов. Неизвестно почему эффекты Robo 4 по нейтрализации VEGF проявляются только при патологическом ангиогенезе в сетчатке и не выявляются в эмбриогенезе. Связывание Slit 2 с Robo 4 напрямую никогда не было доказано, даже с использованием высоко чувствительных методов (Biacore detection method для выявления белок-белковых взаимодействий и аффиности связывания) [Suchting et al., 2005]. Кроме того, на основании рентгено-структруного анализа некоторыми авторами было высказано сомнение в том, что Robo 4 вообще является рецептором Слит белков [Larrivée et al., 2009; Melani and Weinstein, 2010]. В последних обзорах авторы предположили, что Robo 4 взаимодействует с лигандами опосредованно через другие белки. Так, было обнаружено, что в эндотелиальных клетках помимо Robo 4 экспрессируется Robo 1. In vitro было продемонстрировано, что Robo 1 может формировать гетеродимеры с Robo 4 в эндотелиальных клетках, связывание Slit 2 с Robo 1, возможно, активирует в них Robo 4 и таким образом влияет на клеточную миграцию [Larrivée et al., 2009; Melani and Weinstein, 2010]. Другой группой исследователей было обнаружено, что in vitro выделенный и очищенный Slit 2 служит хемоаттрактантом для эндотелиальных и трансформированных клеток, гиперэкспрессирующих Robo 1. Эти же авторы показали, что Robo 1 экспрессируется в опухолевых сосудах in vivo, и что увеличение экспрессии Slit 2 в опухолевых клетках на ксенографтной модели способствует увеличению опухолевого неоангиогенеза [Wang et al., 2003]. Третьей группой исследователей было продемонстрировано, что в эмбриогенезе у мыши именно 78 Robo 4 связывается со Slit 2, что служит негативным сигналом для мигрирующих эндотелиальных клеток и вызывает подавление их миграции [Park et al., 2003]. В одной работе было высказано предположение, что Robo 4 в сосудах не является в собственном смысле слова навигационным рецептором, а его роль сводится к регуляции клеточной специализации - формирование ―tip‖ и ―stalk‖ фенотипа [Jones et al., 2008]. Экспрессия Robo 4 была обнаружена в эндотелиальных ―stalk‖ клетках, в отличие от ―tip‖ клеток, а добаление рекомбинантного Slit 2 подавляло миграцию, формирование капилляро-подобных трубочек и увеличение проницаемости, несмотря на добавление VEGF в среду культивирования. Возможно, Robo 4, таким образом, участвует в поддержании структурной организации сосудов за счет предотвращения активации ―stalk‖ клеток в ответ на увеличение концентрации VEGF. Роль Robo 1 в формировании сосудов в эмбриогенезе до сих пор не исследовалась [Larrivée et al., 2009; Melani and Weinstein, 2010]. Однако в литературе имеются данные, свидетельствующие о роли Robo 1 в опухолевом ангиогенезе. Экспрессия Robo 1 оказывает проангиогенный эффект и способствует опухолевому росту, что было показано на моделях in vitro и in vivo. Введение нейтрализующих Robo 1 антител подавляет формирование мелких сосудов и рост первичного опухолевого узла на ксенографтной модели злокачественной меланомы А375 у мышей [Wang et al., 2003]. На модели химически индуцированной плоскоклеточной карциномы у хомячков было показано, что экспрессия Slit 2 положительно коррелирует с васкуляризацией первичного опухолевого узла. Добавление нейтрализующих Robo 1 антител при этом блокирует опухолевый рост и неоангиогенез. Таким образом, отсутствие выраженного фенотипа у мутантных по Robo или Slit белкам мышей на сегодняшний день не позволяет четко обозначить роль Slit/Robo системы в процессах морфогенеза сосудов. Для выявления навигационной функции Slit/Robo в процессах формирования и роста сосудов необходимы дальнейшие исследования. 1.2.9 Урокиназная система На сегодняшний день к навигационным рецепторам также относят урокиназную систему, включающую урокиназу, еѐ рецептор и ингибиторы. Урокиназа (uPA) или активатор плазминогена урокиназного типа представляет собой протеолитический фермент, превращающий плазминоген в плазмин [Collen et al., 1999]. Урокиназа синтезируется эндотелиальными и ГМК клетками сосудов, эпителиальными клетками, фибробластами, моноцитами/макрофагами, а также клетками злокачественных опухолей 79 различного происхождения [Clowes et al., 1990; Tkachuk et al., 1996; Парфенова и др., 2009]. В структуре урокиназы выделяют три домена: N-концевой домен, подобный эпидермальному фактору роста (GFD, ростовой домен), крингл-домен и С-концевой протеолитический домен (Рисунок 1.9). Структурно ―ростовой‖ домен гомологичен эпидермальному фактору роста. Ростовой домен обеспечивает высокоаффинное связывание урокиназы с ее рецептором uPAR/CD87. Крингл-домен принимает участие в стабилизации комплекса урокиназы с урокиназным рецептором и его взаимодействия с компонентами внеклеточного матрикса [Ткачук и др., 2013]. Крингл-домен также содержит участки связывания с ингибитором активаторов плазминогена PAI-1 и интегринами. Протеолитический домен содержит активный центр урокиназы His-AspSer, именно он ответственен за активацию плазминогена, ряда факторов роста и матриксных металлопротеиназ. Клетки секретируют урокиназу в виде одноцепочечного полипептида с молекулярной массой 54 кДа [Tkachuk et al., 1996], который при взаимодействии с плазминогеном превращает его в плазмин. Плазмин, в свою очередь, является активатором урокиназы и превращает одноцепочечную урокиназу в двухцепочечную форму. Протеолитическая активность двухцепочечной урокиназы в 200 раз выше, чем у одноцепочечной формы [Lijnen et al., 1990]. Урокиназный рецептор состоит из трех гомологичных доменов и заякорен на плазматической мембране через гликозилфосфатидилинозитольный якорь (GPI якорь), что опосредует его высокую латеральную подвижность (Рисунок 1.9). Рецептор урокиназы локализуется в особых участках плазматической мембраны – кавеолах, которые содержат большое количество гликосфинголипидов и холестерола, в этих участках также локализуется большое количество сигнальных молекул, в том числе и Эфрины, и Т-кадгерин, и G-белки [Рубина и Ткачук, 2004; Ткачук и др., 2013]. Своим ростовым доменом урокиназа связывается со специфическим высокоаффинным рецептором (uPAR/CD87) на поверхности клеток [Blasi and Carmeliet, 2002]. Связанная с рецептором одноцепочечная урокиназа активируется плазмином более эффективно, чем свободная. Плазмин действует на матриксные металлопротеазы [Bobik and Tkachuk, 2003; Tkachuk et al., 1996; Vassalli, 1985] и факторы роста [Naldini et al., 1992; Plouet et al., 1997; Saksela and Rifkin, 1990], переводя их из латентного состояния в активное. В сосудистой стенке плазмин расщепляет фибрин, что способствует растворению тромба, а ММП расщепляют белки внеклеточного матрикса и компоненты базальной мембраны, а именно, коллаген, фибронектин и ламинин [Kuzuya and Iguchi., 2003]. Урокиназная система играет важную роль в регуляции направленного движения клеток. Высокая 80 подвижность комплекса урокиназного uPA/uPAR на рецептора обеспечивает лидирующем крае возможность клетки, там, концентрации где необходима протеолитическая активность [Saksela and Rifkin, 1990; Vassalli et al., 1985]. Протеолитическая активность связанной с рецептором урокиназы на лидирующем крае клетки обеспечивает локальный протеолиз и тем самым опоcредует направленное движение клеток. Плазмин-зависимые эффекты урокиназы необходимы для репарации тканей после повреждения, например, для образования рубца в ишемизированном миокарде [Heymans et al., 1999] и регенерации [Ткачук и др., 2013]. Однако роль урокиназы в ремоделировании тканей не ограничивается активацией плазмина и протеолитической активностью, поскольку связывание урокиназы с рецептором активирует внутриклеточные не сигнальные только внеклеточный каскады [Noh et протеолиз, al., 2013]. но и запускает Урокиназа может стимулировать миграцию эндотелиальных клеток, ГМК, эпителиальных клеток и моноцитов [Odekon et al., 1992; Okada et al., 1996; Busso et al., 1994; Resnati et al., 2002], независимо от еѐ протеолитической активности. Связывание урокиназы с uPAR на ГМК и эндотелиальных клетках вызывает активацию Jak/Stat сигнального пути. Janus киназы, Jak1 и Tyk2, образуют комплекс с uPA-uPAR на лидирующем крае клетки, что, в свою очередь, приводит к транслокации факторов транскрипции Stat1, Stat2 и Stat4 в ядро и активации транскрипции генов [Dumler et al., 1999; Парфенова и др., 2009]. Активация миграции клеток происходит в результате сигнального Tyk2/PI3-K/RhoA/Rho киназного пути, что приводит к изменениям клеточного фенотипа и реогранизации цитоскелета [Resnati et al., 2002; Парфенова и др., 2009]. В нашей лаборатории было обнаружено, что крингл-домен урокиназы может опосредовать хемотаксические эффекты uPA на ГМК клетках [Ткачук и др., 2013]. Так, крингл-домен урокиназы, а также рекомбинантная форма урокиназы, лишенная ростового домена и неспособная связывается с «классическим» рецептором uPAR/CD87, вызывают активацию р38 и р42/44 MAP-киназ, активацию малой ГТФазы Rho и миграцию клеток. Несколькими авторами была продемонстирована активация серин-треониновых киназ под действием урокиназы, таких как киназы ERK/MAPK, [Bohuslav et al., 1995; Парфенова и др., 2009; Resnati et al., 2002]. 81 Рисунок 1.9. Взаимодействие урокиназы с рецептором (uPAR/CD87) и крингл связывающим белком (KP- kringleprotein) активирует внутриклеточную сигнализацию, что приводит к адгезии, миграции, пролиферации и дифференцировке клеток. GFD – домен, подобный фактору роста; PD – протеолитический домен; GPI – гликозилфосфатидилинозитольный якорь; VN – витронектин; PAI-1 – ингибитор активаторов плазминогена; scuРA – одноцепочечнаяурокиназа; LRP – рецептор липопротеинов низкой плотности; LRP/α2-MR – протеин, родственный рецептору липопротеинов низкой плотности/рецептор α2-макроглобулина; MAPK – mitogenactivatedproteinkinases; JAK – tyrosineproteinkinases; STAT – ДНК - связывающие активаторы транскрипции; TF – факторы транскрипции; PI3K – фосфоинозитид–3– киназа; FAK – киназа фокальных контактов; NR-TK – нерецепторная тирозинкиназа;Man6P-R – манноза-6-фосфат/рецептор инсулиноподобного фактора роста II; gp130 – сигнальный посредник интерлейкина 6; uPARAP – белок, ассоциированный с урокиназным рецептором (Адаптировано из Ткачук и др., 2013). 82 Поскольку урокиназный рецептор является GPI-заякоренным белком, для активации внутриклеточной сигнализации с участием uPAR/CD87 необходимо образование комплекса урокиназного рецептора с другими трансмембранными белками. В частности, связывание урокиназы может вызывать взаимодействие урокиназного рецептора с интегринами, кавеолином, с рецепторами, сопряжѐнными с G-белками (Gprotein-coupled receptors - GPCRs), и другими белками [Blasi and Carmeliet, 2002]. Так, известно, что uPAR/CD87 взаимодействует с интегринами, такими как лейкоцитарный β2-интегрин Mac-1 (CD11b/CD18), β1-, β3-интегрины, и с рецептором витронектина αvβ5 [Sitrin et al., 2000; Ghosh et al., 2000]. Урокиназа может одновременно связываться с двумя рецепторами на поверхности клетки: с uPAR/CD87 через ростовой домен и с интегрином Mac‑1 через крингл и протеолитический домены. Урокиназа может взаимодействовать не только с uPAR/CD87, но и с рецепторами семейства липопротеинов низкой плотности (LDLR): рецептором α2-макроглобулина белком, родственным рецептору липопротеинов низкой плотности (LRP/α2-MR) и рецептором липопротеинов очень низкой плотности (VLDLR). Эти рецепторы обеспечивают интернализацию урокиназы с поверхности клетки через окаймленные клатрином ямки путем эндоцитоза. Известно, что интернализация урокиназы, а также других лигандов через LRP и ему подобные рецепторы сопровождается активацией сигнальных систем клетки, приводящей к активации внутриклеточных протеинкиназ. Так, LRP рецептор взаимодействует с цитоплазматическим адаптерным белком Dab-1, что приводит к связыванию и активации нерецепторных тирозиновых киназ семейств Src и Abl [Ткачук и др., 2013]. LRP рецептор также принимает участие в активации MAPKзависимого сигнального пути и влияет на адгезию клеток [Gotthardt et al., 2000]. Кроме того, LRP/α2-MR рецептор может взаимодействовать с гетеротримерными G-белками и участвовать в активации протеинкиназы А [Ткачук и др., 2013]. В нашей лаборатории были обнаружены дополнительные участки связывания урокиназы на клетках, один из которых взаимодействует с урокиназой через ее протеолитический домен [Poliakov et al., 2004], а другой связывает крингл домен. Взаимодействие со вторым участком не зависит от присутствия в структуре урокиназы «ростового» домена и опосредует стимулирующие клеточную миграцию эффекты урокиназы [Mukhina et al., 2000]. Так была обнаружена новая мишень связывания урокиназы на поверхности клеток, отличная от uPAR/CD87, - фибулин-5 [Kapustin et al., 2012]. В нашей лаборатории было продемонстировано, что урокиназа может непосредственно взаимодействовать с фибулином-5, интегрин-связывающим белком 83 внеклеточного матрикса, через свой протеолитический домен. Оказалось, что фибулин-5, связываясь с протеолитическим доменом, защищает uPA от действия физиологического ингибитора урокиназы PAI-1 и подавления ее активности. В свою очередь, формирование uPA/PAI-1 комплекса приводит к быстрому эндоцитозу комплекса uPAuPAR с клеточной поверхности и его деградации [Ткачук и др., 2013]. Таким образом, при связывании урокиназы с uPAR клетка с помощью направленного протеолиза локально деградирует внеклеточный матрикс, а крингл-домен урокиназы активирует внутриклеточные сигнальные процессы, которые необходимы для движения клетки [Ткачук и др., 2013]. Недавно в нашей лаборатории был выявлен новый сигнальный путь урокиназы, связанный с активностью еѐ крингл домена и с ее быстрой транслокацией в ядро с помощью нуклеолина. Транслокация урокиназы в ядро не зависит от присутствия ростового домена урокиназы, но невозможна без крингл домена. Урокиназа, проникая в ядро, регулирует транскрипцию генов и вызывает экспрессию гладкомышечного актина [Stepanova et al., 2008]. При повреждении в сосудах происходит опосредованное урокиназой превращение фибробластоподобных клеток в миобласты, клетки мигрируют, пролиферируют, и это важно при развитии констриктивного ремоделирования сосудов [Ткачук и др., 2013]. Данные, полученные в нашей лаборатории, и результаты других исследований свидетельствуют о том, что урокиназная система является мощным стимулятором ангиоартериогенеза. Локальный протеолиз белков внеклеточного матрикса и разрушение базальной мембраны необходимы для того, чтобы эндотелиальные клетки или их циркулирующие в крови предшественники могли мигрировать и формировать новые сосуды [Lamalice et al., 2007]. Урокиназа и плазмин могут активировать и/или высвобождать латентные матриксные металлопротеиназы, а также ангиогенные факторы роста VEGF, bFGF, HGF, TGFβ и PDGF, которые в свою очередь способствуют пролиферации и миграции эндотелиальных клеток, а также их инвазии [Ткачук и др., 2013]. Эндотелиальные клетки, гиперэкспрессирующие урокиназу, характеризуются повышенной инвазивностью in vitro, а добавление антител, блокирующих урокиназный рецептор или протеолитическую активность урокиназы, подавляет миграцию эндотелиальных клеток и формирование ими капилляро-подобных структур [Ткачук и др., 2013]. Экспрессия урокиназного рецептора возрастает на мигрирующих «концевых» клетках растущих сосудов при ангиогенезе [Uhrin and Bress, 2013; Prager et al., 2004]. Результаты исследований, полученные в нашей лаборатории, показывают, что урокиназа и еѐ рецептор экспрессируются на мигрирующих и пролиферирующих клетках сосудов 84 при развитии экспериментального рестеноза и в атеросклеротических бляшках, а внесение урокиназы в стенку поврежденного сосуда стимулирует развитие неоинтимы и неоадвентиции, миграцию, пролиферацию и фенотипическую трансформацию клеток сосудов [Plekhanova et al., 2000; Plekhanova et al., 2001; Plekhanova et al., 2009; Parfyonova et al., 2004]. У трансгенных мышей, нокаутных по гену урокиназы, уровень ремоделирования стенки сосуда и развитие неоинтимы после баллонного повреждения снижен, подавлен ангио-артериогенез в ишемизированной конечности и VEGFстимулированный ангиогенез в периинфарктной зоне [Heymans et al., 1999]. Данные, полученные в нашей лаборатории, показывают, что экспрессия урокиназы при экспериментальной ишемии конечности и инфаркте миокарда стимулирует ангиоартериогенез и восстановление кровотока в той же степени, что и экспрессия основного фактора роста сосудов VEGF [Traktuev et al., 2007]. В нашей лаборатории было обнаружено, что урокиназа стимулирует пролиферацию сосудистых клеток не только через описанные выше механизмы, связанные с активацией факторов роста, но и через активацию внутриклеточных сигнальных систем, ведущих к образованию активных форм кислорода - НАДФН-оксидазы NOX-1 и NOX-4 [Menshikov et al., 2006b].Такие свойства урокиназы и еѐ рецептора делают еѐ важнейшим фактором, регулирующим процессы ремоделирования стенки сосудов после повреждения и процессы ангиогенеза. Хорошо известно, что урокиназная система играет важную роль в развитии опухолей, их васкуляризации и метастазировании [Mekkawy et al., 2014]. Урокиназа стимулирует пролиферацию многих типов клеток, в том числе эпителиальных клеток человека, нормальных и опухолевых клеток почки и клеток меланомы [Kirchheimer et al., 1989; Ткачук и др., 2013]. Формирование комплекса uPA-uPAR и его взаимодействие с интегринами приводит к активации p38 MAPK и пролиферации опухолевых клеток [Aguirre-Ghiso et al., 2001]. На опухолевых клетках была обнаружено, что ростовой домен урокиназы ответственен за индукцию пролиферации остеобластоподобных клеток, клеток меланомы, клеток остеосаркомы человека SaOs-2, и опухолевых клеток рака молочной железы. В литературе имеются данные, свидетельствующие о том, что митогенная активность урокиназы на нормальных и опухолевых клетках активируется через ERK/MAPK сигнальный каскад, для запуска которого необходимы интегрины [Ткачук и др., 2013]. Многие типы клеток, выделенные из злокачественных образований человека, таких как рак желудка, молочной железы, прямой кишки, остеосаркома и рак легкого устойчивы к апоптозу. In vitro при инкубировании MDA-MB-231 клеток рака молочной железы с антителами к uPA, блокирующими взаимодействие uPA с uPAR, наблюдается снижение уровня фосфорилирования ERK/MAР киназ и повышение 85 количества апоптотических клеток [Ma et al., 2001]. В клинических исследованиях показано, что экспрессия урокиназного рецептора характерна для наиболее ―агрессивных‖ клеток опухолей и сосудов, что может являться маркером активного опухолевого ангиогенеза и метастазов [Ткачук и др., 2013]. Таким образом, урокиназная система обладает способностью активировать протеолитические и сигнальные каскады, она выполняет важную функцию в регуляции направленной миграции сосудистых клеток, дифференцировке и пролиферации клеток как в норме, так и при патологии. Урокиназа и еѐ рецептор участвуют в процессах ремоделирования кровеносных сосудов и регуляции ангиогенеза. На сегодняшний день урокиназа является перспективной мишенью для создания лекарств, направленных на профилактику рестенозов, предотвращение негативного ремоделирования артерий, стимуляцию роста сосудов при ишемических заболеваниях и подавление ангиогенеза в онкологии. В настоящее время в нашей лаборатории проводятся исследования с целью выявления возможного участия урокиназной системы в регуляции процессов направленного роста аксонов и миграции клеток в неовной системе на модели эксплантов спинальных ганглиев ex vivo и дифференцировки нейральных клеток in vitro. Таким образом, основные положения об известных на сегодняшний день навигационных молекулах, определяющих процессы морфогенеза в нервной и сосудистой системе, сводятся к следующему: Навигационные молекулы, которые определяют направление роста аксонов нейронов и миграцию нервных клеток в нервной системе, аналогичным образом регулируют миграцию эндотелиальных клеток и формирование сосудистых отростков в процессах ангиогенеза. Основные группы навигационных молекул и их рецепторов представляют собой Эфрины и их рецепторы, Нетрины и их рецепторы, Семафорины и их рецепторы, Слит лиганды и Robo рецепторы, урокиназную систему. Эфрины и их рецепторы играют основную роль в артерио-венозной дифференцировке в процессе формирования сосудов в эмбриогенезе. Семафорины в сосудистой системе опосредуют негативный навигационный сигнал, регулирующий направление миграции эндотелиальных клеток и паттерн формирования сосудов. Нейропилины связываются с Семафоринами в нервной системе и в сосудах. Нейропилины являются ко-рецепторами Плексинов в нервной системе и корецепторами VEGFR2 в сосудах. Связывание Семафоринов с Нейропилинами может ингибировать VEGF сигнализацию, идущую от VEGFR2, и тем самым блокировать стимулиующий сигнал VEGF. Семафорины в сосудах, как и в нервной системе, могут 86 вызывать внутриклеточную сигнализацию через активацию Плексиновых рецепторов, Семафорины при этом могут вызывать сигнализацию независимо от Нейропилинов. Плексины могут опосредовать как проангиогенные эффекты, так и антиангиогенные. Роль Нетринов в процессах ангиогенеза противоречива. Также как это было описано для нервной системы, Нетрины в сосудах могут играть двоякую роль и опосредовать как аттрактивные сигналы, так и сигналы отталкивания в зависимости от конкретного типа клеток и микроокружения. Еще меньше известно о функционировании Слит лигандов и Robo рецепторов и их роли в сосудистой системе, что, возможно, связано с избыточностью в экспрессии Robo рецепторов и различиями в выполняемой функции у разных типов биологических объектов. Урокиназная система активирует протеолитические и сигнальные каскады, что важно для регуляции направленной миграции сосудистых клеток, их дифференцировки и пролиферации клеток как в норме, так и при патологии. Урокиназа и ее рецептор участвуют в процессах ремоделирования кровеносных сосудов и регуляции ангиогенеза. Т-кадгерин является навигационной молекулой, опосредующей негативное регулирование роста аксонов и миграции клеток нервного гребня в нервной системе (см ниже). Т-кадгерин экспрессируется в сосудистых клетках, его экспрессия повышается при различных патологиях. Появляется все больше данных о том, что представители разных групп навигационных рецепторов могут взаимодействовать между собой, что увеличивает разнообразие сигнальных путей, которые при этом активируются, и типов клеточных ответов. 87 1.3 Кадгерины в процессах морфогенеза Кадгерины представляют собой суперсемейство мембранных рецепторов, опосредующих Ca2+-зависимое гомофильное узнавание и межклеточное взаимодействие в тканях взрослого организма и в эмбриогенезе (Рисунок 1.10). В эмбриогенезе кадгерины опосредуют сортинг и сегрегацию клеток, клеточную миграцию, морфогенез и рост аксонов к своим мишеням. Во взрослом организме кадгерины участвуют в организации и поддержании структуры органов и тканей, регулируют обновление клеточного состава, обеспечивают физиологический барьер между контактирующими тканями и селективность транспорта растворѐнных веществ [Gumbiner, 2005; PerezMoreno et al., 2003]. 1.3.1 «Классические» кадгерины Изначально кадгерины были описаны как семейство мембранных рецепторов, опосредующих Са2+-зависимую гомофильную адгезию во всех солидных тканях взрослого организма и в эмбриогенезе. В настоящее время ясно, что «классические» кадгерины представляют собой лишь небольшую группу в суперсемействе кадгеринов, куда также входят десмоглеины и десмоколлины, протокадгерины, белки семейства FAT, 7TM-кадгерины, T-кадгерин и некоторые другие белки [Angst et al., 2001]. Внеклеточная часть кадгеринов достаточно консервативна и представляет собой Са2+связывающие домены, количество которых может варьировать у различных представителей семейства от 5 в случае «классических» кадгеринов до 34 у протокадгеринов [Angst et al., 2001]. Как правило, кадгерины имеют короткую трансмембранную часть, а вариабельная цитоплазматическая часть служит для взаимодействия с многочисленными внутриклеточными белками. «Классические» кадгерины, представляющие лишь небольшую часть молекул, относящихся к семейству кадгеринов, они состоят из пяти внеклеточных доменов (EC1-EC5), трансмембранной и цитоплазматической частей [George and Beeching, 2006; Gumbiner, 2005]. Традиционно «классические» кадгерины подразделяют на два типа: к I типу относятся порядка 30 белков, среди них Е-, М-, N-, Р- и R-кадгерины; ко II типу относятся VE-кадгерин, кадгерин-12 и другие [Suzuki, 1996]. Особенностью кадгеринов I типа является наличие аминокислотной последовательности HAV (His-Ala-Val), которая важна для гомофильного узнавания [Blashuk and Rowlands, 2000]. Считается, что «классические» кадгерины образуют контакты по принципу «застежки-молнии» [Shapiro et al., 1995]. 88 На основе экспериментов, проведенных in vitro на клетках, экспрессирующих Е- и Р-кадгерины, а также E- и N-кадгерины, когда было показано, что экспрессия кадгеринов одного типа обуславливает сегрегацию клеток в культуре [Shan et al., 2000], был сделан вывод о том, что «классические» кадгерины являются молекулами гомофильного узнавания. В то же время, появляется все больше данных о том, что кадгерины, как I типа, так и II типа, способны к гетерофильному взаимодействию как внутри своего типа, так и с кадгеринами другого типа [Shimoyama et al., 2000; Chappuis-Flament et al., 2001; Guthrie, 2002; Niessen and Gumbiner, 2002]. Так, было показано, что клетки в культуре, экспрессирующие N- и R-кадгерины, способны образовывать смешанные агрегаты [Shan et al., 2000]. А Е-кадгерин может замещать N-кадгерин и обеспечивать нормальное развитие сердца у мыши, что позволяет предположить, что в этих условиях определяющим является не тканеспецифическая экспрессия конкретных кадгеринов, а обеспечение кадгерин-опосредованной межклеточной адгезии. В некоторых случаях решающим является не тип экспрессируемых кадгеринов, а уровень их экспрессии. Так, на начальных этапах эмбриогенеза различный уровень экспрессии DE-кадгерина в соседних клетках обеспечивает клеточную сегрегацию и последующие морфогенетические процессы у дрозофилы [Godt and Tepass, 1998]. Первым этапом формирования адгезивных контактов с участием «классических» кадгеринов является латеральная кластеризация кадгериновых молекул с образованием цис-димеров, главную роль при этом играют наиболее аминотерминальные ЕС1 домены; на втором этапе цис-димеры взаимодействуют с аналогичными цис-димерами на соседних клетках. Однако для эффективного гомофильного связывания необходимо еще взаимодействие, как минимум, между ЕС2 и ЕС3 доменами, что предполагает значительное перекрывание между внеклеточными доменами соседних клеток [Chappuis-Flament et al., 2001]. По-видимому, ЕС1 домен играет определяющую роль при формировании цис-димеров, а в транс-взаимодействии между кадгеринами соседних клеток с образованием двумерной решетки клеточных контактов необходимо активное участие остальных доменов [Chappuis-Flament et al., 2001]. Известно, что семейство кадгеринов играет важную роль для роста и созревания сосудов [Cavallaro et al., 2006; George and Beeching, 2006]. Было показано, что в клетках сердечно-сосудистой системы, таких как эндотелий, перициты и гладкомышечные клетки (ГМК), в основном представлены следующие кадгерины: VE-кадгерин, Nкадгерин, R-кадгерин и T-кадгерин (Рисунок 1.1) [Cavallaro et al., 2006; George and Beeching, 2006; Perez-Moreno et al., 2003; Rubina et al., 2005]. VE-кадгерин необходим как для формирования сердца и сосудов в эмбриогенезе, так и для поддержания структуры и 89 функции сосудов во взрослом организме. У эмбрионов экспрессия VE-кадгерина появляется на ранних этапах развития в мезодермальных клетках желточного мешка; также он экспрессируется в прогениторных клетках во время ранней дифференцировки ангиобластов и развития эндокарда [Dejana et al., 2000; Cavallaro et al., 2006]. В литературе существует несколько механизмов, описывающих участие кадгеринов в этих процессах: во-первых, кадгерины обеспечивают локализацию сигнальных и регуляторных молекул в зоне адгезивных контактов, таких как киназы, фосфатазы, Rho ГТФазы и другие; во-вторых, кадгерины участвуют в сигнализации и транскрипции генов, контролируя уровень связанного и свободного катенина; в третьих, кадгерины опосредуют сближение мембран соседних клеток, что важно для формирования плотных и щелевых контактов [Frenzel and Johnson, 1996; Fleming et al., 2000]. Основными белками, взаимодействующими с цитоплазматической частью «классических» кадгеринов, являются катенины и плакоглобины (катенин и р120ctn) (Рисунок 1.10), которые в свою очередь, взаимодействуя с катенинами, винкулином и рядом других белков, связываются с актином [Ozawa et al, 1990; Steinberg and McNutt, 1999]. На каждом этапе образования и функционирования сложного адгезивного комплекса белок-белковые взаимодействия являются предметом регуляции: образование цис-димеров, взаимодействие комлексов кадгерин-катенин, катенин-катенин, катенин-актин, полимеризация актина, а также активация кадгеринов или их переход в неактивное состояние. катенин играет особую роль в регуляции физиологии клетки. Связывание «Armadillo» последовательности необходимо для транспорта катенина вновь с С-концевой синтезированных частью молекул кадгерина кадгерина к плазматической мембране, где происходит дальнейшая сборка кадгериновых контактов. Сродство катенина к кадгерину повышается при фосфорилировании кадгерина по сериновым остаткам и понижается при фосфорилировании катенина по 654 тирозину, что в свою очередь приводит к изменению конформации кадгеринового комплекса и их последующей деградации [Steinberg and McNutt, 1999; Perez-Morenо et al., 2003]. При опухолевом росте уровень тирозинового фосфорилирования в клетке во много раз выше, чем в норме, катенин тоже оказывается гиперфосфорилирован, что способствует увеличению инвазивного потенциала опухолевых клеток [Perez-Morenо et al., 2003]. 90 Рисунок 1.10. Строение «классических» кадгеринов и Т-кадгерина. Некоторые фосфатазы постоянно ассоциированы с адгезивными контактами за счет прямого взаимодействия с кадгеринами (PTP, PTP1B) или катенином (фосфатазыLAR, PTP1B). Мембранные фосфатазы стабилизируют клеточные контакты за счет дефосфорилирования катенина или самих кадгеринов (как в случае PTP), а ингибирование их активности или нарушение взаимодействия фосфатаз с белками кадгеринового комплекса, как в случае фосфатазы PTP приводит к повышению уровня фосфорилирования катенина и дестабилизации контактов. Известно, что VEGFR, связываясь с VEGF, вызывает фосфорилирование VE-кадгерина и дестабилизацию VE-кадгериновых адгезивных контактов, что увеличивает проницаемость эндотелиального барьера и способствует ветвлению сосудов и миграции клеток при ангиогенезе. Недавно была открыта фосфатаза, которая копреципитирует с VE-кадгерином и эффективно его дефосфорилирует. Регуляция баланса между фосфорилированным и дефосфорилированным состоянием катенина или других белков адгезивного комплекса лежит в основе динамичного изменения функции адгезивных контактов [Perez-Morenо et al., 2003]. 91 Взаимодействие между катенином и цитоплазматической частью кадгеринов регулируется Gсубъединицей G-белка. Было показано, что G12/13 связывается с E-, N-кадгеринами и кадгерином 14, причем сайт связывания совпадает с сайтом связывания кадгеринов и РТР1В, что, представляет дополнительные возможности для регуляции активности кадгеринов. Гиперэкспрессия постоянно активной формы G12/13-белка приводит к блокированию взаимодействия катенин с С-концевой частью кадгеринов, что нарушает процессы адгезии в клетках линии НЕК и в эктодерме на поздней стадии гаструляции в эмбрионах Xenopus [Kaplan et al., 2001; Perez-Morenо et al., 2003]. Помимо участия в адгезивных контактах, где катенин находится в связанном состоянии, катенин может образовывать комплекс с TCF/LEF белками, перемещаться в ядро и участвовать в Wnt сигнализации, выполняя функции активатора транскрипции генов, что влияет на пролиферацию клеток [Evers et al., 2000; Lilien et al., 2002]. Р120ctn, также содержит повторы ―armadillo‖ и является ключевым белком, регулирующим функцию кадгеринов, стимулируя их латеральную кластеризацию. Сайт связывания р120ctn находится в околоплазматической части кадгеринов и не перекрывается с сайтами связывания кадгеринов с и катенинами. Фосфорилирование р120ctn по тирозину или серину/треонину стимулирует или подавляет кадгерин-опосредованную межклеточную адгезию, соответственно [Lilien et al., 2002]. В большинстве опухолей эпителиального происхождения экспресиия Е-кадгерина снижена, низкий уровень экспрессии коррелирует с потерей клетками дифференцировки и инвазивностью. Для опухолей пищевода, молочной железы и мочевого пузыря наблюдается еще большая, чем в случае Е-кадгерина, корреляция между низким уровнем экспрессии , катенинов, р120ctn и клеточной дедифференцировкой и метастазированием. Было показано, что нормальная клеточная адгезия в клетках РС3, дефектных по гену катенина, может быть восстановлена после трансфекции кДНК, несущей ген катенина [Evers et al., 2000]. В последнее время все больше исследований посвящено изучению роли белков семейства Rho ГТФаз в регуляции формы клеток и организации цитоскелета. Известно, что Rho стимулирует образование стресс фибрилл, Rac и Cdc42 способствуют образованию ламеллоподий, IQGAP напрямую взаимодействует с катенином и конкурирует с катенином за связывание с цитоплазиатической частью кадгеринов [Angst, 2001; Lilien et al., 2002; Perez-Moreno et al., 2003; Yap and Kovacs, 2003]. Rho ГТФазы функционируют как молекулярные переключатели [Gumbiner, 2000; Yap and Kovacs, 2003; Briggs and Sacks, 2003]. Их участие в клеточной сигнализации 92 обуславливается правильной локализацией во времени и пространстве и способностью в активном состоянии взаимодействовать со своей мишенью. Механизмы передачи сигнала через Rho ГТФазы в различных клетках могут сильно различаться, что обусловлено экспрессией разных кадгеринов и взаимной регуляцией белков внутри семейства Rho ГТФаз [Yap and Kovacs, 2003]. В мигрирующих клетках вновь образующиеся кадгериновые контакты часто локализуются на лидирующем крае, там же где проявляют свою активность Rho ГТФазы. Прижизненные наблюдения с GFP-меченными белками, которые избирательно связываются с активной формой Cdc42, показали, что созреванию клеточных контактов предшествует рекрутирование к плазматической мембране и активация Cdc42 в зоне Екадгериновых контактов, причем связь Cdc42 с кадгериновым комплексом происходит через IQGAP1 [Briggs and Sacks, 2003]. Ингибирование активности Cdc42 препятствует слиянию эпителиальных пластов у дрозофилы в эмбриогенезе. При увеличении проницаемости эндотелиального барьера для его восстановления необходима мембранная активность эндотелиальных клеток и формирование новых адгезивных контактов. Актин-связывающие белки взаимодействуют с комплексом VE-кадгеринкатенин и Cdc42; Cdc42 в свою очередь активирует полимеризацию актина, актин заякоривается на кадгериновый комплекс, а растущие филаменты актина формируют мембранные выросты, необходимые для образования адгезивных контактов [Kouklis et al., 2003]. Генетические и биохимические исследования показали, что в эпителиальных и некоторых других клетках Rac опосредует взаимодействие кадгеринов с динамически активным актином [Kovacs et al., 2002]. Эксперименты по инкубированию клеток на субстрате, содержащем кадгерины, или с «бусами», покрытыми Е-, N- или Скадгеринами, показали, что Rac в первую очередь, в отличие от Rho и Cdc42, рекрутируется в зону формирования новых клеточных контактов. Вместе с Rac в ответ на гомофильное узнавание происходит рекрутирование/активирование белкового комплекса Arp2/3, стимулирующего нуклеацию и полимеризацию актина с последующим расширением зоны вновь формирующихся клеточных контактов. Ингибирование активности Rac блокирует полимеризацию актина в зоне образования новых контактов [Yap and Kovacs, 2003]. В отличие от Rac и Cdc42, для Rho было показано прямое взаимодействие с катенином и р120ctn. В клетках млекопитающих р120ctn ингибирует активность Rho, но активирует Rac и Cdc42. В функциональных тестах с использованием метода РНКинтерференции снижение уровня р120ctn в эмбрионах дрозофилы приводило к 93 ошибочной внутриклеточной локализации Rho и дефектам межклеточной адгезии. Было показано, что очищенные катенин и р120ctn могут напрямую взаимодействовать с Rho, а в эмбрионах, дефектных по гену Rho1 наблюдается нарушение локализации кадгеринов и катенинов с последующими дефектами в формировании нервной трубки [Perez-Mоreno et al., 2003]. Очевидно, что кадгерины представляют собой гликопротеиды, роль которых не ограничивается механическими контактами между соседними клетками. Они участвуют во внутриклеточной сигнализации, регулируя процессы пролиферации, миграции, сортировки клеток, дифференцировки и морфогенеза [Angst et al., 2001; Yap and Kovacs, 2003]. 1.3.2 Экспрессия кадгеринов в нервной системе в эмбриогенезе В эмбриогенезе дифференцировка нервной трубки осуществляется уже на ранних стадиях развития. Еѐ передний конец расширяется, образуя тем самым три первичных мозговых пузыря: первичный передний мозг (prosencephalon), средний мозг (mesencephalon) и первичный задний мозг (rhombencephalon) [Carlson, 1999]. К моменту замыкания задней части нервной трубки по бокам первичного переднего мозга развиваются вторичные выпячивания – глазные пузыри, расширяющиеся латерально. Головной мозг изгибается таким образом, что места изгибов соответствуют границам полостей мозга; два основных изгиба называются головным и затылочным [Gilbert and Singer, 2006]. Затем первичный передний мозг дифференцируется на передний конечный мозг (telencephalon) и промежуточный мозг (diencephalon). Средний мозг на отделы не подразделяется. Первичный задний мозг условно разделяется на будущий продолговатый мозг (myelencephalon) и собственно задний мозг (metencphalon) [Vieira et al., 2010]. Исходно нервная трубка формируется одним слоем псевдомногослойного герминативного нейрального эпителия, который представляет собой быстро делящуюся клеточную популяцию [Fournier-Thibault et al., 2009]. Затем происходит асимметричное деление клеток и миграция клеточных ядер к просвету нервной трубки, что обусловлено градиентным распределением регуляторных факторов [Morrison and Kimble, 2006]. После деления одна или обе дочерние клетки могут терять контакт с вентрикулярной поверхностью и мигрировать наверх. Этот момент является определяющим в дальнейшей судьбе этих клеток: они превращаются либо в нейроны, либо в клетки глии [Gilbert and Singer, 2006]. 94 Периферическую нервную систему образуют клетки нервного гребня, которые остаются между нервной трубкой и вышележащей эктодермой по краям нервных валиков. Фенотип, в который дифференцируются клетки нервного гребня, определяется сигналами, поступающими от соседних клеток [Feng et al., 2009]. При формировании спинного мозга нейроны объединяются в слои или ядра различной формы и размера. Между нейронами устанавливаются связи, в направлении просвета нервной трубки от нейронов отрастают аксоны, образуя бедный клетками краевой слой. Глиальные клетки заключают аксоны этого слоя в миелиновую оболочку. Аксоны спинальных моторных нейронов ствола выходят за пределы нервной трубки и продолжают расти в антерио-постериальном направлении, но на определенном дорзовентральном уровне [Keynes and Stern, 1984]. Этот процесс сопровождается изменениями в экспрессии белков адгезии на поверхности клеток [Varon and Somjen, 1979]. В развивающейся нервной системе во всех ее отделах экспрессируются более 30 различных кадгеринов. Большинство кадгеринов начинает экспрессироваться относительно рано, сразу после сегментации головного мозга. Так, кадгерин-6 экспрессируется в 6-ом ромбомере заднего мозга мышей [Wakazono et al., 1997], в некоторых участках эмбрионального переднего мозга цыпленка экспрессируется Nкадгерин [Ganzler and Redies, 1995]. Экспрессия VE-кадгерина в мозге наблюдается у мышей, начиная с 9 суток эмбрионального развития [Rose et al., 1995]. Подавляющее большинство изученных кадгеринов экспрессируется в определенное время в строго определенных структурах формирующейся центральной нервной системы (ЦНС). Более того границы экспрессии определенных кадгеринов совпадают с границами, формирующимися между специфическими поперечными и продольными структурами ЦНС в раннем эмбриональном развитии. Так, ограниченная во времени и в пространстве экспрессия кадгеринов в определенных отделах эмбрионального головного мозга у мыши была обнаружена для E-кадгерина [Matsunami and Takeichi, 1995], кадгерина-8 [Korematsu and Redies, 1997], и кадгерина-11 [Kimura et al., 1996]. Считается, что кадгерины играют важную роль при развитии ЦНС, контролируя миграцию и избирательную сегрегацию клеток. При формировании спинного мозга нейроны объединяются в слои или ядра различной формы и размера. Мотонейроны, экспрессирующие определенный набор кадгеринов, объединяются в функциональные группы, и именно сочетание различных кадгеринов, их гетерофильные и гомофильные взаимодействия определяют принадлежность к определенному клеточному пулу [Guthrie, 2002]. Экзогенная экспрессия нехарактерных для данной группы мотонейронов 95 кадгеринов ведет к нарушению процессов сегрегации. Более того, было показано, что мотонейроны, их мишени миобласты и Шванновские клетки, с которыми контактируют аксоны, все экспрессируют один и тот же набор кадгеринов [Nakagawa and Takeichi, 1995]; а все афферентные сенсорные нейроны экспрессируют те же кадгерины, что и соответствующие им мотонейроны [Guthrie, 2002]. Как правило, в синаптическом соединении экспрессируется только один тип кадгеринов, где он сначала участвует в формировании синаптического контакта, а потом в поддержании его целостности [Shapiro and Colman, 1998; Bruses, 2000]. Еще один пример участия кадгеринов в процессах морфогенеза представляют собой мигрирующие клетки нервного гребня. Клетки нервного гребня, производные эктодермы, возникают из нервной трубки и мигрируют по строго определенным путям на большие расстояния, где они дифференцируются и дают начало клеткам периферической нервной системы, пигментным клеткам, хрящам и костям черепнолицевого отдела. Перед началом миграции клетки претерпевают эпителиальномезенхимальную трансформацию, изменяют свой фенотип, открепляются от базальной мембраны и мигрируют через окружающий внеклеточный матрикс. При этом экспрессия N-кадгерина, N-САМ и кадгерина 6В подавляется, а кадгеринов 7 и 11 активируется [Nieto, 2001; Takeuchi et al., 2000]. Это означает, что для сегрегации из нервной трубки и последующей направленной миграции в виде отдельных клеток или в виде клеточного агрегата клеткам необходимо изменение набора экспрессируемых кадгеринов. В случае если переключение заблокировано, клетки теряют способность распознавать навигационные сигналы и мигрировать в заданном направлении [Nieto, 2001]. 1.3.3 Роль кадгеринов в развитии сердца и сосудов Сердечно-сосудистая система является первым функциональным органом, развивающимся в эмбриогенезе у млекопитающих [Huber et al., 2004]. Развитие происходит по нескольким параллельным направлениям. Вскоре после стадии гаструляции примордиальная сосудистая система формируется за счет васкулогенеза, во время которого ангиобласты (предшественники эндотелиальных клеток) дифференцируются и сливаются с образованием примитивной сосудистой сети [Carmeliet, 2000]. Затем первичная сосудистая сеть растет и модифицируется за счет формирования новых капилляров в процессе ангиогенеза, превращаясь в крупные и мелкие сосуды, и наполняется кровью [Breier et al., 1996; Risau, 1997; Breier, 2000]. Сердце исходно развивается в области спланхнической мезодермы, позже оно перемещается в область груди. Презумптивные клетки сердца мигрируют из боковых 96 участков мезодермы (на уровне гензеновского узелка). В это время зародыш имеет примитивную организацию и состоит из трех зародышевых листков [Gilbert and Singer, 2006]. На 7 день эмбрионального развития мыши, возникает первичная полоска с узелком на еѐ краниальном конце. На этой стадии, как часть процесса гаструляции, отдельные популяции клеток мигрируют и локализуются справа и слева от средней части диска. Эти области известны как сердце-формирующие регионы [Meilhac et al., 2004]. Описывают две волны миграции клеток из этих регионов. Клетки, мигрирующие в виде первого клона, формируют первичный кардиальный серп. Вторая миграция клеток формирует дополнительную кардиогенную область – вторичный клон сердца. В будущем из клеток первичного кардиального серпа сформируется левый желудочек, а из клеток, происходящих из обоих клонов – камеры предсердия и правый желудочек [Linask and Lash, 1986]. Важную роль в межклеточных взаимодействиях между мигрирующими клетками сердца играет N-кадгерин [Ong et al., 1998]. Предполагается, что N-кадгерин отвечает за связь между соседними миоцитами и за связь эндокарда и миокарда в развивающейся сердечной трубке [Shiraishi et al., 1993]. VE-кадгерин является маркером эндотелиальных клеток и появляется на самых ранних стадиях дифференцировки в мезодермальных клеточных агрегатах, из которых позднее образуются кроветворные островки, далее его экспрессия обнаруживается в гемангиобластах, в формирующихся сосудах и сосудистой системе во взрослом организме [Gory-Faure межклеточную адгезию et в al., 1999]. эндотелии, VE-кадгерин N-кадгерин опосредует опосредует гомофильную взаимодействия эндотелиальных клеток и ГМК в сосудистой стенке [Dejana, 1996]. Нарушение VE- и Nкадгерин-опосредованной адгезии в эмбриогенезе подавляет ангиогенез. Исследования, проведенные на нокаутные мышах по генам VE- и N-кадгерину показали, что VEкадгерин необходим для формирования новых сосудов, а N-кадгерин - для их созревания и формирования полноценной функциональной сосудистой сети. Мутации по VEкадгерину летальны [Gory-Faure et al., 1999]. Помимо своих адгезивных функций VE- и N-кадгерины могут регулировать процессы ангиогенеза за счет прямого взаимодействия с VEGFR-2 и FGF-R [Blashuk and Rowland, 2000]. VEGF может стимулировать тирозиновое фосфорилирование самого VE-кадгерина и белков, с ним ассоциированных:катенин, плакоглобин и р120ctn, что приводит к ослаблению межклеточных контактов [Breier, 2000; Blashuk and Rowland, 2000]. В то же время, в условиях стресса экспрессия VE-кадгерина необходима для проведения сигнала от VEGFR внутрь клетки. Когда эндотелиальные клетки подвергаются растяжению, VEGFR и VE-кадгерин взаимодействуют между собой и компонентами цитоскелета и 97 опосредуют клеточную сигнализацию, необходимую для выживания и ангиогенеза. Антитела к VE- и N-кадгеринам нарушают целостность межклеточных контактов и увеличивают проницаемость эндотелия для различных веществ плазмы крови и нейтрофилов [Blashuk and Rowland, 2000; Gotsch et al., 1997). Эти данные позволяют утверждать, что адгезивные контакты важны не только для поддержания структуры и функции органов и тканей во взрослом организме, но и являются важнейшими регуляторами морфогенетических процессов в эмбриогенезе [Perez-Moreno et al., 2003]. 1.3.4 Т-кадгерин T-кадгерин является нетипичным представителем кадгеринового семейства. В отличие от «классических» кадгеринов, у Т-кадгерина отсутствует трансмембранная и цитоплазматическая часть, и он заякорен на плазматической мембране через гликозилфосфатидилинозитольный (GPI) якорь [Yap and Kovacs, 2003]. Структура внеклеточных доменов Т-кадгерина сходна с «классическими» кадгеринами. Поскольку цитоплазматическая и трансмембранная части является определяющими в обеспечении стабильных межклеточных контактов с участием кадгеринов [Gumbiner, 2005], Ткадгерин, лишѐнный трансмембранной и цитоплазматических частей, вероятно, не участвует в межклеточной адгезии, а играет роль сигнальной молекулы. Т-кадгерин отличается от всех известных кадгеринов тем, что он является GPI-заякоренным белком, благодаря своему GPI якорю он может совершать быстрые латеральные движения в плазматической мембране. Показано, что Т-кадгерин, как и многие другие GPIзаякоренные белки, локализованы в «липидных плотах» - определенных участках плазматической мембраны, обогащенных гликосфинголипидами и холестерином и нерастворимых в неионных детергентах (Рисунок 1.10) [Doyle et al., 1998; Philippova et al, 1998]. Здесь же сконцентрированы многие сигнальные молекулы: Са2+-АТФаза, Са2+каналы, белки семейства Src-киназ, протеинкиназа С, G-белки, кавеолин, урокиназный рецептор и другие [Doyle et al., 1998; Philippova et al., 1998]. По другим данным, Ткадгерин локализуется в ―липидных плотах‖ отличных от кавеол, но возможно, располагающихся в непосредственной близости от них; иммунцитохимический анализ в сочетании с конфокальной и электронной микроскопией показал, что между Ткадгерином и кавеолином в кардиомиоцитах и в ГМК колокализации нет [Doyle et al., 1998; Philippova et al., 2003]. Дойл и соавторы предположили, что Т-кадгерин, располагаясь вблизи кавеол, функционирует как ―губка‖, удерживающая Са 2+ на поверхности плазматической мембраны, и таким образом участвует в регуляции Са 2+зависимой сигнализации и секреции в кавеолах [Doyle et al., 1998]. 98 На моделях in vitro было показано, что Т-кадгерин принимает участие в регуляции роста, жизнеспособности и пролиферации клеток сосудистой стенки. В частности, в культивируемых эндотелиальных клетках экспрессия Т-кадгерина повышается в условиях окислительного стресса, а повышенная продукция свободных радикалов кислорода вызывает повышение экспрессии Т-кадгерина. Эндотелиальные клетки, гиперэкспрессирующие Т-кадгерин, характеризуются сниженным уровнем активации каспаз и повышенной устойчивостью к окислительному стрессу. На основании этих данных было высказано предположение, что сверхэкспрессия Т-кадгерина защищает эндотелиальные клетки от апоптоза, индуцированного стрессом, так как Т-кадгерин активирует PIK3/Akt/mTOR-зависимые пути передачи сигнала, что приводит к подавлению про-апоптотического пути, зависящего от активности р38 МАР-киназы [Joshi et al., 2005]. Для проявления эффектов Т-кадгерина по активации Акt и повышению выживаемости клеток необходимо взаимодействие Т-кадгерина с Grp78, уровень которого, как известно, часто повышен при раке [Philippova et al., 2008; Philippova et al., 2009; Andreeva et al., 2010]. Гиперэкспрессия Т-кадгерина с помощью аденовирусной конструкции в эндотелиальных клетках пупочной вены человека (HUVEC), вызывает изменение клеточного фенотипа и цитоскелета, что приводит к увеличению способности клеток к миграции [Philippova et al., 2005]. Было сделано предположение о том, что гомофильное взаимодействие между молекулами Т-кадгерина на соседних эндотелиальных клетках может приводить к активации малых G-белков, RhoA/ROCK и Rac сигнальных путей с последующей реорганизацией актинового цитоскелета и формированием стресс-фибрилл [Philippova et al., 2005]. Гиперкспрессия Т-кадгерина в гладкомышечных клетках аорты человека и в HUVEC повышает их пролиферативный потенциал [Ivanov et al., 2004b], но то же время, она сопровождается увеличением числа клеток с множественными ядрами. Подавление Т-кадгерина в этих же клетках увеличивает число клеток с множественными центросомами, что предполагает участие Т-кадгерина в регуляции клеточного и центросомального циклов, а возможно, предотвращает апоптоз клеток с нарушениями клеточного деления [Andreeva et al., 2009]. Таким образом, гиперэкспрессия Т-кадгерина in vitro усиливает пролиферацию эндотелиальных клеток и повышает способность HUVEC и других клеток к спонтанной миграции, а также защищает НUVЕС от апоптоза и повышает устойчивость к оксидативному стрессу. В отличие от эндотелиальных клеток, в C6 глиоме [Huang et al., 2003], клетках гепатоцеллюлярной карциномы [Chan et al., 2008], в иммортализованных кератиноцитах [Mukoyama et al., 2005] и в фибробластах от p53(-/-) эмбрионов мыши [Chan et al., 2008] 99 повышенная экспрессия Т-кадгерина подавляет пролиферацию клеток, замедляя переход G2/M. В клетках гепатоцеллюлярной карциномы экспрессия Т-кадгерина также увеличивает чувствительность клеток к TNFα-индуцированному апоптозу [Chan et al., 2008]. Известно, что клетки нейробластом начинают активно делиться в присутствие факторов роста, особенно EGF (фактор роста эпидермиса). Клетки нейробластомы TGW и NH-12, не экспресиирующие Т-кадгерин, теряют способность отвечать усилением пролиферации на добавление EGF после трансфекции кДНК, несущей ген Т-кадгерина [Takeuchi et al., 2000]. На культуре астроцитов показано, что уровень экспрессии Ткадгерина увеличивается при достижении клетками монослоя или в ответ на инкубацию в бессывороточной среде. В нашей лаборатории было показано, что экспрессия Ткадгерина зависит от плотности монослоя (максимальна в конфлюентных культурах), обратимо повышается в условиях депривации в бессывороточной среде, а факторы роста (EGF, PDGF, IGF) дозозависимо подавляют экспрессию Т-кадгерина в культуре ГМК [Kuzmenko et al., 1998]. Эти данные свидетельствуют о том, что Т-кадгерин является сигнальной молекулой, участвующей в регуляции прохождения клеткой клеточного цикла, процессов пролиферации и апоптоза. Экспрессия Т-кадгерина влияет на клеточный цикл, и может модифицировать клеточный ответ на добавление факторов роста, хотя в зависимости от конкретного клеточного контекста клеточный ответ может быть различен. В экспериментах по стимуляции эндотелиальных клеток сосудов и HEK293 клеток, гиперэкспрессирующих Т-кадгерин, с помощью липопротеинов низкой плотности было показано, сигнальный каскад, запускаемый в результате гиперэкспрессии Т-кадгерина приводит к активации фосфолипазы С, образованию IP 3, мобилизации внутриклеточного Са2+, активации тирозиновых киназ Erk1/2 и транслокации в ядро NF-κB [Rubina et al., 2005c]. Хотя Т-кадгерин способен опосредовать слабую Са2+-зависимую гомофильную адгезию клеток в суспензии, подержание стабильной межклеточной адгезии из-за отсутствия трансмембранного и цитоплазматического домена и связи с цитоскелетом, не является основной функцией Т-кадгерина [Vestal and Ranscht, 1992]. In vivo Т-кадгерин локализуется на апикальной поверхности клеток кишечного эпителя цыпленка, а Nкадгерин на базолатеральной, где располагаются межклеточные контакты. При трансфекции культуры клеток MDCK кДНК Т-кадгерина, экспрессия белка Т-кадгерина наблюдается на апикальной поверхности, в то время как N-кадгерин экспрессируется базолатерально [Koller and Ranscht, 1996]. In vitro на модели экспериментальной раны клеточного монослоя иммуноцитохимически показано, что Т-кадгерин локализуется на 100 лидирующем крае мигрирующих клеток (эндотелий, ГМК, линия ECV-304), в то время как VE-кадгерин обнаруживается в зоне контактов [Philippova et al., 2003]. Эти данные хорошо согласуются с представлением о Т-кадгерине как о сигнальной молекуле, регулирующей клеточную адгезию и миграцию. 1.3.5 Навигационная роль Т-кадгерина в нервной системе Т-кадгерин был клонирован из эмбрионального мозга цыплят в начале 90-х годов [Ranscht and Dours-Zimmermann, 1991]. Было обнаружено, что Т-кадгерин экспрессируется в нервной системе в ограниченном количестве клеток в строго определенное время. Иммуногистохимически было показано, что в прорастающих аксонах мотонейронов спинного мозга к своим мышечным мишеням Т-кадгерин экспрессируется в два этапа. На начальных этапах по мере прорастания аксона через спинной мозг к задней конечности цыпленка Т-кадгерин равномерно распределен по всей плазматической мембране, затем, когда происходит разделение аксонов на дорзальные, вентральные и аксоны, иннервирующие мышцы, экспрессия Т-кадгерина снижается. На втором этапе, при формировании синапсов между аксонами и мышцами, экспрессия Ткадгерина опять повышается, причем Т-кадгерин экспрессируется не во всех нейронах, а в некоторых группах - пулах [Guthrie, 2002]. Таким образом, Т-кадгерин направляет рост аксонов мотонейронов и определяет их дальнейшую сортировку на ранних стадиях эмбриогенеза, видимо, позднее Т-кадгерин, определяет взаимодействия нейронов внутри пулов [Fredette and Ranscht, 1994; Guthrie, 2002]. В мезенхиме, где проходит путь, по которому прорастают аксоны, Т-кадгерин экспрессируется в каудальной (задней) части склеротома и на поверхности формирующейся мышцы за исключением будущего синаптического соединения; прорастающие аксоны избегают контакта с этими зонами (Рисунок 1.11). Таким образом, в клетках каудального склетротома и в формирующихся мышцах Т-кадгерин функционирует как молекула-навигатор, определяя путь прорастания аксонов и место формирования синаптичсекого контакта [Fredette and Ranscht, 1994]. Т-кадгерин, в качестве субстрата и как растворимый рекомбинантный белок, ингибирует рост аксонов культивируемых нейронов in vitro, что позволяет предположить гомофильный механизм взаимодействия и негативного регулирования между молекулами Т-кадгерина соседних клеток [Fredette et al., 1996]. Экспрессия Ткадгерина в каудальной части склеротома также определяет миграцию клеток нервного гребня. Клетки нервного гребня мигрируют в вентральном направлении от нервной трубки через сомиты, механизмы данной миграции хорошо описаны. Как только 101 эпидермальные сомиты начинают формировать дерматом (из которого в дальнейшем образуется кожа), миотом (будущие мышцы) и склеротом (будущий позвоночник), большинство мигрирующих в вентральном направлении клеток нервного гребня выбирают направление движения через ростальную часть сомитов каждого склеторотома, избегая при этом каудальной части сомитов [Teillet et al., 1987; Ransch and Bronner, 1991]. Меланоциты кожи исходно также происходят из прогениторных клеток нервного гребня, которые в эмбриогенезе мигрируют из нервной трубки сразу же после закрытия ее концов. Предшественники меланоцитов, происходящие из клеток нервного гребня, во время миграции избегают каудальной части склеротомов, где экспрессирован Т-кадгерин, и мигрируют через ростральную часть, избегая при этом Т-кадгерин позитивную часть склеротомов [Ranscht and Bronner-Fraser, 1991]. Таким образом, в нервной системе Т-кадгерин функционирует как навигационная молекула, вызывающая отталкивание, в результате чего клетки нервного гребня мигрируют по ростральным сомитам, избегая клеток, экспрессирующих Т-кадгерин [Ranscht and Bronner-Fraser, 1991]. Рисунок 1.11. Схема, иллюстрирующая пути ранней миграции прогениторных клеток нервного гребня. Клетки нервного гребня мигрируют из нервной трубки (область отмечена зеленым) и двигаются вентрально (большая изогнутая синяя стрелка) через ростральную часть склеротома сомитов или дорзо-латерально (маленькие синие стрелки) между эктодеромой и дермомиотомом. Вентрально мигрирующие клетки избегают на своем пути каудальной части сомитов и мигрируют только через ростральную часть сомитов [адаптировано Gilbert and Singer, 2006]. 102 Т-кадгерин выявляется не только во время эмбрионального развития, уровень экспресcии Т-кадгерина во взрослом мозге даже выше, чем в эмбриональном [Takeuchi et al., 2000]. Помимо навигационной функции, которую выполняет Т-кадгерин при прорастании аксонов мотонейронов и миграции клеток нервного гребня, Т-кадгерин играет важную роль в регуляции арборизации ретинальных аксонов. Некоторыми авторами было сделано предположение о роли Т-кадгерина в формировании нейромышечных соединений аксонами мотонейронов и в формировании синапсов ретинальными аксонами [Philippova et al., 2009]. При формировании синапсов, Т-кадгерин, в отличие от N- и R-кадгеринов, не концентрируются в зоне синаптического контакта, а экспрессируется на экстрасинаптической поверхности мембраны, точные механизмы его участия в синаптогенезе неизвестны. С использованием методов Нозерн-блота и иммуногистохимии экспрессия Т-кадгерина была обнаружена в ЦНС человека: в коре головного мозга, гипоталамусе, средних отделах мозга, продолговатом мозге, в таикх клетках как пирамидальные и непирамидальные нейроны, астроциты, клетки Пуркинье, корзинчатые клетки и другие [Philippova et al., 2009]. В то же время в спинном мозге уровень экспрессии Т-кадгерина был очень низким или практически отсутствовал, экспрессии Т-кадгерина в клетках олигодендроглии обнаружено не было [Takeuchi et al., 2000]. Во взрослом организме в норме сосудистая система находится в состоянии равновесия, процессы роста или регрессии сосудов четко регулируются. Избыточный или недостаточный ангиогенез, как правило, сопутствует опухолевому росту, патологическому ремоделированию сосудов и различным гиперплазиям тканей. Поскольку рост опухолей связан с рекрутированием новых сосудов, подавление ангиогенеза считается перспективным подходом при терапии раковых заболеваний [Folkman, 1971; Cao, 2005]. В литературе имеются данные, указывающие на взаимосвязь между потерей локуса хромосомы 16q24, содержащей ген Т-кадгерина, или метилированием промотора гена Т-кадгерина и возникновением злокачественных новообразований [Toyooka et al., 2001; Takeuchi et al., 2001]. О роли Т-кадгерина в опухолевой прогрессии в литературе имеются противоречивые данные [Andreeva and Kutuzov, 2010]. Рядом авторов было предположено, что Т-кадгерин функционирует как фактор опухолевой супрессии; снижение его экспрессии в результате аллельной потери или гиперметилирования промотора гена, или в результате других причин связаны с ростом и метастазированием определенных видов опухолей [Takeuchi et al., 2002a; Andreeva and Kutuzov, 2010]. 103 Подавление экспрессии Т-кадгерина, как было показано, коррелирует со злокачественностью фенотипа и онкогенностью раков молочной железы [Riener et al., 2008], легких [Sato et al., 1998] и желчного пузыря [Maruyama et al., 2001]. Однако при других типах опухолей, таких как рак яичников, эндометрия [Suehiro et al., 2008] и остеосаркома [Zucchini et al., 2004], снижение экспрессии Т-кадгерина коррелирует с лучшей выживаемостью пациентов. Повышение экспрессии Т-кадгерина характерно для инвазивных гепатоцеллюлярных характеризующихся высокой карцином степенью [Riou et al., и астроцитом, При опухолевой 2006] злокачественности. неоваскуляризации гепатоцеллюлярной карциномы наблюдается повышение экспрессии Т-кадгерина в эндотелиальных клетках внутриопухолевых капилляров, хотя в окружающей ткани и в ткани печени Т-кадгерин не обнаруживается [Adachi et al., 2006]. Увеличение экспрессии Т-кадгерина в эндотелиальных клетках гепатоцеллюлярной карциномы коррелирует со степенью опухолевой прогрессии [Adachi et al., 2006]. Участие Т-кадгерина в неоангиогенезе меланомы было показано на модели сфероидов in vitro, сформированных опухолевыми клетками при со-культивировании с эндотелиальными клетками: повышение экспрессии Т-кадгерина в эндотелиальных клетках потенцировало неоангиогенез [Ghosh et al., 2007]. Трансфекциия кДНК, содержащей ген Т-кадгерина, существенно ингибирует рост раковых клеток молочной железы в культуре, что также сопровождается изменением их фенотипа с инвазивного на фенотип, характерный для нормальных эпителиальных клеток [Lee, 1996]. Возможно, что противоречивые данные, полученные при изучении роли экспрессии Т-кадгерина в опухолевой прогрессии на системах in vitro и in vivo, обусловлены сложностью и многоплановостью процессов опухолевого роста, включающих как нарушения регуляции пролиферации самих опухолевых клеток, так и взаимодействия между эндотелиальными и стромальными клетками внутри растущей опухоли. В норме в сердечно-сосудистой системе Т-кадгерин экспрессируется в кардиомиоцитах и во всех слоях сосудов: в эндотелиальных клетках, ГМК и перицитах. Накопленные в литературе данные указывают на то, что существует корреляция между повышенной экспрессией Т-кадгерина в клетках сосудов и прогрессированием сердечнососудистых заболеваний, таких как атеросклероз и рестеноз [Ivanov et al., 2001, Kudrjashova et al., 2002]. Кроме того, Т-кадгерин был также обнаружен на микрочастицах в крови пациентов с атеросклерозом, что, по мнению авторов, отражает активацию или повреждение эндотелия [Philippova et al., 2011]. Также было показано [Andreeva et al., 2010], что Т-кадгерин может участвовать в патогенезе инсулинорезистентности в 104 эндотелиальных клетках: Т-кадгерин снижает чувствительность эндотелиальных клеток к инсулину по механизму хронической активации обратной Akt/mTOR-зависимой петли и увеличения деградации субстрата рецептора инсулина. Эти данные позволяют предполагать, что повышенный уровень Т-кадгерина может способствовать активации/дисфункции эндотелиальных клеток, повышению проницаемости эндотелия и повреждению сосудов при описанных патологических состояниях. 1.3.6 «Гормоноподобные» эффекты липопротеидов низкой плотности (ЛНП). Ткадгерин и ЛНП Липопротеиды низкой плотности (ЛНП) представляют собой макромолекулярные комплексы и служат для транспорта холестерина и триглицеридов к периферическим тканям в организме. Известно, что повышенный уровень ЛНП в плазме крови служит фактором риска развития атеросклероза, так как приводит к накоплению холестерина в сосудистой стенке [Goldstein et al., 1985]. Перенос ЛНП происходит путем рецепторопосредованного эндоцитоза, в котором участвует «классический» апо В, Е рецептор [Brown and Goldstein, 1986]. Существуют данные, полученные в нашей и других лабораториях, о том, что ЛНП способны влиять на функциональную активность целого ряда клеток, причем эти эффекты не связаны с эндоцитозом ЛНП [Block et al., 1988; Buhler et al., 1991; Bochkov et al., 1991]. Характерными чертами таких эффектов являются их быстрота и обратимость. Для их развития не требуется длительная преинкубация клеток с липопротеидами, необходимая для изменения внутриклеточного содержания липидов. В частности, было показано, что липопротеиды активируют макрофаги [Kelley et al., 1988], вызывают изменение формы и агрегацию тромбоцитов, секрецию сурфактанта альвеолоцитами [Voyno-Yasenetskaya et al., 1993], миграцию и пролиферацию ГМК в сосудистой стенке [Resink et al., 1995], регулируют тонус сосудов и оказывают ряд других эффектов на клетки крови и сосудистой стенки [Block et al., 1988; Sachinidis et al., 1990]. Такие эффекты сходны с действием гормонов и медиаторов, и поэтому было предложено называть их «гормоноподобными» [Block et al., 1988]. Ряд данных позволяет предположить, что в основе «быстрых» клеточных эффектов липопротеидов лежит активация систем внутриклеточной сигнализации. В пользу этой гипотезы свидетельствуют данные о способности липопротеидов активировать фосфолипазу С и фосфоинозитидный обмен, повышать уровень цитоплазматического Ca2+, а также влиять на активность аденилатциклазы, изменять рН цитоплазмы и стимулировать фосфорилирование специфических клеточных белков [Block et al., 1988; Bochkov et al., 1991; Matsumura et al., 1997; Honda et al., 1999]. 105 В нашей лаборатории было показано, что ЛНП стимулируют фосфоинозититидный обмен, активируют фосфолипазу С и вызывают мобилизацию внутриклеточного Ca2+ в ГМК клетках [Bochkov et al., 1993]. В отличие опосредуемого апо от хорошо изученного В,Е-рецептором, «метаболического» механизмы действия «гормоноподобных» ЛНП, эффектов неизвестны. Характерной особенностью «гормоноподобных» эффектов, вызываемых ЛНП, является их быстрота, насыщаемость и обратимость, что свидетельствует об участие специфического рецептора в передаче сигнала от ЛНП внутрь клетки, однако ни природа предполагаемого рецептора, ни механизмы его сопряжения с системами вторичных посредников до конца не выяснены. Ранее в нашей лаборатории был обнаружен атипичный участок специфического связывания ЛНП на культивируемых ГМК сосудов [Bochkov et al., 1994]. Классический анализ связывания меченных ЛНП с мембранами ГМК сосудов показал, что ГМК в основном экспрессируют низкоаффинный участок специфического связывания ЛНП с Кd 40-50 мкг/мл, в то время как «классический» имеет Кd 1-2 мкг/мл [Tkachuk et al, 1994]. Связывание ЛНП с мембранами ГМК оказалось специфичным для ЛНП (как для нативных, так и модифицированных), Ca2+-зависимым, нечувствительно к присутствию полианионов, поликатионов и антител к апо В, Е рецептору [Bochkov et al., 1994; Bochkov et al., 1996; Tkachuk et al, 1994; Kuzmenko et al, 1994]. Коклюшный токсин ингибировал ЛНП-зависимую мобилизацию Ca2+ и активацию фосфоинозитидного обмена в культивируемых ГМК сосудов, что свидетельствовало об участие G-белков (Go или Gi) в передачи сигнала от ЛНП-рецептора к системе вторичных посредников и об отличие этого связывания от связывания с «классическим» рецептором ЛНП. Для изучения природы этого атипичного участка связывания в нашей лаборатории был использован метод лигандного блоттинга, который ранее был успешно применен для характеристики других рецепторов ЛНП. Используя мембраны ГМК, выделенных из медии аорты человека, были выделены, очищены и охарактеризованы два белка р105 и р130 кДа. Анализ лигандной селективности и чувствительности к различным ингибиторам показал, что р105/р130 является новым ЛНП-связывающим белком, отличным от всех известных липопротеид-связывающих белков и рецепторов [Bochkov et al., 1996]. Параметры связывания меченных ЛНП с белками р105/р130 совпадают с параметрами ЛНП-опосредованной активации систем вторичных посредников [Tkachuk et al, 1994; Kuzmenko et al, 1994; Stambolsky et al., 1999]. Сиквенирование очищенных форм р105/р130 показало, что последовательность этих 106 белков соответствует последовательности Т-кадгерина, причем р105 представляет собой зрелую форму, а р130 – предшественник, содержащий пропептид [Tkachuk et al, 1996]. Связывание Т-кадгерина с ЛНП ингибируется антителами против ЛНП (антитела против апо В белка) [Bochkov et al., 1996]. Таким образом, был получен ряд данных, позволяющих предположить, что Ткадгерин, являясь атипичным представителем семейства кадгеринов, возможно, представляет собой предполагаемый ―сигнальный» ЛНП-рецептор [Tkachuk et al, 1994; Tkachuk et al, 1996; Kuzmenko et al, 1994]. ЛНП не является единственным лигандом для Т-кадгерина. Оказалось, что высокомолекулярные комплексы адипонектина также являются специфичными лигандами Т-кадгерина [Hug et al., 2004]. Адипонектин (30-кДа белок адипоцитов, родственный комплементу) является цитокином, продуцируемым жировой тканью. Этот белок представляет собой уникальный адипоцитарный гормон, который регулирует обмен липидов и глюкозы [Brakenhielm et al., 2004]. Его уровень заметно снижается в плазме крови пациентов с метаболическим синдромом, ожирением, диабетом II и атеросклерозом. Кроме того, адипонектин способен подавлять рост сосудистых клеток и тем самым препятствовать ремоделированию артериальной стенки. Так, инактивация гена адипонектина стимулирует формирование неоинтимы у мышей при экспериментальном повреждении артерий [Parker-Duffen et al., 2013; Parker-Duffen and Walsh, 2014]. По-видимому, в сосудистой стенке адипонектин обладает защитным антиатерогенным действием. В пользу этого предположения свидетельствует способность этого фактора препятствовать связыванию ЛНП с протеогликанами и их накоплению в артериальной стенке [Kobayashi et al., 2005]. Кроме того, адипонектин предотвращает формирование атеросклеротических поражений у мышей с инактивированным геном аро Е [Okamoto et al., 2002]. Помимо Т-кадгерина, существуют еще два мембранных рецептора адипонектина, AdipoRl и AdipoR2, с отдаленной гомологией к 7-доменным рецепторам, сопряженным с G-белками [Hug et al., 2004]. В отличие от AdipoRl и AdipoR2, Т-кадгерин, играет роль рецептора высокомолекулярных изоформ адипонектина, специфичного для сосудистой стенки и сердца [Hug et al., 2004; Parker-Duffen and Walsh, 2014]. Механизмы взаимодействия адипонектина и Т-кадгерина, а также внутриклеточные сигнальные пути, опосредующие эффекты такого связывания, по-прежнему остаются невыясненными, а данные литературы местами противоречивы. В опытах in vitro, по данным одних авторов, адипонектин подавляет индуцированные различными факторами роста пролиферацию и миграцию как ГМК, так 107 и эндотелиальных клеток [Brakenhielm et al., 2004; Wang et al., 2005]. По другим данным, добавление адипонектина в среду культивирования эндотелиальных клеток стимулирует их миграцию и пролиферацию, а подавление экспрессии Т-кадгерина с использованием siRNA в этих клетках нивелирует стимулирующие эффекты адипонектина. На моделях ангиогенеза в хориоаллантоиcной мембране цыпленка и в роговице мыши адипонектин подавляет рост кровеносных сосудов. Это действие адипонектина обусловлено его способностью активировать каспазы -8, -9 и -3 в эндотелиальных клетках в определенных условиях и тем самым вызывать их апоптоз [Brakenhielm et al., 2004]. Взаимодействие адипонектина с Т-кадгерином на клетках сосудистой стенки вызывает активацию NF-В, тогда как в клетках печени и мышц этот цитокин стимулирует cAMP-зависимую протеинкиназу и окисление жирных кислот [Hug et al., 2004]. В то же время в литературе имеются данные о том, что подавление экспрессии адипонектина в экспериментальных условиях приводит к эндотелиальной дисфункции у животных, а его гиперэкспрессия способствует заживлению ран [Parker-Duffen and Walsh, 2014]. На экспериментальной модели ишемии задней конечности с последующей реваскуляризацией у мышей, дефицитных по адипонктину, и у мышей, дефицитных по Т-кадгерину, были обнаружены сходные нарушения кровотока по сравнению с контролем. При добавлении экзогенного адипонектина у мышей, дефицитных по адипонектину, наблюдалось восстановление кровотока до уровня, сравнимого с контролем. Однако у мышей, дефицитных по Т-кадгерину, восстанавливающих эффектов при добавлении экзогенного адипонектина зарегистрировано не было. Авторами был сделан вывод о том, что Т-кадгерин ко-локализуется с адипонектином в скелетных мышцах и опосредует его васкулопротективные эффекты [Parker-Duffen et al., 2013]. Ни мыши, дефицитные по адипонектину, ни мыши, дефицитные по Т-кадгерину, в стандартных условиях не имеют выраженного фенотипа. Однако, как на модели ишемии задней конечности, так и на моделях острой или хронической ишемии миокарда у мышей, дефицитных по адипонектину и по Т-кадгерину, были обнаружены сходные нарушения реваскуляризации тканей. В случае имшемического повреждения сердца и у мышей, дефицитных по адипонектину, и у мышей, дефицитных по Т-кадгерину, наблюдались гипертрофия миокарда и выраженное снижение количества формирующихся капилляров. На модели острой ишемии-реперфузии миокарда у мышей, дефицитных по Т-кадгерину и по адипонектину, размер инфаркта был больше, чем у контрольных мышей. Введение экзогенного адипонектина уменьшало размер инфаркта у 108 мышей, экспрессирующих Т-кадгерин, но не оказывало стимулирующего действия в отсутствии Т-кадгерина. В основе кардиопротективного действия адипонектина лежит активация сердечной AMP киназы (AMP activated protein kinase), которая является нисходящим сигнальным белком-посредником при активации сигнализации с участием адипонектина, и для активации которой необходимо присутствие Т-кадгерина. По мнению авторов, экспрессия Т-кадгерина на эндотелиальных клетках обеспечивает рекрутирование адипонектина из периферической крови и его локализацию на эндотелии, что и обуславливает кардиопротективное действие адипонектина и его положительные эффекты по реваскуляризации ишемизированных конечностей in vivo [Denzel et al., 2010; Parker-Duffen et al., 2013; Parker-Duffen and Walsh, 2014]. 109 Таким образом, из приведенных данных литературы следует, что процессы ангиогенеза и функционирования сосудов находятся под контролем различных факторов роста, хемокинов, цитокинов и навигационных молекул. Многие из этих факторов помимо своих рецепторов взаимодействуют друг с другом, модулируя и модифицируя клеточный ответ. Т-кадгерин является навигационной молекулой, регулирующий рост аксонов и миграцию клеток нервного гребня, что было продемонстрировано на эмбрионах цыпленка и в модельных экспериментах на культуре нормальных и трансформированных клеток. Однако неясно, экспрессируется ли Т-кадгерин в эмбриогенезе млекопитающих, и если да, то каков паттерн его пространственновременной экспрессии. Экспрессируется ли Т-кадгерин в сердечно-сосудистой системе как во взрослом организме человека, или он выполняет роль навигационной молекулы только в нервной системе. Т-кадгерин экспрессируется в сосудах взрослого организма у человека, однако, функция его при этом неизвестна. Влияет ли Т-кадгерин на барьерную функцию эндотелия или нет, и если да, то какова сигнализация, которая активируется с участием Т-кадгерина в эндотелиальных клетках. Неясно, экспрессируется ли Ткадгерин при ангиогенезе в физиологических условиях и при патологии, например, при опухолевом ангиогенезе. Есть данные литературы об изменении экспрессии Т-кадгерина в опухолевых клетках и в сосудах, прорастающих в опухоль, однако, они противоречивы, в литературе нет четкого представления о том, является ли Т-кадгерин фактором, способствующим опухолевой прогрессии или Т-кадгерин – опухолевый супрессор. Экспрессия Т-кадгерина повышается при атеросклерозе, нерешенным в данной проблеме является вопрос о клеточных рецепторах, которые опосредуют «гормоноподобные» эффекты ЛНП. В литературе имеются данные, позволяющие предполагать, что Т-кадгерин может быть рецептором ЛНП, опосредующим их «гормоноподобные» эффекты, однако, доказательств, подтверждающих этот факт в литературе нет, а также неизвестны сигнальные эффекты, которые потенциально опосредуют ЛНП в клетках при связывании с Т-кадгерином. Мы предполагаем, что Т-кадгерин является регулятором функционирования и/или ремоделирования сосудов во взрослом организме, а возможно и навигационной молекулой, регулирующей процессы ангиогенеза. Проверке этих предположений посвящены последующие главы. 110 ГЛАВА 2. МАТЕРИАЛЫ И МЕТОДЫ 2.1 Антитела и реагенты Для выявления экспрессии белков в клетках и тканях методом Вестерн блоттинга и иммунофлуоресцентного окрашивания в сочетании с микроскопическим анализом использовали коммерческие антитела, указанные в Таблице 2.1. Таблица 2.1. Первые антитела, использованные в работе. Антиген CD31 антиген сосудистых клеток Т-кадгерин αSMA антиген гладкомышечных клеток NG2 антиген перицитов vWF сосудистых клеток CD31 антиген сосудистых клеток αSMA антиген гладкомышечных клеток VE-кадгерин Фосфорилированный VEкадгерин Y658 Фосфорилированный VEкадгерин Y731 N-кадгерин Бета-катенин Фосфорилированный бетакатенин Т41 Катенин p120ctn Клатрин PAK1 Фосфорилированный PAK1 T212 ROCK-II Src Специфичность Крысиные против мыши Компания-производитель BD Кроличьи поликлональные против человека Кроличьи поликлональные против мыши Кроличьи поликлональные против мыши Мышиные моноклональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные ProSci Epitomic Chemicon BD BD Epitomic Abcam Abcam Abcam SantaCruz Abcam Abcam SantaCruz Abcam Abcam Abcam SantaCruz Abcam 111 Фосфорилированный Src Y418 p38 Фосфорилированный р38 Т180+Т202 Erk1/2 Фосфорилированный Erk1/2 Т185+Т202 ZO-1 Окклюдин Клаудин-5 Кавеолин-1 EEA1 LAMP1 GAPDH LIMK1 Anti-acetylated α-тубулин с-Myc эпитоп (gp100) Ab-3 PCNA антиген ядер против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Кроличьи поликлональные против человека Мышиные моноклональные против человека Мышиные моноклональные против человека Abcam Sigmaaldrich Abcam Abcam Abcam Invitrogen Abcam Invitrogen BD Abcam Abcam Sigmaaldrich, Santa Cruz Cell Signaling Technology Sigmaaldrich Santa Cruz Lab Vision- Neomarkers DakoCytomation В качестве вторых антител для иммунофлуоресцентного выявления антигенов использовали флуоресцентно-меченные антитела (Molecular Probes); вторые ослиные антитела против мыши и против кролика для Вестерн блоттинга были конъюгированы с пероксидазой хрена HRP (Jackson Immuno-Research Laboratories). В экспериментах ацетилированные также липопротеиды были низкой использованы плотности (ЛНП), следующие реагенты: конъюгированные с флуорохромом AlexaFluor488 (Invitrogen); рекомбинантные домены ЕС1 и ЕС5 Ткадгерина, полученные ранее в нашей лаборатории; краситель для прижизненного выявления лизосом LysotrackerRed® (Invitrogen); фаллоидин для окраски актиновых стресс фибрилл phalloidin AlexaFluor488 (Molecular Probes); реагенты для определения апоптоза и 112 некроза клеток АnV-РЕ (Аннексин V – фикоэритрин, BD Biosciences) и 7-AAD (BD). Реагенты для Вестерн блоттинга были получены от компаний Bio-Rad и Sigmaaldrich; реагенты, используемые для иммуноцитохимического и иммунофлуоресцентного окрашивания, были произведены фирмами Gibco и Electron Microscopy Sciences. Трипсин и желатин были получены от компаний Sigmaaldrich или GIBCO. 2.2 Культивирование эукариотических клеток В работе были использованы следующие типы эукариотических клеток: первичная культура эндотелиальных клеток пуповины человека HUVEC 0-6 пассажа (Cambrex), первичная культура стромальных клеток из подкожной жировой клетки мыши, линия клеток мышиных фибробластов L929 (ATCC №CCL-1) и HIN3T3 (ATCC №CRL-1658), линия клеток эмбрионального почечного эпителия человека HEK293 (ATCC №CRL-1573) и линия опухолевых клеток мышиной меланомы B16F10 (ATCC №CRL-6475). Клетки, если не написано иначе, высевали в культуральные планшеты с плотностью 1х10 4 клеток/см2 и культивировали в инкубаторе при +370С и 5% СО2. Для культивирования клеток L929, HIN3T3 и HEK293 использовали среду Игла, модифицированную Дульбекко (ДМЕМ), содержащую пенициллин (100 ед/мл), стрептомицин (0,1 мг/мл) и 10% фетальную бычью сыворотку (ФБС). Для культивирования клеток B16F10 использовали среду RPMI1640, также содержащую пенициллин (100 ЕД/мл), стрептомицин (0,1 мг/мл), 10% ФБС и 0,2 мМ L-глутамин. Клетки HUVEC культивировали в среде EGM-2 (CC-4176, Lonza Cambrex), содержащей все необходимые ростовые факторы (SingleQuots® Kit, Cambrex), а также пенициллин (100 ед/мл), стрептомицин (0,1 мг/мл) и 10% ФБС, инактивированной при +560С 30 мин. Перед высаживанием клеток HUVEC, поверхность культурального планшета покрывали раствором 0,1% желатина на фосфатно-солевом буфере ФСБ в течение 30 мин при +37ОС. Клетки всех типов пассировали, обрабатывая 0,25% раствором трипсина в 1 мМ ЭДТА. 2.3 Трансфекция эукариотических клеток Клетки B16F10 стабильно трансфицировали полноразмерной кДНК Т-кадгерина человека, клонированной в плазмиду для эукариотической экспрессии рсDNA™3.1 (Invitrogen, США) с устойчивостью к G418 (Geneticin®) (Rubina et al., 2005). Трансфекцию проводили с использованием реагента Lipofectamine™ 2000 (Invitrogen, США) согласно протоколу фирмы-производителя. Путѐм отбора в среде, содержащей селективный 113 антибиотик G418 (Geneticin®, Invitrogen, США) в концентрации 2 мг/мл, получали суммарную популяцию клеток мышиной меланомы B16F10, гиперэкспрессирующих Ткадгерин. Для контрольных экспериментов клетки меланомы трансфицировали контрольной плазмидой рсDNA™3.1/CAT (содержит фрагмент кДНК люциферазы в антисмысловой ориентации) (Invitrogen, США) и путѐм отбора в среде, содержащей селективный антибиотик G418 в концентрации 2 мг/мл, получали суммарную популяцию контрольных клеток. Уровень экспрессии Т-кадгерина в суммарной популяции контрольных клеток мышиной меланомы и клеток, экспрессирующих Т-кадгерин, подтверждали методом Вестерн блоттинга. Для получения клонов меланомы с разным уровнем экспрессии Т-кадгерина клетки после трансфекции рассаживали на 98-луночные планшеты методом предельного разведения и получали клоны клеток в результате селекции на среде, содержащей G418 в концентрации 2 мг/мл. Уровень экспрессии Т-кадгерина в клонах клеток мышиной меланомы подтверждали методом вестерн блоттинга и денситометрическим анализом. Клетки линий L929, HEK293, NIH3T3 трансфицировали плазмидами для эукариотической экспрессии по следующему протоколу. Клетки HEK293 ко- трансфицировали плазмидами pcDNA3.1/T-cad (содержит полноразмерную кДНК Ткадгерина) и pcDNA3.1/GFP (содержит ген зелѐного флуоресцентного белка GFP). В качестве контроля клетки HEK293 ко-трансфицировали плазмидами pcDNA3.1 (содержит фрагмент кДНК люциферазы в анти-смысловой ориентации) и pcDNA3.1/GFP. Селекцию клонов проводили в среде, содержащей антибиотик пуромицин (GIBCO) в концентрации 10 мкг/мл, а также по флуоресценции зелѐного белка. При этом были получены 2 клона HEK293 клеток, ко-экспрессирующие Т-кадгерин и GFP (Th5 и Th8), и клоны контрольных клеток, экспрессирующие GFP. Клоны клеток L929, экспрессирующие ген Т-кадгерина человека (TC3) и контрольную плазмиду (К9) были получены в нашей лаборатории ранее Д. Ивановым. Для ко-экспрессии Т-кадгерина и LIMK1 клетки NIH3T3 ко-трансфицировали плазмидами pcDNA3.1/T-cad и myc-LIMK1 (содержит полноразмерную LIMK1 и mycэпитоп). В качестве контроля NIH3T3 ко-трансфицировали pcDNA3.1 и плазмидой mycLIMK1. Для измерения SRE активности клетки ко-трансфицировали плазмидами pSRE.L (Stratagene) для экспрессии SRE и плазмидой pCMV-Gal (Clontech) для экспрессии bгалактозидазы и контроля трансфекции. Также в зависимости от условий эксперимента клетки ко-трансфицировали плазмидами, несущими мутантные гены неактивных Rho ГТФаз: RhoA (RhoN17), Rac1 (RacN17) и Cdc42 (Cdc42N19), которые были любезно 114 предоставлены лабораторией доктора Т. Войно-Ясенецкой (University of Illinois at Chicago, Chicago, USA). Для положительного контроля в методе измерения SRE активности использовали плазмиду p114RhoGEF (более подробно о порядке трансфекции в методе измерения SRE активности написано в соответствующем разделе). В методе выделения активных Rho ГТФаз для трансфекции NIH3T3 клеток использовали плазмиды, кодирующие активные формы ГТФаз: для RhoA – RhoV12, для Rac1 – RacV12, для Cdc42 – CdcV12. Также использовали плазмиду для гиперэкспрессии Ткадгерина pcDNA3.1/T-cad и контрольную pcDNA3.1. Плазмиды RhoV12, RacV12 и Cdc42V12 также были предоставлены лабораторией доктора Т. Войно-Ясенецкой (University of Illinois at Chicago, Chicago, USA). Для подавления экспрессии Т-кадгерина РНК интерференцией (siRNA), HUVEC 2 пассажа ко-трансфицировали интерферирующими РНК последовательностями (Dharmacon) (siRNA/T-cad) в количестве 50 нМ и 100 нМ на лунку в 24 луночной планшете при плотности клеток 1х105/лунку, и плазмидой pcDNA3.1/GFP для маркѐра положительно прошедшей трансфекции. Контрольные клетки трансфицировали плазмидой pcDNA3.1/GFP. Для гиперэкспрессии Т-кадгерина в эндотелиальных клетках, клетки 2 пассажа ко-трансфицировали плазмидами pcDNA3.1/T-cad и pcDNA3.1/GFP. В качестве контроля клетки ко-трансфицировали плазмидами pcDNA3.1 и pcDNA3.1/GFP. Для трансфекции использовали клетки, достигшие 70% плотности монослоя. Плазмиды (по 5 мкг на 35 мм лунку культуральной планшеты) или siRNA Ткадгерина (50 нМ и 100 нМ) смешивали со 100 мкл бессывороточной среды Opti-MEM (GIBCO). Через 5 мин инкубации в раствор добавляли 20 мкл реагента Lipofectamine2000 (Invirogen) для NIH3T3 и HEK293 клеток, или CytoPure-huv для HUVEC клеток, разведенного предварительно также в 100 мкл Opti-MEM. Полученную смесь плазмид и трансфекционного реагента инкубировали в течение 20 мин при комнатной температуре, после чего раствор ДНК (siRNA/T-cad) и реагента добавляли в среду культивирования и инкубировали в течение ночи. На следующий день меняли среду на стандартную среду с добавками ДМЕМ для NIH3T3 и HEK293 клеток, или среду EGM-2 для HUVEC. Далее клетки использовали в эксперименте или клонировали. Для селекции клонов клетки линии HEK293 высевали в 96-луночные культуральные планшеты и инкубировали в среде ДМЕМ с 10% ФБС и необходимой концентрацией селективного антибиотика. После отбора клонов, клетки наращивали в 100 мм культуральных чашках Петри. Экспрессию Ткадгерина в клетках, содержание фосфорилированной LIMK1, myc эпитопа, активных Rho ГТФаз анализировали с помощью Вестерн блоттинга. 115 2.4 Создание лентивирусных конструкций для трансдукции эндотелиальных клеток HUVEC Лентивирусная конструкция pSIH-H1-Tcad, экспрессирующая Т-кадгерин, была приготовлена с использованием pSIH1-H1-puro лентивирусного вектора (System Bioscience) согласно следующему протоколу. Из вектора был удалѐн ген резистентности пуромицина (puro), при этом клонированная кДНК Т-кадгерина человека (GenBank Accession NoNM_001257.3) была вставлена рядом с ранним промотором цитомегаловируса PCMV. Пустой вектор pSIH-H1-control без каких-либо вставок использовался для контрольной трансдукции. Для РНК интерференции, приводящей к «выключению» экспрессии гена Ткадгерина, использовали лентивирусную конструкцию pSIH-H1-puro-si-Tcad. Эта конструкция экспрессирует короткие шпилечные РНК олигонуклеотиды (shRNA), связывающиеся с последовательностью Т-кадгерина GGTGAGTGTCTTAGCATAT, расположенной на 3‘-нетранслируемой области мРНК Т-кадгерина. Экспрессия shRNA управляется промотором РН1 RNase H1 RNA человека. Для изготовления VSVG-псевдотипичных лентивирусных частиц, линейные клетки для упаковки вирусных частиц НЕК293-TN в количестве 15x106 (System Biosciences) трансфицировали смесью из 6 мкг лентивирусного вектора и 30 мкг плазмиды pPACK (System Biosciences) с использованием трансфекционного реагента Lipofectamine™ 2000 (Invitrogen, США). Спустя 72 часа после трансдукции собирали культуральную среду и центрифугировали в течение 5 часа при 60000 g и температуре +40C. При таких параметрах центрифугирования псевдовирусные частицы остаются в осадке. Далее осадок ресуспендировали в малом объѐме буфера ФСБ, аликвотили, замораживали и хранили при температуре –800C. Для каждой партии псевдовирусных частиц проводили оценку эффективности трансдукции с использованием клеток карциномы человека H1299, а также определяли показатель множественности заражения MOI. Для дальнейших экспериментов использовались частицы с показателем MOI не ниже 0,5. Для лентивирусной трансдукции эндотелиальные клетки 2го пассажа, растущие в 6-луночном планшете (70% монослой), инкубировали в течение 24 часов с добавлением 300 мкл раствора псевдовирусных частиц, растворѐнных в 2 мл свежей среды роста для эндотелиальных клеток, в присутствии 5 мкл/мл полибрена (Sigmaaldrich). Через 24 ч клеткам меняли среду культивирования на свежую среду, и далее клетки использовали в течение 2 недель в экспериментах. Для определения показателя MOI после трансдукции из клеток выделяли геномную ДНК и анализировали методом полимеразной цепной реакции ПЦР с использованием 116 праймеров GGGGACTGGAAGGGCTAATTC и TGCGTCGAGAGAGCTCTGGTT. Праймеры были разработаны для точного распознавания интегрированных копий лентивирусных конструкций. ПЦР проводили при следующих условиях: 31 цикл, денатурация (30 сек при +940C), отжиг праймеров (60 сек при 600C), элонгация (45 сек при 720C). Продукты амплификации разделяли в 1,5% агарозном геле в присутствии 1 мкг/мл бромистого этидия. Количество копий лентивирусного вектора, интегрированного в геном (эквивалентно множественности заражения MOI), оценивали по интенсивности флуоресценции амплифицированных фрагментов ДНК в ультрафиолетовом свете. Для калибровочного стандарта использовали геномную ДНК, выделенную из клеток карциномы легких, трансдуцированных только одной копией лентивирусного вектора. Полученные результаты нормировали с использованием праймеров для гена GAPDH: ACCACAGTCCATGCCATCAC и TCCACCACCCTGTTGCTGTA. Реакцию проводили в течение 25 циклов при тех же условиях. Уровень экспрессии Т-кадгерина в клетках проверяли методом Вестерн блоттинга. 2.5 Создание аденовирусных конструкций для трансдукции эндотелиальных клеток HUVEC Для гиперэкспрессии Т-кадгерина в HUVEC использовали конструкции, созданные на основе экспрессионной системы AdEasyTM (Quantum Biotechnologies). В результате была создана аденовирусная конструкция, позволяющая с высокой эффективностью экспрессировать Т-кадгерин в HUVEC. Система AdEasyTM состоит из двух векторов: вектор pAdEasy, последовательность которого содержит вирусные гены, позволяющие легко трансфицировать и реплицировать вирус в клетках-мишенях; и второй вектор, Transfer Vector (pShuttle-CMV Vector), содержащий сайт множественного клонирования, позволяющий встроить интересующий ген, например, ген Т-кадгерина (pShuttle-CMV-Tcad). В E.сoli штамма BJ5183 ко-трансформация плазмид pAdEasy и pShuttle-CMV-T-cad с последующей селекцией на антибиотике канамицин, позволяет получить одну рекомбинантную вирусную плазмиду pAd-T-cad, кодирующую вирусные гены и ген Ткадгерина. Также был получен контрольный вирус p-Ad на основе рекомбинации плазмид pAdEasy и pShuttle-CMV. Далее в клетках-упаковщиках HEK293А, согласно протоколу изготовителя, амплифицировали контрольный и опытный вирусы в достаточном количестве для последующей трансдукции в HUVEC. Уровень экспрессии Т-кадгерина в трансдуцированных HUVEC проверяли методом иммуноцитохимического окрашивания с использованием первых антител против Т-кадгерина, и вторых антител, конъюгированных 117 с флуорохромом AlexaFluor 488. Визуализацию окрашивания проводили с использованием инвертированного конфокального микроскопа Zeiss модели Axiovert LSM 200М с цифровой камерой AxioCam HRc. Анализ изображения проводили с использованием программ LSM Image Browser (Zeiss) и Meta Morph 5,0 (Meta Imaging Series). 2.6 Вестерн блоттинг (метод иммуноблоттинга) 2.6.1 Приготовление лизатов клеток Все процедуры проводили на льду. Клетки, растущие в культуральных чашках Петри, инкубировали на льду 30 мин, затем промывали холодным буфером ФСБ 3 раза по 5 мин, далее клетки соскребали пластиковым шпателем и центрифугировали при 2000 об/мин при +40С в течение 5 мин. Осадок растворяли в лизирующем буфере. Состав буфера: 100 мМ Тris - НСl, pH8,1, 1% Triton X-100, 20 мМ HEPES, 150 мM NaCl, 1 мM MgCl2, 1 мM CaCl2, ингибиторы протеиназ (PIERCE) 1:100, PMSF 1:100; pH7,5. Образцы инкубировали в лизис-буфере 15 мин, периодически перемешивая. Для удаления нерастворимых клеточных компонентов, Концентрацию полученный белка в лизат центрифугировали полученном лизате 30 мин определяли при методом 12000 об/мин. Бредфорда с использованием БСА в качестве стандарта (Pierce). 2.6.2 Определение концентрации белка в лизатах клеток Определение количества белка в лизатах проводили микрометодом Бредфорда (Bradford, 1976). Лизаты белков разводили в 10000 раз, после чего на 10 мкл пробы добавляли 90 мкл реагента Бредфорда и доводили водой до 1 мл, затем хорошо перемешивали. Через 15 мин проводили измерения на спектрофотометре при длине волны 595 нм. В качестве стандарта использовали коммерческий БСА (Pierce) с концентрацией 0,5 - 2 мкг/мл. Концентрацию белка в пробе определяли по калибровочному графику с использованием программы Sigma Plot. 2.6.3 Вестерн блоттинг Лизаты клеток, полученные как описано выше, смешивали с 6-ти кратным буфером Лаеммли (Laemmli) для нанесения образцов и инкубировали 10 минут при температуре +900С. Образцы наносили на дорожки полиакриламидного геля SDS-PAGE и разделяли в денатурирующих условиях. Процент геля выбирали в зависимости от молекулярных масс исследуемых белков (плотность геля в экспериментах составляла 7,5%-14%). Для идентификации молекулярных масс белков использовали коммерческую смесь лизатов окрашенных белков Kaleidoscope Prestained Standards (Bio-Rad). После этого осуществляли 118 электроблоттинг образцов с геля на PVDF мембрану (Immobilon, Millipore) в течение 30 минут при напряженности поля 25 V. Для блокирования неспецифического связывания вторичных антител мембраны инкубировали в течение 30 минут в блокирующем буфере, содержащем 5% обезжиренное молоко, 0.5% детергента Tween20 в буфере ФСБ, а далее на мембраны последовательно наносили раствор первичных и вторичных антител. Инкубацию с антителами осуществляли в течение 1 ч при комнатной температуре. Для проявления сигнала использовали систему ECL™ Western Blotting Detection Reagents и плѐнку CLXposureTM Film (всѐ от Amersham). Все эксперименты повторяли не менее трѐх раз. Денситометрический анализ результатов Вестерн блоттинга проводили на приборе GS-800 Calibrated Densitometer (Bio Rad) с использованием программного обеспечения Quantity One 4.6 Software (Bio Rad). 2.7 Иммунофлуоресцентное окрашивание эндотелиальных клеток и конфокальная микроскопия Для иммунофлуоресцентного окрашивания эндотелиальные клетки не позднее 4 пассажа высаживали на покровные стекла, предварительно покрытые 0,5% раствором желатина, в концентрации 2x104 клеток/см2 и культивировали до конфлюентого монослоя. Для доступности внутриклеточных антигенов клетки промывали тѐплым буфером HBSS, фиксировали 4% раствором формальдегида и пермеабилизовали в течение 5 минут в 1% растворе детергента Тритон Х-100. Для блокирования неспецифического связывания вторых антител образцы инкубировали в буфере для забивки, содержащем 10% сыворотки донора вторых антител, 1% БСА на буфере ФСБ. После последовательных инкубаций с первыми и вторыми антителами в концентрациях, рекомендованных производителем, покровные стекла заключали в реагент ProLong® Antifade, содержащий DAPI (Invitrogen) для визуализации ядер. Для подавления клатрин-зависимого эндоцитоза VE-кадгерина, клетки перед окрашиванием преинкубировали с раствором диназора (dynasore, Sigmaaldrich) в конечной концентрации 80 мкМ в течение 2 ч. Для визуализации VEкадгерина, локализованного в лизосомах, клетки перед окрашиванием преинкубировали с прижизненным красителем лизосом LysoTracker® RED (Invitrogen, Ex⁄Em: 577⁄590 нм) в течение 60 мин и 120 мин. Для подавления активации малых Rho ГТФаз клетки перед окрашиванием преинкубировали с ингибитором Y27632 в конечной концентрации 10 мкМ (Sigmaaldrich) в течение 12 ч. Изображения окрашенных клеток получали с использованием сканирующего лазерного конфокального микроскопа (модель TCS SP5, Leica), оборудованного Plan-Apo 119 x63, 1.40 NA масляным объективом и 488- и 543- аргоновыми лазерами. Съемку производили при комнатной температуре с использованием иммерсионного масла Leica Type. DAPI, флуоресценция вторых антител была визуализирована с помощью лазеров с длиной волны 405 нм (DAPI), 488 нм (AlexaFluor 488) и 594 нм (AlexaFluor 594), соответственно. Все изображения получали при одинаковых настройках интенсивности лазера и времени экспозиции. Изображения (разрешением 1024x1024 или 2048x2048 пикселей) сохраняли как TIFF файлы и обрабатывали для последующего анализа с помощью программного обеспечения Leica LAS и Photoshop (версия CS5, Adobe). Иммунофлуоресцентное окрашивание, полученное в отдельных каналах, представлено в виде красной и зелѐной флуоресценции, а также полученные изображения представлены в виде наложения в отдельных панелях. Жѐлтое окрашивание свидетельствует о колокализации антигенов. На каждом рисунке представлено характерное окрашивание, полученное в одном из трѐх независимых экспериментов. 2.8 Измерение SRE активности в NIH3T3 клетках Метод SRE (Serum Response Element) применяется для измерения активности Rho ГТФаз [Hill et al., 1995]. Известно, что при активации в цитоплазме Rho белков (RhoA, Rac1 и Cdc42) происходит активация SRE (c-fos Serum Response Element) комплекса в ядре, который формирует тройной комплекс с факторами транскрипции SRF (Serum Response Factor) и TCF (Ternary Complex Factor). C-fos является ранним протоонкогеном, на активность которого, помимо Rho ГТФаз, также влияют факторы роста, сыворотка, MAP киназы [Hill et al., 1995]. В данном методе использовали плазмиду SRE.L, у которой под одним промотором находятся ген SRE и ген люциферазы, что позволяет, измеряя уровень активности люциферазы, оценивать уровень активации Rho ГТФаз. Для оценки влияния гиперэкспрессии Т-кадгерина на активацию Rho ГТФаз клетки NIH3T3 ко- трансфицировали плазмидами pcDNA3.1/T-cad (для гиперэкспрессии Т-кадгерина), SRE.L (Stratagene) и pCMV-b-Gal (плазмида содержит ген b-галактозидазы и используется для нормирования на уровень трансфекции). Контрольные клетки ко-трансфицировали плазмидами pcDNA3.1, SRE.L и pCMV-b-Gal. В качестве положительного контроля использовали клетки, ко-трансфицированные плазмидами pcDNA3.1/T-cad, SRE.L, pCMVb-Gal, и плазмидой, содержащей ген p114RhoGEF. Для подавления Т-кадгеринопосредованной активации SRE клетки NIH3T3 ко-трансфицировали плазмидами pcDNA3.1/T-cad, SRE.L и pCMV-b-Gal, а также одной из плазмид для инактивации Rho ГТФаз: RhoN19, Rac1N17 и Cdc42N19 для RhoA, Rac1 и Cdc42, соответственно. Через 24 ч 120 после трансфекции клетки депривировали в течение 4 ч и добавляли лизирующий буфер (Amersham). В полученных лизатах определяли уровень активности люциферазы (Luc) при длине волны 450 нм, согласно инструкции изготовителя (Promega). Данные нормировали с учетом экспрессии b-галактозидазы (b-Gal). Эксперимент проводили в 5-ти параллелях и повторяли 5 раз. 2.9 Выделение активных Rho ГТФаз (GST pull-down assay) Активные формы малых G белков семейства Rho ГТФаз (RhoA, Rac1 и Cdc42) выделяли с использованием коммерческого набора RhoA/Rac1/Cdc42 activation Assay Combo kit (Cellbiolabs Inc.) согласно инструкции изготовителя. Активные формы RhoA, Rac1, и Cdc42 ГТФаз визуализировали методом электрофореза (в 14% SDS-PAGE полиакриламидном геле) с последующим вестерн блоттингом в присутствии моноклональных антител, специфичных для каждой Rho ГТФазы в отдельности. В качестве положительного контроля использовали коммерческие лизаты, содержащие активные формы RhoA, Rac1 и Cdc42. Общий уровень ГТФаз определяли в лизатах, приготовленных обычным способом. Для выделения активных форм Rho ГТФаз из клеток NIH3T3 клетки предварительно трансфицировали плазмидой pcDNA3.1/T-cad для увеличения экспрессии Т-кадгерина, или контрольной плазмидой pcDNA3.1. Для положительного контроля клетки трансфицировали плазмидами, кодирующими активные формы Rho ГТФаз: для RhoA – RhoV12, для Rac1 – RacV12, для Cdc42 – Cdc42V12. Эти плазмиды были любезно предоставлены лабораторией доктора Т. Войно-Ясенецкой (University of Illinois at Chicago, Chicago, USA). Через 48 ч после трансфекции клетки лизировали, и активные формы Rho ГТФаз выделяли с использованием коммерческого набора RhoA/Rac1/Cdc42 activation Assay Combo kit (Cellbiolabs Inc.) согласно инструкции изготовителя. Количественную оценку содержания активных Rho ГТФаз определяли методом вестерн блоттинга. 2.10 Измерение проницаемости эндотелиального монослоя in vitro Проницаемость эндотелиального монослоя определяли методом, подробно описанным ранее [Semina et al., 2009]. Кратко, эндотелиальные клетки (контрольные, гиперэкспрессирующие Т-кадгерин или после подавления нативной экспрессии) не позднее 2 пассажа высаживали (в концентрации 105 клеток/лунку) в верхнюю камеру двухкамерной системы Трансвелл (размер пор 0,4 мкм, Transwell®, Corning) и культивировали до 121 достижения конфлюентного монослоя не менее 72 ч. После этого в верхнюю камеру системы трансвелл добавляли раствор FITC-декстрана (FITC-dextran, мол. масса 40 кДа, Sigmaaldrich) в конечной концентрации 10 мкг/мл. Через 60 минут из нижней камеры отбирали аликвоты по 20 мкл и измеряли интенсивность флуоресценции FITC на приборе Envision® Multilabel Reader (Perkin Elmer) при длине волны 525 нм. Последующие образцы для анализа отбирали каждые 60 мин. Эксперимент проводили, как минимум в 4 параллелях и повторили 6 раз. Данные представлены как mean ± SEM, p < 0,001. 2.11 Трипсинизация эндотелиальных клеток Для того чтобы разделить в лизатах клеток внутриклеточный и поверхностный пул VE-кадгерина, эндотелиальные клетки, достигшие плотности конфлюентного монослоя промывали, тѐплым буфером Хэнкса (HBSS) и инкубировали в 0,25% растворе трипсин/ЭДТА (GIBCO) при +370C в течение 10 мин для того, чтобы протеолитически удалить VE-кадгерин, находящийся на поверхности клеток. Далее клетки осаждали путѐм центрифугирования в течение 10 минут при 300 об/мин. Осадок клеток промывали укомплектованной средой роста EBM-2 для инактивации трипсина, и далее клетки осаждали центрифугированием, как описано выше. Клеточный осадок растворялся в 2хкратном буфере Лаеммли (Laemmli). Образцы далее анализировали методом вестерн блоттинга. Для контроля использовали лизаты клеток, приготовленные обычным способом. 2.12 Разделение лизатов клеток на субклеточные фракции Субклеточные фракции (цитозольную, мембранную и ядерную) получали с использованием коммерческого набора Qproteome™ Cell Compartment kit согласно протоколу производителя (Qiagen). Кратко, лизаты клеток разделяли на субклеточные фракции с помощью специальных буферов для экстракции цитоплазматических и мембранных белков. Концентрацию белков в каждой субклеточной фракции определяли с использованием коммерческого набора BCA protein assay kit (Pierce Biotechnology, Rockford). Полученные таким образом лизаты, содержащие равное количество общего белка (20 мкг), наносили на дорожки полиакриламидного геля (10% SDS-PAGE). Анализ образцов осуществляли методом вестерн блоттинга. Чистоту и состав фракций подтверждали с использованием антител против маркѐра мембран и ранних эндосомальных белков анти-EEA1 (для определения чистоты мембранной фракции), и маркѐра лизосом анти-LAMP1 (для определения чистоты цитозольной фракции). 122 2.13 Оценка пролиферация эндотелиальных клеток Влияние Т-кадгерина на пролиферацию эндотелиальных клеток (HUVEC) при контактном взаимодействии с другими клетками с высокой экспрессией Т-кадгерина изучали с помощью со-культивирования HUVEC 2 пассажа с клетками эпителия почки эмбриона человека (линия HEK 293) и иммортализованными фибробластами мыши (линия L929), в соотношениях 5:1, 2:1 или 1:1. Клетки высаживали на стекла, покрытые фибронектином (Sigmaaldrich), и культивировали в CO2-инкубаторе в течение 48 ч. Клетки линий HEK 293 и L929 были предварительно трансфицированы плазмидой для экспрессии маркерного белка GFP и флуоресцировали в зеленом канале. Число делящихся клеток определяли с помощью подсчѐта клеток, экспрессирующих ядерный антиген пролиферирующих клеток (PCNA), который выявляли с помощью иммунофлуоресцентного окрашивания антителами мыши против PCNА человека (Dako Cytomation), в сочетании со вторыми антителами против мыши, конъюгированными с флуорохромом AlexaFluor594 (Molecular Probes). Число PCNA-позитивных HUVЕC подсчитывали в 4-5 полях зрения на каждом стекле (при увеличении в 200 раз). Полученное число клеток нормировали на общее количество HUVEC в каждом поле зрения. 2.14 Метод измерения адгезии эндотелиальных клеток Влияние Т-кадгерина на адгезию эндотелиальных клеток изучали на модели прикрепления HUVEC к пластику, покрытому растворами EC1 или ЕС5 рекомбинантных доменов Т-кадгерина, полученных ранее [Ivanov et al., 2004], бычьим сывороточным альбумином (БСА), к монослою контрольных клеток или клеток, гиперэкспрессирующих Ткадгерин. Клетки HUVEC плотностью 8х104/лунку высаживали в лунки 24-луночных планшетов, покрытых 0,2% раствором желатина и содержащих 0,1 мг/мл рекомбинантных доменов Т-кадгерина (ЕС1 или ЕС5) или БСА, и инкубировали в течение 2 ч. По окончании инкубации клетки промывали ФСБ и подсчитывали число адгезировавших клеток. Далее изучали способность эндотелиальных клеток прикрепляться к монослою клеток линии L929, экспрессирующих Т-кадгерин, или контрольных клеток. Для этого клетки линии L929 высевали на 24-луночные культуральные планшеты в концентрации 2х104 или 105 на лунку и культивировали до формирования 50% и 100% монослоя. Далее поверх клеток L929 высаживали эндотелиальные клетки 2 пассажа, предварительно 123 помеченные ацетилированными липопротеидами низкой плотности (ЛНП) плазмы крови человека, конъюгированными с флуорохромом AlexaFluor488 (Ас-LDL-Alexa488, Invitrogen). Эндотелиальные клетки инкубировали с меченными Ас-LDL-Alexa488 в конечной концентрации 0,2 мкг/мл, которые специфически фагоцитировались эндотелиальными клетками, что позволяло прижизненно визуализировать последние в зеленом канале микроскопа и отличать их от фибробластов. Через 12 часов клетки обрабатывали раствором трипсина 0,25% и ЭДТА 0,02% и подсчитывали. Эксперименты проводили в 3 параллелях и повторяли 2 раза. Для этого HUVEC 2 пассажа метили ацетилированными липопротеидами низкой плотности (ЛНП), конъюгированными с флуорохромом AlexaFluor488 (Invitrogen). Клетки подсчитывали в 4-5 полях зрения на каждом стекле (при увеличении в 200 раз). Полученное число клеток нормировали на общее количество HUVEC в каждом поле зрения. Цифровые изображения получали с использованием флуоресцентного микроскопа Axiovert200M (Zeiss) и видеокамеры Axiocam (Zeiss, Германия). Подсчет клеток, флюоресцирующих в зеленом канале микроскопа, производили в 4-5 полях зрения на каждой лунке планшета. 2.15 Метод измерения апоптоза в эндотелиальных клетках Влияние Т-кадгерина на апоптоз эндотелиальных клеток оценивали по активности каспазы-3 в этих клетках с помощью коммерческого набора реагентов для колориметрического измерения активности каспазы-3 (Сaspase-3 Colorimetric Assay, R&D Systems, CN BF3100) согласно инструкции производителя. В качестве положительного контроля для индукции апоптоза в культуру эндотелиальных клеток добавляли 1 нМ стауроспорина (Sigmaaldrich) и культивировали в течение 18 ч, после чего готовили клеточные лизаты обычным способом. Измерение поглощения в полученных растворах проводили при 405 нм. Эксперименты проводили в трех параллелях и повторяли 3 раза. 2.16 Измерение миграции эндотелиальных клеток в камере Бойдена Влияние Т-кадгерина на миграцию HUVEC изучали в камере Бойдена с использованием мембраны с размером пор 8 мкм (Neuro Probe Inc.). Для этого верхнюю часть мембраны инкубировали в 0,2% растворе желатина, содержащем 0,1 мг/мл рекомбинантных ЕС1 или ЕС5 доменов. После инкубации мембраны в течение 12 ч при 124 +370С на неѐ высаживали HUVEC 2 пассажа в количестве 1х105 на лунку. Миграцию HUVEC проводили в течение 4 ч при +370C, используя в качестве аттрактанта среду ДМЕМ с ФБС (в концентрации 1-10%). По окончании миграции мембрану с промигрировавшими клетками окрашивали красителем DIFF-QUICK, сканировали и анализировали полученные изображения с помощью программы Image-J (National Institute of Health). Эксперименты проводили в 5-6 параллелях и повторяли 3 раза. 2.17 Метод изучения формирования капилляро-подобных структур эндотелиальными клетками Для оценки формирования капилляро-подобных структур HUVEC 2 пассажа в количестве 5х104 высевали на 24-луночный планшет, покрытый Матригелем без факторов роста (Matrigel® BD Biosciences), и инкубировали при +370С в течение 24 часов. Сверху в лунки добавляли среду культивирования, содержащую ДМЕМ с 10% FBS (HyClone), 200 мкг/мл ECGF, 5 ЕД/мл гепарина, 1 мМ пирувата натрия (GIBCO BRL), 100 Ед/мл пенициллина и 100 Ед/мл стрептомицина (GIBCO BRL), 20 мМ HEPES (Helicon). Для определения влияния иммобилизованного Т-кадгерина на формирование капилляров, Матригель предварительно до высевания клеток пропитывали 0,1 мг/мл растворами доменов EC1 или EC5 в течение 12 ч. Суммарную длину формирующихся капилляроподобных структур оценивали в 5 случайных полях зрения в каждой лунке, используя программу MetaMorph 5.0 (Universal Imagin). Эксперименты повторяли 3 раза в 3 параллелях. 2.18 Экспериментальные животные Для изучения экспрессии Т-кадгерина в раннем эмбриональном развитии у мыши использовали эмбрионов, полученных от гибридов F1 линий CBA/С57Bl. В работе по изучению влияния Т-кадгерина на рост и метастазирование меланомы использовали мышей гибридной линии BDF1 (самцах) из вивария ГУ РОНЦ им. Н.Н. Блохина РАМН. При изучении влияния Т-кадгерина на васкуляризацию имплантированного подкожно Матригеля использовали бестимусных мышей линии NUDE. Содержание и использование лабораторных животных соответствовало рекомендациям национального совета и законам РФ. В эксперимент были включены 44 животных одинакового возраста. Влияние Т-кадгерина на роста, степень пигментации и васкуляризацию меланомы было также протестировано на хориоаллантоисной мембране куриных эмбрионов. 125 Оплодотворѐнные яйца были предоставлены Петелинской птицефабрикой. В эксперимент были включены 230 куриных эмбрионов. 2.19 Анализ экспрессии Т-кадгерина в раннем эмбриональном развитии у мыши 2.19.1 Получение датированной беременности мыши В работе использовали эмбрионы мышей, полученные от гибридов F1 CBA/С57Bl6. Мышей содержали в стандартных условиях при 14-ти часовом световом дне. В вечернее время суток ссаживали самок с самцами, на следующее утро выявляли спарившихся мышей по наличию вагинальных пробок. День обнаружения вагинальной пробки считался ½ первого дня беременности. Работа сделана в сотрудничестве с д.б.н. Семѐновой М.Л., кафедра эмбриологии биологического факультета МГУ имени М.В.Ломоносова. 2.19.2 Получение эмбрионов мыши на постимплантационной стадии развития Извлечение постимплантационных эмбрионов производили по стандартному протоколу, описанному Манк [Манк М., 1990]. Самку умерщвляли путем дисклокации шейных позвонков, вскрывали брюшную полость, извлекали матку с децидуомами и помещали в чашку Петри с охлажденным фосфатно-солевым буфером ФСБ (Sigmaaldrich). Далее рога матки надрезали по антимезометриальному краю, обнажая децидуальные капсулы с зародышами и отделяли их от мезометриальной стенки. Децидуомы переносили в чистую ч. Петри, содержащую холодный ФСБ. Захватив мезометриальный конец децидуальной капсулы, еѐ надрезали и аккуратно выталкивали эмбрион в раствор. После этого препаровальными иглами эмбрион освобождали из амниотической оболочки и переносили пипеткой в чистую чашку Петри с ФСБ для отмывки. Эмбрионы фиксировали в 4% растворе формальдегида (PRS Panreac) на ФСБ при температуре +40С в течение ночи. 2.19.3 Иммунофлуоресцентное окрашивание мышиных эмбрионов антителами против Т-кадгерина После фиксации эмбрионы промывали 5 раз по 20 мин в ФСБ, содержащем 0,2% детергента Triton X-100 (Sigmaaldrich). Для блокирования неспецифического окрашивания образцы помещали в нормальную неиммунную сыворотку козы (Sigmaaldrich) в разведении 1:10 и инкубировали их при температуре +40С на качалке в течение ночи. Затем эмбрионы инкубировали в растворе моноклональных антителах кролика против Т-кадгерина мыши (BioDesign) в разведении 1:25; в качестве контроля использовали неиммунные иммуноглобулины IgG кролика (Abd Serotec) в концентрации, эквивалентной специфическим антителам. Инкубацию проводили в течение дня при комнатной 126 температуре и при постоянном покачивании. Антители отмывали в 0,2% растворе Triton X100 на ФСБ 3 раза по 20 мин при комнатной температуре и в 4-ой смене раствора на качалке в течение ночи при температуре +4 0С. После отмывки эмбрионы помещали в раствор вторых антител козы, конъюгированных с флуорохромом Alexa Fluor® 594, в которых выдерживали весь день на качалке при комнатной температуре. Ядра клеток докрашивали флуоресцентным красителем DAPI (Sigmaaldrich) в разведении 1:1000 в течение 30 мин, а затем делали троекратную промывку по 20 мин в 0,2% Triton X100 на ФСБ и оставляли отмываться на ночь в том же растворе при температуре +40С на качалке. На следующий день эмбрионы заключали в среду Aqua-Poly/Mount (Polysciences). Готовые образцы анализировали при помощи конфокального мультифотонного микроскопа Leica SP5 и программного обеспечения Leica Application Suite Advanced Fluorescence 2.2.0 (Leica Microsystems). 2.19.4 Трансформация E.coli, амплификация и очистка плазмид для in situ гибридизации Трансформацию компетентных клеток E.coli проводили при температуре +420С в течение 45 сек. Плазмиды для трансформации добавляли в количестве 0,5 мкг ДНК на 100 мкл компетентных клеток. После трансформации к компетентным клеткам добавляли 1 мл среды LB (Luria-Bertani medium) следующего состава: триптон (ДиаМ), дрожжевой экстракт (ДиаМ), NaCl (PRS Panreac), рН=7,4 и оставляли при температуре +370С на 1 час. Затем суспензию трансформированных клеток наносили на залитые агаром (2% агар на LB среде) чашки Петри и помещали в термостат на 16 ч при температуре +37 0С. Не позднее, чем через 24 часа на чашках выбирали крупные, отдельно расположенные колонии, переносили их в 10 мл среды LB, содержащей селективный антибиотик (ампициллин в конечной концентрации 150 мкг/мл) и инкубировали 8 часов при температуре +37 0С при покачивании. Затем образцы среды с выросшими бактериями переносили в 250 мл среды LB, содержащей ампициллин в концентрации 150 мкг/мл, и оставляли на 12-16 часов при температуре +370С при постоянном покачивании (300 об/мин). Очистку и выделение плазмид производили с помощью коммерческого набора EndoFree® Plasmid Maxi Kit (Qiagen) в соответствии с протоколом производителя. 2.19.5 Линеаризация плазмид для in situ гибридизации In situ гибридизацию РНК-содержащими пробами к Т-кадгерину (sense и antisense) и Krox 20 проводили на эмбрионах мыши по методике, разработанной ранее, разработанной 127 в лаборатории профессора Вилкинсона (Development Neurobiology Medical Research Institute, UК) (Wilkinson et al., 1998). Для выявления экспрессии мРНК Т-кадгерина в эмбрионах мыши методом гибридизации in situ использовали плазмиду pFLCI (Imagenes, Germany) со встроенной EST (expressed sequence tag) последовательностью кДНК Т-кадгерина. Для положительного контроля использовали плазмиду Bluescript KS со встроенной EST (expressed sequence tag) последовательностью кДНК Krox20 (Рисунок 3.1). Плазмиды, содержащие EST последовательности, представляют собой короткие cDNA, используемые для выявления экспрессии генов, и доступные в электронной базе данных Gen Bank. Для линеаризации плазмидную ДНК обрабатывали рестриктазами: Bam H1 (Fermentas) для выявления мРНК Krox 20 Not 1 (Fermentas), и Bam H1 для выявления мРНК Т-кадгерина. Состав реакционной смеси для линеаризации: 10-ти кратный буфер, деионизованная вода (Синтол), плазмида (4 мкг), рестриктаза (Not 1 или Bam H1). Смесь инкубировали в течение 12 ч при температуре +370С. После инкубации производили очистку ДНК с использованием коммерческого набора GFX PCR DNA and Gel Purification Kit (GE Healthcare) в соответствии с протоколом производителя. Анализ линеаризованных и нелинеаризованных плазмид осуществляли методом электрофореза в 1,2% агарозном геле при напряженности поля 120 V в течение 30 минут. 2.19.6 Синтез РНК-содержащей пробы для in situ гибридизации Реакционная смесь для синтеза меченной дигоксигенином РНК-содержащей пробы включала: 5-ти кратный транскрипционный буфер, деионизованную, свободную от РНКаз воду, линеаризованную плазмиду, смесь нуклеотидов, меченных DIG (10 мМ АТР, 10 мМ СТР, 10 мМ GТР, 6,5 мМ, UТР, 3,5 мМ DIG-11-UTP, pH 7,5, Roche), ингибитор РНКаз (Merck Biosciences), РНК-полимеразу. Смесь инкубировали в течение 2 ч при температуре +370С. Для синтеза РНК-содержащей пробы с линеаризованной плазмиды, содержащей последовательность Krox 20, использовали РНК-полимеразу Т3 (Fermentas). Для синтеза РНК пробы прямой последовательности Т-кадгерина (отрицательный контроль) с линеаризованной плазмиды использовали Т7 РНК-полимеразу (Promega), а для синтеза обратной РНК пробы для выявления мРНК Т-кадгерина использовали Т3 РНК-полимеразу (Fermentas). Полученные РНК пробы очищали на коммерческой колонке RNAspin Mini (GE Healthcare) в строгом соответствии с протоколом изготовителя. 128 2.19.7. Гибридизация мышиных эмбрионов in situ Гибридизацию с РНК-содержащими пробами (sense и antisense) к Т-кадгерину и Krox 20 проводили на эмбрионах мыши Е8.75, Е9.5 и Е10.5 стадий развития. Для приготовления всех растворов для in situ гибридизации использовали воду, свободную от РНКаз. Для этого дистиллированную воду обрабатывали 0,1% DEPC (diethyl pyrocarbonate, диэтилпирокарбонат, Sigmaaldrich): на 1 литр воды добавляли 1 мл реагента DEPC, инкубировали ночь, а затем автоклавировали 2,5 часа для удаления следов DEPC. Эмбрионы фиксировали в течение ночи в 4% растворе формальдегида (PRS Panreac) на ФСБ, содержащем также 1% Tween-20 (Sigmaaldrich) при температуре +40С, затем отмывали дважды по 5 мин холодным ФСБ и последовательно фиксировали в метаноле с возрастающей концентрацией (25%, 50%, 75%, и дважды 100%). После этого эмбрионы замораживали и хранили для последующих исследований при температуре -200С. Непосредственно перед проведением гибридизации зародышей подвергали регидратации в метаноле с понижающейся концентрацией (75%, 50% и 25%), отмывали 3 раза в ФСБ, обрабатывали протеиназой К при комнатной температуре (Qiagen, в концентрации 10 мкг/мл на ФСБ). Время обработки протеиназой К (10-15мин) зависело от стадии развития мышиного эмбриона. Затем эмбрионы отмывали 2 раза по 5 мин в ФСБ, фиксировали в 4% формальдегиде (PRS Panreac) на ФСБ в течение 20 мин и отмывали 2 раза по 5 мин в ФСБ. Далее эмбрионы постепенно переводили в гибридизационный буфер, содержащий 50% формамид (Sigmaaldrich), 5-ти кратный буфер SSC (исходный раствор 20-ти кратный буфер SSC содержит 3М NaCl, 0,3 M цитрат натрия, рН 7,0), 0.1% Triton X-100 (ДиаЭм), 50 мкг/мл гепарина (Sigmaaldrich), 1 мг/мл РНК из дрожжей IV типа (Sigmaaldrich), 5 мМ ЭДТА (Applichem), 2% блокирующий раствор (Roche) и 0,1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate) (Sigmaaldrich). Эмбрионы инкубировали при комнатной температуре по 10 мин сначала в смеси (1:1) гибридизационного буфера и ФСБ, а затем – в гибридизационном буфере. После этого эмбрионы оставляли в гибридизационном буфере на ночь при температуре +650С. Утром эмбрионы помещали в свежий гибридизационный буфер, добавляли РНК пробу (0,5 мкг пробы на 1 мл буфера) и инкубировали в течение 24 ч при температуре +650С. После гибридизации с пробой эмбрионы сначала отмывали в гибридизационном буфере 3 раза по 30 мин при температуре +650С, а затем в смеси гибридизационного буфера и буфера МАВТ (1:1) в течение 30 мин при температуре +650С. Состав буфера MABT: 100 мМ малеиновая кислота (Sigmaaldrich), pH 7,5, 150 мМ NaCl, 0,1% Tween-20 (Sigmaaldrich). Далее эмбрионы промывали в буфере MABT 3 раза по 10 мин при комнатной температуре, помещали в блокирующий раствор (2% блокирующий раствор (Roche), содержащий 20% сыворотки овцы (Abd Serotec) и 129 MABT) на 2 ч при комнатной температуре, после чего инкубировали в течение 12 ч при +40С с раствором антител против дигоксигенина, конъюгированных с щелочной фосфатазой в разведении 1:200 (Roche), приготовленном на 10% сыворотке овцы. После инкубации с антителами эмбрионы промывали при комнатной температуре 2 раза по 15 мин, и 5 раз по 60 мин в MABT, а затем 2 раза по 5 мин и 1 раз 10 мин в буфере NTMT до появления окрашивания. Состав буфера NTMT на 1 мл: 100 мМ Tris-HCl (Sigmaaldrich), pH 9,5, 50 мМ MgCl2 (Sigmaaldrich), 100 мМ NaCl, 0.1% Tween-20 (Sigmaaldrich), 4,5 мкл NBT (4-nitro-blue tetrasodium chloride, Vector Laboratories) и 3,5 мкл BCIP (5-bromo-4-chloro-3indolylphosphate, Vector Laboratories). Реакцию окрашивания останавливали многократным промыванием в ФСБ, затем эмбрионы фиксировали в 4% формальдегиде (PRS Panreac) на ФСБ в течение 2 ч при комнатной температуре. Съѐмку эмбрионов производили с использованием стереомикроскопа (Olympus SZX 16, камера AxioCam HRc, Zeiss) и программы Axio Vision 3.1. 2.20 Эксплантная модель ex vivo аорты крысы Влияние Т-кадгерина на ангиогенез ex vivo изучали на модели органной культуры сосудистого колечка крысы в Матригеле [Nicosia et al, 1990]. Для этого грудную часть аорты самцов крыс в стерильных условиях тщательно очищали от окружающих тканей, нарезали на колечки длиной 3 мм и помещали в Матригель, не содержащий факторов роста (BD), предварительно заполимеризованный после добавления с ЕС1 или ЕС5 доменами Ткадгерина в концентрации 0,0004–0,02 мг/мл. Экспланты инкубировали в СО2-инкубаторе при температуре +370C в течение 14 дней, регистрируя образование капилляро-подобных структур клетками, мигрирующими из эксплантов в Матригель, через 24 ч. Эксперименты повторяли 4 раза в трѐх параллелях. Зону, занимаемую ветвящимися капиллярами, а также среднюю длину отрастающих капилляров анализировали с помощью программного обеспечения MetaMorph (Universal Imaging) и нормировали на единицу (1 мм) боковой поверхности аорты. 2.21 Подкожная имплантация Матригеля мышам Для исследования влияния Т-кадгерина на рост кровеносных сосудов in vivo была использована модель имплантации Матригеля, содержащего контрольные клетки или клетки, экспрессирующие Т-кадгерин, с целью обеспечения локальной сверхэкспрессии Ткадгерина. Для имплантации использовали клетки линии L929, стабильно 130 экспрессирующие Т-кадгерин человека или контрольную плазмиду (pcDNA 3.1, Invitrogen) [Rubina et al., 2005; Philippova et al., 2003]. Клетки L929 культивировали в среде ДМЕМ, содержащей 10% ФБС, 100 ЕД/мл пенициллина и 100 Ед/мл стрептомицина в клеточном инкубаторе при температуре +370C, 95% воздуха и 5% CO2. Мышам линии NUDE под авертиновым наркозом (2,5% раствор авертина интраперитонеально) подкожно вводили 1,7х106 клеток, смешанных с 400 мкл холодного (+40C) Матригеля (Matrigel® BD). Животных умерщвляли через 3, 7, 10 и 14 дней после имплантации и извлекали Матригели для последующего анализа. Бляшки Матригеля взвешивали, разделяли на две части, в одной из которых оценивали содержание гемоглобина, а вторую замораживали в жидком азоте в среде для замораживания образцов O.C.T. Compound (Sakura Finitec Inc.) для последующего иммунофлуоресцентного анализа. 2.21.1 Оценка содержания гемоглобина Содержание гемоглобина в имплантатах Матригеля оценивали с помощью метода Драбкина, модифицированного для тканей. Для этого фрагменты Матригеля гомогенизировали в 200 мкл 0,9% раствора NaCl и центрифугировали при 5000 об/мин в течение 7 мин. К полученному супернатанту добавляли хлороформ (100 мкл на 50 мкг Матригеля) и центрифугировали при 13 000 об/мин в течение 3 мин. К супернатанту добавляли 150 мкл гемолитика – концентрированного раствора Драбкина, и инкубировали при комнатной температуре в течение 15 мин. Затем раствор центрифугировали в течение 2 мин при 13000 об/мин, объѐм полученного раствора гемцианида доводили до 0,5 мл с помощью 0,9% раствора NaCl и измеряли поглощение при длине волны 594 нм. 2.21.2 Выявление сосудов в Матригеле с помощью иммунофлуоресцентного окрашивания Кровеносные сосуды выявляли с помощью иммунофлуоресцентного окрашивания криосрезов Матригеля (толщиной 6 мкм), используя моноклональные антитела крысы, узнающие маркѐрный антиген эндотелия сосудов CD31 мыши (BD) и вторые антитела осла против крысы, конъюгированные с флуорохромом AlexaFluor 595 (Molecular Probes). Зрелые сосуды оценивали с помощью двойного иммунофлуоресцентного окрашивания криосрезов Матригеля антителами против CD31 мыши и антителами против гладкомышечного актина (aSMA) (Epitomics) или маркера перицитов NG2 (Chemicon). В этом случае использовали вторичные антитела осла против IgG крысы, конъюгированные с флуорохромом AlexaFluor595 (Molecular Probes), и антитела осла против IgG кролика, конъюгированные с флуорохромом AlexaFluor 488 (Molecular Probes). Полученные препараты анализировали при помощи инвертированного флуоресцентного микроскопа 131 Zeiss модели Axiovert 200M. Документирование изображений проводили с помощью цифровой видеокамеры AxioCam HRc и обработки в программе Axiovision (Zeiss). 2.21.3 Подсчѐт сосудов и статистический анализ Размер сосудов и их плотность на окрашенных криосрезах Матригеля оценивали с помощью программного обеспечения MetaMorph (Universal Imaging) и ClickCounter. Плотность сосудов подсчитывали на срезах в 4-5 полях зрения (площадь поля зрения 1,107 мм2) на трѐх случайных срезах для каждой бляшки Матригеля (при увеличении в 100 раз). При этом выделяли капилляры (CD31-положительные образования без просвета или длиной менее 20 мкм), сосуды среднего диаметра (CD31-положительные образования длиной 20–40 мкм) и крупные сосуды (диаметром более 40 мкм); число сосудов в каждой группе подсчитывали отдельно. Полученное число сосудов в каждом поле зрения нормировали на единицу площади среза. 2.22 Приготовление криосрезов тканей человека и животных Для приготовления криосрезов ткань помещали в специальный раствор криопротектора (Tissue Tek, Sakura Finitek Inc.) и постепенно погружали в жидкий азот. Далее замороженные образцы помещали в криохранилище и хранили до использования при температуре -700С. Размораживание ткани не допускалось. Для приготовления криосрезов криостат заранее охлаждали до температуры -200С. Криосрезы ткани толщиной 7 мкм помещали на обработанные предметные стѐкла. 2.23 Иммунофлуоресцентное окрашивание образцов биопсийного материала кожи человека Биопсийный материал кожи человека от 6 здоровых доноров, пациентов с псориазом, 10 пациентов с кератоакантомой, 3 пациентов с кератозом, 30 пациентов с базалиомой, 2 пациентов с плоскоклеточным раком кожи и 2 пациентов с метатипическим раком был получен при проведении совместных исследований с кафедрой кожных и венерических болезней (д.м.н., доцент Хлебникова А.Н., зав. каф. проф. Молочков В.А.) ФППОВ Первый МГМУ им. И.М. Сеченова Минздрава России. Образцы меланомы человека были предоставлены д.м.н. Михайловой И.Н., лаборатория экспериментальной диагностики и биотерапии опухолей Российского онкологического научного центра РАМН имени Н.Н. Блохина. Было исследовано 7 образцов первичной меланомы и 10 образцов висцеральных метастазов меланомы. Образцы тканей, полученных от пациентов в ходе 132 хирургического лечения, замораживали в жидком азоте и хранили при температуре -800C. Криосрезы кожи (толщиной 7 мкм) фиксировали в 4% параформальдегиде в течение 10 минут, отмывали в ФСБ срезы инкубировали в 0,1% растворе БСА (бычий сывороточный альбумин) на ФСБ с 10% нормальной сыворотки осла для блокирования неспецифичного связывания антител. Срезы инкубировали в смеси первых антител против Т-кадгерина (кроличьи против человека, ProSci), маркѐров эндотелиальных клеток - vWF (мышиные иммуноглобулины против антигена человека, BD) или CD31 (мышиные против человека, BD), или маркѐров гладкомышечных клеток сосудов и перицитов – α-актин αSMA (кроличьи против человека, Epitomics). Срезы меланомы и метастазов меланомы инкубировали в смеси первых антител против Т-кадгерина (кроличьи иммуноглобулины против антигена человека, ProSci), маркѐров эндотелиальных клеток - vWF (мышиные иммуноглобулины против антигена человека, BD) или CD31 (мышиные против человека, BD), или маркера меланомы gp100 (Lab Vision- Neomarkers, USA) Ab-3 (Ab-3, представляет собой смесь мышиных антител против человека Ab-1 HMB45 и Ab-2 HMB50, обладающих высокой чувствительностью и выявляющих дифференцирующиеся меланоциты и клетки меланомы). Срезы отмывали и помещали в раствор вторых антител осла против мыши, конъюгированных с AlexaFluor594, или против кролика AlexaFluor488 (Molecular Probes, 10 мкг/мл в ФСБ). Ядра клеток докрашивали флуоресцентным красителем DAPI. Срезы заключали в среду Vectashield mounting media (Vector Inc.). В качестве отрицательного контроля использовали мышиные или кроличьи неспецифичные иммуноглобулины в соответствующих концентрациях. Полученные препараты анализировали с помощью флуоресцентного микроскопа Zeiss Axiovert 200M, оборудованного цифровой видеокамерой CCD Axio Cam HRc и программным обеспечением Axiovision software (Zeiss), Adobe PhotoShop software (Adobe Systems, Mountain View), MetaMorph 5.0 (Universal Imaging). Для каждого поля зрения площадь, занимаемую Т-кадгерин-позитивными клетками меланомы, нормировали на единицу площади, окрашенную DAPI (Sigmaaldrich). Для количественной оценки площадь Т-кадгерин-позитивных областей измеряли в 4-5 полях зрения (площадь поля зрения - 1,107 мм2) на 10 случайных срезах для каждого образца (при увеличении в 100 раз, объектив х10). 2.24 Модель гематогенно метастазирующей в лѐгкие меланомы у мышей in vivo Исследование влияния экспрессии Т-кадгерина в клетках мышиной меланомы B16F10 на рост и васкуляризацию первичного узла in vivo проводили на модели гематогенно метастазирующей в лѐгкие меланомы у мышей. Исследование было проведено 133 совместно с лабораторией комбинированной терапии опухолей Научно-исследовательского института экспериментальной диагностики и терапии опухолей Российского онкологического научного центра РАМН имени Н.Н. Блохина под руководством профессора Трещалиной Е. М. Линия клеток B16F10 характеризуется высокой интенсивностью метастазирования и даѐт макроскопические метастазы, доступные для качественной и количественной оценки. 2.25 Анализ кинетики роста и метастазирования клеток меланомы B16F10 Мышам (самцам линии BDF1) подкожно вводили суспензию клеток высоко метастазирующей в лѐгкие меланомы B16F10 в количестве 1х10 6 клеток в 0,3 мл ростовой среды RPMI1640. При этом использовали клетки клонов меланомы (B16F10 T (-), B16F10 T(+) или B16F10 T(++)) или суспензию клеток суммарной популяции меланомы (контрольной или популяции клеток меланомы, гиперэкспрессирующей Т-кадгерин). Клетки поликлональных культур меланомы (контрольной суммарной популяции и популяции клеток, гиперэкспрессирующей Т-кадгерин) использовали, чтобы подтвердить, что наблюдаемые эффекты Т-кадгерина не связаны с индивидуальными особенностями отобранных клонов, а являются непосредственным следствием увеличения экспрессии Ткадгерина. При этом наблюдали формирование первичного опухолевого узла у мышей. Для каждого типа клеток, когда опухоли достигали размера 50 мм3, мышей, имеющих опухоль, рандомизировали на 2 группы (n = 11 для каждой группы). В первой группе мышей опухоли удаляли хирургически на 10-й день после трансплантации клеток, что приводило к метастазированию меланомы в легкие. Этих мышей умерщвляли на 10-й день после резекции первичного опухолевого узла и проводили забор и замораживание метастазов в легкие для последующего анализа. Мышей из второй группы умерщвляли, когда размер опухоли достигал объема более 1000 мм3, при этом проводили забор первичных узлов для последующего анализа. Размеры опухолей определяли калипером и объѐм рассчитывали по формуле: D1хD2хD3/2, где D1, D2 и D3 представляют собой поперечные диаметры (мм). Извлеченные у мышей легкие взвешивали и определяли стандартный показатель метастазирования - легочный коэффициент (ЛК), который равен отношению средней массы лѐгких в опытной группе к средней массе лѐгких здоровых мышей. Также определяли частоту метастазирования, которая равна отношению количества мышей с метастазами к общему количеству анализируемых мышей и выражается в процентах. Части первичных узлов и легких погружали в среду Tissue-Tek (Sakura Finitek Inc.) для заморозки тканей, замораживали в жидком азоте и хранили при температуре -200С. Полученные криосрезы 134 фиксировали в 4% формальдегиде 10 минут при комнатной температуре, промывали в ФСБ 3 раза по 5 минут и окрашивали гематоксилином Майера. Мышей умерщвляли передозировкой эфирного наркоза, трупы подвергали замораживанию и кремировали в специализированном подразделении РОНЦ имени Н.Н. Блохина с учетом международных рекомендаций по проведению медико-биологических исследований с использованием животных, изложенных в «Европейской конвенции по защите позвоночных животных, используемых для экспериментальных и других научных целей», ЕЭС, Страсбург, 1985 г. [ДИРЕКТИВА 2010, 1993], а также требований Хельсинкской декларации и Всемирной медицинской ассоциации (2000). 2.26 Иммунофлуоресцентное окрашивание криосрезов первичных узлов меланомы Из материала мышиных первичных узлов, замороженных в среде Tissue-Tek, приготавливали срезы толщиной 7 мкм на криостате HM 505E (Microm). Срезы фиксировали в 4% формальдегиде 10 минут, промывали ФСБ и инкубировали 10 минут в 0,2 % растворе Тритона Х100. После инкубации срезы промывали в ФСБ. Далее на препараты наносили нормальную сыворотку крови донора вторых антител (осла) на 30 мин (1:10, Sigmaaldrich), отмывали в ФСБ. Для двойной окраски срезы инкубировали в первых моноклональных антителах крысы против CD31 мыши (1:100, BD Biosciences, кат. № 550274) или против CD90 мыши (1:100, BD Biosciences, кат. № 553000), а затем антитела кролика против Т-кадгерина (1:100). В качестве негативного контроля использовали фракции иммуноглобулинов неимунной сыворотки крысы и кролика. После отмывки в ФСБ, срезы помещали в смесь вторых антител на 60 минут: антител осла против антител крысы, конъюгированных с флуорохромом AlexaFluor488 (Molecular Probes®, Invitrogen) и антител осла против антител кролика, конъюгированных с флуорохромом AlexaFluor594 (Molecular Probes®, Invitrogen). Ядра клеток докрашивали DAPI (Sigmaaldrich). После трехкратной отмывки в ФСБ срезы заключали в среду Mounting Medium Vectashield TM (Vector Laboratories). На срезах, окрашенных антителами против CD31 и Т-кадгерина, производили подсчет сосудов в 4-5 полях зрения, выделяя сосуды среднего диаметра (CD31положительные образования длиной от 20 до 40 мкм), крупные сосуды (диаметром более 40 мкм) и капилляры (диаметром менее 20 мкм, без просвета) с использованием программы AdobePhotoshop 7 (Adobe Systems). Полученное количество сосудов в каждом поле зрения нормировали на единицу площади по DAPI для каждого среза. Статистический анализ 135 данных проводили с использованием программы Statistica 6.0, применялся непараметрический критерий Манна-Уитни. Вклад стромального компонента в прогрессию первичного узла мышиной меланомы анализировали путѐм подсчѐта отношения площади CD90-позитивных областей к общей площади среза в программе Meta Morph 5.0 (Universal Imaging). Для анализа количества очагов некроза срезы фиксировали в 4% формальдегиде 10 минут при комнатной температуре, затем срезы промывали ФСБ 3 раза по 5 минут и окрашивали гематоксилином Майера. После дифференцировки в проточной воде и трехкратной отмывки в ФСБ срезы заключали в среду Mounting Medium VectashieldTM. Полученные препараты анализировали при помощи флуоресцентного микроскопа Leica AF6000. Документирование изображений производили с помощью цифровой видеокамеры Axiocam и обработки в программе Las AF (Leica Application Suite). На препаратах производили подсчѐт отношения количества срезов с очагами некроза к общему количеству срезов. Статистический анализ данных проводился с использованием программы Statistica 6.0, применялся непараметрический критерий Манна-Уитни. 2.27 Модель хориоаллантоиcной мембраны куриного эмбриона in vivo В работе для изучения ангиогенеза in vivo была использована модель хориоаллантоисная мембрана куриного эмбриона по методике, описанной в Angiogenesis Protocols V.46 [Staton et al., 2004]. Эта модель хорошо воспроизводит in vivo многие характеристики опухолей, такие как формирование опухолевой массы, ангиогенез, или неоваскуляризация, инфильтративный рост. До начала проведения эксперимента, оплодотворѐнные куриные яйца хранили при температуре +40С. При проведении эксперимента оплодотворѐнные яйца инкубировали при температуре +370С и 70% влажности в течение 3 дней. На 3 день инкубации яйца протирали 70% этиловым спиртом, с острого конца каждого яйца шприцом удаляли 2-3 мл альбумина для отделения поверхности хориоаллантоиcной мембраны от скорлупы яйца. На 4 день инкубации на тупом конце яйца делали прокол воздушного мешка и выпуск воздуха для увеличения полости под окошечком. В этот же день в скорлупе каждого яйца было сделано окно квадратной формы (2х2 см), раскрывающее хориоаллантоисную мембрану. Скорлупа была удалена пинцетом. Затем окно было закрыто пластырем и яйца инкубировали ещѐ в течение 3 дней. Суммарную популяцию гиперэкспрессирующих Ткадгерин или контрольных клеток мышиной меланомы B16F10 имплантировали в 136 хориоаллантоиcную мембрану куриного эмбриона на 30-31 стадии развития на 7 день инкубации (через окошко в скорлупе) в виде суспензии в ростовой среде (RPMI1640). Среда дополнительно содержала 10% ФБС, 100 Ед/мл пенициллина G, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 2,05 мМ L-глутамина. Для этого клетки, достигшие 90-95% плотности монослоя, снимали с культуральных чашек 0,25% раствором трипсина, а затем ресуспендировали в небольшом объѐме среды. Число собранных клеток определяли с использованием камеры Горяева. Затем приготавливали суспензию клеток в среде в концентрации 1х106 клеток в 500 мкл среды. Введение клеток осуществляли инсулиновым шприцом. Хориоаллантоисную мембрану контрольных и экспериментальных эмбрионов анализировали через 3 дня после операции и фотографировали с использованием стереомикроскопа (Olympus SZX 16, камера Axio Cam HRc, Zeiss) и программы Axio Vision 3.1. 2.28 Оценка пролиферации клеток B16F10 in vitro 2.28.1 Метод подсчѐта абсолютного числа клеток Перед проведением эксперимента клетки B16F10 депривировали в среде RPMI 1640, содержащей 0,1% ФБС, в течение 24 ч. Контрольный клон клеток B16F10 и клоны с разным уровнем экспрессии Т-кадгерина сажали на 6-луночный планшет в количестве 1х105 на лунку в 2 мл ростовой среды RPMI 1640, содержащей 10% ФБС, 100 ЕД/мл пенициллина G, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 0,2 мМ Lглутамина. Каждые 24 ч клетки снимали с одной лунки 6-луночного планшета с помощью 0,25 % раствора трипсина (HyClone) и производили подсчѐт абсолютного числа клеток на приборе Cell Counter (Invitrogen). Общая длительность эксперимента составляла 72 ч. Эксперимент повторяли 5 раз. 2.28.2 Метод измерения клеточного индекса Перед проведением эксперимента клетки B16F10 депривировали в среде RPMI 1640, содержащей 0,1% ФБС, в течение 24 ч. Оценку пролиферации клеток контрольного клона B16F10 и клеток клонов мышиной меланомы с разным уровнем экспрессии Ткадгерина проводили с использованием системы xCELLigence (Roche) на планшетах E-plate (Roche). Перед посадкой клеток в каждую лунку планшета наливали 100 мкл среды RPMI1640, содержащей 10% ФБС, 100 Ед/мл пенициллина G, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 0,2 мМ L-глутамина. Затем планшет помещали на 30 минут в СО2- инкубатор. После этого планшет помещали в камеру RTCA SP Station (Roche) и снимали показатели фона для каждой лунки с помощью анализатора RTCA Analyzer (Roche). Клетки для эксперимента снимали с чашек с использованием HyQtase (HyClone) и 137 число клеток подсчитывали с помощью прибора Cell Counter (Invitrogen). В каждую лунку сажали клетки клонов мышиной меланомы в концентрации 5х103 в 100 мкл среды RPMI1640, содержащей 10% ФБС, 100 Ед/мл пенициллина G, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 2,05 мМ L-глутамина. В каждом эксперименте каждый клон клеток мышиной меланомы сажали в 2 лунки. После посадки клеток планшет помещали в RTCA Station и анализировали динамику роста клонов клеток мышиной меланомы B16F10 в режиме реального времени (первые 5 часов измерения проводили с интервалом 1 мин, последующие 91 ч - с интервалом 10 мин). Анализ полученных результатов проводили с помощью программного обеспечения RTCA Software 1.2.1 (Roche). Эксперимент повторили 3 раза. 2.28.3 Оценка распределения клеток B16F10 по фазам клеточного цикла in vitro Для синхронизации клеточного цикла клетки меланомы B16F10 депривировали в среде RPMI1640, содержащей 0,1% ФБС, в течение 24 ч. После чего клетки клонов мышиной меланомы стимулировали средой RPMI1640, содержащей 10 % ФБС, в течение 48 ч. По истечении 48 ч клетки снимали с чашек 0,25% раствором трипсина, который затем удаляли с помощью центрифугирования. После этого суспензию клеток фиксировали ледяным 70%-ным этанолом в течение 2 ч при температуре -200С и окрашивали в течение 30 мин при комнатной температуре раствором йодистого пропидия (на ФСБ, в конечной концентрации 50 мкг/мл) (Invitrogen), содержащем 200 мкг/мл РНКазы А (Invitrogen) и 0,1 % Тритона Х100. Содержание ДНК в клетках определяли по флуоресценции йодистого пропидия в диапазоне длин волн 600-625 нм (при возбуждении длиной волны 488 нм) методом проточной цитометрии с помощью клеточного сортера MoFlo (DakoCytomation). Эксперимент повторяли 3 раза. Данные проточной цитометрии анализировали с помощью программы для математического анализа распределения клеток по стадиям клеточного цикла Mod Fit LT 3.2 (Verity Software House). Для исключения клеточного дебриса, клеточных агрегатов и нестабильности сигнала исходные данные обрабатывали с использованием двумерных гистограмм: прямого светорассеяния, бокового светорассеяния; ширины импульса, прямого светорассеяния; времени, интенсивности флуоресценции йодистого пропидия, соответственно. Для анализа клеточного цикла использовали одномерные гистограммы распределения клеток по интенсивности флуоресценции йодистого пропидия. Различали три стадии клеточного цикла: G0/G1, S и G2/M. Для расчѐта относительного содержания клеток в этих фазах экспериментальную гистограмму описывали в виде линейной комбинации трѐх модельных функций распределения: для 138 клеток в фазах G0/G1 и G2/M использовали однокомпонентные гауссовы функции, а для фазы S - однокомпонентную трапецеидальную функцию. 2.29 Оценка уровня некроза и апоптоза клеток B16F10 in vitro Клетки контрольного клона B16F10 и клонов мышиной меланомы с разным уровнем экспрессии Т-кадгерина снимали с чашек при помощи 0,25% трипсина, отмывали в буфере для связывания Аннексина V (BD Biosciences) и ресуспендировали в 200 мкл этого же буфера. После этого к клеткам добавляли 5 мкл раствора АnV - РЕ (Аннексин V – фикоэритрин, BD Biosciences) и 10 мкл раствора 7-AAD (BD Biosciences) и инкубировали в течение 15 мин при комнатной температуре в темноте. С помощью проточной цитометрии на приборе FACS Canto II™ (BD Biosciences) определяли количество аннексин V и 7-AAD позитивных клеток; аннексин V - позитивных - 7-AAD - негативных клеток; аннексин V и 7-AAD - негативных клеток. 2.30 Анализ экспрессии генов в клетках клонов B16F10 in vitro 2.30.1 Выделение РНК Все процедуры по выделению РНК и синтезу кДНК проводили на льду в ламинарном боксе. Выделение РНК из клонов клеток мышиной меланомы B16F10 осуществляли с помощью коммерческого набора RNeasy Mini kit (Qiagen) согласно протоколу фирмы-производителя. Во время выделения РНК лизаты клеток обрабатывали ДНКазой I (Qiagen, Германия) в течение 30 мин при комнатной температуре. Концентрацию выделенной тотальной РНК определяли на спектрофотометре NanoDrop 1000 (Thermo Scientific) с использованием программного обеспечения ND-1000 V 3.7.1 (Thermo Scientific). Также с использованием спектрофотометра NanoDrop 1000 осуществляли контроль качества выделенной РНК. Для этого анализировали показатели отношений значений поглощения при длинах волн 260 нм и 280 нм, а также при длинах волн 260 нм и 230 нм. Для последующей процедуры обратной транскрипции отбирали образцы РНК со следующими показателями: 260нм/280нм = 2, 260нм/230нм = 1,8-2,2. 2.30.2 Синтез кДНК и полимеразная цепная реакция с детекцией в режиме реального времени (RT-PCR) На матрице РНК в результате реакции обратной транскрипции синтезировали кДНК. Для этого 1 мкг РНК смешивали с 0,5 мкг неспецифическими праймерами Oligo (dT)18 primer (0,5 мкг/мкл) (Fermentas). Смесь инкубировали 5 минут при температуре +650C. После инкубации к смеси добавляли 4 мкл 5-ти кратного буфера reaction buffer 139 (Fermentas), 0,5 мкл раствора Ribo Lock™ RNase Inhibitor (40 ЕД/мкл) (Fermentas), 2 мкл 10 мМ смеси нуклеотидов dNTP mix (Fermentas). Смесь инкубировали 5 минут при температуре +370C. После инкубации в смесь добавляли 2 мкл M-MulV Reverse Transcriptase (20 ЕД/мкл) (Fermentas). Полученную смесь инкубировали 60 мин при температуре +370C. Останавливали реакцию нагреванием смеси до температуры +70 0C в течение 10 минут. Синтезированную кДНК замораживали и хранили при температуре 200C. Выделение РНК из клеток мышиной меланомы производили в пяти параллелях. Таким образом, в методике RT-PCR использовали по пять проб кДНК каждого клона клеток. Анализировали уровень экспрессии следующих генов: VEGF А, PDGF В, HGF, EGF, bFGF, uPAR, uPA, c-Met (рецептор HGF), MMP2, MMP9. Анализ экспрессии этих генов был проведѐн для контрольного клона клеток мышиной меланомы и клонов мышиной меланомы с разным уровнем экспрессии Т-кадгерина. Также в контрольном клоне и клонах с разным уровнем экспрессии Т-кадгерина мышиной меланомы был произведѐн анализ уровня экспрессии двух генов «домашнего хозяйства»: GAPDH и βактина. Гены «домашнего хозяйства» использовали для сравнительной оценки уровня экспрессии исследуемых генов. Специфические пары праймеры для анализа исследуемых генов были изготовлены фирмой Синтол (Россия) и имели следующие последовательности, представленные в Таблице 2.2. Таблица 2.2. Последовательность использованных праймеров. Название праймеров GAPDH мыши Последовательность праймеров прямой GACCCCTTCATTGACCTCAACTAC обратный TGGTGGTGCAGGATGCATTGCTGA Актин β мыши прямой AGTGTGACGTTGACATCCGTA обратный GCCAGAGCAGTAATCTCCTTCT VEGF А мыши прямой AGAGCAGAAGTCCCATGAAGTGA обратный TCAATCGGACGGCAGTAGCT PDGF В мыши прямой TCTCTGCTGCTACCTGCGTCTGG обратный GTGTGCTCGGGTCATGTTCAAGTC HGF мыши EGF мыши прямой TCATTGGTAAAGGAGGCAGCTATA обратный CTGGCATTTGATGCCACTCTTA прямой CCTGCCCCCTTCCTAGTTTTC 140 обратный CTCCGTTCTGTTGGTCTACCC bFGF мыши прямой CGGCATCACCTCGCTTCC обратный TGCCCAGTTCGTTTCAGTGC uPAR мыши прямой CGTTACCTCGAGTGTGCGTCCTG обратный AGCCTCGGGTGTAGTCCTCATCCT uPA мыши прямой GAATGCGCCTGCTGTCC обратный AGGGTCGCTTCTGGTTGTC c-Met мыши прямой обратный MMP 2 мыши прямой CAACGAGAGCTGTACCTTGACCTTA GCGGGACCAACTGTGCAT AGTTCCCGTTCCGCTTCC обратный GACACATGGGGCACCTTCTG MMP9 мыши прямой CTGGACAGCCAGACACTAAAG обратный CTCGCGGCAAGTCTTCAGAG Для каждого исследуемого гена приготавливали смесь праймеров (прямого и обратного) в конечной концентрации 5 пкмоль/мкл каждого праймера. Для приготовления одной пробы использовали 10 мкл реакционной смеси, содержащей SYBR Green I (Синтол, Россия), 13 мкл ddH2O (Синтол), 1 мкл смеси необходимых праймеров и 1 мкл кДНК клеток мышиной меланомы. Для приготовления NTC (non-template control, отрицательный контроль без добавления матрицы) проб вместо кДНК добавляли 1 мкл ddH2O. Реакцию RT-PCR проводили на приборе Rotor - Gene™ 3000 (Corbett Research). Анализ кривых амплификации производили с помощью программы Rotor – Gene Analysis Software 6.0 (Corbett Research). Температура отжига праймеров к VEGF А, HGF, EGF, uPAR, GAPDH, cMet была равна +600С, к актину β, MMP9 – 610C, к PDGF В, bFGF была равна +58,50C, к uPA и MMP2 была равна +530C. Температура отжига для смеси праймеров на каждый исследуемый ген была предварительно подобрана экспериментально. Для всех проб использовали 40 циклов амплификации. После проведения RT-PCR полученные данные по значениям порогового цикла для каждого гена обрабатывали согласно формуле: N=2-∆C* 1000, ∆С= С1-Сhk Где С1- значение порогового цикла для исследуемого гена, Сhk- среднее значение порогового цикла для двух генов ―домашнего хозяйства‖ (GAPDH и β-актина), N – полученный результат, выраженный в относительных единицах. 141 2.30.3 Синтез кДНК и анализ профиля экспрессии генов с помощью наборов RT2 Profiler PCR Array На матрице РНК с использованием RT2 First Strand Kit (SA Biosciences™) синтезировали кДНК согласно протоколу фирмы-производителя. Для реакции обратной транскрипции брали 1 мкг суммарной РНК. Для проведения реакции ПЦР использовали наборы реагентов RT2 Real-Time™ SYBR Green/Fluorescein PCR master mix (SA Biosciences™, США). Конечный объѐм реакционной смеси, согласно инструкции производителя, составлял 25 мкл. Для анализа влияния экспрессии Т-кадгерина на изменение профиля экспрессии анализируемых генов в клетках клонов мышиной меланомы B16F10 были использованы системы анализа экспрессии генов RT2 Profiler PCR Array (SA Biosciences™), адаптированные для проведения ПЦР на приборе IQ5 (Bio Rad). При этом была проанализирована экспрессия следующих генов: внеклеточный матрикс и молекулы адгезии, ангиогенные факторы и ингибиторы ангиогенеза, хемокины и их рецепторы. Для нормализации результатов исследования использовались 5 генов «домашнего хозяйства»: B2M, HPRT1, RPL13A, GAPDH и β-актин. 2.31 Концентрирование кондиционированной среды клеток B16F10 Клетки меланомы B16F10 контрольного клона и клонов с разным уровнем экспрессии Т-кадгерина высаживали на 100 мм чашки Петри в 7 мл среды RPMI1640, содержащей 10% ФБС, 100 Ед/мл пенициллина G, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 0,2 мМ L-глутамина. Через 48 ч 4 мл ростовой среды от каждого клона B16F10 наносили на колонку Centricon 30 кДа (Millipore, США) и центрифугировали при 4000 об/мин 20 минут при комнатной температуре. В полученный концентрат добавляли коктейль ингибиторов протеиназ Protease Inhibitor Cocktail (1:100), замораживали и хранили при температуре -200С. 2.32 Оценка инвазивного потенциала клеток B16F10 in vitro За 24 ч до эксперимента клетки клонов мышиной меланомы B16F10 депривировали в среде RPMI1640, содержащей 0,1% ФБС. После депривации подсчитывали общее число клеток в популяции с помощью прибора Cell Counter (Invitrogen) и готовили суспензию клеток с концентрацией 3х105/мл. Затем пробирку с клетками переносили в штатив на +4 0C и смешивали с 5 мкл Матригеля, лишѐнного факторов роста GFR Matrigel™ (Growth Factor Reduced Matrigel, BD) в соотношении 1:1. На 60 мм чашках Петри из 0,2 мкл полученной смеси формировали капли (Рисунок 2.1). После полимеризации первой капли GFR 142 Матригеля с клетками сверху наносили 5 мкл Матригеля, содержащего факторы роста Matrigel™ (BD Biosciences), для формирования второй внешней капли. После полимеризации чашки Петри заливали средой RPMI1640, содержащей 10% ФБС. Оценивали инвазивный потенциал клеток, выползающих из Матригеля, лишенного факторов роста, активно деградирующих матрикс и мигрирующих по градиенту хемоаттрактантов в Матригель с факторами роста. Рисунок 2.1. Схема эксперимента по миграции клеток в двойной капле Матригеля для оценки инвазивного потенциала клеток клонов мышиной меланомы B16F10 in vitro. Полученные капли фотографировали через 24 и 48 ч с помощью микроскопа Leica AF6000 (Leica Microsystems) с объективом х5. В программе Las (Leica Application Suite) AF (Leica Microsystems) измеряли диаметры капли (D1 и D2) (Рисунок 2.2) и подсчитывали число клеток, инвазировавших из GFR Матригеля в Матригель, содержащий факторы роста. D2 Граница капли D1 Рисунок 2.2. Капля GFR Матригеля с клетками клонов мышиной меланомы B16F10 до начала эксперимента. 143 2.33 Получение мезенхимных стромальных клеток жировой ткани (МСК-ЖТ) мыши МСК-ЖТ мыши были выделены из подкожного жира области бѐдер самцов линии CBA/C57BL. Жировую ткань помещали в буфер Хэнкса (HBSS, HyClone), содержащий 500 Ед/мл пенициллина G и 500 мкг/мл стрептомицина, после чего переносили в чашку Петри и измельчали ножницами в стерильных условиях до получения однородной суспензии. После измельчения жировую ткань обрабатывали коллагеназой I типа (200 ед/мл, Worthington) и протеазой (30 ед/мл, Worthington) в ростовой среде ДМЕМ (HyClone) в течение часа при температуре +370С и постоянном перемешивании. Затем к суспензии добавляли равный объѐм среды ДМЕМ, содержащей 10% ФБС, и центрифугировали 10 минут при 200 g. Осадок, в котором находилась фракция стромально-васкулярных клеток, ресуспендировали в ростовой среде ДМЕМ c 10% ФБС, после чего фильтровали через фильтр с диаметром пор 40 мкм. Полученную суспензию центрифугировали 5 мин при 200 g. Для лизиса эритроцитов к осадку добавляли 9 мл стерильной дистиллированной воды и инкубировали 2 мин до покраснения. Затем добавляли 1 мл стерильного 10-ти кратного буфера ФСБ (Sigmaaldrich) и центрифугировали 5 минут при 200 g. Полученный осадок ресуспендировали в среде роста ДМЕМ, подсчитывали число клеток в камере Горяева и высаживали в чашки Петри в концентрации 104 клеток/мл. Для экспериментов использовали клетки 2 пассажа. 2.34 Влияние кондиционированной среды клеток B16F10 на пролиферацию и миграцию МСК-ЖТ in vitro 2.34.1 Влияние кондиционированной среды клеток B16F10 на пролиферацию МСКЖТ МСК-ЖТ высаживали в концентрации 104/мл на 24-луночный планшет в 500 мкл среды RPMI1640, содержащей 0,1% ФБС. В этот же день в верхнюю камеру вставки трансвелл Transwell® (Millipore) сажали клетки клонов мышиной меланомы B16F10 в концентрации 5х103/мл в 200 мкл среды RPMI1640, содержащей 10% ФБС. В качестве контроля в верхнюю камеру вместо клонов меланомы сажали фибробласты NIH/3T3 в той же концентрации, также в качестве контроля в верхнюю камеру вносили 200 мкл среды RPMI1640, содержащей 10% ФБС (Рисунок 2.3). В эксперименте использовали вставку с мембраной, диаметр пор в которой составлял 0,4 мкм. На следующий день заменяли среду во вставках с посаженными на них клетками меланомы или NIH/3T3 на RPMI1640, содержащую 0,1% ФБС, и помещали в 24-луночный планшет с посаженными клетками 144 МСК-ЖТ. Инкубировали 48 ч, после чего клетки МСК-ЖТ в нижней камере промывали ФСБ, фиксировали 4% формальдегидом (PRS Panreac) и окрашивали гематоксилином Майера (Dako). На окрашенных препаратах осуществляли подсчѐт количества клеток в 4-5 полях зрения, что отражало влияние кондиционированной среды от клеток клонов мышиной меланомы B16F10 на пролиферацию клеток МСК-ЖТ. Рисунок 2.3. Схема эксперимента по изучению влияния среды роста от клеток клонов меланомы B16F10 на пролиферацию МСК-ЖТ. 2.34.2 Влияние среды от клеток клонов B16F10 на миграцию МСК-ЖТ В эксперименте использовали вставку Transwell® (Millipore) с мембраной, диаметр пор в которой составлял 8 мкм. Мембрану вставки перед экспериментом покрывали коллагеном I типа (0,1 мкг/мл) (Имтек, Россия). Клетки МСК-ЖТ сажали в концентрации 5х104/мл в верхнюю камеру вставки в 200 мкл среды RPMI1640, содержащей 0,1% ФБС. В этот же день в лунки 24-луночного планшета (в дальнейшем нижяя камера трансвелл) высаживали клетки клонов мышиной меланомы B16F10 в концентрации 105/мл в 500 мкл среды RPMI1640, содержащей 10% ФБС. В качестве клеточного контроля в нижнюю камеру высаживали фибробласты NIH/3T3 в той же концентрации, что и клоны меланомы (Рисунок 2.4); в качестве положительного контроля использовали среду RPMI1640, содержащую 10% ФБС. На следующий день среду в нижних камерах с посаженными на них клетками меланомы B16F10 или NIH/3T3 меняли на среду RPMI1640, содержащую 0,1% ФБС, и переносили вставки с посаженными клетками МСК-ЖТ в 24-луночный планшет. Инкубировали 48 ч, после чего промигрировавшие клетки МСК-ЖТ на нижней стороне мембраны вставки промывали буфером ФСБ, фиксировали 4% формальдегидом и 145 окрашивали гематоксилином Майера. На окрашенных мембранах проводили подсчѐт количества клеток в 4-5 полях зрения, что отражало влияние кондиционированной среды от клеток клонов мышиной меланомы B16F10 или NIH/3T3 на миграцию клеток МСК-ЖТ. Рисунок 2.4. Схема эксперимента по изучению влияния среды роста от клеток клонов меланомы B16F10 на миграцию МСК-ЖТ. 2.35 Статистическая обработка результатов Полученные результаты анализировали с помощью программного обеспечения Statistica 6.0. Данные представлены как mean ± SEM. Данные считались достоверными при p<0.05. 2.36 Иммуногистохимическое окрашивание криосрезов аорты человека Биопсийные образцы аорты больных атеросклерозом сосудов и здоровых доноров были любезно предоставлены профессором Зотиковым А.Е. из отделения сердечнососудистой хирургии Института хирургии им. А.В.Вишневского. Из образцов ткани получали криосрезы по методике, описанной выше. После размораживания срезы фиксировали в 4% растворе формальдегида. Для блокирования неспецифического связывания вторых антител образцы инкубировали в 10% растворе сыворотки донора вторых антител, содержащей также 1% раствор БСА на буфере ФСБ. В качестве вторых антител для иммуногистохимического производства Jackson ImmunoResearch. окрашивания использовали антитела 146 После промывки образцы помещали в полимерную среду для заключения образцов Aqua POLY/MOUNT и оставляли полимеризоваться в темноте в течение 1-2 часов при комнатной температуре. Окрашивание визуализировали с использованием микроскопа Zeiss модели DM 6000. Обработку полученных изображений осуществляли в программе Adobe PhotoShop software (Adobe Systems, Mountain View). 2.37 Выделение и йодирование липопротеидов низкой плотности Липопротеиды низкой плотности ЛНП (р 1,019-1,023 г/мл) выделяли из свежей плазмы здоровых доноров методом препаративного ультрацентрифугирования в градиенте плотности NaBr по методу Havel [Havel et al., 1955]. Для йодирования ЛНП радиоактивным йодом 125I по тирозиновым остаткам белков использовали монохлорид йода [Goldstein et al., 1985]. Количество используемых реагентов рассчитывали по следующей формуле: А мг ЛНП (по белку) в В мкл NaCl (150 мМ)/ Na2-ЭДТА(0,24 мМ) + В мкл 1М глицинового раствор + А/1,5 мКи Na125I (2000 Ки/мМ) + 13хА мкл IСlр-р. Все процедуры выполняли на льду под тягой за свинцовым экраном. Радиоактивные отходы складывали в свинцовый контейнер. Рабочий раствор йодистого хлора готовили по формуле: ICl р-р = 1 V IClstock + 12,5 V 2 M NaCl В пробирку добавляли раствор ЛНП, глицина, Na125I, и потом добавляли раствор йодистого хлора IСlр-р. Тщательно перемешивали и инкубировали на льду 45 мин, перемешивая каждые 5-10 мин. Наносили смесь на колонку PD-10, предварительно промытую раствором NaCl/ЭДТА (рН 7,4), элюировали раствором NaCl/ЭДТА (нанося порциями по 0,5 мл) и собирали меченные ЛНП (125I-ЛНП). Аликвоту меченых ЛНП отбирали для определения концентрации белка (по методу Лоури-Петерсону). Для определения удельной активности 10 мкл меченных ЛНП разбавляли в 800 мкл раствора NaCl/ЭДТА, затем пробу делили на три аликвоты. Далее в образцы для осаждения белка вносили равное количество раствора NaCl/ЭДТА, 100 мкл 0,2% раствора БСА (с конечной концентрацией 10 мг/мл), и 100 мкл 50% трихлоруксусной кислоты ТХУ. Образцы 147 перемешивали и инкубировали 10 мин при комнатной температуре, после чего центрифугировали в течение 5-10 мин при 10000-13000 об/мин. Супернатант переносили в новые пробирки. Удельную радиоактивность в осадке и супернатанте считали на γсчетчике. 2.38 Определение поверхностного связывания 125I-ЛНП с клетками Клетки культивировали до состояния монослоя. Перед экспериментом чашки с клетками помещали на лѐд и промывали 3 раза холодной средой ДМЕМ, содержащей 0,1% БСА и 20 мМ HEPES. Затем к клеткам добавляли по 1 мл среды (ДМЕМ, 0,2% БСА и 20 мМ HEPES) содержащей 0, 5, 10, 25, 50, 75 и 100 мкг/мл меченных ЛНП. Для определения неспецифического связывания добавляли 50 кратный избыток немеченых ЛНП. Клетки инкубировали на льду 4 ч, через каждые 15-20 мин аккуратно перемешивая. Среду, содержащую не связавшиеся ЛНП, аспирировали и затем клетки промывали 3 раза ФСБ c 0,2% БСА. Промытые клетки лизировали раствором, содержащим 1% SDS и 0,1 М NaOH. Затем лизаты переносили во флаконы, и с помощью γ-счетчика определяли связанную с клетками радиоактивность. Данные, полученные по специфическому связыванию меченных 125 I-ЛНП с клетками, анализировали с помощью линейной и нелинейной регрессии с использованием программы Graph Pad Prism 4.0 для определения числа участков связывания (Bmax) и константы диссоциации (Kd). 2.39 Определение концентрации внутриклеточного Ca2+ Работа проводилась совместно с Федеральным государственным бюджетным научным учреждением «Научный центр здоровья детей», лабораторией Клеточных и молекулярных технологий. Для измерения внутриклеточного Ca2+ ([Ca2+]in) клетки высаживали на покровные стекла, которые предварительно были покрыты поли-L-лизином (Диаэм, Россия) для улучшения клеточной адгезии. Плотность посадки клеток подбирали таким образом, чтобы к моменту начала измерений плотность клеток составляла 70-80% от монослоя. За сутки до начала проведения эксперимента в клетках меняли среду на бессывороточную. Измерение [Ca2+]in проводили методом флуоресцентной микроскопии с использованием двухволнового зонда FURA-2АМ [Beck et al., 2004]. Перед экспериментом клетки инкубировали в культуральной среде с зондом для измерения [Ca2+]in. После этого клетки промывали 3-4 раза солевым раствором Хэнкса (HBSS, Gibco) для измерения 148 кальция следующего состава (в мМ): NaCl - 130, KCl - 5, СаCl2 - 1,8, MgCl2 - 1,0, Na2HPO4х12H2O - 0,6, KH2PO4 - 0.4, HEPES - 20, глюкоза - 5, pH 7,4 – для отмывки от не связавшегося зонда, и выдерживали в нѐм 15-20 минут в темном месте для полной деэтерификации молекул зонда [Grynkewicz et al., 1985]. Затем стекло с клетками приклеивали к перфузионной камере (объѐм — 0,2 мл), смонтированной на столике инвертированного микроскопа (Axiovert 200, Zeiss), который совмещѐн со спектрофлуориметром (Spex). Система для анализа изображения представлена на Рисунке 2.5. 5 4 2 3 6 9 1 7 8 Рисунок 2.5. Схема установки для измерения [Ca2+] in в клетках. 1. Источник света (ксеноновая лампа); 2. шторка (shutter), регулирующая время экспозиции (30-50 мсек через каждые 10 сек); 3. Колесо с фильтрами, пропускающими возбуждающий флуоресценцию свет с длинами волн ex = 340 нм и ex = 380 нм (excitation filter wheel); 4. Объектив х20 (high NA objective); 5. Объект (перфузионная камера с приклеенным стеклом с культурой клеток); 6. Дихроический луче-расщепитель (dichroic beamsplitter); 7. Колесо с фильтром для эмиссии λem = 510 нм (emission filter wheel). 8. Видеокамера для анализа изображения (CCD camera); 9. Монитор ЭВМ [Chiesa et al., 2001]. Вся система позволяла одновременно регистрировать флуоресценцию от нескольких клеток (система анализа изображения «Имидж», Диаморф, Россия) при освещении попеременно двумя лучами света с разными длинами волн. Источником света служила ксеноновая лампа мощностью 450 Вт, работающая от стабилизированного источника питания. Экспозиция (фаза чоппера) длилась 0,5-2 сек, с интервалом (темновая фаза) в 10 сек во избежание «выгорания» зонда. Интенсивность флуоресценции 149 регистрировали, используя соответствующий для FURA-2AM фильтр, пропускающий свет с эмиссионной длиной волны 510 нм. Видеокамера (Cool Snapfx CCD camera, Roper Sci Inc.) регистрировала изображение от клеток, а компьютерная программа AnalySys позволяла получать цифровую запись эксперимента и еѐ обрабатывать. Обработку изображений проводили в программе Graph Pad Prism 4.0. Для измерения [Ca2+]in клетки инкубировали в течение 40 мин в CO2 инкубаторе при температуре +370С в присутствии 4 мкМ ацетоксиметильного эфира высокоаффинного зонда Fura-2АМ в конечной концентрации 5 мкМ с константой диссоциации 225 нМ [Grynkewicz et al., 1985]. Спектр флуоресценции этого липофильного зонда смещается к более коротким волнам при увеличении связывания молекул зонда с Са2+ (Рисунок 2.6). Рисунок 2.6. Спектр возбуждения индикатора для Са2+ - Fura-2АМ в растворе с различной концентрацией свободного Са2+ (от 0 до 39,8 мкМ). При связывании с Са2+ спектр смещается к более коротким волнам. По оси абсцисс: длина волны возбуждающего света в нм; по оси ординат: изменение интенсивности свечения в усл. ед. (fluorescence excitation). Длина волны = 360 нм – изобестическая точка, в которой свечение не зависит от концентрации Са2+ и определяется только концентрацией индикатора. Эмиссия – 510 нм. Для возбуждения флуоресценции использовали волны длиной ex= 340 и 380 нм, для эмиссии - фильтр, вырезающий 505 10 нм. В тех случаях, когда приведена шкала [Ca2+]i, калибровку проводили по формуле [Kiedrowski et al., 1999]: [Ca2+]i = [(RRmin)/(RmaxR)] x Kd x S 150 где R – это найденное отношение интенсивностей флуоресценции зонда при 340 нм и 380 нм F340/F380 в каждой точке кривой; Kd – это константа диссоциации комплекса FURA-2AM-Ca2+, равная 225 нМ; S – отношение интенсивности флуоресценции зонда в свободном и связанном от 2+ Ca виде при 380 нм (S = Fmin (при 380 нм)/Fmax (при 380 нм)). Измерение минимального (Rmin) и максимального (Rmax) уровня свечения производили путем последовательной инкубации клеток в следующих калибровочных растворах: для измерения Rmax – 130 мM KCl, 5 мM NaCl, 0 Mg2+, 5 мM CaCl2, 5 мM HEPES, 5 мМ Ca2+ /H+ ионофора иономицина, для измерения Rmin – бескальциевый HBSS, содержащий 5 мМ ЭГТА. Результаты обрабатывали с использованием статистических программ: Graph Pad Prism 3.00 и Statistics 6.0. 2.40 Оценка влияния липопротеидов низкой плотности на миграцию L929 клеток Изучение миграции клеток проводили с использованием микрокамеры Бойдена. За 24 ч до начала эксперимента клетки L929, ТС3 или К9, переводили на бессывороточную среду ДМЕМ. В нижние ячейки микрокамеры Бойдена помещали раствор ЛНП в концентрации 60 мкг/мл. В качестве контролей использовали среду ДМЕМ, содержащую раствор ЛНП (60 мкг/мл), преинкубированный в течение 1 часа с козьими антителами против ЛНП (на рисунке представлено как Fab, 100 мкг/мл), а также ДМЕМ, содержащую Fab (100 мкг/мл) [Bochkov et al., 1996], которые помещали в нижнюю камеру. Для отрицательного контроля использовали бессывороточную среду ДМЕМ, содержащую 1% БСА. Верхние и нижние ячейки камеры разделяли полупроницаемой мембраной (Neuro Probe Inc.) с размером пор 8 мкм. Мембрану покрывали раствором коллагена (50 г/мл). Камеру с клетками помещали в СО2-инкубатор при температуре +370С на 10 часов. Непромигрировавшие клетки счищали шпателем с верхней стороны мембраны. Промигрировавшие клетки на нижней стороне мембраны фиксировали в 96% этаноле в течение 5 мин, и окрашивали красителем Diff-Quik® Staining Kit (Dade Behring GmbH, Marburg, Germany). Далее мембрану сканировали и анализировали при помощи программы Scion Image software (Scion Corporation). Эксперименты проводили в 5-6 параллелях и повторили 3 раза. 151 ГЛАВА 3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Т-кадгерин, впервые был обнаружен более 20 лет назад в эмбриональном мозге цыпленка [Ranscht and Dours-Zimmermann, 1991] в развивающейся нервной системе он функционирует как «молекула отталкивания». В нашей лаборатории было выявлено, что Ткадгерин экспрессируется во взрослом организме не только в нервной системе, но и в сердечно-сосудистой [Ivanov et al., 2001; Kudrjashova et al., 2002]. Известно, что в эмбриогенезе формирование нервной и сердечно-сосудистой систем происходит параллельно, в результате чего крупные артерии и нервы во многих случаях располагаются в непосредственной близости друг от друга. В ряде случаев, причиной параллельного роста сосудов и нервов является секреция нервными клетками ангиогенных факторов, а клетками сосудов – нейротрофических факторов [Carmeliet, 2003]. В мировой литературе имеется достаточно большой массив данных о механизмах регуляции направленного роста аксонов и миграции нервных клеток [Poliakov et al., 2004; Weinstein, 2005; Eichmann et al., 2005]. В то же время данные о факторах и механизмах, регулирующих направленный рост сосудов, весьма немногочисленны. Участие в регуляции развития нервной и сосудистой систем было установлено для таких белков, как семафорины и их рецепторы (плексин и нейропилин), нейтрин и его рецепторы (DCC/неогенин и Unc5), слит-лиганды и их рецепторы (Robo), а также для ряда других белков [Weinstein, 2005]. Данных об экспрессии Т-кадгерина в сердечно-сосудистой системе в эмбриогенезе в литературе до сих пор нет. Неизвестно, насколько Т-кадгерин важен для регуляции траектории роста сосудов при формировании органов и тканей. Механизм, по которому Т-кадгерин осуществляет регуляцию роста нейронов в нервной системе в эмбриогенезе, заключается в гомофильном узнавании Т-кадгеринов и в последующем «отталкивании». Мы предположили, что сходный механизм мог бы иметь место и при регуляции роста кровеносных сосудов. Возможно, в зрелых сосудах Т-кадгерин играет роль фактора, обуславливающего целостность сосудистой стенки, препятствуя как прорастанию сосудов в Т-кадгерин-положительные участки, так и миграции клеток, несущих Т-кадгерин, в сосудистую стенку; при неоангиогенезе Т-кадгерина мог бы выполнять функцию «отталкивания», тем самым регулируя траекторию роста вновь формирующихся сосудов. Таким образом, полученные ранее в нашей лаборатории результаты и данные литературы послужили основой настоящего исследования, которое посвящено выявлению роли Т-кадгерина и механизмов его участия в функционировании зрелых сосудов, регуляции направленного роста кровеносных сосудов, формирующихся de novo, в норме и при патологии. 152 3.1 Выявление экспрессии Т-кадгерина в эмбриогенезе у мыши Для подтверждения высказанных предположений в первую очередь, используя методы in situ гибридизации и иммунофлуоресцентного окрашивания целых эмбрионов в сочетании с конфокальной микроскопией, мы оценили экспрессию Т-кадгерина в раннем эмбриональном развитии у мыши. 3.1.1 Синтез РНК пробы Для изучения экспрессии Т-кадгерина в период эмбрионального развития мыши использовали метод in situ гибридизации целых эмбрионов мыши (whole mount in situ hybridization detection of mRNA). Для этого проводили гибридизацию эмбрионов мыши с РНК пробой (antisense), которую синтезировали на матрице последовательности гена Ткадгерина. РНК проба antisense (обратная последовательность) связывается с мРНК в клетках эмбрионов мыши, что позволяет выявлять экспрессию тестируемых белков на уровне транскрипции. Sense проба (прямая последовательность) не связывается с мРНК, поскольку не является комплементарной к мРНК в клетках эмбрионов и использовалась в качестве отрицательного контроля. Положительным контролем служила РНК проба, синтезированная на матрице последовательности гена Krox20, уровень экспрессии которого в клетках центральной нервной системы эмбрионов мыши на этих стадиях высок. В работе были использованы плазмиды, содержащие EST последовательности, доступные в электронной базе данных Gen Bank (expressed sequence tag), которые представляют собой короткие cDNA последовательности, используемые для выявления экспрессии генов или определения последовательности гена. Для выявления экспрессии мРНК Т-кадгерина и Krox20 была проведена трансформация E.сoli и их наращивание с последующим выделением и очисткой векторов. Для синтеза РНК проб был сначала проведен аналитический анализ с использованием небольшого количества кДНК Т-кадгерина и Кrox20, при этом кольцевые плазмиды были линеаризованы по сайтам, доступным для соответствующих полимераз (Т7 и Т3). Для рестрикции были использованы рестриктазы: Sal1, Not1, Xho1, BamH1. Линеаризацию подтверждали электрофорезом в агарозном геле (Рисунок 3.1). 153 Рисунок 3.1. Карта плазмидных векторов pFLCI, содержащая кДНК Т-кадгерина и Bluescript Ks, содержащая Krox20, со встроенными EST последовательностями для in situ гибридизации. Схема синтеза РНК проб. Рисунок 3.2. Электрофореграмма рестрикционного анализа линеаризованных плазмид с использованием электрофоретического разделения образцов ДНК в агарозном геле. Последовательность нанесения образцов: Krox20 plasmid – исходная плазмида с геном Krox20; Krox20/Sal1 – линеаризованная плазмида после обработки рестриктазой Sal1; Krox20/Xho1 – линеаризованная плазмида после обработки рестриктазой Xho1; Krox20/Not1 – линеаризованная плазмида после обработки рестриктазой Not1; Krox20/BamH1 – линеаризованная плазмида после обработки рестриктазой BamH1; Tcad plasmid - исходная плазмида с геном Т-кадгерина; T-cad/Sal1 – линеаризованная плазмида после обработки рестриктазой Sal1; T-cad/Not1 – линеаризованная плазмида после обработки рестриктазой Not1; T-cad/Xho1 – линеаризованная плазмида после обработки рестриктазой Xho1; T-cad/BamH1 – линеаризованная плазмида после обработки рестриктазой BamH1. 154 Основываясь на результатах рестрикционного анализа и карт плазмидных ДНК, для линеаризации плазмид с целью дальнейшего синтеза мРНК Т-кадгерина (с обратной и прямой последовательностью РНК) были выбраны рестриктазы Not1 и BamH1 (Рисунок 3.1 и Рисунок 3.2). Рестриктазы Sal1 и Xho1 режут плазмиду более чем по одному сайту, и эти сайты рестрикции находятся внутри последовательности, кодирующей Т-кадгерин, что делает нецелесообразным их использование. Для линеаризации с последующим синтезом мРНК Кrox20 была выбрана рестриктаза BamH1, поскольку, как и в случае плазмиды, содержащей кДНК для Т-кадгерина, BamH1 была единственной рестриктазой, которая разрезала исходную плазмиду по одному сайту. После рестрикционного анализа была проведена линеаризация всей плазмидной ДНК для синтеза Кrox20 (4 мкг) и Т-кадгерина (8 мкг) с целью дальнейшего синтеза РНК проб. Качество линеаризованной плазмиды проверяли ДНК электрофорезом в агарозном геле (Рисунок 3.3). Линеаризованную ДНК очищали от компонентов рестрикционной смеси и использовали для синтеза проб РНК. Т3 РНК-полимераза была использована для синтеза РНК-содержащей пробы с обратной последовательностью для Т-кадгерина и Krox20 (положительный контроль); Т7 РНК-полимераза - для синтеза пробы Т-кадгерина в прямой последовательности (отрицательный контроль). Наличие РНК после синтеза РНКсодержащих проб подтверждали электрофорезом в агарозном геле (Рисунок 3.4). После получения пробы были очищены и использованы для in situ гибридизации эмбрионов мыши. 155 Рисунок 3.3. Контроль линеаризации плазмид, содержащих гены Krox20 и Ткадгерина (T-cad), после обработки рестриктазами BamH1 и Not1 методом гельэлектрофореза. Krox20 plasmid – исходная плазмида с геном Krox20; Krox20/BamH1 – линеаризованная плазмида после обработки рестриктазой BamH1; T-cad plasmid исходная плазмида с геном Т-кадгерина; T-cad/BamH1 – линеаризованная плазмида после обработки рестриктазой BamH1; T-cad/Not1 – линеаризованная плазмида после обработки рестриктазой Not1. На дорожках геля с образцами необработанных плазмид Krox20 plasmid и T-cad plasmid присутствуют исходная и суперскрученная формы плазмиды; на дорожках геля с образцами плазмид после обработки рестриктазами присутствует только одна форма линеаризованной плазмиды. Рисунок 3.4. Анализ синтеза проб РНК генов Krox20 и Т-кадгерина (Т7 – отрицательный контроль, Т3 – проба для Т-кадгерина) методом гель-электрофореза. 156 3.1.2 Экспрессия Т-кадгерина в эмбриональном головном мозге у мыши Экспрессию Т-кадгерина оценивали с использованием методов in situ гибридизации и иммунофлуоресцентного окрашивания целых эмбрионов мыши на стадиях Е8.75 – Е11.5 (суток эмбрионального развития). На ранних этапах эмбрионального развития (эмбрионы Е8.75, Е9.5, Е10.5) для выявления экспрессии Т-кадгерина использовали метод in situ гибридизации. Экспрессия мРНК Т-кадгерина была обнаружена в формирующемся головном мозге, начиная со стадии Е8.75, в частности, в промежуточном мозге и в переднем мозге – во внутренней выстилке полости конечного мозга (telencephalon) (Рисунок 3.5). Также мРНК Т-кадгерина выявлялась в области глазных пузырей – в месте перехода промежуточного мозга в развивающиеся зрительные бугры. В отрицательном контроле (sense проба) наблюдали неспецифическое фоновое диффузное окрашивание в переднем мозге, отличное от специфического окрашивания, выявленного при использовании пробы для выявления в обратной ориентации (antisense). На стадии Е9.5 экспрессия мРНК Т-кадгерина была выявлена в переднем мозге, в утолщающейся обонятельной плакоде, в основании глазных пузырей, в области теменного изгиба, в месте перехода продолговатого мозга в спинной – в области затылочного изгиба (Рисунок 3.6). Возможно, экспрессия Т-кадгерина в основании формирующихся глазных пузырей связана с эпителизацией структур будущих глазных бокалов, или Т-кадгерин участвует в формировании сосудистой оболочки глаза. Однако для определения конкретной роли Т-кадгерина в формировании этих структур необходимы дополнительные исследования. Известно, что в области изгибов головного мозга на этой стадии развития происходит активное формирование и рост кровеносных сосудов [Vasudevan and Bhide, 2008]. Мы предположили, что экспрессия Т-кадгерина в этих областях формирующегося мозга связана с интенсивными процессами ангиогенеза и возможным участием этого белка в процессах регуляции направленного роста кровеносных сосудов так же, как это происходит при росте аксонов мотонейронов к своим мишеням в нервной системе. 157 Рисунок 3.5. In situ гибридизация эмбрионов мыши на стадии Е8.75. Экспрессия мРНК Т-кадгерина: 1 – в области среднего мозгового пузыря; 2 – в основании формирующегося глазного пузыря; 3 – во внутренней выстилке конечного мозга; 4 – в продолговатом мозге. 5, 6 – слуховые пузырьки. Отсутствие специфического окрашивания в отрицательном контроле. Увеличение в 3.2, 5 и 6 раз. Рисунок 3.6. In situ гибридизация эмбрионов мыши на стадии Е9.5. Окрашивание участков головного мозга соответствует локализации мРНК Т-кадгерина. Экспрессия наблюдается в основании формирующихся глазных пузырей; в области теменного и затылочного изгибов. Маленькие стрелки указывают на основание формирующегося глазного пузыря. Увеличение в 3.2, 5 и 6 раз. 158 На стадии Е10.5 наблюдалась интенсивная экспрессия мРНК Т-кадгерина в среднем мозге в формирующейся эпендимной крыше промежуточного мозга (диенцефалон, diencephalon) и его латеральных областях (Рисунок 3.7 и Рисунок 3.8). Также наблюдали специфическое окрашивание в области сосудистого сплетения конечного мозга (telencephalon) (Рисунок 3.7). Окрашенные области морфологически соответствовали областям формирования сосудистых сплетений в стенках формирующейся системы желудочков мозга. В отрицательном контроле специфического окрашивания не обнаружили. В положительном контроле наблюдали характерное для экспрессии гена Krox 20 окрашивание (Рисунок 3.7 и Рисунок 3.8). Экспрессию белка Т-кадгерина детектировали методом иммунофлуоресцентного окрашивания антителами против Т-кадгерина целых эмбрионах мыши в сочетании с конфокальной микроскопией. Рисунок 3.7. In situ гибридизация эмбрионов мыши на стадии Е10.5. Интенсивная экспрессия Т-кадгерина в формирующейся крыше мозга, в латеральных областях диенцефалона. Стрелкой указано специфическое окрашивание сосудистого сплетения в области конечного мозга. Отсутствие специфического окрашивания в отрицательном контроле. Увеличение в 3.2, 5 и 6 раз. 159 -1- -2- -3- Рисунок 3.8. In situ гибридизация эмбрионов мыши на стадии Е10.5. 1 – специфическое окрашивание в крыше мозга в области затылочного изгиба, во внутренней выстилке конечный мозга; 2 – отсутствие специфического окрашивания в отрицательном контроле; 3 – окрашивание структур центральной нервной системы в положительном контроле. Увеличение в 3.2 раза. Экспрессия белка Т-кадгерина выявлялась, начиная со стадии Е9.5, при этом специфическое окрашивание наблюдалось в выстилках развивающегося головного мозга (Рисунок 3.9), в том числе в основании формирующихся глазных пузырей, что соответствует ранее полученным методом in situ гибридизации данным. Экспрессия Ткадгерина в формирующемся глазном бокале указывает на возможное участие этого белка в развитии сосудистой оболочки глаза. Рисунок 3.9. Иммунофлуоресцентное окрашивание эмбрионов мыши на стадии Е9.5 (слева) и Е12.5 (справа). Специфическое окрашивание (красная флуоресценция) соответствует экспрессии Т-кадгерина в выстилках мозга (стадии Е9.5 и Е12.5); экспрессия Т-кадгерина в формирующемся глазном пузыре у эмбриона Е9.5. Синяя флуоресценция соответствует ядрам, докрашенным DAPI. Увеличение в 5 раз. 160 Высокий уровень экспрессии Т-кадгерина во внутренней выстилке головного мозга был выявлен, начиная с Е11.5 стадии. А именно, интенсивное специфическое окрашивание наблюдалось в области промежуточного мозга, формирующегося глазного бокала, а также в области среднего и заднего мозга (Рисунок 3.10). -1- -2- -3- Рисунок 3.10. Иммунофлуоресцентное окрашивание эмбрионов мыши на стадии Е11.5. Специфическое окрашивание (красная флуоресценция) соответствует экспрессии белка Т-кадгерина. Синяя флуоресценция соответствует ядрам, докрашенным DAPI. 1 – специфическое окрашивание в области промежуточного мозга, а также в области формирующегося глазного бокала; 2 – специфическое окрашивание в области среднего и заднего мозга; 3 – тот же участок, что и на 2 – на другом уровне оптической плоскости. Увеличение в 5 раз. Мы предположили, что Т-кадгерин участвует в формировании системы желудочков головного мозга, а именно – в формировании сосудистых сплетений в стенках желудочков, поскольку известно, что именно здесь на этом этапе эмбрионального развития происходит активное формирование сосудов мозга [Vasudevan and Bhide, 2008]. Таким образом, использование методов in situ гибридизации и иммунофлуоресцентного окрашивания в сочетании с конфокальной микроскопией целых эмбрионов мыши позволило выявить стадию, на которой экспрессируется Т-кадгерин, и морфологические области его экспрессии. Экспрессия Т-кадгерина на уровне мРНК была обнаружена, начиная со стадии Е8.75, в разных отделах формирующегося головного мозга. Экспрессия белка Т-кадгерина была выявлена, начиная со стадии Е9.5. Наибольшая экспрессия Т-кадгерина при этом наблюдалась во внутренней выстилке головного мозга, что позволило высказать предположение об участии Т-кадгерина в формировании сосудистых сплетений в стенках желудочков в развивающемся головном мозге. 161 3.1.3 Экспрессия Т-кадгерина в эмбриональном сердце Экспрессия Т-кадгерина в эмбриональном сердце на уровне белка была выявлена, начиная со стадии Е11.5. На стадиях Е8.75, Е9.5, Е10.5 ни экспрессии мРНК Т-кадгерина, ни экспрессии белка Т-кадгерина в формирующемся сердце обнаружено не было (Рисунок 3.11). Экспрессия Т-кадгерина, выявленная в эмбриональном сердце на стадии Е11.5 (Рисунок 3.12), что, по всей видимости, связана с активными процессами формирования и роста сердца и его отделов и процессами ангиогенеза [Burggren and Keller, 1997]. Предположительно, между стадиями Е10.5 – Е11.5 эмбрионального развития мыши происходит активация синтеза мРНК Т-кадгерина, после чего происходит быстрое и интенсивное накопление белка Т-кадгерина. Вполне вероятно, что Т-кадгерин также принимает участие в установлении синаптических контактов в формирующейся проводящей системе сердца. Полученные данные свидетельствуют о том, что начало экспрессии Т-кадгерина в развивающемся головном мозге и сердце у мыши совпадает с активизацией процессов формирования и роста сосудов за счет васкуло- и ангиогенеза в сердечно-сосудистой системе и головном мозге в эмбриогенезе [Gilbert and Singer, 2006]. Эти результаты позволили выдвинуть предположение о возможной роли Т-кадгерина как молекулырегулятора процессов формирования и роста сосудов. Для проверки предположения о том, что Т-кадгерин действительно участвует в этих процессах и выявления возможных механизмов его функционирования был использован ряд экспериментальных подходов. 162 Рисунок 3.11. Отсутствие мРНК Т-кадгерина в формирующемся сердце эмбрионов мыши на стадиях Е8.75 – Е10.5. Стрелки указывают на область формирующегося сердца. Увеличение в 5 и 6 раз. -1- -2- Рисунок 3.12. Иммунофлуоресцентное окрашивание эмбрионов мыши на стадии Е11.5. Специфическое окрашивание (красная флуоресценция) соответствует экспрессии белка Т-кадгерина. Синий цвет соответствует клеточным ядрам. 1 – специфическое окрашивание соответствует экспрессии Т-кадгерина в области сердца; 2 – контрольное окрашивание антителами к иммуноглобулину G. Увеличение в 5 раз. 163 3.2 Т-кадгерин и опухолевая прогрессия Согласно исследованию, проведенному в 187 странах в период с 1980 по 2010, ежегодно от рака кожи (статистика на 2010 год) умирает 80 000 человек; 49 000 смертей из них связано с меланомой кожи и 31 000 с немеланомными раками кожи [Lozano, 2013; Green, 2012]. На ранних этапах диагностики меланома может быть успешно удалена хирургическим путем, и в 80% случаев такое лечение является успешным [Gray-Schopfer et al., 2007]. Однако на стадии метастазирования меланома уже резистентна к существующим в настоящее время методам лечения и имеет плохой прогноз. Несмотря на то, что большинство опухолевых образований кожи сохраняют сходство с нормальной тканью, из которой они происходят, существуют некоторые виды опухолей с сильно измененной структурой, что в свою очередь создает трудности для постановки патологоанатомического диагноза. В отличие от доброкачественных опухолей, которые в большинстве случаев остаются на месте их образования и формируют компактную массу опухолевых клеток, злокачественные опухоли кожи состоят из клеток, способных проникать через базальную мембрану и метастазировать в другие органы через кровь и лимфатические сосуды [Quinn and Perkins, 2010]. Одним из важнейших вопросов для диагностики, прогноза и лечения новообразований кожи является дифференциальный диагноз между доброкачественными и злокачественными опухолями кожи. Считается, что для активно растущих и метастазирующих опухолей характерна потеря функциональности межклеточных контактов и нарушение межклеточных взаимодействий [Hanahan and Weinberg, 2000]. Так, агрессивные и злокачественные опухоли кожи, например, карциномы, характеризуются снижением или полным исчезновением экспрессии Е-кадгерина с поверхности клеток, которое происходит параллельно с увеличением экспрессии N-кадгерина [Frixen et al., 1991; Kurzen et al., 2003; Gloushankova, 2008; Berx and Van Roy, 2009] и/или белка десмосом десмоглеина [Gloushankova, 2008; Berx and Van Roy, 2009]. В настоящее время известно множество молекулярных маркеров, помогающих определить степень агрессивности и инвазивности новообразований кожи. Важными прогностическими факторами являются изменение экспрессии р53 [Verdolini et al., 2001], увеличение экспрессии некоторых транскрипционных факторов [Keehn et al., 2004], матричных металлопротеиназ (MMPs, ММП) [Verdolini et al., 2001], белков mib1, Ki-67 [Oh and Penneys, 2004], c-kit и p63 [Winters et al, 2008], увеличение фосфорилирования регуляторных белков и увеличение количества рецепторов факторов роста [Weigelt et al., 2005]. 164 Данная область требует дальнейших исследований, позволяющих понять биологию опухолевой прогрессии и помогающих развитию новых возможностей для терапии. 3.2.1 Т-кадгерин и опухолевый рост Данных о биологической роли и возможных механизмах участия Т-кадгерина в малигнизации и прогрессии опухолей мало и, в целом, они достаточно противоречивы. Данных о механизмах участия Т-кадгерина в опухолевом росте и метастазировании еще меньше. В ряде работ было сделано предположение о том, что Т-кадгерин является опухолевым супрессором, поскольку снижение его экспрессии в результате аллельной потери или гиперметилирования промотора гена, или других причин, связаны с ростом и метастазированием определенных видов опухолей [Takeuchi et al., 2002a и Andreeva and Kutuzov, 2010]. Так, подавление экспрессии Т-кадгерина коррелирует со злокачественностью фенотипа и онкогенностью раков молочной железы [Riener et al., 2008], лѐгких [Sato et al., 1998] и желчного пузыря [Maruyama et al., 2001]. Однако существует целый ряд работ, в которых показано что не всегда опухолевая прогрессия коррелирует со снижением экспрессии Т-кадгерина. Так, повышение экспрессии Ткадгерина при раке яичников, эндометрия [Suehiro et al., 2008] и остеосаркоме [Zucchini et al., 2004] положительно коррелирует с выживаемостью пациентов. Следует отметить, что в литературе встречается несколько походов к изучению роли Т-кадгерина в опухолевой прогрессии. В то время как некоторые авторы исследовали корреляцию между гиперметилированием промотора Т-кадгерина/аллельной потерей и изменением экспрессии Т-кадгерина, другие изучали влияние ре-экспрессии Т-кадгерина в опухолевых клетках на их злокачественные свойства с последующим введением в организм на животных моделях in vivo. Так, уменьшение роста опухолей при ре-экспрессии Ткадгерина наблюдается в случае введения клеток меланомы человека бестимусным мышам [Kuphal et al., 2009], гепатоцеллюлярной карциномы [Chan et al., 2008] и карциномы молочной железы [Lee et al., 1998]. Трансфекция клеток молочной железы геном Ткадгерина приводит к подавлению их пролиферации в культуре, что также сопровождается трансформацией фенотипа из злокачественного в нормальный [Lee, 1996]. Повышение экспрессии Т-кадгерина в клетках нейробластомы приводит к подавлению инвазии клеток и потери их способности пролиферировать при добавлении эпидермального фактора роста (EGF) [Takeuchi et al., 2000]. Гиперэкспрессия Т-кадгерина в клетках глиомы С6 сопровождается уменьшением способности их к миграции и подавлением роста и 165 пролиферации клеток в результате блокады клеточного цикла на стадии G 2 [Huang et al., 2003]. Об экспрессии Т-кадгерина в новообразованиях и предраковых состояниях кожи в литературе существуют отдельные разрозненные данные. В нормальной коже человека Ткадгерин экспрессируется в кератиноцитах, меланоцитах и фибробластах дермы, а также в сосудах дермы [Kuphal et al., 2009], максимальная его экспрессия наблюдается в кератиноцитах базального клеточного слоя [Zhou et al., 2002]. При солнечном (актиническом) кератозе наибольшая экспрессия Т-кадгерина наблюдается в атипичных кератиноцитах, в то время как при болезни Боуэна экспрессия Т-кадгерина сильно изменяется и, в общем и целом, уменьшена по сравнению с нормальной кожей [Pfaff et al., 2010]. Экспрессия Т-кадгерина снижается при псориазе [Zhou et al., 2002] и базальноклеточной карциноме кожи [Takeuchi et al., 2002a], а при инвазивной плоскоклеточной карциноме кожи полностью отсутствует в связи с нарушением метилирования и делецией гена [Takeuchi et al., 2002b]. В иммортализованной клеточной линии кератиноцитов, полученной из плоскоклеточной карциномы, искусственное увеличение экспрессия Т-кадгерина приводит к снижению пролиферативной активности этих клеток [Mukoyama et al., 2005]. Роль Т-кадгерина в прогрессии меланомы изучалась в нескольких работах, результаты которых противоречивы. В то время как зрелые меланоциты экспрессируют Ткадгерин [Kuphal et al., 2009; Bosserhoff et al., 2011], предшественники меланоцитов, клетки нервного гребня, его не экспрессируют. При выборе траектории миграции в эмбриогенезе клетки нервного гребня избегают каудальной части склеротома, где экспрессируется Ткадгерин, и мигрируют через ростральную часть [Ranscht and Bronner-Fraser, 1991]. Было высказано предположение о том, что Т-кадгерин, являясь навигационным рецептором, обеспечивает топографическое картирование для мигрирующих меланоцитов, а или также что де-дифференцировка предшественников опухолевая трансформация меланоцитов может сопровождаться потерей экспрессии Т-кадгерина. 3.2.2 Строма играет важную роль в регуляции роста и прогрессии опухолей В нормальной ткани фибробласты стромы ответственны за синтез, продукцию и ремоделирование внеклеточного матрикса, а также за секрецию растворимых паракринных факторов, регулирующих фенотип клеток, их пролиферацию, миграцию, выживаемость и апоптоз [Tlsty and Coussens 2006; Klopp et al., 2011]. Фибробласты, обнаруживаемые в опухолях, частично происходят из мезенхимных стволовых клеток, которые могут быть 166 расположены регионально (резидентные или ткане-специфические МСК), или относятся к циркулирующей популяции мезенхимных клеток костного мозга [Spaeth et al., 2008, Mishra et al., 2008]. Такие фибробласты, выделенные из злокачественных опухолей, имеют измененный фенотип, в большинстве случаев обусловленный нарушенной продукцией белков внеклеточного матрикса и факторов роста [Bauer et al., 1979; Knudson et al., 1984; Tlsty and Coussens 2006], усиленной пролиферацией и различными нарушениями гистологии сформированной ими ткани [Rasmussen and Cullen, 1998; van den Hooff, 1988]. Стромальные клетки вместе с клетками опухолей регулируют опухолевый неоангиогенез частично путем локального изменения баланса между растворимыми и нерастворимыми молекулами с про- и антиангиогенными свойствами [Takeuchi and Ohtsuki, 2001; Wyder et al., 2000; Cavallaro et al., 2006]. Известно, что стромальные клетки также способны самостоятельно секретировать множество проангиогенных факторов, таких как VEGF, FGF, PDGF-β, и стромальный фактор роста-1 (stromal derived factor-1 SDF-1) [Rubina et al., 2005; Efimenko et al., 2009]. Перечисленные цитокины стимулируют миграцию и пролиферацию эндотелиальных и гладкомышечных клеток в опухолях, усиливая ангиогенез [Potapova et al., 2007; Kinnaird et al, 2004]. К другим факторам роста, ответственным за влияние мезенхимных стромальных клеток на сосуды опухолей, относят фактор роста гепатоцитов HGF, циклооксигеназу, инсулино-подобный фактор роста IGF-1, фактор роста из тромбоцитов PDGF-BB, и фактор роста опухолей альфа – TGFβ [Beckermann et al., 2008]. Таким образом, опухолевые фибробласты не просто являются пассивным компонентом, поддерживающим транформированные клетки в структуре новообразованной ткани, а способны активно участвовать в процессах регуляции пролиферации, роста, дифференцировки и ее васкуляризации. 3.2.3 Т-кадгерин и васкуляризация опухолей Опухоли способны расти до размеров 1-2 мм3 без неоваскуляризации, получая все необходимые питательные вещества путем диффузии. Опухоли, которые не способны стимулировать рост новых сосудов и капилляров, не превышают эти размеры [Folkman, 2008]. В том случае, если опухоль продолжает расти, а васкуляризации не происходит, ткань подвергается некрозу или апоптозу [Holmgren et al., 1995; Parangi et al., 1996]; рост новых сосудов в опухоль напротив, стимулирует рост опухоли и метастазирование. Зависимость роста и прогрессии опухолей от неоангиогенеза лежит в основе перспективной стратегии противоопухолевой терапии [Folkman, 1995; Hanahan and Folkman, 1996; Risau, 167 1997; Folkman, 2008]. Для целого ряда опухолей было показано, что по сравнению с цитостатиками, которые влияют на пролиферацию самих опухолевых клеток, антиангиогенный подход вызывает быстрое и резкое подавление роста сосудов в целом ряде онкологических заболеваний, приводя к последующей массовой гибели клеток опухолей [Denekamp, 1993; Chaplin et al., 1996]. Опухолевый неоангиогенез происходит вследствие повышения экспрессии клетками опухоли или стромальными клетками проангиогенных факторов, таких, например, как VEGF, или снижения экспрессии антиангиогенных факторов, или и того и другого одновременно [Hasan et al., 2002]. При нормальном ангиогенезе сначала происходит миграция и пролиферация эндотелиальных клеток, а затем гладкомышечные клетки и перициты стабилизируют вновь сформированные сосуды. В процессе опухолевого неоангиогенеза происходит направленная миграция эндотелиальных клеток через базальную мембрану и миграция периваскулярных стромальных клеток к клеткам опухоли, продуцирующим специфичные ангиогенные факторы. Однако, вновь сформированные сосуды опухолей обычно представляют собой нестабильные тонкостенные капилляры или синусоиды, состоящие из одного слоя эндотелиальных клеток с базальной мембраной и предрасположенные к спонтанным геморрагиям и тромбозам [Carmeliet 2000; Yancopoulos et al. 2000; Jain and Carmeliet, 2001]. Известно, что такие представители суперсемейства кадгеринов как VE- и Nкадгерин играют важную роль в организации сосудистой сети: VE-кадгерин опосредует межклеточные взаимодействия между эндотелиальными клетками и необходим для выживания и обеспечения барьерной функции эндотелия, а N-кадгерин – участвует в стабилизации сосудов за счет взаимодействия эндотелиальных клеток с муральными (гладкомышечными клетками и перицитами) [Cavallaro et al., 2006]. Мы предположили, что Т-кадгерин, еще один представитель суперсемейства кадгеринов, также является перспективной мишенью для опухолевой терапии за счет своей способности подавлять рост кровеносных сосудов [Rubina et al., 2005]. Данные об экспрессии Т-кадгерина при опухолевом неоангиогенезе весьма немногочисленны и противоречивы. Известно, что экспрессия Т-кадгерина изменена в кровеносных сосудах метастазов в легких карциномы Льюиса и в сосудах F9 эндодермальной тератокарциномы, а также в сосудах при раке простаты PC-3 и А673 рабдомиосаркомы [Riou et al., 2006; Wyder et al., 2000]. Инактивация гена Т-кадгерина уменьшает рост опухоли молочной железы и подавляет ее васкуляризацию, что было показано на трансгенной мышиной модели рака молочной железы (mouse mammary tumor virus (MMTV) - polyoma virus middleT (PyV-mT)) [Hebbard et al., 2008]. В то же время в случае гепатоцеллюлярной карциномы человека экспрессия Т- 168 кадгерина повышается в эндотелиальных клетках капилляров опухоли, что коррелирует с ее ростом и метастазированием [Adachi et al., 2006]. Повышенная экспрессия Т-кадгерина в эндотелиальных клетках стимулирует опухолевый ангиогенез на сфероидной модели сокультивирования эндотелиальных клеток и клеток меланомы in vitro [Ghosh et al., 2007]. Таким образом, поскольку в ряде работ было продемонстрировано, что потеря экспрессии Т-кадгерина в кератиноцитах коррелирует с агрессивностью и опухолевой прогрессией было сделано предположение о том, что Т-кадгерин является возможным эндогенным регулятором пролиферации кератиноцитов. В то же время известно, что в норме активно пролиферирующие кератиноциты базального слоя эпидермиса также экспрессируют Т-кадгерин, что свидетельствует о том, что механизмы участия Т-кадгерина в регуляции физиологических процессов в нормальной коже и процессов роста и инвазии опухолей сложнее, чем просто влияние на пролиферацию клеток. Мы предположили, что Ткадгерин в коже не только регулирует пролиферацию кератиноцитов, но и играет роль в межклеточных взаимодействиях между кератиноцитами, мезенхимными клетками и сосудами, прорастающими в подлежащую строму в норме и при патологии. И, поскольку Ткадгерин является навигационной молекулой, регулирующей рост сосудов и нервов, сравнивая образцы новообразований, мы нормальной кожи, ожидали увидеть доброкачественных изменение и экспрессии злокачественных Т-кадгерина в неопластических клетках и в прорастающих в опухоли сосудах. 3.2.4 Потеря экспрессии Т-кадгерина в кератиноцитах и нарушение его экспрессии в сосудах коррелирует с процессами озлокачествления немеланомных новообразований кожи человека Для выявления роли Т-кадгерина в патогенезе опухолевых образований кожи был проведѐн сравнительный анализ экспрессии Т-кадгерина в образцах нормальной кожи, образцах немеланомного рака кожи и связанных с ним предраковых состояниях. Криообразцы кожи от здоровых пациентов, пациентов с псориазом, кератоакантомой в стадии роста и стадии стабилизации, кератозом, поверхностными, нодулярными и инфильтрирующими формами базальноклеточного рака (базалиомы), плоскоклеточным раком и метатипическим раком были окрашены антителами против Т-кадгерина и маркеров клеток сосудов и проанализированы с использованием флуоресцентного микроскопа соответствующим программным обеспечением. Подробная классификация предраковых состояний и опухолей кожи описана в книге «Дерматология» под редакцией проф. ГалилОглы, проф. Молочкова В.А., проф. Сергеева Ю.В. 169 3.2.4.1 Нормальная кожа В нормальной коже наибольшая экспрессия Т-кадгерина наблюдалась в базальных кератиноцитах, стромальных клетках и во всех кровеносных сосудах в подлежащей дерме, что было обнаружено при окрашивании антителами против эндотелиального маркера vWF и маркера ГМК сосудов – α-актина (Рисунок 3.13). Полученные нами данные согласуются с результатами других исследований [Takeuchi et al., 2002b]. Также была обнаружена экспрессия Т-кадгерина в волосяных фолликулах и в сальных железах (Рисунок 3.13). В отличие от других публикаций [Zhou et al., 2002; Pfaff et al., 2010] в настоящем исследовании была выявлена экспрессия Т-кадгерина и в супрабазальных слоях эпидермальных кератиноцитов, хотя и слабее, чем в базальном слое. 170 Рисунок 3.13. Двойное иммунофлуоресцентное окрашивание образцов нормальной кожи антителами к Т-кадгерину (красная флуоресценция) и эндотелию vWF (зелѐная флуоресценция) (A, E) или эндотелию vWF (красная флуоресценция) и α-актину (зелѐная флуоресценция) (C). Рисунки A, B и C - параллельные криосрезы одного образца ткани. Ядра докрашены DAPI (синяя флуоресценция). На фотографии B показан фазовый контраст. Экспрессия Т-кадгерина обнаруживается в базальных кератиноцитах, супрабазальных слоях, стромальных клетках, волосяных луковицах и сальных железах. Т-кадгерин обнаруживается также в крупных и малых сосудах (жѐлтая флуоресценция), ко-локализация Т-кадгерина и vWF показана стрелками. Стабильные сосуды визуализируются в дерме и имеют двойное окрашивание на vWF и α-актин; стромальные клетки экспрессируют α-актин (C). Масштаб 100 мкм. На рисунке D представлена схема, показывающая стабильную экспрессию Т-кадгерина в базальных слоях кожи (красный цвет) и в клетках стромы, умеренную экспрессию Т-кадгерина в супрабазальных слоях (розовый цвет) и экспрессию Т-кадгерина во всех vWFэкспрессирующих сосудах (жѐлтый цвет). F - подписи к схеме. 171 3.2.4.2 Псориаз Псориаз – хроническое заболевание, при котором кератиноциты пролиферируют значительно быстрее, чем в нормальной коже, и в последующем смещаются на поверхность с формированием четко ограниченных эритематозных бляшек, варьирующих по размеру [Godic, 2004]. В патогенезе псориаза отражается аномальная пролиферация и дифференцировка кератиноцитов и сниженный уровень апоптоза. Механизм, приводящий к повышенной пролиферации кератиноцитов, остается неясным. Предполагается, что это заболевание генетически детерминировано, а также имеется аутоиммунный компонент [Godic, 2004]. Однако псориаз является уникальным заболеванием, так как, несмотря на повышенную скорость пролиферации кератиноцитов, она все-таки контролируется. В недавних исследованиях было показано снижение экспрессии Т-кадгерина в эпидермальных кератиноцитах псориатических бляшек по сравнению с нормальной кожей. В исследованиях этих авторов экспрессия Т-кадгерина была ограничена кератиноцитами базальноклеточного слоя [Zhou et al., 2003]. Напротив, наши данные указывают на одинаковую экспрессию Т-кадгерина как в псориатической, так и в нормальной коже (Рисунок 3.13 и Рисунок 3.14). Время экспозиции для образцов было четко фиксировано, и интенсивность иммунофлуоресценции для обоих типов образцов была одинакова. Возможно, сохранение экспрессии Т-кадгерина в гиперпролиферирующих кератиноцитах лишний раз подтверждает тот факт, что псориаз представляет собой доброкачественный процесс, хотя в отдельных случаях характеризующийся достаточно тяжѐлым течением. 172 Рисунок 3.14. Двойное иммунофлуоресцентное окрашивание образа псориаза антителами к Т-кадгерину (красная флуоресценция) и маркѐру эндотелия сосудов vWF (зелѐная флуоресценция) (A). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B показано окрашивание гематоксилином параллельного криосреза образца ткани. Стабильная экспрессия Т-кадгерина обнаруживается в базальных кератиноцитах, кератиноцитах гиперпролиферативного супрабазального слоя, стромальных клетках и кровеносных сосудах эпидермального слоя кожи. Т-кадгерин также обнаруживался во всех мелких и крупных сосудах (жѐлтая флуоресценция); стрелками показана ко-локализация Т-кадгерина и маркѐра эндотелия vWF. Масштаб 100 мкм. На рисунке (C) схематично показана стабильная экспрессия Т-кадгерина в базальном слое (красный цвет) и умеренная экспрессия Т-кадгерина в супрабазальном слое кератиноцитов и клетках стромы (розовый цвет); все кровеносные сосуды экспрессируют Т-кадгерин (жѐлтый цвет). 173 В псориатической коже экспрессия Т-кадгерина обнаруживалась в кератиноцитах всех слоев, причѐм базальные кератиноциты больше экспрессировали Т-кадгерина, чем кератиноциты гиперпролиферативного супрабазального слоя. Также отмечалась экспрессия Т-кадгерина в стромальных клетках и во всех кровеносных сосудах подлежащей дермы. Следует отметить, что по сравнению с нормальной кожей, при псориазе наблюдалось аномальное расположение кровеносных сосудов в эпидермальном слое, чего никогда не наблюдается в нормальной коже, в которой сосуды располагаются в подлежащей дерме (Рисунок 3.13 и Рисунок 3.14). Данные сосуды, что было подтверждено окрашиванием на маркер эндотелия vWF, равномерно экспрессировали Т-кадгерин (Рисунок 3.14). 3.2.4.3 Актинический (солнечный) кератоз (АК) АК обычно характеризуется наличием на коже роговых чешуек, папул на эритематозном фоне, размер которых обычно не превышает 1-2 см в диаметре. Исторически, АК относят к предраковым состояниям [Quinn and Perkins, 2010]. Многие авторы считают, что АК является «предраковой эпителиальной опухолью», способной перерождаться в плоскоклеточный рак [Jorizzo et al., 2004; Epstein, 2004; Yu et al., 2003]. Другие авторы настаивают на том, что нет пато- и биологических различий между АК и плоскоклеточным раком, и АК является одним из вариантов плоскоклеточного рака кожи. Без должного лечения АК переходит в плоскоклеточный рак, способен метастазировать и вызывать летальные исходы [Freeman et al., 1984; Lebwohl et al., 2004; Ackerman and Mones, 2006]. На Рисунке 3.15 представлено окрашивание гематоксилином и иммунофлуоресцентное окрашивание образцов АК антителами против Т-кадгерина, vWF и α-актина. Область повреждения характеризуется наличием атипичных кератиноцитов в базальном слое эпидермиса и уменьшенным количеством зрелых кератиноцитов. Зона АК располагается под эпидермисом и четко отделена от последнего. Т-кадгерин равномерно экспрессируется в кератиноцитах всех слоев, включая супрабазальные слои, отмечается незначительное увеличение окраски в базальном слое (Рисунок 3.15), что согласуется с данными литературы [Pfaff et al., 2010]. 174 Рисунок 3.15. Двойное иммунофлуоресцентное окрашивание образцов солнечного кератоза с использованием антител к Т-кадгерину (красная флуоресценция) и vWF (зелѐная флуоресценция) (А); или vWF (зелѐная флуоресценция) и α-актину (красная флуоресценция) (С). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B представлена окраска гематоксилином параллельного криосреза образца ткани. Экспрессия Т-кадгерина обнаруживается в базальных кератиноцитах, включая супрабазальные слои, с незначительным увеличением в базальном слое. Экспрессия Ткадгерина обнаруживается не во всех vWF-позитивных сосудах. Стрелками показаны сосуды, ко-экспрессирующие Т-кадгерин и vWF; vWF-позитивные сосуды, не экспрессирующие Т-кадгерин, показаны треугольной стрелкой. Масштаб 100 мкм. На рисунке D схематично изображена стабильная экспрессия Т-кадгерина в клетках базального слоя (красный цвет) и умеренная экспрессия Т-кадгерина в супрабазальном слое и клетках стромы (розовый цвет). Кровеносные сосуды, ко-экспрессирующие Ткадгерин и vWF, показаны жѐлтым, сосуды без Т-кадгерина – зелѐным. 175 Экспрессия Т-кадгерина обнаруживается также в кровеносных сосудах дермы (в эндотелиальных и муральных клетках сосудистой стенки), и в стромальных клетках в непосредственной близости от атипичных кератиноцитов. Однако не все сосуды, несущие vWF, экспрессируют Т-кадгерин, что ставит АК в один ряд со злокачественным повреждениям кожи, таким как плоскоклеточный рак (см. ниже). 3.2.4.4 Кератоакантома (КА) Быстрорастущая доброкачественная опухоль кожи, способная к медленному спонтанному регрессу. КА состоит из ороговевших чешуйчатых клеток, происходящих из волосяного фолликула, и гистопатологически напоминает плоскоклеточный рак [Quinn and Perkins, 2010]. КА подвергается регрессии и представляет собой «биологически доброкачественный» вариант плоскоклеточного рака [Zalaudek et al., 2009]. В своем развитии КА проходит три этапа: рост, стабилизация и инволюция. Первая фаза характеризуется быстрым увеличением в размерах в течение нескольких недель или месяцев, после чего происходит стабилизация процесса и спонтанный регресс за 4-6 месяцев. Однако иногда КА может вырастать до 5 см и становиться локально агрессивной [Quinn and Perkins, 2010]. На Рисунке 3.16 и Рисунке 3.17 представлены КА в стадиях роста и стабилизации, соответственно. В целом, согласно нашим гистологическим данным, клетки при КА зрелые, а эпителий дифференцируется от базального слоя к поверхностным кератиноцитам. Однако можно наблюдать анормальное и преждевременное образование кератина (дискератоз), что проявляется в появлении одиночных ороговевших клеток или кератиновых зерен. Строма несет сосуды и инфильтрирована. На стадии стабилизации Т-кадгерин обнаруживается в кератиноцитах всех слоев и расположенных рядом кровеносных сосудах, что подтверждается двойным окрашиванием антителами против Т-кадгерина, маркера эндотелия vWF и маркера ГМК сосудистой стенки – α-актина (Рисунок 3.17). Обращает на себя внимание тот факт, что часть кровеносных сосудов на стадии роста КА не экспрессирует Т-кадгерин (Рисунок 3.16), а также наличие нехарактерной локализации кровеносных сосудов в гипертрофированном слое эпидермиса и вокруг ороговевших клеток на месте будущих гранул кератина (Рисунок 3.16). Такое нетипичное расположение кровеносных сосудов и снижение экспрессии Т-кадгерина при КА в стадии роста, возможно, отражает высокую скорость роста и локальную агрессивность опухоли, поскольку в стадии стабилизации расположение кровеносных сосудов и экспрессия Ткадгерина сравнимы с таковыми в нормальной коже. 176 Рисунок 3.16. Двойное иммунофлуоресцентное окрашивание образцов кератоакантомы в стадии роста с использованием антител к Т-кадгерину (красная флуоресценция) и vWF (зелѐная флуоресценция) (А). Ядра докрашены DAPI (синяя флуоресценция). На фотографии В представлено изображение параллельного криосреза образца ткани, полученное с помощью фазового контраста. Экспрессия Т-кадгерина обнаруживается в кератиноцитах всех слоѐв. Не все сосуды экспрессируют Т-кадгерин. Стрелками показаны сосуды, ко-экспрессирующие Т-кадгерин и vWF; сосуды, не экспрессирующие Т-кадгерин, показаны треугольной стрелкой. Масштаб 100 мкм. На рисунке С схематично изображена стабильная экспрессия Т-кадгерина в клетках базального слоя (красный цвет) и умеренная экспрессия Т-кадгерина в супрабазальном слое (розовый цвет). Кровеносные сосуды, ко-экспрессирующие Т-кадгерин и vWF, показаны жѐлтым, сосуды без Т-кадгерина – зелѐным. 177 Рисунок 3.17. Двойное иммунофлуоресцентное окрашивание образцов кератоакантомы в стадии стабилизации с использованием антител к Т-кадгерину (красная флуоресценция) и vWF (зелѐная флуоресценция) (А); или vWF (зелѐная флуоресценция) и α-актину (красная флуоресценция) (С). Ядра докрашены DAPI (синий цвет). На фотографии B представлена окраска гематоксилином параллельного криосреза образца ткани. Экспрессия Т-кадгерина обнаруживается в кератиноцитах всех слоѐв. В большинстве сосудов наблюдается ко-экспрессия Т-кадгерина и vWF (А, показано стрелками). Масштаб 100 мкм. На рисунке D схематично изображена стабильная экспрессия Т-кадгерина в клетках базального слоя (красный цвет), умеренная экспрессия Т-кадгерина в супрабазальном слое (розовый цвет). Кровеносные сосуды, ко-экспрессирующие Т-кадгерин и vWF, показаны жѐлтым цветом. 178 3.2.4.5 Базальноклеточный рак (базалиома) Базалиома – наиболее распространенная злокачественная опухоль кожи, происходящая из клеток базального слоя эпидермиса и его придатков, а именно, из стволовых клеток волосяного фолликула или из межфолликулярного эпидермиса [Quinn and Perkins, 2010; Wang et al., 2011]. Типичная базалиома обладает низкой скоростью роста и редко метастазирует. На ранних стадиях опухоль обычно небольшого размера, прозрачная или жемчужного цвета и покрыта эпидермисом. Некоторые опухоли растут очень медленно и в клиническом смысле являются доброкачественными. Это верно для двух типов базалиомы: поверхностной и нодулярной [Quinn and Perkins, 2010]. Базалиома на более поздней стадии очень разнообразны по внешнему виду и форме. По-настоящему метастазирующие базалиомы встречаются редко. В этом случае базалиома растет в виде округлых масс небольшого размера, не имеет палисадного слоя клеток или структурированной стромы и способна агрессивно инвазировать в подлежащую дерму и более глубокие слои [Quinn and Perkins, 2010]. Несмотря на высокий митотический индекс, базалиома характеризуется медленным темпом роста в связи с высоким уровнем апоптоза. Вокруг опухоли часто наблюдается умеренное воспаление [Quinn and Perkins, 2010]. Самой распространенной формой базальноклеточной карциномы является нодулярная, при которой опухолевые клетки растут в виде округлой массы в дерме. «Частокол» клеток между опухолью и стромой четко определяется. При инфильтрирующей форме базалиомы опухолевые клетки располагаются нерегулярными группами в дерме, инфильтрируя строму. Определить границу области поражения невозможно. При инфильтрирующей форме базалиомы клетки различаются размерами и организованы в небольшие группы. «Частокол» призматических клеток на периферии часто отсутствует. Поверхностная форма базалиомы характеризуется ростом базальных клеток в виде тяжей, направленных от эпидермиса в поверхностный слой дермы [Rippey, 1998]. Нодулярная и поверхностная формы базальноклеточной карциномы являются наименее злокачественными, в то время как инфильтрирующая форма наиболее часто обладает агрессивным ростом [Maloney, 1995]. Нами была исследована экспрессия Т-кадгерина в образцах поверхностных, нодулярных, инфильтрирующих форм базалиомы. Поверхностная базалиома характеризовалась ярко выраженной экспрессией Т-кадгерина в клетках опухоли и в 179 окружающей строме. Большинство сосудов ко-экспрессировали маркѐры эндотелиальных клеток vWF и Т-кадгерин, однако, в образцах также присутствовали сосуды, в которых не наблюдалась экспрессия Т-кадгерина (Рисунок 3.18). Нодулярная форма базалиомы имела «классические» черты, свойственные данной опухоли, включая большие ограниченные «частоколом» клеток узлы опухоли и ретракцию стромы. В нодулярном типе базалиомы экспрессия Т-кадгерина была гетерогенна: часть опухолевых узлов были позитивны по Ткадгерину, в части опухолевых узлов Т-кадгерин был экспрессирован слабо или вообще не выявлялся (Рисунок 3.19). Полученные нами данные коррелируют с данными некоторых авторов [Buechner et al., 2009], но не совпадают с данными Такеучи и коллег, которые обнаружили, что экспрессия Т-кадгерина снижена в образцах базалиомы [Takeuchi et al., 2002]. В большинстве образцов нодулярной базалиомы сосуды ко-экспрессировали Ткадгерин и vWF. В то же время в некоторых образцах этого типа базалиом была обнаружена аберрантная экспрессия Т-кадгерина и маркеров сосудистых клеток: в некоторых vWF-позитивных сосудах экспрессия Т-кадгерина отсутствовала, в то время как были обнаружены структуры, морфологически сходные с сосудами, которые экспрессировали α-актин, но не экспрессировали vWF (Рисунок 3.19). В образцах инфильтрующей базалиомы клетки опухоли формировали участки различного размера (хотя часто мелкие узлы) с нечетко выявляемыми границами (Рисунок 3.20). «Частокол» клеток был мало выражен, и не наблюдалось ретракции стромы. Ткадгерин был обнаружен не во всех клетках опухоли и окружающей стромы, в то время как в сосудах наблюдалось нарушение экспрессии Т-кадгерина и маркеров сосудов: часть vWFпозитивных сосудистых структур не экспрессировала Т-кадгерин. 180 Рисунок 3.18. Двойное иммунофлуоресцентное окрашивание образца поверхностной формы базалиомы антителами против Т-кадгерина (красная флуоресценция) и маркѐра эндотелиальных клеток vWF (зелѐная флуоресценция) (A). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B показано окрашивание гематоксилином параллельного криосреза образца ткани. Экспрессия Т-кадгерина обнаруживается в опухолевых клетках и клетках окружающей опухоль стромы. Не все кровеносные сосуды, экспрессирующие vWF, экспрессируют также Т-кадгерин. Колокализация Т-кадгерина и vWF показана стрелками; сосуды, не экспрессирующие Ткадгерин, показаны треугольными стрелками. Масштаб 100 мкм. 181 Рисунок 3.19. Двойное иммунофлуоресцентное окрашивание образцов нодулярной формы базалиомы с использованием антител к Т-кадгерину (красная флуоресценция) и vWF (зелѐная флуоресценция) (А, D), или vWF (красная флуоресценция) и α-актину (зелѐная флуоресценция) (C). Ядра докрашены DAPI (синяя флуоресценция). На фотографии E показана окраска гематоксилином параллельного криосреза ткани, представленного на фотографии D. На фотографии B показан фазовый контраст параллельного криосреза образца ткани, представленного на фотографиях А и С. Часть опухолевых узлов имеет выраженную экспрессию Т-кадгерина (А), в то время как в крупном опухолевом узле в центре и в отдельных опухолевых клетках, экспрессия Ткадгерина отсутствует (D). Большинство кровеносных сосудов ко-экспрессируют Ткадгерин и маркѐр эндотелиальных клеток vWF (A и D, стрелки). В то же время, часть образцов опухоли имеют аберрантную экспрессию Т-кадгерина и маркѐра эндотелия: часть vWF-позитивных сосудов не экспрессирует Т-кадгерин (A и D, треугольные стрелки). Тем не менее, обнаруживаются структуры, морфологически схожие с сосудами и экспрессирующие α-актин, но не экспрессирующие маркѐр эндотелия vWF (C, стрелки). Масштаб 100 мкм. 182 Рисунок 3.20. Двойное иммунофлуоресцентное окрашивание образцов инфильтрирующей формы базалиомы с использованием антител к Т-кадгерину (красная флуоресценция) и маркѐра эндотелиальных клеток vWF (зелѐная флуоресценция) (А); или vWF (красная флуоресценция) и α-актина (зелѐная флуоресценция). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B представлена окраска гематоксилином параллельного криосреза того же образца ткани. Экспрессия Т-кадгерина обнаруживается в опухолевых клетках и клетках окружающей стромы. Часть vWFпозитивных кровеносных сосудов, расположенных в строме, не экспрессирует Ткадгерин (A, стрелки); в то время как в структурах, морфологически схожих с сосудами и содержащих α-актин, не обнаруживается экспрессия маркѐра эндотелиальных клеток vWF (C, треугольные стрелки). Масштаб 100 мкм. На рисунке D стабильная экспрессия Т-кадгерина показана красным цветом и умеренная - розовым. Часть клеток, расположенных в узлах базалиомы содержат Т-кадгерин, в то время как в других клетках экспрессия Т-кадгерина потеряна. Кровеносные сосуды, коэкспрессирующие Т-кадгерин и vWF, показаны жѐлтым цветом, а сосуды без Ткадгерина – зелѐным. 183 Таким образом, при исследовании образцов базалиомы, характеризующихся малой скоростью роста и низкой инвазивностью, экспрессия Т-кадгерина была обнаружена в кератиноцитах, строме и в окружающих кровеносных сосудах в 90% образцов. Следует отметить, что в некоторых образцах нодулярной и инфильтрующей форм базалиом опухолевые клетки не экспрессировали Т-кадгерин, а в сосудах, прорастающих в эти опухоли, была обнаружена аберрантная экспрессия Т-кадгерина и маркеров сосудистых клеток. 3.2.4.6 Плоскоклеточный рак или плоскоклеточная карцинома (ПР) Плоскоклеточный рак кожи представляется собой злокачественное новообразование, происходящее из кератиноцитов эпидермиса и его придатков. К плоскоклеточному раку кожи относится множество подтипов опухолей: от медленнорастущих до очень агрессивных с большим метастатическим потенциалом [Cassarino et al., 2006a, Cassarino et al., 2006b]. Было предложено несколько классификаций плоскоклеточного рака, основанных на степени злокачественности: с низкой, умеренной и высокой способностью к метастазированию [Cassarino et al., 2006a, Cassarino et al., 2006b; Yanofsky et al., 2011]. Согласно последней классификации гистопатологически плоскоклеточный рак может быть разделен на три категории: актинический или солнечный кератоз (СК) и ПР in situ (Болезнь Боуэна) – схожие типы предракового состояния, являющиеся результатом чрезмерной инсоляции; инвазивный ПР, чистоклеточный ПР, веретеноклеточный (саркоматоидный) ПР и ПР с единичными клеточными инфильтратами – подтипы опухолей, возникающие при инвазивной прогрессии СК и ПР in situ; de novo ПР, лимфоэпителиомоподобная карцинома кожи и веррукозная карцинома – очень редкие варианты ПР, не связанные с инсоляцией или актиническим кератозом [Yanofsky et al., 2011]. Инвазивные ПР являются самыми распространенными и делятся на три гистологические группы в зависимости от ядерной атипии и ороговения (высокодифференцированные, умеренно- и низкодифференцированные). В отличие от клеток высокодифференцированных форм инвазивных ПР, клетки низкодифференцированных опухолей при ПР характеризуются крупными плеоморфными ядрами, с высокой степенью атипии и частыми митозами. Образование кератина этими клетками снижено. Низкодифференцированные инвазивные ПР глубоко инвазируют в подлежащую дерму и более глубокие слои. Данные типы опухолей в клиническом проявлении достаточно агрессивны, часто метастазируют и дают рецидивы. Третий тип ПР, умеренно дифференцированная форма инвазивного ПР объединяет черты двух других 184 [Yanofsky et al., 2011]. Считается, что низкодифференцированные ПР обладают самой высокой способностью к метастазированию [Lund et al., 1984; Dinehart and Pollack, 1989; Dinehart et al., 1997]. В оценке злокачественности опухолей важным является наличие периневрального или периваскулярного распространения. Инвазия в капилляры и нервы указывает на более агрессивный характер опухоли и коррелирует с повышенной частотой метастазов, возможностью рецидива и летальный исход [Yanofsky et al., 2011]. В нашем исследовании в образцах умеренно- и низкодифференцированных форм инвазивного ПР опухолевые клетки были обнаружены в глубоких слоях подлежащей дермы (Рисунок 3.21). В некоторых случаях в части атипичных клеток в отдельно располагающихся опухолевых «гнездах» и в стромальных клетках выявлялось слабое окрашивание на Т-кадгерин. Однако в базалоидных клетках большинства опухолевых «гнезд» экспрессия Т-кадгерина была подавлена. В активированной строме были выявлены клетки позитивные по экспрессии α-актина. Полученные нами данные о потере экспрессии Т-кадгерина в опухолевых клетках ПР согласуются с результатами других авторов [Takeuchi et al., 2002b; Zhou et al., 2003; Mukoyama et al., 2005; Pfaff et al., 2010] и указывают на то, что снижение экспрессии Ткадгерина в результате аберрантного метилирования гена Т-кадгерина, или в результате его делеции, коррелирует со степенью агрессивности и инвазивности ПР. Тем не менее, наши результаты показывают, что экспрессия Т-кадгерина изменяется не только в атипичных кератиноцитах. Среди других нарушений можно отметить аберрантную экспрессию маркѐров сосудов и Т-кадгерина: в части vWFпозитивных сосудов Т-кадгерин не обнаруживался; в то же время были выявлены капилляро-подобные структуры, не несущие vWF, но экспрессирующие Т-кадгерин и αактин. Следует отметить, что в некоторых образцах ПР были обнаружены области, экспрессирующие CD31/vWF, но морфологически отличающиеся от сосудов (Рисунок 3.21). 185 Рисунок 3.21. Двойное иммунофлуоресцентное окрашивание образцов низкодифференцированной формы плоскоклеточного рака с использованием антител к Т-кадгерину (красная флуоресценция) и vWF (зелѐная флуоресценция) (А); или маркѐра эндотелиальных клеток CD31 (красная флуоресценция) и α-актину (зелѐная флуоресценция) (С). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B представлена окраска гематоксилином параллельного криосреза образца ткани. Экспрессия Т-кадгерина частично обнаруживается в клетках изолированных опухолевых “гнѐзд”, а также в клетках стромы (А), тем не менее, в большинстве базалоидных клеток в изолированных опухолевых “гнѐздах” экспрессия Т-кадгерина не наблюдается (А). Выявлены структуры, морфологически сходные с сосудами и не экспрессирующие CD31 антиген (показаны звѐздочкой, С). Масштаб 100 мкм. На схеме D красным цветом показана стабильная экспрессия Т-кадгерина, розовым – умеренная экспрессия Т-кадгерина. В большинстве опухолевых клеток, расположенных в малых “гнѐздах”, экспрессии Т-кадгерина нет. Сосуды, ко-экспрессирующие Т-кадгерин и CD31 показаны жѐлтым цветом, сосуды, не экспрессирующие Т-кадгерин, показаны зелѐным цветом. Структуры, морфологически схожие с сосудами, и экспрессирующие α-актин, показаны фиолетовым цветом 186 3.2.4.7 Метатипический рак (МР) Термин метатипический рак или базально-плоскоклеточная карцинома (БПКК) относится к опухолям, несущим признаки как базально-клеточного, так и плоскоклеточного рака [Garcia et al., 2009; Cassarino et al., 2006a; Cassarino et al., 2006b]. Диагностическая важность этого типа рака заключается в том, что МР исключительно агрессивен и метастазирует значительно чаще, чем базалиома или ПР [Smith and Irons, 1983; Banks et al., 1992; Farmer and Helwig, 1980; Winzenburg et al., 1998]. Диагноз МР основывается на гистологических критериях, предложенных Ваином с соавторами [Wain et al., 1986]. МР характеризуется формирование агрегатов опухолевых клеток, без характерного клеточного «частокола» и глубоко проникающих в окружающую плотную волокнистую строму. Окружающая строма фиброзирована в связи с отложением внеклеточного гиалина в непосредственной близости от опухолевых агрегатов [Sarbia et al., 1997; Zbaren et al., 2004]. На Рисунке 3.22 представлено иммуногистохимическое окрашивание образцов МР антителами против Т-кадгерина, маркеров эндотелиальных клеток - vWF и гладкомышечных клеток сосудов/перицитов – α-актина. Опухолевые клетки формировали агрегаты различных размеров и обычно не экспрессировали Т-кадгерин; в некоторых клетках по периферии агрегатов была обнаружена слабая экспрессия Т-кадгерина. Опухолевые массы были окружены активированной стромой, несущей Т-кадгерин. Большинство сосудов вокруг опухоли ко-экспрессировали маркер эндотелиальных клеток – vWF и Т-кадгерин или vWF и α-актин (Рисунок 3.22), напоминая капилляры и стабильные кровеносные сосуды нормальной кожи. В то же время были обнаружены структуры, морфологически напоминающие кровеносные сосуды и экспрессирующие Т-кадгерин или α-актин, но не несущие классического маркера эндотелиальных клеток vWF. 187 Рисунок 3.22. Двойное иммунофлуоресцентное окрашивание образцов метатипического рака антителами против Т-кадгерина (красная флуоресценция) и vWF (зелѐная флуоресценция) (А); и антителами к маркѐру эндотелиальных клеток CD31 (красная флуоресценция) и α-актину (зелѐная флуоресценция) (С). Ядра докрашены DAPI (синяя флуоресценция). На фотографии B представлена окраска гематоксилином параллельного криосреза образца ткани. Опухолевые клетки образуют узлы различных размеров с редкой экспрессией Т-кадгерина, однако, клетки некоторых опухолевых агрегатов, а также некоторые клетки, расположенные на периферии агрегатов, сохраняют экспрессию Т-кадгерина (А). Большинство сосудов, окружающих опухолевые агрегаты, ко-экспрессируют Т-кадгерин и vWF (показаны стрелками). В тоже время, часть образцов опухоли имеют аберрантную экспрессию Т-кадгерина и маркѐров сосудистых клеток: часть структур, морфологически сходных с сосудами, экспрессируют Т-кадгерин и не экспрессируют CD31 (отмечено звездочкой на С). Масштаб 100 мкм. На рисунке D схематически красным цветом показана стабильная экспрессия Т-кадгерина, розовым – умеренная экспрессия Т-кадгерина. В большинстве опухолевых клеток, растущих в агрегатах, экспрессия Т-кадгерина утеряна. Сосуды, коэкспрессирующие Т-кадгерин и vWF показаны жѐлтым цветом, сосуды, не экспрессирующие Т-кадгерин, показаны зелѐным цветом. Структуры, морфологически схожие с сосудами, и экспрессирующие α-актин, показаны фиолетовым цветом 188 Таким образом, полученные данные свидетельствуют о том, что первоначальное снижение экспрессии Т-кадгерина в кератиноцитах и кровеносных сосудах при предраковых состояниях кожи, а в последующем прогрессирующее снижение вплоть до полного исчезновения экспрессии Т-кадгерина в опухолевых клетках и в сосудах различных типов раков, которое сопровождается нарушением экспрессии маркеров, характерных для эндотелиальных и муральных клеток, коррелирует со злокачественной трансформацией новообразований кожи. Полученные в данной работе результаты подтверждают тот факт, что потеря экспрессии Т-кадгерина в кератиноцитах эпидермиса биологически связана со злокачественной трансформацией нормальных клеток эпидермиса, что согласуются с данными литературы. Мы предположили, что Т-кадгерин участвует в поддержании целостности ткани в нормальных условиях путем регуляции миграции клеток в ткани и ограничения перемещения клеток между слоями в ткани. Возможно, именно поэтому экспрессия Ткадгерина наиболее выражена в кератиноцитах базального слоя, прикреплѐнных к базальной мембране и разграничивающих эпидермис и дерму. В нормальной коже активная пролиферация кератиноцитов наблюдается только в базальном слое эпидермиса, что коррелирует с максимальной экспрессией Т-кадгерина в этих клетках. При созревании эпидермальные кератиноциты отрываются от базальной мембраны и перемещаются на поверхность кожи, не пересекая слой с максимальной экспрессией Т-кадгерина. Это позволяет высказать гипотезу о том, что Т-кадгерин выполняет барьерную функцию и определяет направление миграции дифференцирующихся кератиноцитов к поверхности кожи, а не в сторону дермы. При предраковых состояниях кожи, таких как КА в стадии стабилизации, псориаз, солнечный кератоз и поверхностная базалиома, которые характеризуются медленной или контролируемой скоростью роста неопластических клеток, а также сохраняют, в большинстве случаев, связь с базальной мембраной, паттерн экспрессии Т-кадгерина аналогичен таковому в нормальной коже. Экспрессия Т-кадгерина снижена или отсутствует при малигнизации, в случае метатипического рака, плоскоклеточного рака и в некоторых вариантах агрессивной базалиомы, характеризующихся высоким пролиферативным, инвазивным и метастатическим потенциалом, где кератиноциты имеют тенденцию расти в малых агрегатах и не связаны с базальной мембраной. Возможно, что Т-кадгерин выступает как регулятор пролиферации и миграции кератиноцитов в нормальной коже и как опухолевый супрессор при предраковых состояниях, который ограничивает пролиферацию, регулирует 189 миграцию и взаимодействие кератиноцитов разных слоев между собой и стромой. При раке степень агрессии опухолевых клеток коррелирует с потерей ими экспрессии Т-кадгерина. В образцах нормальной кожи и в образцах кожи предраковых состояний, таких как псориаз, КА в стадии стабилизации и поверхностная базалиома, все сосуды равномерно экспрессируют маркер эндотелиальных клеток vWF и Т-кадгерин. Хотя АК некоторыми авторами относится к предраковому состоянию, по нашим данным, потеря экспрессии Ткадгерина кровеносными vWF-позитивными сосудами свидетельствует о начальных процессах малигнизации и позволяет отнести актинический кератоз в одну группу злокачественных новообразований кожи вместе с плоскоклеточным раком. При КА в стадии роста часть кровеносных сосудов также не экспрессирует Т-кадгерин и прорастают в слой гипертрофированных и быстро пролиферирующих кератиноцитов, что отражает агрессивное поведение данной опухоли на данной стадии. В образцах МР, ПР и базалиомы наблюдалась гетерогенность в экспрессии маркеров сосудов (vWF и маркера гладкомышечных клеток сосудов/перицитов α-актина) и Т-кадгерина, что коррелирует со степенью их злокачественности. Таким образом, потеря экспрессии классических маркеров эндотелиальных клеток (vWF), экспрессия α-актина в сосудисто-подобных структурах и гетерогенность в экспрессии Т-кадгерина в опухолевых сосудах коррелировали с гистологическими признаками и инвазивным фенотипом более агрессивных опухолей, таких как МР, ПР и базалиома. Возможно, высокий уровень экспрессии Т-кадгерина в клетках нормальной кожи и доброкачественных опухолях по принципу контактного торможения предотвращает избыточный рост сосудов, которые также экспрессируют Ткадгерин. Опухолевая трансформация сопровождается нарушением экспрессии Т-кадгерина не только в клетках опухоли, но и в клетках сосудов, что вызывает аберрантную васкуляризацию опухолевых узлов и окружающей стромы. Невозможно точно предсказать, как будет прогрессировать каждое конкретное новообразование кожи, и станет ли оно, в конечном счѐте, раком. Результаты данного исследования свидетельствуют о том, что наличие изменений в экспрессии Т-кадгерина в кровеносных сосудах при предраковых состояниях, возможно, является одним из прогностически неблагоприятных факторов агрессивности и инвазивности данного неопластического образования. 190 3.2.4.8 Потеря экспрессии Т-кадгерина в опухолевых клетках и нарушение его экспрессии в сосудах коррелирует с опухолевой прогрессией меланомы Меланоциты – это специальные пигментированные клетки, которые располагаются в коже, внутреннем ухе, пигментированной части эпителия сетчатки глаза, а также сосудистого слоя глаз. В коже меланоциты располагаются в базальном слое (stratum basale) эпидермиса и в волосяных фолликулах [Gray-Schopfer et al., 2007]. Меланоциты продуцируют меланин, обуславливающий цвет кожи и волос, и выполняющий защитную функцию базальных кератиноцитов от ультрафиолета [Gray-Schopfer et al., 2007]. Во время воздействия ультрафиолетовых лучей кератиноциты продуцируют растворимые факторы, которые, наряду с межклеточными контактами, контролируют пролиферацию, дифференцировку и миграцию меланоцитов [Gray-Schopfer et al., 2007]. В то же время, меланоциты являются предшественниками одной из самых опасных форм рака – меланомы. Появляющаяся как доброкачественный невус, меланома может быстро прогрессировать в злокачественную стадию [Bar-Eli, 1997]. При этом меланоциты формируют невус, начинают пролиферировать и мигрировать либо только в эпидермисе, либо глубже, проникая в дерму [Gray-Schopfer et al., 2007]. Невусы могут прогрессировать и формировать меланому, для которой характерно внутриэпидемальное распространение со спорадической микроинвазией в подлежащую дерму (фаза радиального роста). Меланома в фазе радиального роста может трансформироваться в вертикально растущую меланому (фаза вертикального роста) с высоким инвазивным потенциалом. В конечном счете, меланома после фазы вертикального роста может прогрессировать и формировать метастазы [Clark et al., 1984; Miller et al., 2006]. Описаны несколько клинических типов меланомы: поверхностно растущая меланома, нодулярная меланома, акральная лентигинозная меланома, и меланома сосудистой оболочки глаза [Clark et al., 1984; Ishihara et al., 2001; Kuchelmeister et al., 2000]. Исходно меланоциты кожи происходят из клеток нервного гребня, мигрируют в вентральном направлении от нервной трубки через сомиты. Известно, что ключевую роль в миграции предшественников меланоцитов из нервной трубки играет регуляция экспрессии и активности различных кадгеринов (Е-кадгерин, N-кадгерин), а также N-CAM в зоне формирующегося нервного гребня [Nakagawa and Takeichi, 1995]. В нормальной множественные коже базальные кератиноциты образуют Е-кадгерин-содержащие адгезивные контакты, с меланоцитами что позволяет им контролировать пролиферацию и правильное расположение меланоцитов в коже [Haas and Herlyng, 2005; Haass et al., 2005]. Потеря экспрессии Е-кадгерина является одним из ранних событий в опухолевой прогрессии меланомы и коррелирует с увеличением экспрессии N- 191 кадгерина, что повышает ее инвазивный потенциал [Johnson, 1999; Hsu et al., 2000; Mc Gary et al., 2002; Li et al., 2002; Haass and Herlyn, 2005]. Предполагается, что клетки меланомы формируют N-кадгериновые адгезивные контакты с фибробластами стромы, эндотелиальными клетками сосудов и соседними клетками меланомы. N-кадгеринсодержащие контакты позволяют клеткам меланомы ползти по дермальным фибробластам и трансмигрировать через стенки сосудов в кровь [Li and Herlyn, 2000]. Было показано, что VE-кадгерин также играет важную роль в инвазии клеток меланомы и метастазировании [Voura et al., 1998 Hendrix et al., 2001; Hendrix et al., 2003]. В норме VE-кадгерин экспрессируется исключительно в эндотелиальных клетках и обеспечивает барьерную функцию эндотелия [Dejana, 1996]. Однако клетки агрессивных форм меланомы кожи и сосудистой оболочки глаза также могут экспрессировать VE-кадгерин, что облегчает их взаимодействие с эндотелиальными клетками и инвазию в просвет сосуда [Hendrix et al., 2001]. Экспрессия VE-кадгерина лежит в основе явления сосудистой мимикрии, при которой клетки меланомы сами формируют характерную сеть сосудистых каналов и обеспечивают опухоль питательными веществами и кислородом [Hendrix et al., 2001; Hendrix et al., 2003]. В то время как зрелые меланоциты экспрессируют Т-кадгерин [Kuphal et al., 2009; Bosserhoff et al., 2011], предшественники меланоцитов, клетки нервного гребня его не экспрессируют. Экспрессия Т-кадгерина также снижена в меланоцитах, которые дедифференцируются в меланобластоподобные клетки в культуре, а также не определяется примерно в 80% клеточных линий меланомы человека [Kuphal et al., 2009; Bosserhoff et al., 2011]. В то время как Т-кадгерин экспрессируется в доброкачественных невусах, его экспрессия не обнаруживается в большинстве образцов ткани первичной меланомы человека, метастазах меланомы в лимфатические узлы и внутренние органы, что указывает на возможную роль Т-кадгерина в прогрессии меланомы [Kuphal et al., 2009]. Кроме того, экспрессия Т-кадгерина путем стабильной трансфекции в клетках меланомы человека приводит к замедлению роста опухоли и уменьшению миграционной и инвазивной способности этих клеток у бестимусных мышей линии NUDE (Kuphal et al., 2009). Однако в 20% клеточных линий меланом человека по-прежнему сохраняется экспрессия Т-кадгерина на значительном уровне [Kuphal et al., 2009; Bosserhoff et al., 2011], и эти клеточные линии обладают высоким инвазивным и метастатическим потенциалом, что показано на моделях in vitro и in vivo. Эти данные свидетельствует о том, что, возможно, роль Т-кадгерина в опухолевом росте и прогрессии меланомы сложнее, чем первоначально предполагалось. Для изучения роли Т-кадгерина в опухолевом росте и прогрессии меланомы, в настоящей работе мы проведи сравнительный анализ экспрессии Т-кадгерина в 192 меланоцитах кожи человека в норме, в клетках меланомы и в кровеносных сосудах образцов первичных меланом и метастазов, полученных от больных. В дальнейшем было проведено исследование для выявления роли Т-кадгерина в опухолевой прогрессии меланомы на экспериментальных моделях. 3.2.4.9 Экспрессия Т-кадгерина в меланоме человека В образцах нормальной кожи Т-кадгерин экспрессируется в базальных кератиноцитах, дифференцирующихся кератиноцитах, в клетках стромы, в сосудах подлежащей дермы и во всех меланоцитах разной степени зрелости (Рисунок 3.23). Иммунофлуоресцентное окрашивание антителами gp100 свидетельствует о том, что дифференцирующиеся меланоциты локализованы рядом со слоем базальных кератиноцитов эпидермиса, а отростки меланоцитов располагаются над кератиноцитами (Рисунок 3.24). Экспрессию Т-кадгерина в дифференцирующихся меланоцитах выявляли двойным иммунофлуоресцентным окрашиванием (gp100 и Т-кадгерин) и наложением зеленой и красной флуоресценции (Рисунок 3.24). Сопоставление фазово-контрастного изображения и двойного флуоресцентного окрашивания показало, что зрелые меланоциты, не экспрессирующие gp100, экспрессируют Т-кадгерин. Поскольку ранее проведенные исследования показали, что при опухолевой прогрессии экспрессия Т-кадгерина может снижаться или отсутствовать, мы проанализировали экспрессию Т-кадгерина в образцах первичной меланомы (Рисунок 3.25) и в метастазах (Рисунок 3.26), причѐм один из исследованных образцов как первичной опухоли, так и метастазов через несколько месяцев после первичного забора биопсии был получен у одного и того же пациента. Окрашивание тканей с последующим анализом изображений показало, что, в случае первичной меланомы человека 60% срезов содержали клетки меланомы, равномерно экспрессирующие Т-кадгерин (Рисунок 3.27). При этом 30% срезов содержали области с гетерогенным, мозаичным паттерном экспрессии Т-кадгерина, где некоторые клетки меланомы экспрессируют Т-кадгерин, а некоторые нет; на оставшихся 10% срезов клетки меланомы не экспрессировали Т-кадгерин. 193 Рисунок 3.23. Двойное иммунофлуоресцентное окрашивание образца нормальной кожи антителами к Т-кадгерину (красная флуоресценция) и маркѐру сосудов vWF (зелѐная флуоресценция). Ядра докрашены DAPI (синяя флуоресценция). В норме Т-кадгерин обнаруживается в базальных кератиноцитах, супрабазальных слоях, клетках стромы и в сосудах. Стрелкой показана ко-локализация Т-кадгерина и маркѐра сосудистых клеток vWF. Масштаб 50мкм. Рисунок 3.24. Двойное иммунофлуоресцентное окрашивание образцов нормальной кожи антителами к Т-кадгерину (красная флуоресценция) (A, B, C) и маркѐру дифференцирующихся меланоцитов gp100 (зелѐная флуоресценция) (A, B). Ядра докрашены DAPI (синяя флуоресценция). На рисунке D представлено фазово-контрастное изображение. На рисунках B, C и D представлено иммунофлуоресцентное окрашивание параллельных криосрезов одного образца ткани. Экспрессия Т-кадгерина (красная флуоресценция) наблюдается в меланоцитах разной степени дифференцировки: от незрелых меланоцитов (зелѐная флуоресценция, показанная стрелками на А и В) до дифференцированных меланоцитов (зрелые меланоциты, не экспрессирующие gp100, указанные жирной стрелкой на рисунке С и D). Масштаб на А -100 мкм, на В, С и D – 20 мкм. 194 Рост и прогрессирование опухолей требует кровоснабжения. Как уже упоминалось выше, существует три механизма, с помощью которых солидные опухоли получают кровь. Во-первых, опухоль и окружающие еѐ стромальные клетки секретируют ангиогенные факторы, которые стимулируют рост кровеносных сосудов и прорастание их в ткань опухоли из предсуществующих сосудов путем ангиогенеза [Folkman et al., 1971; Folkman, 1995]. Кроме того, циркулирующие эндотелиальные предшественники из периферической крови могут встраиваться в области ангиогенеза, индуцированного ишемией или гипоксией (Asahara et al., 1997). Помимо этих двух механизмов, агрессивные первичные и метастатические меланомы способны формировать микроциркуляторные каналы (лакуны), которые выстланы линейно расположенными клетками опухоли, экспрессирующими VEкадгерин [Hendrix et al., 2001; Maniotis et al., 1999]. Это явление было названо «васкулогенной мимикрией» [Maniotis et al., 1999]. В нашей работе было показано, что в первичных опухолевых узлах и в метастазах человека активно образуются лакуны, полость которых выстланы Т-кадгерин-экспрессирующими клетками меланомы (Рисунок 3.25 и Рисунок 3.26). Механизмы такой «васкулогенной мимикрии» мало изучены, однако, очевидно, что поскольку «васкулогенная мимикрия» является дополнительным источником кровоснабжения для растущих и прогрессирующих опухолей, биохимические и клеточные механизмы их формирования могут оказаться полезными при разработке новых методов диагностики и лечения меланомы. Полученные в настоящем исследовании результаты указывают на то, что области первичных меланом, в которых опухолевые клетки не экспрессируют Т-кадгерин, более васкуляризированы, чем области с высокой экспрессией Т-кадгерина. 80% кровеносных сосудов, которые были положительно окрашены на фактор Вон Виллебранда, также экспрессировали Т-кадгерин (Рисунок 3.25), в остальных кровеносных сосудах экспрессии Т-кадгерина выявлено не было (Рисунок 3.25 и Рисунок 3.26). Кроме vWF-позитивных кровеносных сосудов в первичных меланомах и висцеральных метастазах, были обнаружены и сосудистые каналы, полость которых была выстлана клетками, не экспрессирующими маркеров эндотелиальных клеток. Мы впервые обнаружили, что стенки этих каналов состояли из линейно расположенных клеток, экспрессирующих Т-кадгерин. 195 Рисунок 3.25. Двойное иммунофлуоресцентное окрашивание образцов первичной меланомы антителами к Т-кадгерину (зелѐная флуоресценция на А, В и С) и маркѐру сосудов vWF (красная флуоресценция на А и В) или антигену меланоцитов gp100 (красная флуоресценция на С). В первичных узлах меланомы наблюдается гетерогенная экспрессия Т-кадгерина в опухолевых клетках и в сосудах. Наряду с областями, в которых клетки меланомы тотально экспрессируют Т-кадгерин (зеленая флуоресценция на А и В), встречаются зоны с мозаичной экспрессией Т-кадгерина или с отсутствием его экспрессии. На рисунке А стрелками показаны сосуды, экспрессирующие Т-кадгерин (желтая флуоресценция). Сосуды, не экспрессирующие Ткадгерин указаны стрелками на рисунке В. Ядра докрашены DAPI (синяя флуоресценция). Масштаб 50 мкм 196 Рисунок 3.26. Двойное иммунофлуоресцентное окрашивание образцов первичной меланомы (А) и метастазов (В и С) антителами к Т-кадгерину (зелѐная флуоресценция) и маркѐру сосудов vWF (красная флуоресценция). На рисунке А тонкая стрелка указывает на большую лакуну первичного узла меланомы, полость которой выстлана клетками, экспрессирующими Т-кадгерин. Сосуды первичных узлов меланомы и метастазов гетерогенны по экспрессии Т-кадгерина и маркеров эндотелия. Сосуды, экспрессирующие vWF и не экспрессирующие Т-кадгерин, расположены по периметру лакуны и показаны толстой стрелкой. В метастазах меланомы встречаются vWFпозитивные сосуды, как экспрессирующие Т-кадгерин (толстые стрелки на С), так и не экспрессирующие Т-кадгерин (тонкие стрелки на С). Ядра докрашены DAPI (синяя флуоресценция). Масштаб 50 мкм. 197 При прогрессировании заболевания экспрессия Т-кадгерина в меланоме человека значительно снижается. В то время как на 60% исследованных срезов опухолевые клетки в образцах первичной меланомы экспрессировали Т-кадгерин, в 30% случаев экспрессия Ткадгерина была мозаична, а в 10% полностью отсутствовала. В случае метастазов 60% срезов содержали клетки меланомы с гетерогенной экспрессией Т-кадгерина (мозаичный паттерн), а количество Т-кадгерин-положительных клеток было снижено до 30%, в 10% полностью отсутствовало (Рисунок 3.27). Соотношение сосудов с разным уровнем экспрессии Т-кадгерина 100% Т-кад+ 90% 80% Т-кад+/Т-кад- 70% Т-кад- 60% 50% 40% 30% 20% 10% 0% Первичный узел Метастазы Рисунок 3.27. Снижение экспрессии Т-кадгерина при опухолевой прогрессии меланомы. В первичных узлах меланомы в 60% случаях наблюдается равномерная экспрессия Т-кадгерина в клетках меланомы (Т-кад+), в 30% экспрессия Т-кадгерина мозаична (Т-кад+/Т-кад-), и около 10% исследованных срезов не содержат Т-кадгеринпозитивных клеток меланомы (Т-кад-). В метастазах около 60% образцов имеют мозаичную экспрессию Т-кадгерина (Т-кад+/Т-кад-), лишь в 30% образцов экспрессия Ткадгерина ярко выражена). В 10% исследованных образцов метастазов Т-кадгерин не выявлен (Т-кад-). Похожий паттерн экспрессии был описан выше для предраковых состояний и для немеланомных онкологических заболеваний кожи: при озлокачествлении экспрессия Ткадгерина в трансформированных клетках постепенно снижалась. Наблюдалось нарушение экспрессия Т-кадгерина в кровеносных сосудах, что коррелировало с гистологическими и инвазивными свойствами более агрессивных опухолей. Для оценки потенциальной роли Т-кадгерина в прогрессировании и росте меланомы, мы использовали хорошо описанную ранее модель меланомы B16F10 в BDF1 мышах. Результаты этого исследования приведены в следующем разделе. 198 3.3 Т-кадгерин и меланома По данным литературы мышиная меланома B16F10 с метастазами в легкие в мышах BDF1 является адекватной моделью для оценки эффективности противораковой терапии на преклинических стадиях, а также моделью, используемой для прогнозирования и скрининга противораковых препаратов в случае диссеминированной меланомы человека [Teicher and Andrews, 2004]. 3.3.1 Анализ экспрессии Т-кадгерина в клетках мышиной меланомы В16F10 В результате стабильной трансфекции контрольной плазмидой или плазмидой, содержащей кДНК для экспрессии Т-кадгерина, и последующей селекции на среде, содержащей селективный антибиотик G418, было получено 2 суммарные популяции клеток мышиной меланомы B16F10: суммарная популяция, клетки которой гиперэкспрессируют Т-кадгерин (B16F10 T (+) сумм), и контрольная суммарная популяция, клетки которой не экспрессируют Т-кадгерин (B16F10 T (-) сумм). Уровень экспрессии Т-кадгерина в суммарных популяциях клеток мышиной меланомы B16F10 определяли методом вестерн блоттинга (Рисунок 3.28). Клетки B16F10 T(-) сумм и B16F10 T(+) сумм далее использовали для исследования влияния экспрессии Т-кадгерина на неоваскуляризацию первичного узла меланомы на мышах BDF1, а также рост опухолевой массы на модели хориоаллантоисной мембраны куриного эмбриона. В результате стабильной трансфекции контрольной плазмидой или плазмидой, содержащей кДНК Т-кадгерина, селекции на среде, содержащей селективный антибиотик G418, и последующего клонирования было получено 6 клонов клеток мышиной меланомы B16F10 с разным уровнем экспрессии Т-кадгерина и один контрольный клон мышиной меланомы, клетки которого не экспрессируют Т-кадгерин (Рисунок 3.29). Уровень экспрессии Т-кадгерина в полученных клонах B16F10 определяли с помощью метода Вестерн блоттинга. Для количественной оценки уровня экспрессии Т-кадгерина в полученных клонах был проведен денситометрический анализ трѐх независимых результатов вестерн блоттинга. В результате было обнаружено, что уровень экспрессии Т-кадгерина в контрольном клоне составил 0,002 ОЕ, тогда как в клонах, трансфицированных плазмидой, содержащей кДНК Т-кадгерина, этот уровень варьировал от 3,3 до 34 ОЕ (Рисунок 3.30). 199 Рисунок 3.28. Анализ экспрессии Т-кадгерина в полученных суммарных популяциях клеток мышиной меланомы B16F10 методом Вестерн блоттинга. Клетки HUVEC 2 пассажа использовали в качестве положительного контроля. Рисунок 3.29. Анализ экспрессии Т-кадгерина в полученных клонах клеток мышиной меланомы B16F10 методом Вестерн блоттинга. В лизатах клонов клеток (клоны 1, 3, 4, 6) обнаруживается высокая экспрессия зрелого Т-кадгерина (105 кДа), в лизатах клона 5 обнаруживается умеренная экспрессия зрелого Т-кадгерина. В клоне 2 обнаруживаются следовые количества Т-кадгерина; контрольный клон не экспрессирует Т-кадгерин. 200 Опираясь на данные денситометрического анализа, для дальнейших экспериментов из полученной панели клонов было выбрано 3 клона: контрольный клон B16F10 T(-), клетки которого не экспрессируют Т-кадгерин, клон 2 B16F10 T(+) с низким уровнем экспрессии Т-кадгерина и клон 1 B16F10 T(++) с высоким уровнем экспрессии Т-кадгерина (Рисунок 3.31). В дальнейшем эксперименты по исследованию влияния Т-кадгерина на рост первичного узла и метастазирование на мышах BDF1 были выполнены с использованием этих трѐх клонов мышиной меланомы. Купхал с коллегами обнаружили, что клоны клеток, трансфицированные Ткадгерином, могут терять экспрессию Т-кадгерина при длительном культивировании, в том числе и при культивировании в селективной среде, содержащей антибиотик [Kuphal et al., 2009]. В связи с этими мы проверяли экспрессию Т-кадгерина в полученных суммарных популяциях и клонах мышиной меланомы методом вестерн блоттинга перед началом и в конце каждого эксперимента. В том числе, экспрессию Т-кадгерина в клетках меланомы тестировали, выделяя клетки из первичного меланомного узла мыши в культуру. Однако потери или изменения уровня экспрессии Т-кадгерина в клонах или суммарных популяциях мышиной меланомы B16F10 обнаружено не было. 201 Рисунок 3.30. Денситометрический анализ результатов, полученных методом вестерн блоттинга при изучении экспрессии Т-кадгерина в клонах мышиной меланомы B16F10, выполненный с помощью программного обеспечения Quantity One 4.6. ОП – оптическая плотность, площадь – площадь одного пикселя. Т-кадгерин (105 кДа) Актин (42 кДа) B16F10 T (-) B16F10 T (+) B16F10 T (++) Рисунок 3.31. Отобранные для дальнейших экспериментов клоны мышиной меланомы B16F10: B16F10 T (-) (контрольный клон), B16F10 T (+) (клон 2) и B16F10 T (++) (клон 1). 202 3.3.2 Влияние экспрессии Т-кадгерина на пролиферацию клеток мышиной меланомы B16F10 in vitro Анализ пролиферации клеток клонов мышиной меланомы B16F10 и построение кривых роста in vitro проводили двумя методами: подсчѐтом абсолютного числа клеток с помощью прибора Cell Counter (Рисунок 3.32 А) и измерением клеточного индекса в режиме реального времени с помощью прибора xCELLigence (Рисунок 3.32 Б). На основании данных, полученных при измерении клеточного индекса на приборе xCELLigence, были высчитаны времена удвоения для каждого клона мышиной меланомы B16F10. Данные представлены в Таблице 3.1. Таблица 3.1. Время удвоения популяций клеток контрольного клона и экспрессирующих Т-кадгерин клонов мышиной меланомы B16F10. Клон клеток Время удвоения, час B16F10 T (-) 31,7 B16F10 T (+) 29,8 B16F10 T (++) 28,4 Двумя методами анализа пролиферации было показано, что in vitro скорость роста клона B16F10 с высокой экспрессией Т-кадгерина достоверно превышает скорость роста клона с низкой экспрессией Т-кадгерина и контрольного клона (p<0,05). Было также обнаружено, что скорость роста клона с низкой экспрессией Т-кадгерина превышает скорость роста контрольного клона, однако, эти различия не достоверны (p>0,05). Таким образом, на основании данных in vitro, можно заключить, что экспрессия Ткадгерина в клетках мышиной меланомы B16F10 приводит к увеличению пролиферативной активности клеток за счѐт сокращения времени удвоения популяции клонов клеток, экспрессирующих Т-кадгерин. 203 Рисунок 3.32. Кривые роста контрольного клона B16F10 и клонов мышиной меланомы B16F10 с разным уровнем экспрессии Т-кадгерина: А - подсчѐт абсолютного числа клеток с помощью прибора Cell Counter в течение 72 часов (n=5), Б - измерение клеточного индекса на приборе xCELLigence в течение 96 часов (n=3). 204 3.3.3 Влияние экспрессии Т-кадгерина на распределение клеток клонов B16F10 по фазам клеточного цикла in vitro Из данных литературы известно, что экспрессия Т-кадгерина может вызывать задержку в G2 фазе клеточного цикла [Huang et al., 2003]. В связи с этим было проанализировано распределение клеток контрольного клона и клонов мышиной меланомы B16F10 с экспрессией Т-кадгерина по фазам клеточного цикла c помощью метода проточной цитометрии (Рисунок 3.33). Для этого клетки контрольного клона и экспрессирующих Т-кадгерин клонов мышиной меланомы B16F10 окрашивали йодистым пропидием, по флуоресценции которого определяли содержание в них ДНК. В ходе проведенного анализа не было выявлено достоверных различий между клонами мышиной меланомы по количеству клеток, находящихся на каждой из трѐх исследованных фаз клеточного цикла (G0/G1, S, G2/M). Таким образом, экспрессия Ткадгерина не приводит к изменению структуры клеточного цикла в клетках клонов мышиной меланомы B16F10. 3.3.4 Влияние экспрессии Т-кадгерина на апоптоз и некроз клеток мышиной меланомы B16F10 in vitro Количество клеток на различных стадиях апоптоза и некроза в культуре контрольного клона и клонов мышиной меланомы с разным уровнем экспрессии Ткадгернна анализировали методом проточной цитометрии. Для этого клетки окрашивали аннексином V, конъюгированным с фикоэритрином, а также красителем 7-AAD, который позволяет распознавать клетки, находящиеся на ранних стадиях апоптоза. Клетки позитивные и по аннексину V, и по 7-AAD считали клетками мѐртвыми или находящимися на поздних стадиях апоптоза. Клетки, позитивные по Аннексину V, но негативные по 7AAD считали клетками, находящимися на ранних стадиях апоптоза. Клетки, негативные по аннексину V и 7-AAD считали живыми. Было обнаружено, что процент мѐртвых клеток и клеток на ранних стадиях апоптоза достоверно не различается между клонами меланомы (Рисунок 3.34). 205 100 Процент клеток 90 80 70 60 50 40 30 20 10 0 G0/G1 B16F10 T (-) S B16F10 T (+) G2/M B16F10 T (++) Процент мёртвых клеток и клеток на ранних стадиях апоптоза Рисунок 3.33. Распределение клеток контрольного клона и экспрессирующих Ткадгерин клонов мышиной меланомы B16F10 по фазам клеточного цикла (n=3, p>0,05). 6 5 4 3 2 1 0 B16F10 T (-) B16F10 T (+) B16F10 T (++) Рисунок 3.34. Процент мѐртвых клеток и клеток на ранних стадиях апоптоза в культуре клонов мышиной меланомы B16F10 (n=3, р>0,05). 206 3.3.5 Влияние экспрессии Т-кадгерина на рост, метастазирование и васкуляризацию первичного узла меланомы B16F10 на модели гематогенно метастазирующей в лѐгкие меланомы у мышей Для исследования влияния Т-кадгерина на рост, метастазирование и васкуляризацию первичного узла мышиной меланомы B16F10 in vivo была выбрана модель гематогенно метастазирующей в лѐгкие меланомы у мышей BDF1. На этой модели были проанализированы характеристики роста первичного узла (размер первичного узла, васкуляризация первичного узла и вклад стромального компонента в рост первичного узла), а также оценена частота метастазирования. 3.3.5.1 Влияние экспрессии Т-кадгерина на рост первичного узла мышиной меланомы B16F10 in vivo Влияние Т-кадгерина на рост первичного узла оценивали путѐм анализа кинетики роста опухолей после подкожного введения клеток мышиной меланомы мышам (7 измерений размера опухоли в течение эксперимента). Наиболее быстрый рост первичного узла наблюдали при введении клеток меланомы, экспрессирующих Т-кадгерин (высокий и низкий уровень экспрессии), по отношению к первичным узлам, образованным контрольными клетками (p<0,05) (Рисунок 3.35). К 15-му дню эксперимента средний размер первичного узла, образованного клетками меланомы с высоким уровнем экспрессии Т-кадгерина, достигал 778,2 мм3, с низким уровнем экспрессии Т-кадгерина - 695,5 мм3, тогда как для первичных узлов, образованных контрольными клетками меланомы, - 117,75 мм3. Статистически значимое различие в размере опухолей, сформированных контрольным клоном и Т-кадгерин экспрессирующими клонами, наблюдалось на 8-, 12-, 15- и 18-й день эксперимента (р<0,05). Первые мыши, объем первичных опухолевых узлов у которых достигал 1000 мм3 и более в группах с введенными клонами B16F10 T(+) и B16F10 T(++), были зарегистрированы на 15-й день, тогда как в контрольной группе - на 21-й день. Достоверное увеличение скорости роста наблюдалось также в случае первичных узлов, образованных поликлональной линией с гиперкспрессирующей Т-кадгерин B16F10 (T+) по сравнению с первичными узлами, образованными контрольной поликлональной линией B16F10 (T-) (p<0,05) (Рисунок 3.36). 207 * Рисунок 3.35. Динамика роста подкожных первичных узлов, образованных клонами клеток мышиной меланомы B16F10 у мышей BDF1 (n=11, *р<0,05). B16F10 T() - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. Рисунок 3.36. Динамика роста подкожных первичных узлов, образованных поликлональными культурами мышиной меланомы B16F10 (контрольной суммарной популяцией B16F10(T-) или суммарной популяцией клеток, гиперэкспрессирующей Ткадгерин B16F10(T+)) у мышей BDF1 (n=11, *р<0,05). – контрольная популяция, популяция клеток, гиперэкспрессирующих Т-кадгерин . 208 3.3.5.2 Влияние экспрессии Т-кадгерина на частоту очагов некроза на срезах первичных узлов меланомы В16F10 Гистологический анализ показал, что первичные опухоли у мышей, сформированные клонами меланомы с разным уровнем экспрессии Т-кадгерина, заметно отличаются. При окраске гематоксилином и анализе криосрезов первичных узлов были обнаружены обширные области некротического поражения. Анализ отношения количества срезов с очагами некроза к общему количеству срезов для каждого клона показал, что значительную долю первичных узлов, образованных Т-кадгерин-позитивными клетками, занимают очаги некроза, тогда как в первичных узлах, образованных контрольными клетками количество таких очагов достоверно меньше (p<0,05) (Рисунок 3.37). 3.3.5.3 Влияние экспрессии Т-кадгерина на васкуляризацию первичных узлов мышиной меланомы Увеличение некрозов в первичном узле меланомы может быть связано, прежде всего, с его недостаточным кровоснабжением. Для подтверждения этого предположения криосрезы первичных узлов меланомы окрашивали антителами к CD31, маркѐру эндотелиальных клеток (Рисунок 3.38). На окрашенных срезах производили подсчѐт числа сосудов различного диаметра: крупных сосудов, сосудов среднего диаметра и капилляров. Было обнаружено, что количество сосудов среднего диаметра и капилляров на срезах первичных меланомы, узлов, достоверно образованных меньше, чем в экспрессирующими Т-кадгерин случае узлов, контрольными клетками (p<0,05) (Рисунок 3.39). первичных клетками образованных 209 Рисунок 3.37. Частота встречаемости очагов некроза на криосрезах первичных узлов меланомы B16F10 у мышей BDF1 (n=150, *р<0,05). B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. Рисунок 3.38. Иммунофлуоресцентное окрашивание криосрезов первичных узлов мышиной меланомы B16F10 антителами против CD31. B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. Масштаб – 100 мм. 210 Рисунок 3.39. Васкуляризация первичных узлов меланомы B16F10 у мышей BDF1 сосудами различного диаметра, (n=150, *р<0,05). B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. 211 3.3.5.4 Экспрессия Т-кадгерина в клетках меланомы вызывает активацию стромы и увеличивает вклад стромального компонента в формирование первичных узлов мышиной меланомы B16F10 Поскольку известно, что мезенхимные стромальные клетки могут вносить существенный вклад в опухолевый рост и прогрессию, в настоящем исследовании мы провели оценку вклада стромального компонента в формирование первичного узла in vivo путем окрашивания криосрезов на маркер активированной стромы CD90 (Thy-1) [True et al., 2010] (Рисунок 3.40). На полученных срезах с помощью программы MetaMorph оценивали отношение площади CD90-позитивных областей к общей площади среза. Было обнаружено, что на срезах первичных узлов, образованных экспрессирующими Т-кадгерин клетками мышиной меланомы, CD90 позитивные области занимают достоверно большую площадь, чем в случае первичных узлов, сформированных контрольными клетками (p<0,05) (Рисунок 3.41). Рисунок 3.40. Иммунофлуоресцентное окрашивание криосрезов первичных узлов мышиной меланомы B16F10 антителами против CD90. B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. Масштаб – 100 мкм. 212 Рисунок 3.41. Площадь CD90 позитивных областей на срезах первичных узлов меланомы у мышей BDF1, (n=150, *р<0,05). B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. 3.3.5.5 Изучение влияния экспрессии Т-кадгерина на метастазирование клеток меланомы in vivo Одной из ключевых характеристик опухолевой прогрессии является метастазирование. Для оценки параметров метастазирования мышей умерщвляли, извлекали лѐгкие, взвешивали и тестировали их на наличие метастазов. В качестве параметров оценивали частоту метастазирования (отношение числа мышей с метастазами к общему числу анализируемых мышей) и лѐгочный коэффициент (отношение средней массы лѐгких в опытной группе к средней массе лѐгких здоровых мышей). Криосрезы образцов ткани легких окрашивали гематоксилином и эозином для визуализации легочных метастаз (Рисунок 3.42). Было обнаружено, что в группе мышей, которым были введены клетки меланомы B16F10 с высоким уровнем экспрессии Т-кадгерина, частота метастазирования и показатель лѐгочного коэффициента были выше, чем в контроле (Таблица 3.2). Таблица 3.2. Частота метастазирования клонов мышиной меланомы B16F10 с разным уровнем экспрессии Т-кадгерина, (n=11). Клон клеток Число мышей с метастазами / общее число мышей Процент мышей с метастазами Средняя масса лѐгких, г Лѐгочный коэффициент B16F10 T (-) B16F10 T (+) B16F10 T (++) 1/11 2/11 6/11 9,1 % 18,2 % 54,5 % 157 156 160 1 1 1,1 213 Рисунок 3.42. Окрашивание гематоксилином срезов первичных узлов меланомы (верхняя панель) и метастазов в легкие (нижняя панель), образованных клонами клеток мышиной меланомы B16F10 у мышей BDF1. Масштаб 100 мкм. 214 3.3.6 Влияние среды роста от клеток клонов B16F10 на пролиферацию и миграцию МСК-ЖТ in vitro 3.3.6.1 Влияние среды роста от клеток клонов B16F10 на миграцию МСК-ЖТ Для выявления механизмов стимулирующего влияния экспрессии Т-кадгерина в клетках меланомы на стромальные клетки проводили исследование влияния среды роста от клеток клонов мышиной меланомы на миграцию МСК-ЖТ. Для этого использовали систему Transwell®, содержащих вставку с полупроницаемой мембраной с диаметром пор 8 мкм. Мембрана предварительно была покрыта коллагеном. В качестве контроля использовали среду роста от эмбриональных фибробластов NIH/3T3 и стандартную среду RPMI1640, содержащую 10% ФБС. В ходе эксперимента было обнаружено, что кондиционирования среда клеток клонов меланомы B16F10 вне зависимости от экспрессии Т-кадгерина стимулирует миграцию МСК-ЖТ по сравнению со средой роста эмбриональных фибробластов мыши NIH3T3 и стандартной средой с 10% сывороткой для культивирования клеток (p<0,05) (Рисунок 3.43). Однако при гиперэкспрессии Т-кадгерина в клетках меланомы стимулирующий эффект среды культивирования от этих клеток на миграцию МСК-ЖТ увеличивается в 1,4 раза по сравнению с контрольным клоном (p<0,05). Так как было показано, что в кондиционированной среде от клеток CHO, трансфицированных кДНК Т-кадгерина, содержится Т-кадгерин [Vestal and Ranscht, 1992], мы проверили среду роста от клонов мышиной меланомы B16F10 и фибробластов NIH/3T3 на присутствие растворимой формы Т-кадгерина. Анализ методом вестерн блоттинга показал, что Т-кадгерина в кондиционированной среде роста от всех используемых типов клеток нет (Рисунок 3.44). Клетки NIH/3T3, используемые в качестве контроля, и клетки МСК-ЖТ были также проверены на наличие экспрессии Т-кадгерина методом иммуноблоттинга. Экспрессии Т-кадгерина в этих клетках обнаружено не было (Рисунок 3.44). 215 Рисунок 3.43. Влияние кондиционированной среды роста от клеток клонов мышиной меланомы B16F10 на миграцию МСК-ЖТ. * - достоверные различия по отношению к контрольному клону клеток, ** - достоверные различия по отношению к клонам меланомы, р<0,05. Рисунок 3.44. Анализ экспрессии Т-кадгерина (T-cadherin) в лизатах клеток HUVEC, NIH/3T3 (3T3), МСК-ЖТ и в кондиционированных средах от клеток клонов меланомы B16F10 методом Вестерн блоттинга. Клетки HUVEC 2 пассажа использовали в качестве положительного контроля. 130 kDa – предшественник Т-кадгерина, 105 kDa – зрелый Т-кадгерин. Данные нормировали на актин (actin, 45 kDa). B16F10 T(-) контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. 216 3.3.6.2 Исследование влияния среды роста от клеток клонов B16F10 на пролиферацию МСК-ЖТ Исследование влияния среды роста от клеток клонов мышиной меланомы B16F10 на пролиферацию МСК-ЖТ проводили с использованием системы Transwell®, содержащей полупроницаемую мембрану с диаметром пор 0,4 мкм. В качестве контроля использовали кондиционированную среду роста от эмбриональных фибробластов NIH/3T3 и стандартную среду культивирования, содержащую 10% ФБС. Было обнаружено, что кондиционированная среда от Т-кадгерин-экспрессирующих клонов имеет тенденцию стимулировать пролиферацию клеток МСК-ЖТ по сравнению с контрольным клоном, однако, достоверных отличий в пролиферации МСК-ЖТ при этом обнаружено не было (p>0,05) (Рисунок 3.45). Достоверные различия были обнаружены только между кондиционированными средами от клонов клеток мышиной меланомы вне зависимости от экспрессии Т-кадгерина и стандартной средой для культивирования клеток с добавлением 10% сыворотки (p<0,05), что свидетельствует в принципе о стимулирующем влиянии кондиционированной среды от клеток меланомы на пролиферацию МСК-ЖТ. Таким образом, при бесконтактном со-культивировании in vitro высокий уровень экспрессии Т-кадгерина в клетках мышиной меланомы B16F10 приводит к стимуляции миграции стромальных клеток из подкожной жировой клетчатки (МСК-ЖТ), но не влияет на их пролиферацию. Этот эффект не связан с секрецией или слущиванием Т-кадгерина с поверхности мембраны клеток меланомы. Рисунок 3.45. Влияние среды роста от клонов клеток мышиной меланомы B16F10 на пролиферацию клеток МСК-ЖТ. N=4, *р<0,05. B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. 217 3.3.7 Исследование влияния экспрессии Т-кадгерина на инвазивный потенциал клеток клонов B16F10 in vitro Исследование влияния Т-кадгерина на инвазивный потенциал клеток клонов мышиной меланомы B16F10 проводили с использованием специально разработанной нами в настоящей работе модели двух капель Матригеля: на каплю Матригеля без факторов роста (GFR Матригель), содержащую клетки мышиной меланомы (контрольный клон или клон, экспрессирующий Т-кадгерин), сверху наслаивали каплю Матригеля, содержащего факторы роста (Рисунок 3.46). Поскольку инвазия клеток меланомы из GFR Матригеля в полный Матригель сопровождается не только миграцией клеток, но и протеолитическим расщеплением окружающего матрикса, что приводит к размыванию границы между двумя Матригелями и изменению размеров капли, мы оценивали число клеток, инвазировавших в полный Матригель, а также размеры внутренней капли после 48 часов инкубации. Было обнаружено, что через 48 часов размеры капель Матригеля с Т-кадгерин гиперэкспрессирующими клетками были достоверно больше по сравнению с размерами капель, в которые были помещены контрольные клетки или клетки с низким уровнем экспрессии Т-кадгерина (p<0,05) (Рисунок 3.47). Также было обнаружено, что количество клеток клона мышиной меланомы с высоким уровнем экспрессии Т-кадгерина, инвазировавших из GFR Матригеля в полный Матригель, достоверно превышает аналогичный показатель для контрольного клона и клона с низким уровнем экспрессии Т-кадгерина (p<0,05) (Рисунок 3.48). Рисунок 3.46. Изучение инвазивного потенциала клеток меланомы B16F10 на модели двух капель Матригеля. GFR Matrigel without growth factors+melanoma cells – Матригель без факторов роста, содержащий клетки меланомы; Matrigel with growth factors – Матригель, содержащий факторы роста; RPMI1640+10%FBS – среда роста клеток меланомы, содержащая 10% сыворотку; Boundary drop – границы капли Матригеля. D1 – размер капли по горизонтали; D2- размер капли по вертикали. 218 Рисунок 3.47. Диаметры капель Матригеля, содержащих клетки мышиной меланомы (n=3, *р<0,05). D1 – размер капли по горизонтали; D2- размер капли по вертикали. B16F10 T(-) - контрольный клон, B16F10 T(+) - клон с невысокой экспрессией Т-кадгерина; B16F10 T(++) - клон с высокой экспрессией Т-кадгерина. Рисунок 3.48. Количество клеток мышиной меланомы B16F10, инвазировавших в полный Матригель через 48 ч, на модели двух капель Матригеля in vitro (n=3, *р<0,05). 219 3.3.8 Исследование влияния Т-кадгерина на экспрессию цитокинов, факторов роста и матриксных металлопротеиназ в клетках B16F10 in vitro Исследование влияния Т-кадгерина на экспрессию цитокинов, факторов роста и матриксных металлопротеиназ в клетках меланомы проводили методом ПЦР в режиме реального времени (RT-PCR), при этом изучали экспрессию VEGF А, PDGF В, EGF, bFGF, HGF, uPAR, uPA, c-Met (HGFR), MMP2, MMP9. Цитокиновый профиль, экспрессию белков внеклеточного матрикса и молекул адгезии, ангиогенных факторов и ингибиторов ангиогенеза, хемокинов и их рецепторов исследовали с использованием коммерческих наборов RT2 Profiler PCR Array. С помощью RT-PCR было проведено сравнение клеток клонов мышиной меланомы по уровню экспрессии некоторых ангиогенных факторов. По уровню экспрессии генов VEGF А (Рисунок 3.49), PDGF В (Рисунок 3.50), bFGF (Рисунок 3.51), uPA (Рисунок 3.52), uPAR (Рисунок 3.53), MMP2 (Рисунок 3.54) достоверных различий между контрольным клоном и клонами c разным уровнем экспрессии Т-кадгерина обнаружено не было. Экспрессии мРНК EGF, HGF и MMP ни в контрольном клоне, ни в клонах меланомы, экспрессирующей Т-кадгерин, обнаружено не было. Однако было обнаружено, что гиперэкспрессия Т-кадгерина в клетках меланомы приводит к значительному увеличению экспрессии в этих клетках c-Met, рецептора HGF (Рисунок 3.55). С помощью коммерческого набора RT2 Profiler PCR Array (ангиогенные факторы и ингибиторы ангиогенеза) было обнаружено, что экспрессия Т-кадгерина в клетках меланомы B16F10 приводит к сильному увеличению (более чем в 4 раза) экспрессии 3-х генов: CXCL10, heparanase и Foxf1a (Таблица 3.3, цвета на градиенте соответствуют разнице в экспрессии). Экспрессия ингибиторов ангиогенеза, таких как chromogranin A и procollagen XVIIIα1 выявляется исключительно в клонах меланомы, экспрессирующих Ткадгерин, и не детектируется в контрольном клоне. Экспрессия Т-кадгерина приводит к небольшому увеличению (более чем в 2 раза) экспрессии 2-х генов ингибиторов ангиогенеза: аngiopoietin 2 и stabilin 1 (ингибитор ангиогенеза), но вызывает значительное подавление экспрессии Tie 1 – стимулятора ангиогенеза. Также было показано, что экспрессия TGFα присутствует только в контрольном клоне, тогда как в экспрессирующих Т-кадгерин клонах мРНК TGFα не обнаруживается. Экспрессия в клетках мышиной меланомы B16F10 остальных 75 генов, изучаемых с помощью RT2 Profiler PCR Array, не зависит от экспрессии Т-кадгерина. 220 Рисунок 3.49. Экспрессия VEGF A в клонах мышиной меланомы B16F10 (n=5), р>0,05. B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Ткадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. Относительные единицы 0,6 0,5 0,4 0,3 0,2 0,1 0 B16F10 T- B16F10 T+ B16F10 T++ Рисунок 3.50. Экспрессия PDGF B в клонах мышиной меланомы B16F10 (n=5), р>0,05. B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Ткадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. 221 Рисунок 3.51. Экспрессия bFGF в клонах мышиной меланомы B16F10 (n=5, р>0,05). B16F10 (T-) - контрольный клон, B16F10 (T+) - клон с невысокой экспрессией Ткадгерина; B16F10 (T++) - клон с высокой экспрессией Т-кадгерина. 5 Относительные единицы 4,5 4 3,5 3 2,5 2 1,5 1 0,5 0 B16F10 T- B16F10 T+ B16F10 T++ Рисунок 3.52. Экспрессия uPA в клонах мышиной меланомы B16F10 (n=5), р>0,05. B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Т-кадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. Относительные единицы 6 5 4 3 2 1 0 B16F10 T- B16F10 T+ B16F10 T++ 222 Рисунок 3.53. Экспрессия uPAR в клонах мышиной меланомы B16F10 (n=5), р>0,05. B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Ткадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. Относительные единицы 0,6 0,5 0,4 0,3 0,2 0,1 0 B16F10 T- B16F10 T+ B16F10 T++ Рисунок 3.54. Экспрессия MMP2 в клонах мышиной меланомы B16F10 (n=5), р>0,05. B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Ткадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. Рисунок 3.55. Экспрессия c-Met (HGFR) в клонах мышиной меланомы B16F10 (n=5, *р<0,05). B16F10 T- - контрольный клон, B16F10 T+ - клон с невысокой экспрессией Т-кадгерина; B16F10 T++ - клон с высокой экспрессией Т-кадгерина. 223 Таблица 3.3. Сравнение экспрессии генов, участвующих в процессах ангиогенеза, в клетках клонов B16F10. Показаны различия в экспрессии по отношению к контрольному клону. -10 -5 0 5 10 Изменение экспрессии (число раз в сравнении с контролем) B16F10 T (+) B16F10 T (++) Ген Функция 2 2 Angiopoietin 2 (Angpt 2) стимулятор/ингибитор ангиогенеза дестабилизирует сосуды 2 3 Stabilin 1 (Stab 1) негативно влияет на ангиогенез 5 7,8 Chemokine (C-X-C motif) ligand 10 (CXCL 10) ингибитор ангиогенеза 8,7 6,3 Heparanase (Hpse) негативно влияет на ангиогенез 4 5,5 Forkhead box F1a (Foxf1a) позитивно влияет на ангиогенез -10 -11 Tyrosine kinase receptor 1 (Tie 1) стимулятор ангиогенеза С помощью коммерческого набора RT2 Profiler PCR Array (хемокины и их рецепторы) было обнаружено, что экспрессия Т-кадгерина в клетках B16F10 приводит к сильному увеличению экспрессии гена CXCR7 (более чем в 10 раза) и CXCL10 (более чем в 10 раза) (Таблица 3.4, цвета на градиенте соответствуют разнице в экспрессии). Небольшое увеличение экспрессии также наблюдалось для генов CCL5, CMTM3, Nfkb1 (p105), CXCL11 и CCRL1. Экспрессия 3 генов CCL7, Gdf 5 и interleukine 8rα была выявлена исключительно в клонах меланомы с экспрессией Т-кадгерина и не детектировалась в контрольном клоне, в то время как мРНК interleukin 16, interleukin 4 и Il8rb детектировалась исключительно в контрольном клоне. Также было обнаружено, что экспрессия Т-кадгерина приводит к подавлению экспрессии CCL9. Экспрессия остальных 72 генов, исследуемых с помощью RT2 Profiler PCR Array, не изменяется в зависимости от экспрессии Т-кадгерина. 224 Таблица 3.4. Сравнение экспрессии генов хемокинов и их рецепторов в клетках клонов меланомы B16F10. Показаны различия в экспрессии по отношению к контрольному клону. -10 -5 0 5 10 Изменение экспрессии (число раз в сравнении с контролем) B16F10 T (+) Ген Функция B16F10 T (++) 1,8 2,5 Chemokine (C-X-C motif) Хемоаттрактант для строligand 11 (CXCl 11) мальных и иммунных клеток 2,4 2,2 CKLF-like MARVEL Хемоаттрактант для строtransmembrane domain мальных и иммунных клеток containing 3 (CMTM3) 3,3 2,4 Chemokine (C-C motif) Хемоаттрактант для строligand 5 (CCl5) мальных и иммунных клеток 2 3,2 Nuclear factor of kappa Хемоаттрактант для строlight chain gene enhancer мальных и иммунных клеток in B cells, p105 (Nfkb 1) 1,5 2 2,9 15,3 Chemokine (C-X-C motif) Хемоаттрактант для строligand 10 (CXCL10) мальных и иммунных клеток 5,7 17,6 Chemokine C-X-C motif Хемоаттрактант для строreceptor 7 (CXCR7) мальных и иммунных клеток -1,4 -3,1 Chemokine (C-C motif) Хемоаттрактант для строligand 9 (CCL9) мальных и иммунных клеток С помощью Chemokine С-С motif receptor-like 1) (CCRL1) коммерческого набора RT2 Profiler Рецептор хемокинов PCR Array (молекулы внеклеточного матрикса и молекулы адгезии) было обнаружено, что экспрессия Ткадгерина в клетках мышиной меланомы приводит к сильному увеличению (более чем в 10 раз) экспрессии одного гена - cadherin 1 (Е-кадгерин) (Таблица 3.5, цвета на градиенте соответствуют разнице в экспрессии). Также экспрессия Т-кадгерина вызывает увеличение более чем в 2 раз экспрессии 5 генов: MMP14, procollagen Iα1, integrin α5, integrin β3 и integrin αE и fibronectin 1, небольшому увеличению (менее чем в 2 раза) экспрессии integrin αV, а также небольшому снижению экспрессии TGFbi. Экспрессия PEСAM1 (CD31), Adamts 2 и laminin α3 была выявлена исключительно в клонах меланомы с экспрессией Ткадгерина и не детектировалась в контрольном клоне. Также было обнаружено, что экспрессия Т-кадгерина приводит к подавлению экспрессии 2-х генов: Adamts 5 и laminin 225 β3. Экспрессия же остальных 70 генов, исследуемых с помощью RT 2 Profiler PCR Array, не зависит от экспрессии Т-кадгерина. Таблица 3.5. Сравнение экспрессии генов молекул внеклеточного матрикса и клеточной адгезии в клетках клонов мышиной меланомы B16F10. Показаны различия в экспрессии по отношению к контрольному клону. -10 -5 0 5 10 Изменение экспрессии (число раз по сравнению с контролем) B16F10 T (+) Ген Функция B16F10 T (++) 1,1 2,3 Integrin beta 3 (Itgb3) опухолевая адгезия 1,3 2,6 Integrin alpha 5 (Itga5, fibronectin receptor alpha) опухолевая адгезия 1,8 2,5 Integrin alpha V (Itgav) опухолевая адгезия 1 3,3 Integrin alpha E, epithelial-associated (ItgE) опухолевая адгезия 1,2 3,9 2,3 2 Transforming growth factor, beta induced (Tgf bi) опухолевая адгезия 4 15 Cadherin 1 (Cdh1) (E-cadherin) нормальная адгезия 1,3 3,2 Matrix metallopeptidase 14 (MMP14) опухолевая прогрессия -2,3 -2,1 A distintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 5 (Adamts 5) нормальный ангиогенез Fibronectin 1 нормальная и опухолевая адгезия Данные ПЦР анализа и результаты, полученные с использованием коммерческого набора RT2 Profiler PCR Array, были частично подтверждены с помощью метода вестерн блоттинга в сочетании с денситометрией, а также иммунофлуоресцентным окрашиванием с использование конфокального микроскопа при одинаковых настройках лазера и времени экспозиции. Было обнаружено, что экспрессия Т-кадгерина вызывает изменение на уровне продукции белка следующих ангиогенных факторов и ингибиторов ангиогенеза: снижение 226 Tie 1, увеличение chromogranin A, аngiopoietin 2 и heparanase (Рисунок 3.56), что соответствует ранее полученным данных по экспрессии мРНК этих белков. Экспрессия Т-кадгерина вызывает увеличение экспрессии на уровне белка хемокина CXCR7 и хемоаттрактанта Nfkb1 (p105), а также Е-кадгерина (cadherin 1) (Рисунок 3.57) и интегринов integrin α5, integrin β3 и integrin αV, что соответствует ранее полученным данным по экспрессии мРНК этих белков (Рисунок 3.58). 227 C Рисунок 3.56. Анализ экспрессии ангиогенных факторов и ингибиторов ангиогенеза в клонах меланомы B16F10 в зависимости от экспрессии Т-кадгерина. А – иммунофлуоресцентное окрашивание клеток B16F10 антителами к Тie 1 (верхняя панель, красная флуоресценция) и антителами к heparanase (нижняя панель, красная флуоресценция). Ядра клеток докрашены DAPI. B – анализ экспрессии Tie 1, Angiopoetin 228 2, heparanase и chromogranin A методом вестерн блоттинга в лизатах клеток. С – денситометрический анализ результатов Вестерн блоттинга. N=3, p<0,05. Данные нормированы по уровню экспрессии GAPDH. Показан воспроизводимый результат трѐх независимых экспериментов. Т- - контрольный клон, Т+ -клон с низкой экспрессией Ткадгерина; Т++ - клон с высокой экспрессией Т-кадгерина. 229 C Рисунок 3.57. Анализ экспрессии хемокинов и их рецепторов в клонах меланомы B16F10 в зависимости от экспрессии Т-кадгерина. А – иммунофлуоресцентное окрашивание клеток B16F10 антителами к CXCR7 (красная флуоресценция). Ядра клеток докрашены DAPI. B – анализ экспрессии CXCR7 и Nfkb 1 методом Вестерн блоттинга в лизатах клеток. С – денситометрический анализ результатов вестерн блоттинга. N=3, p<0,05. Данные нормированы по уровню экспрессии GAPDH. Показан воспроизводимый результат трѐх независимых экспериментов. Т- - контрольный клон, Т+ -клон с низкой экспрессией Т-кадгерина; Т++ - клон с высокой экспрессией Т-кадгерина. 230 231 C Рисунок 3.58. Анализ экспрессии интегринов и Е-кадгерина в клонах меланомы B16F10 в зависимости от экспрессии Т-кадгерина. А – иммунофлуоресцентное окрашивание клеток B16F10 антителами к integrin b3 (верхняя панель, красная флуоресценция), integrin a5 (средняя панель, красная флуоресценция), cadherin 1 (средняя панель, красная флуоресценция) и integrin av (нижняя панель, зеленая флуоресценция). Ядра клеток докрашены DAPI. B – анализ экспрессии integrin b3, integrin a5 и cadherin 1 методом вестерн блоттинга в лизатах клеток. С – денситометрический анализ результатов Вестерн блоттинга. N=3, p<0,05. Данные нормированы по уровню экспрессии GAPDH. Показан воспроизводимый результат трѐх независимых экспериментов. Т- контрольный клон, Т+ -клон с низкой экспрессией Т-кадгерина; Т++ - клон с высокой экспрессией Т-кадгерина. Таким образом, при экспрессии Т-кадгерина в клетках меланомы B16F10 не изменяется экспрессия основных факторов роста, белков урокиназной системы и матриксных металлопротеиназ, влияющих на ангиогенез (VEGF А, PDGF В, bFGF, uPА, uPAR и MMP2), однако, происходит увеличение экспрессии ряда ингибиторов ангиогенеза (CXCL10, angiopoietin 2, procollagen type XVIII alpha 1, chromogranin A) и подавление экспрессии некоторых стимуляторов ангиогенеза (TGFα и Tie-1). Кроме того, при этом в клетках меланомы значительно увеличивается экспрессия c-Met, рецептора HGF, и ряда 232 хемокинов, ответственных за привлечение мезенхимных стромальных клеток и клеток иммунной системы (CXCR7, CXCL10, CCL5, CXCL11, CCRL1, CCL7), а также Е-кадгерина и проонкогенных интегринов (integrin α5, integrin αV, integrin αE, integrin β) и MMP14. Экспрессия PECAM 1 (CD31), Adamts 2 и laminin α3 была выявлена исключительно в клонах меланомы с экспрессией Т-кадгерина. Данные по экспрессии мРНК были подтверждены на уровне экспрессии белков: экспрессия Т-кадгерина вызывает снижение ангиогенного фактора Tie 1, увеличение ингибиторов ангиогенеза chromogranin A, аngiopoietin 2 и heparanase, увеличение хемокина CXCR7, хемоаттрактанта Nfkb1 (p105), Екадгерина (cadherin 1) и интегринов integrin α5, integrin β3 и integrin αV. 3.3.9 Влияние экспрессии Т-кадгерина на неоангиогенез в меланоме В16F10 на модели хориоаллантоисной мембраны куриного эмбриона На модели хориоаллантоисной мембраны куриного эмбриона было проанализировано количество сосудов, подрастающих к опухолевым массам, сформированным суммарной популяцией Т-кадгерин экспрессирующих (B16F10 T (+) сумм) и контрольных (B16F10 T (-) сумм) клеток мышиной меланомы. Подсчѐт числа подрастающих сосудов осуществлялся с использованием стереомикроскопа без дополнительного окрашивания. Средний показатель числа сосудов, подрастающих к опухолевым массам, сформированным экспрессирующими Т-кадгерин клетками мышиной меланомы, равен 3,1; аналогичный же показатель для опухолевых масс, сформированных контрольными клетками, равен 7,1 (Рисунок 3.59). Различия между показателями статистически значимы (p<0,01). Таким образом, на модели хориоаллантоисной мембраны куриного эмбриона с использованием суммарной популяции контрольных клеток меланомы и клеток, экспрессирующих Т-кадгерин, были подтверждены данные о том, что экспрессия Ткадгерина в клетках меланомы подавляет их способность стимулировать неоангиогенез и прорастание сосудов в опухоль. Число подрастающих сосудов 233 9 8 7 6 5 4 3 2 1 0 * B16F10 Т (-) сумм B16F10 Т (+) сумм Рисунок 3.59. Число сосудов, подрастающих к опухолевым массам, сформированным экспрессирующими Т-кадгерин (B16F10 T (+) сумм) и контрольными (B16F10 T (-) сумм) клетками мышиной меланомы. N=4, *р<0,01. 234 3.3.10 Обсуждение результатов Влияние Т-кадгерина на рост и опухолевую прогрессию было исследовано в целом ряде работ [для обзора см. Andreeva and Kutuzov, 2010], однако, однозначного понимания роли Т-кадгерина в этих процессах нет. В одних исследованиях было показано, что Ткадгерин подавляет рост и способность к инвазии опухолевых клеток [Takeuchi et al., 2000; Huang et al., 2003; Mukoyama et al., 2005; Pfaff et al., 2010], а в других - стимулирует их пролиферацию и метастазирование [Gutmann et al., 2001; Riou et al., 2006]. Кроме того, в недавнем исследовании Пфафф и соавторов было показано, что как подавление, так и гиперэкспрессия Т-кадгерина в клетках плоскоклеточной карциномы может приводить к одному и тому же результату - усилению опухолевого роста [Pfaff et al., 2011]. Меланоциты нормальной кожи экспрессируют Т-кадгерин, однако, трансформация меланоцитов может сопровождаться потерей экспрессии Т-кадгерина, и как было показано ранее около 80% клеточных линий меланомы человека не экспрессируют этот белок [Kuphal et al., 2009; Bosserhoff et al., 2011]. В настоящей работе для выявления возможной роли Т-кадгерина в опухолевом росте и метастазировании в модельных экспериментах in vivo и in vitro мы экспрессировали Т-кадгерин в мышиной меланоме B16F10, которая исходно не экспрессирует Т-кадгерин. На первом этапе двумя методами in vitro было показано, что экспрессия Т-кадгерина, приводит к увеличению пролиферативного потенциала этих клеток. В основе увеличения пролиферативной активности Т-кадгерин экспрессирующих клеток меланомы, по всей видимости, лежит механизм сокращения времени удвоения популяции (продолжительности клеточного цикла), по сравнению с клетками контрольного клона. Из данных литературы известно, что экспрессия Т-кадгерина может вызывать задержку в G2 фазе клеточного цикла в клетках глиомы C6 или в G2/M фазе в клетках плоскоклеточной карциномы кожи in vitro [Huang et al., 2003; Mukoyama et al., 2005; Chan et al., 2008]. В настоящей работе мы показали, что усиление пролиферации Т-кадгерин экспрессирующих клеток не сопровождается ни задержкой в какой-либо из фаз клеточного цикла, ни изменением структуры клеточного цикла, а также не связано с апоптотической гибелью клеток. Возможно, что различие в данных о влиянии Т-кадгерина на клеточный цикл обусловлено разными вариантами регуляции в опухолевых клетках различного происхождения. Для оценки влияния экспрессии Т-кадгерина в клетках мышиной меланомы на рост, метастазирование и васкуляризацию опухоли была выбрана модель гематогенно метастазирующей меланомы B16F10 у мышей. Эта модель является хорошо описанной в литературе и активно применяемой для исследования различных аспектов опухолевой 235 прогрессии и тестирования противоопухолевых препаратов [Guo et al., 2001; Gabri et al., 2006; Scutti et al., 2011; Teng et al., 2011]. На этой модели было обнаружено, что экспрессия Т-кадгерина в клетках клонов меланомы приводит к увеличению скорости роста первичного узла, по сравнению с первичными узлами, образованными контрольными клетками. Увеличение скорости роста первичных узлов Т-кадгерин-позитивными клетками может быть объяснено увеличением пролиферации этих клеток, что подтверждают полученные нами данные in vitro. При анализе криосрезов первичных узлов было обнаружено, что в узлах, образованных Т-кадгерин экспрессирующими клетками, увеличивается количество очагов некроза по сравнению с контрольными узлами. В то же время in vitro разницы по проценту мѐртвых клеток и клеток, находящихся на разных стадиях апоптоза, между контрольными клетками и клетками, экспрессирующими Т-кадгерин обнаружено не было. Известно, что значительные области опухолей могут отмирать при недостаточном кровоснабжении. В связи этим мы провели сравнительный анализ неоваскуляризации первичных узлов, образованных клетками с разным уровнем экспрессии Т-кадгерина. С помощью иммунофлуоресцентного окрашивания срезов меланомы антителами к маркѐру эндотелиальных клеток CD31 было показано, что количество сосудов среднего диаметра и капилляров достоверно снижено в первичных узлах, образованных Т-кадгерин- позитивными клетками по сравнению с контрольными узлами. Таким образом, экспрессия Т-кадгерина в клетках меланомы приводит к подавлению неоваскуляризации первичного узла у мышей, образованного этими клетками, что, возможно, и является причиной формирования многочисленных очагов некроза. В данной работе подавляющее влияние экспрессии Т-кадгерина в опухолевых клетках на неоангиогенез было также подтверждено на модели хориоаллантоисной мембраны куриного эмбриона. Полученные данные по подавлению неоваскуляризации первичных опухолевых узлов меланомы при экспрессии Т-кадгерина в опухолевых клетках согласуются с данными о подавлении роста сосудов в Матригель, полученными на модели подкожного введения Матригеля с фибробластами L929, экспрессирующими Т-кадгерин (см. раздел 3.4). В основе наблюдаемых эффектов лежит механизм гомофильном взаимодействии между молекулами Т-кадгерина, экспрессированными на поверхности эндотелиальных клеток, прорастающих в ткань сосудов, и молекулами Т-кадгерина на поверхности клеток стромы, в которую врастают сосуды. Это взаимодействие приводит к подавлению миграции эндотелиальных клеток и ингибированию неоангиогенеза. Возможно, обнаруженное нами подавление неоангиогенеза в первичном узле меланомы у мышей, а также на модели хориоаллантоисной мембраны куриного эмбриона при экспрессии Т-кадгерина в клетках 236 меланомы также обусловлено влиянием Т-кадгерина на миграцию эндотелиальных клеток. В литературе имеются данные, которые косвенным образом подтверждают высказанную нами гипотезу о механизме подавления ангиогенеза с участием Т-кадгерина. Так, неоваскуляризация гепатоцеллюлярной карциномы сопровождается повышением экспрессии Т-кадгерина в эндотелиальных клетках капилляров, прорастающих в опухоль, хотя в окружающей ткани опухоли или в ткани нормальной печени Т-кадгерин не обнаруживается [Adachi et al., 2006]. Увеличение экспрессии Т-кадгерина в эндотелиальных клетках гепатоцеллюлярной карциномы, по мнению авторов, коррелирует со степенью опухолевой прогрессии [Adachi et al., 2006]. Возможно, что Т-кадгерин при этом выполняет навигационную функцию и участвует в регуляции роста сосудов в опухоль, отсутствие экспрессии Т-кадгерина в окружающей опухолевой ткани не препятствует активному неоангиогенезу и коррелирует с опухолевой прогрессией. При анализе профиля экспрессии ангиогенных факторов в клетках меланомы были обнаружено, что в клетках, экспрессирующих Т-кадгерин, увеличивается экспрессия ингибиторов ангиогенеза: stabilin 1, heparanase, Foxfl1a, CXCL10/IP-10 [Angiolillo et al., 1995; Strieter et al., 1995; Bodnar et al., 2006] и angiopoietin 2 [Maisonpierre et al., 1997; Cao et al., 2007], снижается экспресия стимулятора ангиогенеза Tie 1 и полностью исчезает TGF. Экспрессия Т-кадгерина в клетках меланомы не только количественно изменяет мРНК ряда белков, участвующих в ангиогенезе, но активирует экспрессию генов procollagen type XVIII alpha 1, предшественника ингибитора ангиогенеза - эндостатина [O´Reilly et al., 1997; Xu et al., 2011] и chromogranin A, предшественника ингибитора ангиогенеза вазостатина-1 [Belloni et al., 2007; Veschini et al., 2011]. Эндостатин специфически ингибирует пролиферацию эндотелиальных клеток, вызывая накопление клеток в G0-G1 фазах клеточного цикла, а также снижает экспрессию в них CD105 и CD146 [Xu et al., 2011]. Вазостатин-1 ингибирует миграцию и пролиферацию эндотелиальных клеток [Belloni et al., 2007]. Кроме того, на модели аденокарциномы молочной железы было показано, что вазостатин-1 подавляет неоваскуляризацию опухоли [Veschini et al., 2011]. CXCL10 не оказывает влияние на пролиферацию эндотелиальных клеток, однако, подавляет их миграцию [Bodnar et al., 2006]. Действие ангиопоэтина 2 может быть как про-, так и антиангиогеннным в зависимости от наличия VEGF. Секреция ангиопоэтина 2 способствует нарушению адгезивного взаимодействия между эндотелиальными клетками и перицитами, что приводит к дестабилизации сосуда и облегчает миграцию эндотелиальных клеток. Однако при достаточном количестве VEGF-А дестабилизирующий эффект ангиопоэтина 2 усиливает ангиогенное действие VEGF [Holash et al., 1999]. В то же время системная гиперэкспрессия ангиопоэтина 2 в клетках карциномы толстого кишечника 237 приводит к массовой регрессии сосудов независимо от присутствия VEGF [Cao et al., 2007]. Таким образом, при экспрессии Т-кадгерина в клетках мышиной меланомы изменяется соотношение факторов, подавляющих и активирующих ангиогенез, в пользу первых, что, возможно, обуславливает снижение способности этих клеток стимулировать неоангиогенез в опухоли. По-видимому, в основе механизма подавления неоваскуляризации первичного узла меланомы сосудами среднего диаметра и капиллярами с участием Т-кадгерина лежит несколько процессов: стимуляция экспрессии некоторых ингибиторов ангиогенеза, подавления экспрессии ряда стимуляторов ангиогенеза, а также контактное ингибирование в результате гомофильного связывания и последующего «отталкивания». На in vivo модели гематогенно метастазирующей в лѐгкие меланомы в настоящей работе было обнаружено, что экспрессия Т-кадгерина в клетках меланомы приводит к активации клеток стромы и привлечению их в первичные узлы, образованные экспрессирующими Т-кадгерин клетками. Это было показано с использованием иммунофлуоресцентной окраски криосрезов первичных узлов антителами к CD90 – маркѐру активированной стромы в опухолях [True et al., 2010]. Эти данные были подтверждены экспериментами in vitro с использованием метода бесконтактного сокультивирования. Под влиянием кондиционированной среды роста от экспрессирующих Ткадгерин клеток мышиной меланомы происходит усиление миграционной активности мезенхимных стромальных клеток, выделенных из жировой ткани мыши. При анализе экспрессии генов хемокинов в клетках клонов мышиной меланомы с помощью коммерческих наборов PCR Array было обнаружено увеличение экспрессии в Т-кадгеринпозитивных клетках меланомы ряда хемокинов и их рецепторов: CXCR7, CXCL10, CCL5, CXCL11, CCL7 и некоторых других. Далее эти данные были частично подтверждены на уровне экспрессии белков. Кроме того, экспрессия Т-кадгерина в клетках меланомы не только количественно изменяет мРНК ряда хемокинов и рецепторов хемокинов, являющихся хемоаттрактантами для стромальных и иммунных клеток, но и активирует экспрессию генов CCL7, Gdf 5 и interleukin 8r, экспрессия которых не детектировалась в контрольном клоне. Хемокины – семейство белков, которые способствуют клеточной активации и миграции. Свои функции хемокины осуществляют при связывании с семидоменными G-белок-ассоциированными рецепторами [Rollins, 1997; Rossi and Zlotnik, 2000]. Основная роль хемокинов заключается в привлечении лейкоцитов, однако, известно, что стромальные клетки обладают схожими с иммунными клетками механизмами ответа, и способны мигрировать в место поражения или воспаления по градиенту хемоаттрактанта [Spaeth et al., 2008; Xu et al., 2010; Ito, 2011]. В частности, было показано, что CXCL10 и CXCL11 значительно увеличивают миграцию МСК человека [Kalwitz et al., 2010]. 238 Возможно, что хемокины, секретируемые Т-кадгерин экспрессирующими клетками меланомы, участвуют в активации и миграции МСК из окружающей ткани, что вносит существенный вклад в рост первичного узла и частично компенсирует подавление неоангиогенеза и некроз в опухоли. Хорошо известно, что МСК секретируют большое количество факторов роста и цитокинов, и их продукция увеличивается в условиях гипоксии [Rubina et al., 2005; Martin-Rendon, 2007]. Среди них такой фактор роста как HGF/SF, являющийся стимулятором синтеза ДНК, роста нормальных меланоцитов человека и клеток меланомы [Halaban et al., 1993]. c-Met является рецептором HGF и состоит из двух полипептидных цепей: α и β, расположенных в плазматической мембране. В литературе существуют данные, что c-Met может транслоцироваться в ядро в некоторых опухолевых клетках после активации своим лигандом HGF [Gomes et al., 2008; Matteucci et al., 2009]. Ранее было обнаружено, что повышенная экспрессия протоонкогена c-Met в клетках меланомы человека тесно коррелирует с опухолевой прогрессией меланомы из фазы радиального роста в фазу вертикально роста [Natali et al., 1993]. В настоящем исследовании количественный ПЦР анализ показал, что контрольные клетки меланомы экспрессируют c-Met на низком уровне и не экспрессируют HGF. Экспрессия Т-кадгерина приводит к 6-кратному увеличению содержания мРНК c-Met в меланоме, но при этом мРНК HGF не обнаруживается. По нашим данным, при экспрессии Т-кадгерина клетки меланомы секретируют большое количество хемокинов, привлекающих МСК в область первичного опухолевого узла. Ранее в нашей лаборатории было показано, что МСК-ЖТ мыши экспрессируют HGF, причем его экспрессия значительно увеличивается при гипоксии [Efimenko et al., 2009]. Мы предполагаем, что, приходя в опухоль, МСК оказываются в гипоксических условиях, что стимулирует в них высокую экспрессию HGF [Rubina et al., 2005]. HGF, связываясь со своим рецептором c-Met на клетках меланомы, запускает сигнальный каскад, приводящий, по данным литературы, к стимуляции роста меланомы и увеличению ее инвазивного потенциала [Matsumoto et al., 1991; Halaban et al., 1993; Kenessey e al., 2010]. Кроме того, было показано, что экспрессия с-Met не детектируется в доброкачественных меланоцитарных новообразованиях, тогда как его экспрессия наблюдается в клетках меланомы, особенно метастазирующей [Natali et al., 1993; Lee et al., 2011]. Ингибирование активности рецептора с-Met с помощью селективного ингибитора SU11274 приводит к снижению пролиферации и миграции клеток меланомы, а также подавлению роста первичного узла [Kenessey et al., 2010]. В связи с этими данными подавление активности с-Met рассматривается исследователями в качестве мишени для терапии меланомы [Abdel-Rahman et al., 2010; Kenessey et al., 2010]. Полученные данные о возможной активации сигнального пути HGF/c-Met и коррелирующего с ним увеличении 239 инвазивной и метастатической активности клеток меланомы свидетельствуют о возможных компенсаторных механизмах, возникающих в ответ на антиангиогенные эффекты Ткадгерина. Подобный феномен необходимо учитывать в дальнейшем при разработке антиангиогенных подходов для опухолевой терапии. Таким образом, мы предполагаем, что экспрессия Т-кадгерина в клетках мышиной меланомы приводит к секреции хемоаттрактантов этими клетками, что вызывает привлечение и миграцию МСК в первичный узел меланомы и его рост. Данные литературы свидетельствуют о том, что экспрессия c-Met коррелирует с метастазированием меланомы, и позволяют предполагать, что сигнальный путь HGF/c-Met является важным механизмом, обуславливающим опухолевую прогрессию. На животной модели гематогенно метастазирующей в лѐгкие меланомы было обнаружено, что высокий уровень экспрессии Т-кадгерина в клетках меланомы приводит к увеличению их метастатической активности in vivo. Увеличение протеолитических и инвазивных способностей у клеток меланомы с высоким уровнем экспрессии Т-кадгерина было подтверждено также и in vitro с использованием специально разработанного метода двух капель Матригеля. При анализе экспрессии генов молекул адгезии и внеклеточного матрикса в клетках клонов меланомы с помощью коммерческих наборов PCR Array в Ткадгерин содержащих клетках было обнаружено увеличение экспрессии ряда проонкогенных интегринов: integrin α5, integrin αV, integrin αE, integrin β3, а также MMP14. Далее эти результаты были частично подтверждены на уровне экспрессии белков. Наши данные коррелируют с данными литературы о том, что экспрессия Т-кадгерина может приводить к изменению адгезивных свойств кератиноцитов и инвазивного потенциала клеток плоскоклеточной карциномы за счѐт влияния на экспрессию интегрина β1 [Mukoyama et al., 2007]. Следует отметить, что экспрессия PECAM 1 (CD31 – маркер эндотелиальных клеток) была выявлена исключительно в клонах меланомы с экспрессией Т-кадгерина. Известно, что агрессивные формы меланомы могут сами формировать сеть сосудистых каналов (сосудистая мимикрия) [Hendrix et al., 2001; Hendrix et al., 2003] и обеспечивать опухоль питательными веществами и кислородом. Возможно, появление PECAM 1 в Т+ и Т++ клонах меланомы отражает компенсаторный механизм, при котором экспрессия Т-кадгерина, вызывая снижение экспрессии ангиогенных факторов и увеличение экспрессии ингибиторов ангиогенеза, одновременно повышает способность клеток меланомы к сосудистой мимикрии. Предположительно, обнаруженное повышение экспрессии интегринов и MMP14 в клетках мышиной меланомы, экспрессирующих Т-кадгерин, может быть еще одной причиной увеличения метастазирования этих клеток in vivo. В литературе есть данные о 240 том, что активация MMP14 в клетках меланомы приводит к увеличению инвазии этих клеток на коллагеновом матриксе [Hotary et al., 2003]. Кроме того, экспрессия MMP14 в клетках меланомы человека коррелирует со снижением продолжительности жизни пациентов [Kondratiev et al., 2008]. В экспериментальных условиях гиперэкспрессия β3 субъединицы интегрина в клетках меланомы на ранней стадии радиального роста индуцирует переход в фазу вертикального роста, характеризующуюся усиленным ростом, инвазией и метастазированием [Hsu et al., 1998]. Кроме того, в клеточных линиях метастазирующей меланомы наблюдается увеличение экспрессии интегрина αvβ3 в 5001000 раз по сравнению с меланоцитами. Такое увеличение экспрессии αvβ3 интегрина повышает способность клеток меланомы к инвазии и миграции через внеклеточный матрикс, состоящий из ламинина, фибронектина, витронектина [Danen et al., 1994; Filardo et al., 1995; Pinon and Wehrle-Haller, 2011]. Было показано, что α5 субъединица интегрина также вовлечена в процесс метастазирования меланомы B16F10: нейтрализация этой субъединицы блокирующими антителами приводит к значительному подавлению метастатического потенциала этих клеток и их адгезии на фибронектине in vitro [Qian et al., 2005]. Таким образом, в настоящей работе раскрыты механизмы влияния Т-кадгерина на опухолевую прогрессию на модели сингенной меланомы B16F10 у мышей BDF1, которая представляет собой модель агрессивного опухолевого роста. Суммируя полученные в работе результаты в виде схемы (Рисунок 3.60), можно сделать вывод о том, что Т-кадгерин является важным регулятором прогрессии меланомы. Экспрессия Т-кадгерина вызывает подавление неоангиогенеза в первичном узле, что, по всей видимости, обусловлено активацией экспрессии ингибиторов ангиогенеза и снижением экспрессии ряда активаторов ангиогенеза в клетках меланомы B16F10. Определенный вклад, по-видимому, в подавление неоангиогенеза в опухоли под влиянием экспрессии Т-кадгерина вносит описанный ранее механизм контактного ингибирования прорастания сосудов в ткани, клетки которой экспрессируют Т-кадгерин. В результате из-за недостаточного кровоснабжения в гипоксических условиях значительные участки опухоли подвергаются некрозу. Несмотря на выраженный антиангиогенный эффект Т-кадгерина, полученные результаты свидетельствуют о том, что Т-кадгерин оказывает стимулирующее влияние на опухолевую прогрессию, предположительно за счѐт повышения пролиферативного потенциала клеток меланомы и повышения способности их к инвазии. В основе этого, по-видимому, лежит увеличение экспрессии ряда хемокинов Т-кадгерин-позитивными клетками. При достижении достаточных размеров первичный узел становится очагом привлечения МСК, приходящих в меланому по градиенту секретируемых хемокинов. Приходящие в опухоль 241 МСК под влиянием гипоксии секретируют HGF, тем самым активируя сигнальный каскад с участием рецептора c-Met, экспрессия которого повышена в клетках меланомы, экспрессирующих Т-кадгерин. Запуск сигнального пути c-Met/HGF приводит к увеличению пролиферации, инвазии и метастазирования меланомы. Дополнительный вклад в увеличение инвазии и метастазирования вносит повышенный уровень экспрессии в клетках меланомы проонкогенных интегринов и ММР14, вызванное также экспрессией Ткадгерина. Рисунок 3.60. Возможная схема участия Т-кадгерина в прогрессии меланомы B16F10. Объяснения в тексте. 242 3.4 Выявление роли Т-кадгерина в процессах ангиогенеза Первой среди экспериментальных моделей, использованных для выявления возможной роли Т-кадгерина в ангиогенезе, была in vivo модель подкожной имплантации Матригеля, содержащего контрольные или экспрессирующие Т-кадгерин L939 фибробласты. 3.4.1 Введение клеток, экспрессирующих Т-кадгерин, локально подавляет ангиогенез в бляшках Матригеля Модель подкожной имплантации Матригеля предполагает подкожное введение мышам линии NUDE Матригеля, содержащего контрольные фибробласты, не экспрессирующие Т-кадгерин, или клетки, с высоким уровнем экспрессии Т-кадгерина. При температуре +40С Матригель представляет собой вязкую жидкость, при повышении температуры Матригель полимеризуется с формированием плотных бляшек. Матригель перемешивали с клетками на льду и вводили мышам подкожно. Бляшки Матригеля извлекали на 3-, 7-, 10- и 14-е сутки после инъекции и оценивали степень васкуляризации Матригеля сосудами, отрастающими от a. Subclavia. Во всех случаях имплантаты Матригеля с фибробластами L939, экспрессирующими Т-кадгерин, макроскопически отличались от контрольных Матригелей: были меньшего размера и имели более жѐлтый оттенок (Рисунок 3.61). На 14-й день бляшки с клетками, экспрессирующими Т-кадгерин, были достоверно меньше по весу (0,28 г ± 0,02 г в Матригелях с Т-кадгерин позитивными клетками; 0,45 г ± 0,02 г в контроле, p<0,01), что обусловлено ярко выраженной разницей в степени васкуляризации между Матригелями с контрольными и экспрессирующими Ткадгерин клетками. Содержание гемоглобина, нормированное на вес Матригеля, также было существенно ниже в бляшках с Т-кадгерин экспрессирующими клетками (0,74 ± 0,04 в Матригелях с Т-кадгерин позитивными клетками; 1,17 ± 0,08 в контроле, p<0,05). Разница в содержании гемоглобина, отражающая уровень кровотока в бляшке, была очевидной уже к 10-му дню, что по данным литературы соответствует времени, необходимому для прорастания сосудов и образования капиллярной сети в Матригеле [Passaniti et al., 1992; Stieger et al., 2006]. Анализ динамики прорастания сосудов (с 3 по 14-й день) показал, что содержание гемоглобина в бляшках Матригеля с клетками, гиперэкспрессирующими Т-кадгерин, было ниже во всех исследованных точках (Рисунок 3.62). Таким образом, результаты изучения динамики васкуляризации Матригеля показали, что введение Т-кадгерин экспрессирующих клеток в Матригель, подавляет прорастание в него сосудов. 243 Рисунок 3.61. Бляшки Матригеля. A. – Макроскопический вид бляшек Матригеля, содержащих L939 клетки: контрольные (Т-) или Т-кадгерин экспрессирующие клетки (Т+). Б. – Содержание гемоглобина в имплантатах Матригеля, оценѐнное методом Драбкина на 3-, 7- и 10-е сутки. Данные представлены как среднее ± стандартная ошибка среднего (N=14, p<0.05). Рисунок 3.62. Влияние экспрессии Т-кадгерина на содержание гемоглобина в имплантах. Уровень гемоглобина в имплантатах, содержащих клетки, гиперэкспрессирующие Т-кадгерин, во всех случаях был ниже, чем в контрольных имплантатах. Данные представлены как среднее ± стандартная ошибка среднего (N=14, p<0,05). 244 3.4.2 Экспрессия Т-кадгерина подавляет образование капилляров, но не влияет на формирование зрелых сосудов Для выяснения на какую стадию формирования сосудов (на образование и рост, или на созревание) влияет экспрессия Т-кадгерина в окружающей ткани, мы использовали метод иммунофлуоресцентного окрашивания. Криосрезы Матригеля с разных частей бляшки фиксировали и окрашивали антителами против маркера эндотелиальных клеток CD31. Далее на срезах Матригеля был проведен анализ плотности сосудов, их размер и качественный состав. В зависимости от размера все сосуды были условно разделены на три группы: крупные сосуды с диаметром более 40 мкм и/или просветом, сосуды среднего размера с диаметром от 20 до 40 мкм и капилляры с диаметром менее 20мкм. Разницы по плотности крупных сосудов в Матригеле с Т-кадгерин гиперэкспрессирующими клетками и в контроле не наблюдалось (1,25 ± 0,22 по сравнению с контролем 1,27 ± 0,33; p>0,05). В то время как в случае сосудов среднего диаметра (91,5 ± 7,2 по сравнению с контролем 207,6 ± 9,7; p<0,05) и капилляров (Рисунок 3.63 и Рисунок 3.64) плотность сосудов была достоверно ниже в Матригелях с Т-кадгерин гиперэкспрессирующими клетками по сравнению с контролем. В Матригелях с контрольными клетками большинство сосудов экспрессировали Т-кадгерин. Однако в Матригелях, где клетки экспрессировали Ткадгерин, количество Т-кадгерин позитивных сосудов было снижено по сравнению с контролем (Рисунок 3.64). Таким образом, экспрессия Т-кадгерина в Матригеле влияет на прорастание сосудов в бляшку, вызывая достоверное снижение плотности сосудов среднего размера и капилляров. Одной из возможных причин снижения плотности сосудов в бляшках Матригеля с Т-кадгерин экспрессирующими клетками может быть негативное влияние Т-кадгерина на стабилизацию новообразующихся сосудов. Для ответа на вопрос, влияет ли локальная экспрессия Т-кадгерина в Матригеле на созревание прорастающих в него сосудов, была проведена оценка плотности зрелых сосудов, окруженных перицитами/гладкомышечными клетками. Зрелые сосуды определяли методом двойного иммунофлуоресцентного окрашивания антителами против маркера эндотелиальных клеток CD31 в сочетании с антителами против -гладкомышечного актина или антителами против NG2-антигена – маркера перицитов [Gerhardt and Betsholtz, 2003; Bergers and Song, 2005]. 245 Рисунок 3.63. Васкуляризация Матригелей. A, Б. – Иммунофлуоресцентное окрашивание криосрезов Матригелей антителами против маркера эндотелиальных клеток CD31 (красная флуоресценция). Крупные сосуды отмечены большими стрелками, мелкие сосуды – двойными стрелками, капилляры – маленькими стрелками. Масштабный отрезок – 100 мкм. В, Г. – Двойное иммунофлуоресцентное окрашивание антителами против Т-кадгерина (зелѐная флуоресценция) и антителами против маркера эндотелиальных клеток CD31 (красная флуоресценция) на срезах Матригеля контрольных бляшек (В) и Матригелей с Т-кадгерин экспрессирующими клетками (Г). Жѐлтый цвет соответствует сосудам, в которых наблюдается экспрессия Ткадгерина, красный – сосудам, в которых нет Т-кадгерина. Ядра докрашены DAPI. Д. – Плотность капилляров определяли по количеству CD31-позитивных сосудов в поле зрения, нормированному на единицу площади по DAPI. Е. – Долю капилляров, экспрессирующих Т-кадгерин, определяли как количество двойных позитивных сосудов при окрашивании антителами против Т-кадгерина и CD31 по отношению к общему количеству CD31-позитивных сосудов. Данные представлены как среднее ± стандартная ошибка среднего (N=42, p<0,05). Масштаб – 20 мкм. 246 Разницы между плотностью зрелых сосудов (CD31/актин или CD31/NG2 позитивные сосуды) в контрольных Матригелях и в Матригелях с Т-кадгерин позитивными клетками обнаружено не было (Рисунок 3.64). Эти данные позволяют утверждать, что экспрессия Т-кадгерина в клетках стромы Матригеля in vivo, достоверно подавляет прорастание сосудов в бляшку, скорее всего на начальные этапы ангиогенеза, но не влияет на процесс их созревания. Для выявления возможных механизмов ингибирующего влияния Т-кадгерина на прорастание сосудов были использованы клеточные модели in vitro и эксплантные модели ex vivo. Влияние Т-кадгерина на адгезию, пролиферацию, апоптоз и миграцию эндотелиальных клеток in vitro изучали с использованием рекомбинантных очищенных доменов Т-кадгерина (ЕС1 - аминотерминальный домен и ЕС5 – C-концевой домен). Рекомбинантные белковые фрагменты активно используются в экспериментах in vitro для воспроизведения различных эффектов кадгеринов на адгезию, пролиферацию и сортинг клеток [Staton et al., 2004; Bergers and Song, 2005]. 247 Рисунок 3.64. Стабильные сосуды и капилляры в бляшках Матригелей. A, Б – Двойное иммунофлуоресцентное окрашивание антителами против -гладкомышечного актина (зеленая флуоресценция, a-SMA) и антителами против маркера эндотелиальных клеток CD31 (красная флуоресценция) на криосрезах Матригеля. Ядра докрашены DAPI. В, Г - Двойное иммунофлуоресцентное окрашивание криосрезов Матригеля антителами против маркера эндотелиальных клеток CD31 (красная флуоресценция) и маркера перицитов NG2 (зеленая флуоресценция). Д – Плотность распределения различных сосудов в Матригеле (CD31-позитивных, -SMA позитивных и -SMA/CD31 двойных позитивных сосудов), нормированная на единицу площади по DAPI. Е – Плотность двойных позитивных CD31/NG2 сосудов на срезах Матригеля, нормированная на единицу площади по DAPI. Данные представлены как среднее ± стандартная ошибка среднего (N=42, p<0,05). Масштаб – 20 мкм. 248 3.4.3 Рекомбинантный аминотерминальный ЕС1 домен Т-кадгерина подавляет ангиогенез in vitro Для подтверждения предположения о том, что Т-кадгерин является ингибитором ангиогенеза, были использованы модели формирования и роста капилляро-подобных трубочек в Матригеле сосудистыми клетками, эндогенно экспрессирующими Т-кадгерин. Рекомбинантные домены Т-кадгерина (аминотерминальный EC1 и С-концевой примембранный EC5) были выделены и очищены в нашей лаборатории ранее [Ivanov et al., 2004]. EC1 или EC5 домены (иммобилизация доменов Т-кадгерина в Матригеле) добавляли в Матригель и оценивали формирование капилляро-подобных (микрососудистых) структур. Известно, что EC1 домен кадгеринов имеет определяющее значение для межклеточного узнавания и формирования контактов, в то время как EC5 домен часто используется в качестве негативного контроля [Bergers and Song, 2005]. При иммобилизации ЕС1 домена на поверхности Матригеля регистрировали значительное снижение формирования трубочек по сравнению с контрольным ЕС5 доменом (Рисунок 3.65). Аналогичные результаты были получены при изучении влияния рекомбинантных доменов Т-кадгерина на формирование капилляро-подобных структур на модели сосудистого колечка крысы ex vivo. Было обнаружено, что иммобилизация ЕС1 домена в Матригеле снижала рост и ветвление капилляров в 2-3 раза по сравнению с контрольным ЕС5 доменом. Этот эффект ЕС1 домена Т-кадгерина наблюдался на протяжении всего эксперимента (с 3-го по 14-й день) и был дозозависимым (Рисунок 3.66). Таким образом, ЕС1 домен, имитирующий присутствие Т-кадгерина в Матригеле, значительно подавляет ангиогенез. Следует отметить, что ингибирующий эффект Т-кадгерина реализуется только если сосудистые клетки мигрируют и формируют капилляро-подобные трубочки на субстрате, содержащем активный ЕС1 домен. Культивирование HUVEC, экспрессирующих Ткадгерин, в присутствии растворимых ЕС1 и ЕС5 доменов Т-кадгерина в среде в концентрации 0,1 мг/мл не влияет на формирование капиллярных трубочек на Матригеле in vitro (Рисунок 3.65). Полученные результаты свидетельствуют о том, что растворенные фрагменты Ткадгерина не оказывают никакого влияния на рост и ветвление капилляров, в то время как иммобилизованный ЕС1 домен Т-кадгерина, по-видимому, имитирует гомофильное взаимодействие между клетками [Gerhardt and Betsholtz, 2003] и значительно подавляет ангиогенез на моделях in vitro и ex vivo. 249 Рисунок 3.65. Формирование капилляро-подобных структур эндотелиальными клетками в Матригеле, содержащем EC1 или EC5 домены Т-кадгерина. A – HUVEC в Матригеле с ЕС5 доменом. Б – HUVEC в Матригеле с ЕС1 доменом. В – Средняя длина капилляро-подобных структур. Масштаб – 50 мкм. Данные представлены как среднее ± стандартная ошибка среднего (N=12, p<0,05). 250 Рисунок 3.66. ЕС1 домен Т-кадгерина подавляет рост капилляро-подобных структур на модели сосудистого колечка ex vivo. A– Вид сосудистого колечка в Матригеле с отрастающими сосудами через 3 и 14 дней инкубации. Б – Средняя длина капиллярных трубочек, оцененная в 12 лунках в 4 независимых экспериментах. В – Дозовая зависимость подавления роста сосудов в присутствие доменов Т-кадгерина, выраженная в процентном отношении к контролю. Масштаб – 650 мкм. Данные представлены как среднее ± стандартная ошибка среднего; N=3, p<0, 001. 251 3.4.4 Т-кадгерин подавляет миграцию эндотелиальных клеток, но не влияет на их адгезию, пролиферацию и апоптоз С целью выявления механизмов ингибирующего влияния Т-кадгерина на ангиогенез in vivo было проведено исследование по изучение способности эндотелиальных клеток пролиферировать, адгезировать, мигрировать или вступать в апоптоз в присутствии Т-кадгерина в условиях in vitro. Влияние Т-кадгерина на пролиферацию HUVEC оценивали при со- культивировании с клетками HEK293. Для того, чтобы различить эндотелиальные клетки от HEK293, использовали HEK293, предварительно трансфицированные плазмидой для экспрессии маркерного белка GFP (флуоресценция в зелѐном канале) (Рисунок 3.67). Пролиферацию эндотелиальных клеток оценивали по количеству клеток, находящихся в клеточном цикле и способных вступать в митоз; такие клетки экспрессируют белок PCNA (Proliferating Cell Nuclear Antigen). При со-культивировании HUVEC с клетками HEK293, экспрессирующими Т-кадгерин или контрольными, достоверной разницы в количестве PCNA-позитивных эндотелиальных клеток не наблюдалось (Рисунок 3.68). Аналогичные результаты были получены при со-культивирования HUVEC с клетками L929. При изучении адгезии HUVEC на пластике, покрытом рекомбинантными ЕС1 или ЕС5 доменами или альбумином, достоверной разницы в количестве прикрепившихся эндотелиальных клеток зарегистрировано не было (данные не представлены). Для того чтобы оценить влияние Т-кадгерина на способность эндотелиальных клеток адгезировать на монослое клеток, клетки L929, гиперэкспрессирующие Т-кадгерин, или контрольные культивировали на пластиковых планшетах до формирования монослоя, а затем сверху высаживали HUVEC. Для визуализации эндотелиальных клеток HUVEC предварительно инкубировали в присутствии ЛНП, конъюгированных с флуорохромом (Ac-LDL-AlexaFluor 488). Эндотелиальные клетки специфически эндоцитировали ЛНП, что позволяло прижизненно их визуализировать в зеленом канале микроскопа. Адгезировавшие эндотелиальные клетки подсчитывали в 5 полях зрения, полученных с каждой лунки планшета. Количество адгезировавших эндотелиальных клеток не зависело от экспрессии Т-кадгерина в клетках L929, а изменялось пропорционально площади, занятой клетками L929 (Рисунок 3.69 и Рисунок 3.70). 252 Рисунок 3.67. Иммунофлуоресцентное окрашивание клеток антителами против PCNA при совместном культивировании HUVEC и НЕК293. А - Клетки НЕК293, гиперэкспрессирующие Т-кадгерин, экспрессируют GFP и флюоресцируют в зеленом канале. Для окрашивания против PCNA использовали вторые антитела, конъюгированные с флуорохромом AlexaFluor 594 (красная флуоресценция). Ядра клеток докрашены DAPI. Б - Клетки HUVEC. Для окрашивания против PCNA использовали вторые антитела, конъюгированные с флуорохромом AlexaFluor 594. Ядра клеток докрашены DAPI. Масштаб - 100 мкм. Рисунок 3.68. Количество пролиферирующих HUVEC, нормированное на единицу площади поля зрения, занятой HUVEC, не отличалось при со-культивировании HUVEC с клетками НЕК293 Т+ (Т-кадгерин гиперэкспрессирующими), НЕК293 Т- (контрольными) и в контроле (только HUVEC). N=3, p>0,05. 253 Рисунок 3.69. Влияние экспрессии Т-кадгерина в клетках L929 вызывать адгезию HUVEC. А, В, Д – фазово-контрастное изображение; Б, Г, Е - флуоресцентное изображение в зелѐном канале. Клетки L929 (контрольные Т-, или экспрессирующие Ткадгерин Т+) культивировали на чашках Петри до формирования 100% монослоя, затем на слой клеток L929 высаживали HUVEC, предварительно проинкубированные с мечеными Ac-LDL-AlexaFluor488. Б, Г – соответствующие изображения только эндотелиальных клеток, адгезировавших к монослою клеток L929.Д, Е – культура клеток HUVEС после инкубации с Ac-LDL-AlexaFluor 488. Масштаб- 100 мкм. 254 Рисунок 3.70. Экспрессия Т-кадгерина в клетках L9929 не влияет на адгезию к ним HUVEC. Данные представлены как среднее ± стандартная ошибка среднего,N=3, p>0,05. Рисунок 3.71. Влияние Т-кадгерина на миграцию эндотелиальных клеток. Миграция HUVEC в камере Бойдена через мембрану, покрытую рекомбинантными доменами Т-кадгерина. EC1 – аминотерминальный домен, EC5 – С-концевой домен (контроль), ФТС- фетальная телячья сыворотка. Данные представлены как среднее ± стандартная ошибка среднего; p<0,05. 255 В литературе описаны данные о 50%-м подавлении адгезии эндотелиальных клеток при высаживании их на клетки L929, гиперэкспрессирующие Т-кадгерин. Этот эффект наблюдался после фиксации клеток L929 параформальдегидом с последующим высаживанием на них HUVEC [Ivanov et al, 2004]. Мы изучали влияние живых нефиксированных клеток L929 с высоким уровнем экспрессии Т-кадгерина на адгезию HUVEC и не получили достоверных данных о подавлении адгезии HUVEC. Более того, культивирование НUVEC на пластике, покрытом ЕС1 доменом Ткадгерина (в концентрации 0,1 мг/мл), не вызывало активацию каспазы-3, ключевого участника апоптоза в HUVEC (данные не представлены). Полученные данные позволяют утверждать, что обнаруженные ингибиторные эффекты Т-кадгерина на процессы ангиогенеза не связаны с подавлением адгезии, пролиферации или активацией апоптоза в эндотелиальных клетках. Анализ миграции эндотелиальных клеток в камере Бойдена через мембрану, покрытую доменами Т-кадгерина показал, что ЕС1 домен, участвующий в межклеточном узнавании, подавляет миграцию HUVEC в 2 раза по сравнению с контрольным ЕС5 доменом (Рисунок 3.71), что позволяет предполагать, что Т-кадгерин опосредует свое негативное влияние на рост сосудов через подавление миграции эндотелиальных клеток. По-видимому, ЕС1 домен, обеспечивающий гомофильное узнавание, имитирует межклеточное взаимодействие между клетками, на поверхности которых экспрессируется Т-кадгерин, и подавляет процессы, связанные с миграцией эндотелиальных клеток: формирование капиллярных трубочек, их рост и ветвление. 256 3.4.5 Обсуждение результатов Во взрослом организме в норме сосудистая система находится в состоянии равновесия, процессы роста или регрессии сосудов четко регулируются. Избыточный или недостаточный ангиогенез, как правило, сопутствует опухолевому росту, патологическому ремоделированию сосудов и различным гиперплазиям тканей. Известно, что Т-кадгерин эндогенно экспрессируется в эндотелиальных, гладкомышечных клетках и перицитах сосудов [Ivanov et al., 2001], а его экспрессия в сосудистой стенке повышается и коррелирует с патологическим ангиогенезом при атеросклерозе и рестенозе [Ivanov et al., 2001; Kudrjashova et al., 2002]. Данные, полученные в настоящем исследовании, свидетельствуют о том, что Ткадгерин ингибирует ангиогенез на моделях in vivo и in vitro на уровне подавления миграции эндотелиальных клеток и формирования мелких сосудов и капилляров. Повидимому, в основе такого подавления миграции лежит гомофильное взаимодействие между молекулами Т-кадгерина [Ivanov et al., 2004]. Подобный механизм гомофильного взаимодействия и контактного ингибирования был уже описан для Т-кадгерина в нервной системе ранее. В эмбриогенезе Т-кадгерин выполняет функцию навигационного рецептора и регулирует миграцию клеток нервного гребня и траекторию роста аксонов к своим мишеням [Ranscht and Dours-Zimmermann, 1991; Fredette and Ranscht, 1994; Fredette et al., 1996]. В литературе имеются данные о том, что одни и те же молекулы могут осуществлять функцию навигационных рецепторов в нервной и сосудистой системе, более того существует данные о взаимной регуляции прорастания нервов и сосудов [Carmeliet, 2003]. Известно, по крайней мере, четыре семейства таких молекул, выполняющих навигационную функцию и опосредующих процессы «отталкивания» или адгезии клеток: нетрины и их рецепторы DCC/неогенины и Unc5, слит лиганды и их Робо рецепторы, семафорины и их рецепторы плексины и нейропилины, эфрины и их рецепторы [Weinstein, 2005; Eichmann et al., 2005]. Эфрины и их рецепторы – пара навигационных молекул, осуществляющих регуляцию роста нервов и сосудов в эмбриогенезе и при опухолевом ангиогенезе [Poliakov et al., 2004; DavyandSoriano, 2005]. Эфрины класса А являются, как и Т-кадгерин, GPI-заякоренными молекулами [Poliakov et al., 2004; Davy and Soriano, 2005]. Результатом взаимодействия эфринов со своими рецепторами является либо адгезия клеток, либо их «отталкивание». Механизм передачи сигнала внутрь клетки через GPI-заякоренные эфрины до конца не ясен, но, во всей видимости, происходит кластеризация молекул эфринов в липидных плотах и/или участвуют адапторные белки. Ещѐ меньше известно о 257 механизмах внутриклеточной сигнализации и белках-партнерах Т-кадгерина, но, возможно, они аналогичны эфринам А класса, поскольку сходное строение позволяет предполагать аналогичные механизмы активации внутриклеточной сигнализации. Данные, полученные в настоящей работе, позволяют утверждать, что Т-кадгерин является еще одной молекулой с навигационной функцией, регулирующей направление роста не только нервов, но и сосудов через окружающие ткани к своим мишеням (Рисунок 3.72). Данные in vitro по миграции эндотелиальных клеток в камере Бойдена, при формировании капилляров эндотелиальными клетками в Матригеле или на модели сосудистого колечка с использованием рекомбинантного ЕС1 домена, ответственного за гомофильное узнавание между молекулами Т-кадгерина, позволяют говорить о том, что Ткадгерин участвует в навигации прорастающих сосудов по такому же принципу. In vivo экспрессия Т-кадгерина клетками L929 в Матригеле создает микроокружение с высоким содержанием Т-кадгерина для прорастающих в бляшку сосудов, что тормозит начальные этапы ангиогенеза, но не влияет на созревание сосудов. В нашей работе влияния Т-кадгерина на пролиферацию, адгезию и апоптоз эндотелиальных клеток in vitro выявлено не было. В то же время, в литературе имеются данные о том, что гиперэкспрессия Т-кадгерина с использованием вирусной трансдукции может усиливать пролиферацию эндотелиальных клеток [Ivanov et al., 2004; Philippova et al., 2006]. Кроме того, вирусная или плазмидная гиперэкспрессия Т-кадгерина защищает эндотелиальные клетки от апоптоза и повышает их устойчивость к оксидативному стрессу in vitro, а также повышает способность эндотелиальных клеток и фибробластов к спонтанной миграции [Philippova et al., 2003; Joshi et al., 2005]. Эти данные, полученные на культивируемых клетках, коррелируют с данными in vivo по экспрессии Т-кадгерина в сосудах некоторых опухолей. Так, при формировании лѐгочных метастаз карциномы Левиса (Lewis carcinoma) было отмечено повышение уровня экспрессии Т-кадгерина в сосудах, прорастающих в опухоль. Однако следует подчеркнуть, что при этом в окружающей ткани экспрессия Т-кадгерина отсутствует [Wyder et al., 2000]. 258 Рисунок 3.72. Схематическое изображение роста сосудов, избегающих на своем пути тканей, в которых клетки экспрессируют Т-кадгерин. Аналогичным образом экспрессия Т-кадгерина в эндотелиальных клетках капилляров, васкуляризирующих гепатоклеточную карциному, повышается по сравнению с сосудами нормальной печени, в то время как в окружающей ткани опухоли Т-кадгерин не обнаруживается [Adachi et al., 2006]. Все эти факты, казалось бы, свидетельствуют о том, что увеличение экспрессии Т-кадгерина в эндотелиальных клетках in vitro или в сосудах, прорастающих в опухоль, коррелирует с повышенным уровнем ангиогенеза. Однако в описанных работах не учитывалась роль стромы и еѐ возможного влияния на процессы ангиогенеза. В отличие от других авторов, в данной работе была использована модель подкожной имплантации Матригеля, в которой клетки стромы создают микроокружение с высоким содержанием Т-кадгерина для прорастающих сосудов. В этих условиях мигрирующие эндотелиальные клетки, эндогенно экспрессирующие Т-кадгерин, контактируют со стромальными клетками, экспрессирующими Т-кадгерин, что приводит к контактному торможению и подавлению прорастания кровеносных сосудов в Матригель. С нашими данными in vivo хорошо согласуются результаты, полученные на модели 259 сосудистого колечка ex vivo, где наблюдается дозовая зависимость подавления роста капилляров в присутствии ЕС1 домена в Матригеле, что говорит о специфичности наблюдаемых эффектов Т-кадгерина. Предполагается, что гомофильное взаимодействие между молекулами Т-кадгерина на поверхности контактирующих клеток подавляет инвазию эндотелиальных клеток в строму Матригеля. Эта гипотеза получила дальнейшее подтверждение при изучении качественного и количественного состава прорастающих сосудов в Матригель. Присутствие Т-кадгерина в Матригеле не влияло на количество крупных сосудов, в то время как плотность мелких сосудов и капилляров была значительно ниже в Матригеле с Т-кадгерин позитивными клетками, что позволяет предполагать, что присутствие Т-кадгерина на клетках стромы влияет на начальные стадии ангиогенеза. Анализ качественного состава сосудов в бляшках показал, что большое количество сосудов, прорастающих в Матригель с Т-кадгерин позитивными стромальными клетками, сами не экспрессируют Т-кадгерин. Таким образом, экспрессия Т-кадгерина в строме может оказывать влияние на качественный состав сосудов, инфильтрирующих ткань, либо за счет регуляции направления роста сосудов, либо за счет регуляции экспрессии в прорастающих сосудах Т-кадгерина. Гипотеза о навигационной роли Т-кадгерина, гомофильном взаимодействии и контактном ингибировании роста сосудов с участием Ткадгерина объясняет, почему подавление экспрессии Т-кадгерина в строме или опухолевых клетках, таких опухолей как базальноклеточный рак, немелкоклеточная карцинома легкого, рак яичников, рак поджелудочной железы и некоторые другие коррелирует с высокой васкуляризацией опухолей и негативным прогнозом и прогрессией заболевания [Kawakami et al., 1999; Takeuchi et al., 2002; Hibi et al., 2004; Sakai et al., 2004; Kim et al., 2005; Mukoyama et al., 2005], а Т-кадгерину приписывается роль онкосупрессора [Wyder et al., 2000; Takeuchi et al., 2000; Takeuchi and Ohtsuki, 2001]. 260 3.5 Т-кадгерин и проницаемость сосудов. Сигнальные эффекты Т-кадгерина Эндотелий является основным регулятором сосудистого гомеостаза, поскольку выполняет барьерную функцию и способен отвечать на различные физические и химические сигналы путѐм выработки большого спектра факторов, регулирующих тонусов сосудов, клеточную адгезию, миграцию гладкомышечных клеток и их пролиферацию, воспаление в сосудистой стенке и тромбообразование [Deanfield et al., 2007]. Эндотелиальная дисфункция проявляется во многих патологических состояниях, таких как атеросклероз, гипертония и сахарный диабет. На физиологическом уровне эндотелиальная дисфункция характеризуется изменением фенотипа эндотелиальных клеток, нарушением межклеточных взаимодействий и, как следствие, увеличением проницаемости эндотелия [Frey et al., 2009]. VE-кадгерин играет ключевую роль в формировании адгезивных контактов в эндотелии, что обеспечивает поддержание целостности эндотелиального монослоя и выполнение им барьерной функции [Vestweber, 2008]. VE-кадгерин участвует в межклеточной адгезии путем гомофильного Ca2+зависимого взаимодействия между внеклеточными доменами VE-кадгеринов на соседних клетках. Как и «классические» кадгерины, VE-кадгерин взаимодействует через цитоплазматическую часть с p120ctn, бета-катенином, альфа-катенином, плакоглобином с актиновым цитоскелетом и промежуточными филаментами, что обеспечивает прочную межклеточную адгезию [Dejana, 1996; Dejana et al., 2008; Vestweber, 2008]. Большинство состояний, при которых увеличивается проницаемость эндотелия, связано с разборкой кадгерин-цитоскелетного комплекса или с нарушением его нормальной организации, что приводит к увеличению межклеточных промежутков и сокращению клеток [Dejana et al., 2008]. В нескольких публикациях была показана корреляция между изменением стабильности VE-кадгериновых контактов и уровнем фосфорилирования белков кадгеринкатенинового комплекса [Dejana et al., 2008; Vestweber, 2008]. Такие увеличивающие проницаемость эндотелия факторы как гистамин [Andriopoulou et al., 1999], фактор некроза опухолей TNFα [Angelini et al., 2006], фактор активации тромбоцитов (platelet-activating factor, PAF) [Hudry-Clergeon et al., 2005], VEGF [Esser et al.,1998] или адгезия лейкоцитов к эндотелиальным клеткам за счет взаимодействия с адгезивные молекулы ICAM1 [Allingham et al., 2007; Turowski et al., 2008; Albelda et al., 1991] обычно вызывают фосфорилирование VE-кадгерина или его адаптерных белков, таких как p120ctn, бета-катенин, альфа-катенин и плакоглобин. Механизмы фосфорилирования VE-кадгерина и связанных с ним белков остаются до конца не выясненными. Известно, что тирозиновые киназы семейства Src, богатая 261 пролином тирозиновая киназа PYR2, агенты, вызывающие активацию сигнальных путей с участием Rho ГТФаз или влияющие на полимеризацию актина, а также увеличивающие концентрацию внутриклеточного кальция, вызывают фосфорилирование белков кадгеринового комплекса и увеличение проницаемости [Allingham et al., 2007, Weis et al., 2005, Turowski et al., 2008]. Ряд предварительных результатов показал, что Т-кадгерин, экспрессируемый эндотелиальными клетками, также может влиять на проницаемость эндотелия [Andreeva et al., 2009], но точные механизмы Т-кадгерин-опосредованной регуляции проницаемости эндотелия остаются невыясненными. 3.5.1 Распределение экспрессии Т-кадгерина во фракциях эндотелиальных клеток после лентивирусной трансдукции Для изучения роли Т-кадгерина в регуляции барьерной функции эндотелия в настоящей работе была создана лентивирусная конструкция, обеспечивающая эффективное подавление нативной экспрессии Т-кадгерина (si-Т-cad), а также лентивирусная конструкция для гиперэкспрессии Т-кадгерина (Т-cad) в эндотелиальных клетках HUVEC. В качестве контроля (control) использовали HUVEC, трансдуцированные лентивирусом, содержащем кДНК антисмысловой последовательности люциферазы. Содержание Ткадгерина в общем лизате HUVEC, а также в мембранной, цитоплазматической и ядерной фракциях оценивали после фракционирования лизатов с использованием коммерческого набора Qproteome™ Cell Compartment kit методом вестерн блоттинга с последующим денситометрическим анализом. Чистоту субклеточных фракций, полученных с помощью набора Qproteome™ Cell Compartment kit, а также содержание Т-кадгерина в субклеточных фракциях подтверждали методом вестерн блоттинга (Рисунок 3.73). Через 72 ч после лентивирусной трансдукции методом вестерн блоттинга оценивали содержание Т-кадгерина в общем лизате, мембранной, цитозольной и ядерной фракциях. Чистоту фракций подтверждали с использованием антител к маркерам различных внутриклеточных компартментов: анти-EEA1 (для выявления мембранной фракции и фракции белков ранних эндосом) и анти-LAMP1 (маркер лизосом для выявления цитоплазматических белков). Мембранная и ядерная фракция не содержали LAMP1, а в цитоплазматической он присутствовал, в то время как в мембранной и цитоплазматической присутствовал EEA1, отсутствовавший в ядерной фракции. 262 Рисунок 3.73. Анализ чистоты субклеточных фракций после фракционирования. Чистоту и состав клеточных фракций, полученных после фракционирования лизатов HUVEC (T-cad, si-T-ca и control клеток) оценивали методом вестерн блоттинга. (A) Анализ мембранной (мембр. фракц), цитоплазматической (цитоп. фракц) и ядерной (ядерн. фракц) фракций антителами против ранних эндосомальных белков EEA1. Мембранная и цитозольная фракции содержат белки ранних эндосом, в то время как ядерная фракция нет. (Б) Анализ мембранной (мембр. фракц), цитоплазматической (цитоп. фракц) и ядерной (ядерн. фракц) фракций антителами против лизосомальных маркеров LAMP1. Лизосомальные белки содержит только цитоплазматическая фракция. Показан воспроизводимый результат трѐх независимых экспериментов. 263 После трансдукции контрольным лентивирусом (control) в тотальном лизате Ткадгерин обнаруживается в двух формах: в виде зрелого белка (молекулярный вес 105 kD) и в форме предшественника (молекулярный вес 130 kD) (Рисунок 3.74 А, Б). В мембранной фракции Т-кадгерин присутствует преимущественно в зрелой форме, но также обнаруживается предшественник Т-кадгерина; в ядерной и цитоплазматической фракциях содержатся обе формы Т-кадгерина: зрелая форма и форма предшественника (Рисунок 3.74 В). Трансдукция HUVEC лентивирусной конструкцией T-cad приводит к значительному увеличению экспрессии Т-кадгерина (Т-cad) в общих лизатах, причѐм повышается содержащие обеих форм Т-кадгерина: зрелой формы и предшественника. Увеличение содержания Т-кадгерина (также обеих форм) обнаруживалось как в мембранной, так и в цитоплазматической и ядерной фракциях (Рисунок 3.74 В). Подавление нативной экспрессии Т-кадгерина (si-T-cad) в HUVEC приводит к полному исчезновению предшественника Т-кадгерина, как видно из анализа тотальных лизатов; однако, сохраняются следовые количества зрелой формы белка Т-кадгерина. Отсутствие незрелой формы и наличие зрелой формы Т-кадгерина в общих лизатах si-T-cad клеток показывает, что после подавления экспрессии мРНК Т-кадгерина синтез новых молекул Т-кадгерина практически останавливается. После фракционирования обе формы Т-кадгерина практически исчезают из цитоплазматической и ядерной фракций, в то время как зрелая форма ещѐ обнаруживается в мембранной фракции (Рисунок 3.74 В). Денситометрический анализ полученных изображений с помощью программы ImageJ подтвердил результаты Вестерн блоттинга (Рисунок 3.74 Б, Г, Д, Е). Таким образом, лентивирусная трансдукция HUVEC T-cad вызывает увеличение экспрессии Т-кадгерина обеих форм и во всех субклеточных фракциях: на мембране, в цитоплазме и ядре. При подавлении нативной экспрессии Т-кадгерина si-T-cad в HUVEC, в первую очередь, исчезает форма предшественника Т-кадгерина, что отражает процесс подавления синтеза мРНК с помощью siРНК. 264 Рисунок 3.74. Экспрессия Т-кадгерина после лентивирусной трансдукции. Эндотелиальные клетки HUVEC 2 пассажа трансдуцировали лентивирусной конструкцией для повышения экспрессии Т-кадгерина (Т-кад), подавления (si-Т-кад) или контрольной конструкцией (control). Экспрессию Т-кадгерина анализировали методом вестерн блоттинга с помощью антител против Т-кадгерина (А, В). Результаты подтверждали денситометрическим анализом (Б, Г, Д, Е), данные нормировали по содержанию GAPDH. (А) - Экспрессия Т-кадгерина в общих лизатах, (В) – экспрессия Ткадгерина в субклеточных фракциях. Т-cad – HUVEC, трансдуцированные лентивирусом для гиперэкспрессии Т-кадгерина, si-Т-cad - HUVEC, трансдуцированные лентивирусом для подавления экспрессии Т-кадгерина, control - HUVEC, трансдуцированные контрольным лентивирусом. 130 kD- предшественник Т-кадгерина, 105 kD – зрелый Ткадгерин. Представлены воспроизводимые результаты трех независимых экспериментов, N=3, *р<0,05. 265 3.5.2 Экспрессия Т-кадгерина регулирует проницаемость монослоя эндотелия Для оценки влияния экспрессии Т-кадгерина на барьерную функцию эндотелия измеряли проницаемость эндотелиального монослоя для молекул FITC-декстрана. В эксперименте использовали трансфицированные нетрансфицированные контрольным лентивирусом HUVEC (control), и (mock), HUVEC, лентивирусом для гиперэкспрессии Т-кадгерина (T-cad), а также лентивирусом для подавления нативной экспрессии Т-кадгерина (si-T-cad). Клетки высаживали в верхнюю мембрану Трансвелл в равном количестве и культивировали в полноценной среде роста до образования плотного монослоя не менее 72 ч. Перед началом каждого эксперимента оценивали плотность и целостность монослоя с помощью светового микроскопа. Раствор FITC-декстрана добавляли в верхнюю камеру трансвелл. Аликвоты сред из нижних камер Трансвелл отбирали и анализировали каждые 60 мин (Рисунок 3.75). При изучении проницаемости эндотелия было обнаружено, что гиперэкспрессия Ткадгерина в HUVEC значительно увеличивает проницаемость эндотелиального монослоя по сравнению с контролем (control и mock), при этом через 6 часов разница в проницаемости между control и T-cad клетками составила 1,42 раза (р<0,001). В монослое клеток при подавлении экспрессии Т-кадгерина (si-Т-cad) проницаемость для молекул FITC-декстрана уменьшилась в 1,47 раз по сравнению с контролем (control) (р<0,001). Таким образом, данные результаты позволяют утверждать, что экспрессия Т-кадгерина влияет на барьерную функцию эндотелия и проницаемость для макромолекул in vitro: повышенная экспрессия Т-кадгерина увеличивает проницаемость эндотелия, в то время как подавление экспрессии наоборот, уменьшает. 266 Рисунок 3.75. Влияние экспрессии Т-кадгерина на проницаемость эндотелиального монослоя. FITC-декстран в конечной концентрации 10 мкг/мл вносили в верхнюю камеру трансвелл. Образцы аликвот из нижней камеры отбирали каждые 60 мин и измеряли флуоресценцию FITC при длине волны 525 нм. Гиперэкспрессия Ткадгерина приводит к повышению проницаемости эндотелиального монослоя. В монослое эндотелиальных клеток, гиперэкспрессирующих Т-кадгерин (T-cad), проницаемость для FITC-декстрана через 6 часов становится в 1,42 раза больше, чем в контроле (control). Проницаемость эндотелия подавленной экспрессией Т-кадгерина (siT-cad) напротив, снижена в 1,47 раза по сравнению с контролем. Mock – нетрансфицированные HUVEC. Данные представлены как среднее ± SEM (* р <0,001, N = 24). 267 3.5.3 Гиперэкспрессия Т-кадгерина вызывает образование межклеточных щелей и исчезновение VE-кадгерина из межклеточных контактов в эндотелиальных клетках Известно, что не только экспрессия VE-кадгерина, но его правильная локализация на мембране и целостность VE-кадгериновых адгезивных контактов необходимы для поддержания барьерной функции эндотелия [Vestweber, 2008; Komarova and Malik, 2010]. Мы предположили, что Т-кадгерин может регулировать проницаемость эндотелия, изменяя экспрессию и/или локализацию VE-кадгерина в клетках. Экспрессию VE-кадгерина в контрольных клетках (control), гиперэкспрессирующих Т-кадгерин (T-cad), а также в клетках после подавления экспрессии Т-кадгерина (si-Т-cad) оценивали вестерн блоттингом и последующим денситометрическим анализом. При этом оценку экспрессии VE-кадгерина проводили как в общем лизате клеток, так и в субклеточных фракциях (Рисунок 3.76 А). Локализацию VE-кадгерина выявляли методом иммунофлуоресцентной окраски клеток антителами к VE-кадгерину с последующим анализом на конфокальном микроскопе (Рисунок 3.77). Было обнаружено, что через 72 ч после трансдукции HUVEC содержание VE-кадгерина в общем лизате в Т-cad клетках достоверно снижено по сравнению с контролем (control) и si-Т-cad (Рисунок 3.76 А, верхняя панель и Рисунок 3.76 Б). Для того чтобы оценить содержание VE-кадгерина в цитоплазме, клетки перед лизированием обрабатывали трипсином согласно протоколу, описанному в разделе Материалы и методы. При трипсинизации клеток происходит разрушение и/или удаление мембранно-связанных поверхностных белков, в частности VE-кадгерина, в то время как внутриклеточный пул VE-кадгерина остаѐтся интактным [Xiao et al., 2003a]. Кроме того, содержание VEкадгерина было использованием проанализировано коммерческого в набора клеточных фракциях Qproteome™ Cell после разделения Compartment kit. с Было обнаружено, что при гиперэкспрессии Т-кадгерина происходит небольшое снижение содержания VE-кадгерина на мембране (Рисунок 3.76 Г), и увеличение его содержания в цитоплазме (Рисунок 3.76 А, В, Д) по сравнению с содержанием VE-кадгерина в контрольных клетках (control) и в клетках с подавленной экспрессией Т-кадгерина (si-Tcad) (Рисунок 3.76). Напротив, в клетках с подавленной экспрессией Т-кадгерина (si-Т-cad) содержание VE-кадгерина увеличивается в мембранной фракции (мембр. фракц на Рисунок 3.76 А), и значительно снижается в цитоплазматической фракции. Денситометрический анализ полученных изображений подтвердил данные вестерн блоттинга (Рисунок 3.76 Б-Д). Полученные результаты свидетельствуют о том, что повышенная экспрессия Т-кадгерина вызывает уменьшение содержания VE-кадгерина в мембранах клеток, в то время как подавление экспрессии Т-кадгерина, наоборот, приводит к увеличению локализации VEкадгерина на мембране клеток. 268 Рисунок 3.76. Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает накопление VE-кадгерина в цитоплазме. (А) Экспрессию VE-кадгерина в эндотелиальных клетках (Т-cad, si-Т-cad или контрольных control) определяли методом вестерн блоттинга с последующим денситометрическим анализом (Б-Д). Чтобы различить поверхностный и внутриклеточный пул VE-кадгерина, мембранный VEкадгерин протеолитически удаляли обработкой клеток трипсином. Гиперэкспрессия Ткадгерина (T-cad) вызывает увеличение содержания внутриклеточного VE-кадгерина (цитопл. фракц) по сравнению с контролем (control). В клетках с подавленной экспрессией Т-кадгерина (в si-Т-кад) содержание внутриклеточного VE-кадгерина снижено по сравнению с контролем. Субклеточное фракционирование подтвердило уменьшение содержания VE-кадгерина в мембране (мембр. фракц) и увеличение его в цитоплазме Т-cad клеток. В si-Т-кад клетках количество VE-кадгерина значительно увеличивалось в мембранной фракции и практически не определялось в цитозольной. Данные нормированы по уровню GAPDH. Представлен воспроизводимый результат гистограммы денситометрического анализа трѐх независимых экспериментов (N=3, * р <0,05). Результаты подтверждали денситометрическим анализом (Б, В Г, Д), данные нормировали по содержанию GAPDH. 269 Для визуализации локализации VE-кадгерина в клетках была проанализирована его экспрессия в зоне межклеточных контактов с помощью двойной иммунофлуоресцентной окраски антителами против Т-кадгерина и VE-кадгерина. Окрашивание эндотелиальных клеток с различной экспрессией Т-кадгерина проводили без пермеабилизации для сохранения целостности цитоплазматических мембран. Анализ полученных изображений осуществляли на конфокальном микроскопе при одинаковых настройках интенсивности лазера и времени экспозиции. При этом было обнаружено, что при гиперэкспрессии Ткадгерина в HUVEC происходит нарушение VE-кадгериновых адгезивных контактов: в клетках Т-cad окрашивание VE-кадгерина в мембранах неравномерное и носит сетчатый характер. Исчезновение VE-кадгерина из зон межклеточных контактов сочетается с формированием щелей или промежутков между клетками (стрелки на Рисунке 3.77), в то время как в случае si-Т-cad клеток щелей в зоне адгезивных VE-кадгериновых контактов между клетками не обнаружено. Напротив, подавление нативной экспрессии Т-кадгерина приводит к утолщению зоны VE-кадгериновых контактов, что согласуется с предыдущими данными Вестерн блоттинга и денситометрии (Рисунок 3.76 Б). Помимо анализа состояния VE-кадгериновых адгезивных контактов в качестве контроля была проведена оценка влияния экспрессии Т-кадгерина на экспрессию и локализацию N-кадгерина и белков, участвующих в формировании плотных контактов в эндотелиальных клетках. Согласно данным Вестерн блоттинга экспрессия Т-кадгерина не оказывает влияния на экспрессию и/или локализацию N-кадгерина в различных клеточных компартментах, а также на уровень экспрессии белков, участвующих в формировании плотных контактов, таких как окклюдина, клаудина-5 и ZO-1 (Рисунок 3.78 A, Б). 270 Рисунок 3.77. Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает разборку адгезивных контактов. Двойное иммунофлуоресцентное окрашивание эндотелиальных клеток осуществляли антителами против Т-кадгерина (зеленая флуоресценция) и VE-кадгерина (красная флуоресценция). Изображения были получены с помощью конфокального микроскопа при одинаковых настройках интенсивности лазера и времени экспозиции. Ядра клеток окрашены DAPI (синяя флуоресценция). Стрелками указаны щели в зоне VE-кадгериновых адгезивных контактов. Окрашивание VEкадгерина в зоне межклеточных контактах в Т-сad клетках значительно слабее, чем в контрольных и si-T-cad клетках. Масштаб- 20 мкм. 271 Рисунок 3.78. Т-кадгерин не влияет на экспрессию N-кадгерина, окклюдина, клаудина-5 и ZO-1 в эндотелиальных клетках. (A) Экспрессию и мембранную локализацию N-кадгерина анализировали методом Вестерн блоттинга с использованием антител против N-кадгерина. Для сравнения содержания N-кадгерина на поверхности мембраны и в цитоплазме оценивали количество N-кадгерина в общих лизатах и в лизатах после трипсинизации. (B) Экспрессия окклюдина, клаудина-5 и ZO-1 анализировалась Вестерн блоттингом. Данные нормированы по уровню GAPDH. Control – эндотелиальные клетки, трансдуцированные контрольным лентивирусом, T-cad эндотелиальные клетки, трансдуцированные лентивирусом для гиперэкспрессии Ткадгерина, si-T-cad - эндотелиальные клетки, трансдуцированные лентивирусом для подавления экспрессии Т-кадгерина. Представлен воспроизводимый результат трѐх независимых экспериментов. 272 Таким образом, полученные результаты позволяют сделать предположение, что увеличение экспрессии Т-кадгерина в эндотелиальных клетках приводит к селективному снижению общего содержания VE-кадгерина в клетках, при этом большая часть VEкадгерина обнаруживается не на мембране клеток, а в цитоплазме, что сопровождается нарушением межклеточных контактов и формированием промежутков между эндотелиальными клетками и, как следствие, повышением проницаемости эндотелиального монослоя. Подавление экспрессии Т-кадгерина вызывает противоположные эффекты – увеличение содержания VE-кадгерина на мембране и уменьшение его в цитоплазматической фракции, формирование более прочных адгезивных контактов по сравнению с контролем и с Т-кадгерин-экспрессирующими клетками. Мы предположили, что экспрессия Т-кадгерина повышает проницаемость эндотелия, вызывая интернализацию VE-кадгерина путѐм эндоцитоза с его последующей деградацией в цитоплазме. 3.5.4 Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает клатринзависимую интернализацию VE-кадгерина и деградацию в лизосомах По данным ряда исследований, основным путѐм интернализации VE-кадгерина является клатрин-зависимый путь [Xiao et al., 2003a; Xiao et al., 2003b; Gavard and Gutkind, 2006]. Для того чтобы проверить гипотезу о том, что повышенная экспрессия Т-кадгерина приводит к клатрин-зависимому эндоцитозу VE-кадгерина, был использован метод двойного иммунофлуоресцентного окрашивания для выявления возможной ко-локализации VE-кадгерина и клатрина. Изображения высокого разрешения были получены с помощью конфокального микроскопа при одинаковых настройках интенсивности лазера и времени экспозиции. Как показано на Рисунке 3.79, в Т-кадгерин гиперэкспрессирующих клетках VEкадгерин ко-локализуется с клатрином на поверхности эндотелиальных клеток (клатринпозитивные участки окрашивания указаны белыми стрелками на Рисунке 3.79 и вынесены в отдельную панель снизу), а также в цитоплазме; в то время как в контрольных клетках и клетках после подавления экспрессии Т-кадгерина ко-локализация VE-кадгерина и клатрина менее выражена. Данные иммунофлуоресцентного окрашивания подтверждены денситометрическим анализом (Рисунке 3.79 Б). 273 Рисунок 3.79. Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает клатрин-опосредованную интернализацию VE-кадгерина. (А) Конфокальные изображения контрольных клеток (control), Т-кадгерин гиперэкспрессирующих (T-cad) клеток и клеток с подавленной экспрессией Т-кадгерина (si-T-cad) окрашенных двойной иммуноокраской антителами против клатрина (зелѐная флуоресценция) и VE-кадгерина (красная флуоресценция). Ядра клеток окрашены DAPI (синяя флуоресценция). Для доступности цитоплазматических антигенов клетки подвергали пермеабилизации Тритоном Х-100. Жѐлтая флуоресценция соответствует ко-локализации VE-кадгерина и клатрина (белые стрелки, также представлены на нижней панели). Изображения получены на конфокальном микроскопе высокого разрешения с одинаковыми настройками интенсивности лазера и времени экспозиции. Стрелками и отдельными сносками на нижней панели указаны зоны ко-локализации клатрина и VE-кадгерина на мембране. Масштаб 20 мкм. (Б) Денситометрический анализ ко-локализации VEкадгерина и клатрина. Представлен воспроизводимый результат трѐх независимых экспериментов (N=3, * р <0,05). 274 Для подтверждения того, что экспрессия Т-кадгерина вызывает эндоцитоз VEкадгерина по клатрин-зависимому пути в эндотелиальных клетках, был применѐн ингибиторный анализ с помощью диназора (dynasore) [Macia et al., 2006]. Диназор – избирательный ингибитор активности ГТФ-зависимого белка динамина, который участвует в формировании клатриновых пузырьков, но не влияет на организацию цитоскелета и активность малых ГТФаз [Macia et al., 2006]. Эксперименты с использованием диназора (в конечной концентрации 80 мкM) показали значительное уменьшение окрашивания на клатрин и VE-кадгерин в цитоплазме и практически полное отсутствие ко-локализации клатрина и VE-кадгерина в мембранах эндотелиальных клеток в T-cad, control, si-Т-cad клетках (Рисунок 3.80 А, выноски на нижней панели). Результаты, полученные с использованием ингибиторного анализа, подтверждают гипотезу о том, что повышенная экспрессия Т-кадгерина вызывает эндоцитоз VE-кадгерина по клатрин-зависимому пути. Несмотря на то, что клатрин-зависимая интернализация VE-кадгерина является основным путѐм эндоцитоза VE-кадгерина с поверхности эндотелиальных клеток, в литературе есть данные, что кавеолин-1 может также вносить свой вклад в интернализацию классических кадгеринов [Xiao et al., 2003a, Gavard and Gutkind 2006]. В частности, было показано, что тромбин-индуцированная разборка адгезивных VE-кадгериновых контактов опосредуется активацией белка кавеолина-1 и, как следствие, интернализация VEкадгерина происходит через кавеолин-зависимый путь эндоцитоза [Kronstein et al., 2012]. Мы проанализировали возможную ко-локализацию VE-кадгерина и кавеолина-1 в эндотелиальных клетках в норме, при гиперэкспрессии Т-кадгерина, а также при подавлении Т-кадгерина. Оказалось, что ко-локализации VE-кадгерина и кавеолина-1 не наблюдается ни в одном типе клеток. Следовательно, нет оснований полагать, что экспрессия Т-кадгерина активирует кавеолин-опосредованный эндоцитоз VE-кадгерина в эндотелиальных клетках (Рисунок 3.81). 275 Рисунок 3.80. Диназор подавляет клатрин-зависимый эндоцитоз VE-кадгерина в эндотелиальных клетках. (А) Конфокальные изображения высокого разрешения контрольных клеток (control), Т-кадгерин гиперэкспрессирующих клеток (T-cad), и клеток с подавленной экспрессией Т-кадгерина (si-T-cad), окрашенных с использованием антител против клатрина (зеленая флуоресценция) и VE-кадгерина (красная флуоресценция). Ядра клеток окрашены DAPI (синяя флуоресценция). Для доступности цитоплазматических антигенов клетки подвергали пермеабилизации Тритоном Х-100. Для подавления клатрин-зависимого эндоцитоза клетки преинкубировали с диназором (80 мкM) в течение 2 часов перед окраской. Изображения получены на конфокальном микроскопе высокого разрешения с одинаковыми настройками интенсивности лазера и времени экспозиции. На нижней панели показана зона мембраны с возможной колокализацией клатрина и VE-кадгерина. Масштаб 20 мкм. (Б) Денситометрический анализ ко-локализации VE-кадгерина и клатрина. Представлен воспроизводимый результат трѐх независимых экспериментов (N=3, *, **р <0,05). 276 Рисунок 3.81. Гиперэкспрессия Т-кадгерина в эндотелиальных клетках не вызывает интернализацию VE-кадгерина по кавеолин-зависимому пути. Двойное иммунофлуоресцентное окрашивание эндотелиальных клеток антителами против VEкадгерина (зеленая флуоресценция) и кавеолина-1 (красная флуоресценция) показало отсутствие ко-локализации этих антигенов. Ядра клеток окрашены DAPI (синяя флуоресценция). Для доступности цитоплазматических антигенов клетки перед окрашиванием пермеабилизовали Тритоном Х-100. Изображения получены с помощью конфокального микроскопа при одинаковых настройках интенсивности лазера и времени экспозиции. Масштаб 10 мкм. Control – клетки, трансдуцированные контрольным лентивирусом, T-cad - клетки, трансдуцированные лентивирусом для гиперэкспрессии Т-кадгерина, si-T-cad - клетки, трансдуцированные лентивирусом для подавления нативной экспрессии Т-кадгерина. Представлен воспроизводимый результат трѐх независимых экспериментов. 277 Для того чтобы проследить дальнейшую судьбу VE-кадгерина в цитоплазме после его эндоцитоза с поверхности мембраны, а также ответить на вопрос о возможной деградации VE-кадгерина в лизосомах, эндотелиальные клетки были преинкубированы с Lysotracker RED® - флуоресцентным маркѐром лизосом, а затем окрашены антителами к VE-кадгерину. При этом в случае если VE-кадгерин после эндоцитоза подвергается дальнейшей деградации в лизосомах, ожидали, что он будет ко-локализоваться с флуоресцентным красителем Lysotracker RED® [Kandimalla et al., 2009]. Контрольные (control), гиперэкспрессирующие Т-кадгерин (T-cad) и клетки после подавления экспрессии Т-кадгерина (si-T-cad) преинкубировали с Lysotracker RED® (в концентрации 75 нМ) в течение 60 и 120 минут. Далее клетки окрашивали антителами против VE-кадгерина и оценивали возможную ко-локализацию VE-кадгерина и Lysotracker RED® с использованием конфокального микроскопа. Прижизненная инкубация клеток с маркѐром лизосом (в течение 120 мин) с последующим окрашиванием антителами показала, что колокализация VE-кадгерина с маркером лизосом была более выражена в цитоплазме T-cad клеток, чем в контрольных control, и особенно si-Т-cad клетках (Рисунок 3.82). Жѐлтая окраска гранул (наложение красной и зеленой флуоресценции) в цитоплазме эндотелиальных клеток, гиперэкспрессирующих Т-кадгерин, указывает на то, что VEкадгерин (зелѐная флуоресценция) локализуется в цитоплазме клеток в лизосомах (красная флуоресценция) (Рисунок 3.82). То есть под влиянием гиперэкспрессии Т-кадгерина происходит увеличение интернализации VE-кадгерина с поверхности цитоплазматической мембраны внутрь клеток с его последующей деградацией в лизосомах. Таким образом, полученные нами данные по изучению роли Т-кадгерина в экспрессии и локализации VE-кадгерина в эндотелии указывают на то, что при повышении экспрессии Т-кадгерина увеличивается клатрин-зависимый эндоцитоз VE-кадгерина с последующей его деградацией в лизосомах. 278 Рисунок 3.82. Гиперэкспрессия Т-кадгерина в эндотелиальных клетках вызывает накопление VE-кадгерина в лизосомах. Контрольные (control), гиперэкспрессирующие Ткадгерин (T-cad) или после подавления экспрессии Т-кадгерина (si-T-cad) эндотелиальные клетки преинкубировали с лизосомальным красителем Lysotracker RRED® (красная флуоресценция) в течение 60 минут (А) и 120 минут (Б), затем окрашивали антителами против VE-кадгерина (зелѐная флуоресценция). Ядра клеток окрашены DAPI (синяя флуоресценция). Для доступности цитоплазматических антигенов клетки перед окрашиванием пермеабилизовали Тритоном Х-100. Жѐлтая флуоресценция соответствует ко-локализации VE-кадгерина и лизосомального красителя. Локализация VE-кадгерина в лизосомах обнаружена во всех T-cad клетках в отличие от контрольных control и si-Т-cad клеток. Изображения получены с помощью конфокального микроскопа при одинаковых настройках интенсивности лазера и времени экспозиции. Масштаб 10 мкм. 279 3.5.5 Гиперэкспрессия Т-кадгерина увеличивает уровень фосфорилирования VEкадгерина по тирозину Y731 в эндотелиальных клетках Внутриклеточный домен VE-кадгерина имеет несколько тирозиновых остатков, по которым возможно его фосфорилирование [Esser et al., 1998, Andriopoulou et al., 1999]. Фосфорилирование VE-кадгерина по тирозиновым остаткам приводит к нарушению взаимосвязи между VE-кадгерином и адаптерными белками и, как следствие, с цитоскелетом. Все эти процессы коррелирует с разрушением межклеточных адгезивных контактов и снижением барьерной функции эндотелия [Dejana et al., 2008, Vestweber, 2008]. Наши результаты свидетельствуют о том, что под влиянием гиперэкспрессии Ткадгерина в эндотелиальных клетках происходит клатрин-зависимый эндоцитоз VEкадгерина и его последующая деградация в лизосомах. Этому процессу предшествует разрушение адгезивных контактов и формирование щелей между эндотелиальными клетками. На основании этих результатов было сделано предположение о том, что увеличение экспрессии Т-кадгерина может приводить фосфорилированию VE-кадгерина по каким-то тирозиновым остаткам, его эндоцитозу и деградации. Внутриклеточный домен VE-кадгерина имеет два остатка тирозина в 731 (Y731) и 658 (Y658) положении, по которым осуществляется преимущественное фосфорилирование этого белка. При этом участок Y731 обеспечивает взаимодействие VE-кадгерина с бета-катенином, в то время как Y658 – опосредует связывание VE-кадгерина и катенина p120ctn [Lampugnani et al., 2002, Allingham et al., 2007, Turowski et al., 2008]. Для ответа на вопрос, существует ли связь между Т-кадгерин-зависимой интернализацией VE-кадгерина и фосфорилированием VE-кадгерина по этим тирозиновым остаткам, лизаты control, T-cad и si-T-cad клеток анализировали методом Вестерн блоттинга с использованием специфичных антител против цитоплазматического домена VEкадгерина, фосфорилированного по Y658 или Y731 участкам. Было обнаружено, что гиперэкспрессия Т-кадгерина приводит к избирательному увеличению фосфорилирования VE-кадгерина по Y731 участку, в то время как фосфорилирование по Y658 участку не изменяется (Рисунок 3.83). В si-Т-cad клетках уровень фосфорилирования цитоплазматического домена VE-кадгерина по Y731участку снижается, а по Y658 не изменяется. Полученные результаты указывают на то, что изменение экспрессии Ткадгерина приводит к изменению уровня фосфорилирования VE-кадгерина по тирозину в 731 положении (сайт взаимодействия с бета-катенином): гиперэкспрессия к увеличению фосфорилирования, подавление экспрессии к снижению фосфорилирования. 280 Рисунок 3.83. Гиперэкспрессия Т-кадгерина вызывает увеличение фосфорилирования VE-кадгерина по участку тирозина Y731, но не по Y658. Количество фосфорилированного VE-кадгерина анализировали в лизатах контрольных (control), гиперэкспрессирующих Т-кадгерин (T-cad) клеток и клеток без Т-кадгерина (si-T-cad). Использовали метод Вестерн блоттинга с антителами против фосфорилированной формы VE-кадгерина по Y731 и по Y658 участкам. В качестве контроля содержания общего белка использовали антитела к GAPDH. Известно, что бета-катенин играет двойную роль в клетках: с одной стороны, бетакатенин связывает кадгерины с цитоскелетом, с другой стороны, при разрушении кадгеринового комплекса свободный бета-катенин может накапливаться в цитоплазме или транслоцироваться в ядро [Behrens et al., 1996, Mukherjee et al., 2006]. В цитоплазме бетакатенин может подвергаться фосфорилированию и последующей убиквитин-зависимой деградации в протеосомах, в то время как в ядре он участвует в регуляции экспрессии генов [Gumbiner, 1995]. В ряде опубликованных работ говорится о том, то помимо бета-катенина другой катенин, p120ctn, также участвует в регуляции стабильности связи кадгеринового комплекса с цитоскелетом и предотвращает деградацию кадгеринов в лизосомах [Davis et al., 2003, Xiao et al., 2003a, Xiao et al., 2003b]. Мы оценивали влияние экспрессии Ткадгерина на локализацию и фосфорилирование этих двух белков: бета-катенина и р120ctn. При анализе субклеточных фракций методом вестерн блоттинга, а также последующим денситометрическим обсчѐтом не было выявлено статистически значимых различий в содержании бета-катенина в ядерной и цитозольной фракции клеток как при гиперэкспрессии Т-кадгерина (T-cad), так и в контроле (control) (Рисунок 3.84 А-Г). Однако следует отметить, что наблюдалась тенденция к увеличению содержания бета-катенина в ядерной фракции в T-cad клетках (Рисунок 3.84 Г). Достоверной разницы в содержании мембанно-связанного бета-катенина и фосфорилированного бета-катенина, который может подвергаться деградации в протеосомах, в control, T-cad и si-T-cad клетках обнаружено не было (Рисунок 3.84 Д, Е). 281 Рисунок 3.84. Экспрессия Т-кадгерина в эндотелиальных клетках не влияет на общее содержание и локализацию бета-катенина в субклеточных компартментах, а также на уровень его фосфорилирования бета-катенина в общих лизатах. (A) Экспрессия бета-катенина после субклеточного фракционирования методом Вестерн блоттинга с последующим денситометрическим анализом (Б, В, Г) в цитоплазматической, мембранной и ядерных фракциях контрольных (control), Ткадгерин гиперэкспрессирующих (T-cad) клеток, и клеток после подавления экспрессии Т-кадгерина (si-T-cad). (Д) Уровень фосфорилированного бета-катенина определяли в общих лизатах контрольных, Т-cad и si-Т-cad клеток антителами против фосфорилированного бета-катенина в Т41 положении методом вестерн блоттинга с последующим денситометрическим анализом (Е). (* р˃0,05). Данные нормированы по общему содержанию бета-катенина. 282 Интересно, что в то время как количество p120ctn в цитозольной, мембранной и ядерной фракции контрольных и Т-кадгерин гиперэкспрессирующих клеток не отличалось, в мембранной фракции клеток, в которых экспрессия Т-кадгерина была подавлена, количество белка p120ctn было значительно выше, чем в контроле (Рисунок 3.85). Мы предположили, что при подавлении экспрессии Т-кадгерина p120ctn может участвовать в стабилизации VE-кадгериновых адгезивных контактов и тем самым улучшать барьерную функцию эндотелия. Гиперэкспрессия Т-кадгерина в свою очередь приводит к повышению проницаемости эндотелия за счет увеличения фосфорилирования VE-кадгерина по тирозину в 731 положении, нарушению связывания цитоплазматического домена VEкадгерина с бета-катенином, интернализации VE-кадгерина по клатрин-зависимому пути и деградации его в лизосомах. Рисунок 3.85. Подавление экспрессии Т-кадгерина в эндотелиальных клетках вызывает увеличичение содержания p120ctn в мембране клеток. (А) Экспрессия p120ctn в субклеточных фракциях была оценена Вестерн блоттингом с последующим денситометрическим анализом (Б-Г). Подавление экспрессии Т-кадгерина приводит к достоверному увеличению p120ctn в мембранной фракции по сравнению с контрольными (control) и гиперэкспрессирующими Т-кадгерин (T-cad) клетками. Уровень белка p120ctn в цитоплазматической и ядерной фракции Т-cad клеток соизмерим с таковым в контрольных клетках. Представлен воспроизводимый результат гистограммы трѐх независимых экспериментов (N=3, * р <0.05). 283 3.5.6 Т-кадгерин активирует SRE в фибробластах NIH3T3 Ранее в работе наших коллег из Базеля было обнаружено, что гиперэкспрессия Ткадгерина может приводить к активации и стабилизации белка бета-катенина, который при этом не деградирует в протеосомах, а транслоцируется в ядро и участвует в регуляции активности генов [Joshi et al., 2007]. Поскольку в настоящей работе нам не удалось выявить достоверного изменения активации бета-катенина (в виде увеличения содержания бетакатенина в ядерной фракции), мы предположили, что изменение проницаемости эндотелия в ответ на гиперэкспрессию Т-кадгерина может быть связано с активацией других сигнальных путей, например, с активацией малых Rho ГТФаз. Известно, что малые Rho ГТФазы (RhoA, Rac1 и Cdc42) регулирует проницаемость эндотелия за счѐт фосфорилирования белков, участвующих в формировании адгезивных контактов, а также за счѐт полимеризации/деполимеризации цитоскелета и активности актомиозинового комплекса [Essler et al., 1998; Ponimashkin et al., 2002]. Способность Ткадгерина активировать малые G белки семейства Rho ГТФаз была подтверждена в модельных экспериментах на линейных фибробластах NIH3T3. Мы измеряли активность SRE элемента в клетках NIH3T3, гиперэкспрессирующих Т-кадгерин и в контрольных клетках. SRE (Serum Response Element), или s-fos SRE относится к ранним протоонкогенам, на активность которых влияет активация Rho ГТФаз в цитоплазме клетки. Для гиперэкспрессии Т-кадгерина линию мышиных фибробластов NIH3T3 трансфицировали плазмидой, кодирующей кДНК Т-кадгерина человека (NIH3T3/T-cad), или контрольной плазмидой (NIH3T3). Оценку гиперэкспрессии Т-кадгерина в NIH3T3 проводили методом вестерн блоттинга (Рисунок 3.86), который показал, что через 48 ч после трансфекции плазмидой pcDNA3.1/T-cad, клетки NIH3T3/T-cad гиперэкспрессируют зрелый Т-кадгерин молекулярной массой 105 кДа, тогда как в клетках, трансфицированных контрольной плазмидой NIH3T3 экспрессия Т-кадгерина не обнаруживается. Для измерения активности SRE NIH3T3 клетки, гиперэкспрессирующие Ткадгерин, и контрольные клетки, ко-трансфицировали плазмидами pSRE.L (содержит под одним промотором ген SRE и ген люциферазы Luc) и плазмидой pCMV-β-Gal (содержит ген бета-галактозидазы b-Gal и используется для нормирования трансфекции). В качестве положительного контроля клетки NIH3T3 ко-трансфицировали плазмидами pcDNA3.1, pSRE.L, pCMV-β-Gal а также плазмидой, кодирующей p114RhoGEF (фактор обмена гуанидина для Rho ГТФаз). 284 130 кДа 95 кДа HUVEC NIH3T3 NIH3T3/T-cad Рисунок 3.86. Оценка экспрессии Т-кадгерина в клетках NIH3T3. Клетки NIH3T3 трансфицировали плазмидой для увеличения экспрессии Т-кадгерина (pcDNA3.1/T-cad), или контрольной плазмидой (pcDNA3.1). Через 48 ч после трансфекции в лизатах определяли уровень Т-кадгерина методом Вестерн блоттинга. Клетки NIH3T3/T-cad экспрессируют зрелый Т-кадгерин (105 кДа), контрольные клетки NIH3T3 не экспрессируют Т-кадгерин. В качестве положительного контроля использовали клетки HUVEC, которые в норме экспрессируют как зрелый Т-кадгерин, так и его предшественник (130 кДа). 70 * Luc/ b-Gal, OD 60 50 40 30 20 10 0 F 7 19 EF 17 3 .1 cad GE N1 oG /T4 2N c1N ho NA ho h c 1 a . R D R R d c 3 R + C 14 p 14 d+ NA p1 cad d+ p1 -ca d+ -ca /TT pcD a 1 / T . c 1 / 3 3. 3 .1 NA .1/T NA NA A3 D pcD N c pcD p pcD Рисунок 3.87. Гиперэкспрессия Т-кадгерина в NIH3T3 клетках активирует SRE. Для измерения SRE активности клетки NIH3T3 ко-трансфицировали плазмидами pSRE.L, pCMV-b-Gal и pcDNA3.1/T-cad или контрольной pcDNA3.1. Ко-экспрессия плазмид pcDNA3.1/T-cad и RacN17 или Cdc42N19 вызывает подавление активации SRE. Ко-экспрессия плазмид pcDNA3.1/T-cad и RhoN17 не влияет на активность SRE. Коэкспрессия p114RhoGEF и Т-кадгерина (pcDNA3.1/T-cad+p114RhoGEF) достоверно активирует SRE в гораздо большей степени, чем при экспрессии каждой из плазмид по отдельности. Данные получены из 5-ти независимых экспериментов, повторенных в 5ти параллелях (N=25, р<0,01). 285 Было обнаружено, что гиперэкспрессия Т-кадгерина в NIH3T3 клетках вызывает значительную активацию SRE, и уровень активации SRE соизмерим с уровнем активации SRE в положительном контроле, с использованием плазмиды p114RhoGEF (Рисунок 3.87). При этом ко-экспрессия Т-кадгерина и p114RhoGEF приводит к активации SRE в большей степени, чем при раздельной экспрессии этих плазмид в клетках, что предполагает наличие общего механизма активации Rho ГТФаз. Как известно, на активность SRE влияют все Rho ГТФазы: RhoA, Rac1 и Cdc42 [Ponimashkin et al., 2002]. Для того чтобы оценить специфическое влияние гиперэкспрессии Т-кадгерина на активность каждой из этих ГТФаз, были использованы плазмиды, специфически подавляющие активацию Rho ГТФаз в клетках: RhoN17, RacN17 и Cdc42N19 для RhoA, Rac1 и Cdc42 соответственно. Эти плазмиды экспрессируют белки антагонисты, конкурирующие за связывание с факторами обмена гуанидина Rho ГТФаз, блокируя тем самым их активацию. При ко-экспрессии Т-кадгерина и RacN17 или Cdc42N19 происходило частичное угнетение Т-кадгерин зависимой активации SRE (Рисунок 3.87). Однако ко-экспрессия Т-кадгерина и RhoN17 не подавляла активацию SRE, которая, в данном случае, сравнима с положительным контролем (p114RhoGEF). Такое избирательное подавление SRE может предполагать, что Т-кадгерин при его гиперэкспрессии в линейных фибробластах NIH3T3 клетках, активирует Rac1 и Cdc42, но не затрагивает RhoA сигнальный путь. 286 3.5.7 Т-кадгерин активирует Rac1 и Cdc42, но не влияет на активность RhoA в фибробластах NIH3T3 Для дальнейшего доказательства гипотезы о том, что Т-кадгерин избирательно активирует Rac1 и Cdc42 ГТФазы и не влияет на активацию RhoA, были выделены активные формы Rho ГТФаз из цитоплазмы NIH3T3 клеток, гиперэкспрессирующих Ткадгерин, и контрольных. В качестве положительного контроля использовали клетки, трансфицированные плазмидами, кодирующими гены доминантно-активных форм Rho ГТФаз: для RhoA -RhoV12, для Rac1 - RacV12, для Cdc42 - Cdc42V12. При выделении активных форм Rho ГТФаз методом осаждения с использованием сефарозы, было обнаружено (Рисунок 3.88), что гиперэкспрессия Т-кадгерина активирует Rac1 и Cdc42, уровень которых в клетках, гиперэкспрессирующих Т-кадгерин сравним с уровнем активных Rac1 и Cdc42 в клетках, трансфицированных для положительного контроля плазмидами RacV12 и Cdc42V12 (Рисунок 3.88 А и Рисунок 3.88 Б). При этом гиперэкспрессия Т-кадгерина не влияет на активность RhoA (Рисунок 3.88 В). Гиперэкспрессия Т-кадгерина в NIH3T3 клетках не влияет на общий уровень экспрессии этих ГТФаз, а лишь избирательно их активирует, что подтверждается одинаковым уровнем общего количества каждой из ГТФаз в тотальном лизате клеток до и после экспрессии Ткадгерина (Рисунок 3.88 А, Б, В - нижние полосы). Таким образом, на основании полученных результатов, можно сделать вывод о том, что в фибробластах NIH3T3 Т-кадгерин избирательно активирует Rac1 и Cdc42 ГТФазы, но не влияет на активность RhoA. 287 А активный Rac1 общий Rac1 Б активный Cdc42 общий Cdc42 В pcDNA3.1 pcDNA3.1/T-cad Cdc42V12 pcDNA3.1 pcDNA3.1/T-cad RhoV12 активный RhoA общий RhoA Рисунок 3.88. Т-кадгерин активирует Rac1 и Cdc42. Клетки NIH3Т3 трансфицировали плазмидой для гиперэкспрессии Т-кадгерина (pcDNA3.1/T-cad) или контрольной (pcDNA3.1). В качестве положительного контроля использовали плазмиды: для RhoA – RhoV12, для Rac1 – RacV12, для Cdc42 – CdcV12. Через 48 ч после трансфекции определяли уровень активных Rho ГТФаз методом вестерн блоттинга; А, Б - уровень активных Rac1 и Cdc42 определяли по количеству белка, связавшегося с PBD-Rac1 и PBD-Cdc42, соответственно (верхняя полоса), уровень общего Rac1 и Cdc42 определяли в общем лизате клеток (нижняя полоса); В - уровень активного RhoA определяли по количеству белка, связавшегося с RBD-RhoA (верхняя полоса), уровень общего RhoA определяли в общем лизате (нижняя полоса). 288 3.5.8 Т-кадгерин активирует малые Rho ГТФазы RhoA, Rac1 и, Cdc42 и усиливает сборку стресс-фибрилл актина в эндотелиальных клетках Для оценки возможного влияния малых Rho ГТФаз на Т-кадгерин-опосредованные эффекты в эндотелиальных клетках были выделены активные формы Rho ГТФаз из лизатов контрольных клеток (control), Т-кадгерин гиперэкспрессирующих клеток (T-cad), а также клеток после подавления нативной экспрессии Т-кадгерина (si-Т-cad). Было обнаружено, что повышенная экспрессия Т-кадгерина в HUVEC приводит к активации RhoA, Rac1 и Cdc42, в то время как общий уровень RhoA, Rac1 и Cdc42 не изменяется (Рисунок 3.89). Примечательно, что увеличения активности RhoA и Cdc42 в контрольных и si-Т-cad клетках обнаружено не было (Рисунок 3.89). Таким образом, активация малых Rho ГТФаз при гиперэкспрессии Т-кадгерина была продемонстрирована на двух типах клеток, на линейных фибробластах, и на эндотелиальных клетках HUVEC. Рисунок 3.89. Т-кадгерин активирует малые Rho ГТФазы в эндотелиальных клетках. Активные формы Rho ГТФаз были выделены методом сефарозного осаждения из общих лизатов контрольных (control), Т-кадгерин гиперэкспрессирующих клеток (Tcad) и клеток после подавления нативной экспрессии Т-кадгерина (si-T-cad) и проанализированы методом вестерн блоттинга. Гиперэкспрессия Т-кадгерина вызывает активацию RhoA, Rac1 и Cdc42 в отличие от контрольных и si-T-cad клеток, в которых активации Rho ГТФаз не наблюдалось. Общий пул RhoA, Rac1 и Cdc42 остается неизменным. Представлен воспроизводимый результат трѐх независимых экспериментов. 289 3.5.9 Т-кадгерин фосфорилирует LIMK1 и влияет на полимеризацию микротрубочек в эндотелиальных клетках Микротрубочки - компоненты цитоскелета, представляющие собой филаменты, состоящие из α- и β-субъединиц тубулина с диаметром 25 нм. Микротрубочки – динамические структуры, изменение организации микротрубочек влияет на организацию и динамику других компонентов цитоскелета в клетке, вызывая формирование стрессфибрилл, изменение формы эндотелиальных клеток и проницаемости эндотелия [Verin et al., 2001]. Серин-треониновая киназа LIMK1 является важным связующим звеном между Rho ГТФазами и микротрубочками в эндотелиальных клетках. LIMK1 в неактивной форме ко-локализуется с микротрубочками, но при фосфорилировании (активации) LIMK1 происходит диссоциация комплекса LIMK-МТ, что приводит к дестабилизации микротрубочек и дальнейшим изменениям клеточного фенотипа: полимеризации стрессфибрилл актина и сокращению клетки (Рисунок 3.90) [Gorovoy et al., 2005]. Наши результаты свидетельствуют о том, что Т-кадгерин активирует Rho ГТфазы Rac1 и Cdc42. Мы предположили, что гиперэкспрессия Т-кадгерина может вызывать фосфорилирование LIMK1, деполимеризацию микротрубочек и формирование стресс-фибрилл. Рисунок 3.90. Серин-треониновая киназа LIMK1 является связующим звеном между Rho ГТФазами и микротрубочками, и участвует в регуляции полимеризации как микротрубочек, так и актинового цитоскелета (пояснения в тексте). 290 Чтобы оценить влияние Т-кадгерина на активность LIMK1, клетки NIH3T3 котрансфицировали плазмидами pcDNA3.1/T-cad или контрольной pcDNA3.1, и плазмидой для увеличения экспрессии LIMK1 киназы, содержащей myc эпитоп (myc-LIMK1). При анализе методом иммуноблоттинга с использованием моноклональных антител против фосфорилированной LIMK1 было показано, что через 48 ч после трансфекции в NIH3T3 клетках, гиперэкспрессирующих Т-кадгерин, активность LIMK1 увеличивается. При этом общее количество LIMK1 в контрольных клетках и клетках, гиперэкспрессирующих Ткадгерин, не изменяется, о чем свидетельствует одинаковое количество Myc в контрольных клетках и клетках, гиперэкспрессирующих Т-кадгерин. Однако содержание фосфорилированной формы LIMK1 в Т-кадгерин гиперэкспрессирующих клетках увеличивается по сравнению с контролем (Рисунок 3.91). p-LIMK1 myc pcDNA3.1 myc-LIMK1 pcDNA3.1/T-cad myc-LIMK1 Рисунок 3.91. Т-кадгерин фосфорилирует LIMK1 киназу. Клетки NIH3Т3 котрансфицировали плазмидой pcDNA3.1/T-cad для гиперэкспрессии Т-кадгерина, и плазмидой myc-LIMK1, кодирующей LIMK1 и myc эпитоп. В контроле клетки котрансфицировали pcDNA3.1 и myc-LIMK1. Через 48 ч методом вестерн блоттинга измеряли количество фосфорилированной LIMK1 (p-LIMK) с использованием антител против p-LIMK1. Эффективность трансфекции плазмидой myc-LIMK1 оценивали с использованием антител против myc эпитопа. Влияние экспрессии Т-кадгерина на организацию микротрубочек в эндотелиальных клетках оценивали двумя способами: гиперэкспрессией Т-кадгерина с использованием плазмидной трансфекции (pcDNA3.1/T-cad), и подавлением нативной экспрессии Т-кадгерина РНК интерференцией с использованием коротких РНК олигонуклеотидов против мРНК Т-кадгерина (siRNA/T-cad). Эффективность подавления нативной экспрессии Т-кадгерина в эндотелиальных клетках оценивали методом иммуноблоттинга. Через 48 ч после трансфекции эндотелиальных клеток siRNA/T-cad, эффективное подавление наблюдалось при дозировке siRNA/T-cad 100 нМ по сравнению с siRNA/T-cad 50 нМ (Рисунок 3.92). При этом экспрессия зрелого Т-кадгерина (105 кДа) и его предшественника (130 кДа) практически полностью подавлялась. 291 130 кДа 105 кДа siRNA/T-cad 50 нМ - 100 нМ Рисунок 3.92. Подавление нативной экспрессии Т-кадгерина в эндотелиальных клетках. Для оценки дозозависимого эффекта подавления экспрессии Т-кадгерина HUVEC 2 пассажа трансфицировали РНК олигонуклеотидами (siRNA/T-cad) в концентрации 50 нМ и 100 нМ, и через 48 ч оценивали уровень экспрессии Т-кадгерина вестерн блоттингом. В качестве контроля использовали HUVEC, нативно экспрессирующие Т-кадгерин. 130 кДа – предшественник Т-кадгерина, 105 кДа – зрелый Т-кадгерин. Влияние Т-кадгерина на микротрубочковый цитоскелет в эндотелиальных клетках оценивали методом иммунофлуоресцентного окрашивания с использованием антител против ацетилированного Эндотелиальные клетки α-тубулина, выявляющего ко-трансфицировали стабильные плазмидами микротрубочки. pcDNA3.1/T-cad и pcDNA3.1/GFP для гиперэкспрессии Т-кадгерина и визуализации трансфицированных клеток (клетки, экспрессирующие GFP, также гиперэкспрессируют Т-кадгерин). Интенсивность окрашивания на α-тубулин в GFP-экспрессирующих HUVEC оценивали в условиях временной трансфекции. Для подавления экспрессии Т-кадгерина, эндотелиальные клетки ко-трансфицировали плазмидой pcDNA3.1/GFP и siRNA/T-cad (в количестве 100 нМ); в клетках, экспрессирующих GFP, экспрессия Т-кадгерина была подавлена. Результаты иммунофлуоресцентного окрашивая эндотелиальных клеток, полученные с использованием конфокальной микроскопии, свидетельствуют о том, что гиперэкспрессия Т-кадгерина вызывает уменьшение количества стабильных ацетилированных по α-тубулину микротрубочек, а подавление экспрессии Т-кадгерина вызывает противоположный эффект – увеличивает их количество. На Рисунке 3.93, на верхней панели, представлен характерный паттерн окрашивания: три гиперэкспрессирует клетки HUVEC, Т-кадгерин, а две одна из другие которых не экспрессирует трансфицированы. GFP При и этом флуоресценция в красном канале соответствует окрашиванию на ацетилированный αтубулин микротрубочек. В клетке, гиперэкспрессирующей Т-кадгерин, интенсивность окрашивания меньше, чем в соседних клетках с эндогенной экспрессией Т-кадгерина. На нижней панели представлены эндотелиальные клетки, в которых экспрессия Т-кадгерина была подавлена с использованием siRNA/T-cad в концентрации 100 нМ. В клетке, которая 292 экспрессирует GFP, и экспрессия Т-кадгерина в которой снижена, интенсивность окрашивания на ацетилированный α-тубулин выше, чем в окружающих не трансфицированных клетках. Полученные данные свидетельствуют о том, что Т-кадгерин влияет на организацию микротрубочек: гиперэкспрессия Т-кадгерина вызывает снижение количества стабильных микротрубочек и как следствие их деполимеризацию, а подавление экспрессии Т-кадгерина, наоборот, увеличивает их стабильность. GFP α-тубулин наложение pcDNA3.1/GFP pcDNA3.1/T-cad pcDNA3.1/GF siRNA/T-cad Рисунок 3.93. Т-кадгерин влияет на стабилизацию микротрубочек в эндотелиальных клетках. HUVEC 2 пассажа ко-трансфицировали плазмидами pcDNA3.1/T-cad (для гиперэкспрессии Т-кадгерина) и pcDNA3.1/GFP (зеленая флуоресценция на верхней панели), или siRNA/T-cad (для подавления экспрессии Ткадгерина) и pcDNA3.1/GFP (зеленая флуоресценция на нижней панели). Иммунофлуоресцентное окрашивание HUVEC антителами против α-тубулина (красная флуоресценция). Масштаб -10 мкм. 293 3.5.10 Т-кадгерин влияет на полимеризацию стресс-фибрилл актина в эндотелиальных клетках Семейство Rho-ассоциированных киназ, ROCK-II и PAK1, являются непосредственными нисходящими мишенями Rho ГТФаз (ROCK-II активируется RhoA, в то время как PAK1 является мишенью для активированных Rac1 и Сdc42). Известно, что активные ROCK-II и PAK1 вызывают полимеризацию стресс-фибрилл актина, что приводит к сокращению клеток и, таким образом, увеличению проницаемости сосудистой стенки [Gorovoy et al., 2005, Stockton et al., 2007]. Результаты же наших экспериментов показывают, что гиперэкспрессия Т-кадгерина чѐтко коррелирует с увеличением содержания ROCK-II и повышением уровня фосфорилирования PAK1 по сравнению с контролем (Рисунок 3.94 А и Рисунок 3.94 Б). В si-Т-cad HUVEC содержание ROCK-II и фосфорилирование PAK1 снижено по сравнению с контрольными клетками (Рисунок 3.94 А и Рисунок 3.94 Б). Результаты, полученные при измерении Т-кадгерин-зависимой активации Rho ГТФаз и их нисходящих сигнальных посредников ROCK-II и PAK1, были в дальнейшем подтверждены окрашиванием на актиновый цитоскелет, свидетельствующим о функциональной реорганизации актиновых филаментов. Как показано на Рисунке 3.95, повышенная экспрессия Т-кадгерина в эндотелиальных клетках приводит не только увеличению содержания ROCK-II, но и к полимеризации актиновых стресс-фибрилл (окраска на фаллоидин), в то время как при подавлении нативной экспрессии Т-кадгерина (si-Т-cad) стресс-фибриллы актина менее выражены, чем в контрольных эндотелиальных клетках (control) (Рисунок 3.95). 294 Рисунок 3.94. Т-кадгерин активирует ROCK-II и PAK1 в эндотелиальных клетках Уровни активации нисходящих мишеней Rho ГТФаз ROCK-II (А) и PAK1 (Б) были оценены методом вестерн блоттинга в общих лизатах контрольных (control), Ткадгерин гиперэкспрессирующих клеток (T-cad) и клеток после подавления нативной экспрессии Т-кадгерина (si-T-cad). По сравнению с control, гиперэкспрессия Т-кадгерина (T-cad) приводит к увеличению содержания ROCK-II и фосфорилирования PAK по треонину в 212 положении (T212). В si-T-cad клетках содержание ROCK-II и уровень фосфорилирования PAK1 снижено по сравнению с control. Представлен воспроизводимый результат трѐх независимых экспериментов. 295 Рисунок 3.95. Гиперэкспрессия Т-кадгерина вызывает реорганизацию цитоскелета эндотелиальных клеток. Конфокальные изображения высокого разрешения контрольных (control), гиперэкспрессирующих Т-кадгерин клеток (T-cad) и клеток без Т-кадгерина (si-T-cad), окрашенных антителами против ROCK-II (красная флуоресценция) и фаллоидином для выявления актиновых стресс-фибрилл (зелѐная флуоресценция). Ядра клеток докрашены DAPI. Для доступности цитоплазматических антигенов клетки пермеабилизовали Тритоном Х-100. Гиперэкспрессия Т-кадгерина вызывает изменение формы клетки и усиливает сборку стресс-фибрилл актина, характеризующуюся формированием толстых фибрилл актина по оси клеток. В Т-cad клетках окраска ROCK-II была более выражена, чем в контрольных клетках. Экспрессия ROCK-II и количество стресс-фибрилл актина в si-T-cad клетках снижено по сравнению с control и T-cad клетками. Масштаб 25 мкм. 296 Для проверки предположения о том, что активация ROCK-II, которую вызывает Ткадгерин, необходима для полимеризации актиновых стресс-фибрилл был проведен ингибиторный анализ. Y27632 – ингибитор Rho киназы ROCK-II, избирательно подавляющий еѐ активность. Было обнаружено, что Y27632 вызывает разборку актиновых стресс-фибрилл в эндотелиальных клетках, гиперэкспрессирующих Т-кадгерин (Рисунок 3.96), что позволяет предполагать, что ROCK-II действует как мишень RhoA и опосредует эффекты Т-кадгерина на организацию актинового цитоскелета. Для оценки специфичности эффектов Т-кадгерина на активацию Rho ГТФаз и полимеризацию актинового цитоскелета, был также проанализирован уровень фосфорилирования киназ Src, p38 и ERK1/2. Методом вестерн блоттинга какого-либо значимого влияния изменения экспрессии Т-кадгерина на активацию/фосфорилирование этих киназ выявлено не было (Рисунок 3.97). Таким образом, полученные данные свидетельствуют о том, что гиперэкспрессия Т-кадгерина в эндотелиальных клетках снижает барьерную функцию эндотелия за счет активации клатрин-зависимого эндоцитоза VE-кадгерина и его деградации в лизосомах, активации Rho ГТФаз и их нисходящих сигнальных посредников ROCK-II и PAK1 и увеличения полимеризации актиновых стресс-фибрилл. 297 Рисунок 3.96. Ингибитор Rho киназы Y27632 блокирует Т-кадгеринзависимые эффекты в эндотелиальных клетках. Контрольные (control), Т-кадгерин гиперэкспрессирующие (T-cad) эндотелиальные клетки и клетки после подавления экспрессии Т-кадгерина (si-T-cad) инкубировали с ингибитором ROCK-II Y27632 (в конечной концентрации 10 мкM) в течение 10 часов перед иммунофлуоресцентным окрашиванием. Эндотелиальные клетки окрашены антителами против ROCK-II (красная флуоресценция) и фаллоидином (для окраски актиновых стресс-фибрилл, зелѐная флуоресценция). Ядра клеток докрашены DAPI. Для доступности цитоплазматических антигенов клетки пермеабилизовали Тритоном Х100. Добавление Y27632 вызывает разборку актиновых стресс-фибрилл в control, T-cad и si-T-cad. Масштаб 25 мкм. 298 Рисунок 3.97. Экспрессия Т-кадгерина не влияет на фосфорилирование p38, ERK1+2 или Src в эндотелиальных клетках. Control – контрольные эндотелиальные клетки, T-cad - Т-кадгерин гиперэкспрессирующие эндотелиальные клетки, si-T-cad эндотелиальные клетки после подавления экспрессии Т-кадгерина. (A) Фосфорилирование белка p38 по треонину (положение T180) или тирозину (положение Y182) оценивали вестерн блоттингом в общих лизатах клеток с использованием специфичных антител к фосфорилированному p38 в участках треонина 180 и тирозина 182. Результаты нормализовали по общему содержанию белка p38. (Б) Фосфорилирование ERK1/2 по треонинам в положениях T185 и T202 оценивали Вестерн блоттингом в общих лизатах клеток с использованием специфичных антител к фосфорилированной форме киназы ERK1/2 по этим участкам. Результаты нормализовали по общему содержанию ERK1/2. (В) Фосфорилирование Src по тирозину в положении Y418 оценивали Вестерн блоттингом в общих лизатах клеток с использованием специфичных антител к фосфорилированной форме киназы Src по тирозину Y418. Результаты нормализовали по общему содержанию киназы Src. При изменении экспрессии Т-кадгерина в эндотелиальных клетках различий в уровне фосфорилирования p38, ERK1/2 или Src выявлено не было. Представлен воспроизводимый результат трѐх независимых экспериментов. 299 3.5.11 Обсуждение результатов Накопленные в литературе данные указывают на то, что существует корреляция между повышенной экспрессией Т-кадгерина в клетках сосудов и прогрессированием сердечно-сосудистых заболеваний, таких как атеросклероз и рестеноз [Ivanov et al., 2001, Kudrjashova et al., 2002]. Кроме того, Т-кадгерин был также обнаружен на микрочастицах в крови пациентов с атеросклерозом, что, по мнению авторов, отражает активацию или повреждение эндотелия [Philippova et al., 2011]. Также было показано [Andreeva et al., 2010], что Т-кадгерин может участвовать в патогенезе инсулинорезистентности в эндотелиальных клетках: Т-кадгерин снижает чувствительность эндотелиальных клеток к инсулину по механизму хронической активации обратной Akt/mTOR-зависимой петли и увеличения деградации предполагать, что субстрата повышенный рецептора уровень инсулина. Эти Т-кадгерина данные может позволяют способствовать активации/дисфункции эндотелиальных клеток, повышению проницаемости эндотелия и повреждению сосудов при описанных патологических состояниях. В настоящей работе описываются новые биохимические механизмы регуляции проницаемости эндотелия с участием Т-кадгерина. Повышенная экспрессия Т-кадгерина снижает барьерную функцию эндотелия за счѐт активации каскадного пути с участием Rho ГТФаз и их нисходящих сигнальных посредников, фосфорилирования VE-кадгерина с последующим клатрин-зависимым эндоцитозом и деградацией в лизосомах и реорганизацией актинового и микротрубочкового цитоскелета. Подавление экспрессии Ткадгерина вызывает противоположные эффекты. Полученные результаты согласуются с данными, опубликованными ранее другими авторами. В обзоре Андреевой и соавторов было высказано предположение о том, что Т-кадгерин участвует в регуляции барьерной функции эндотелия. Исследователи показали, что Т-кадгерин участвует в уменьшении трансэндотелиального сопротивления при стимуляции тромбином или сывороткой [Andreeva et al., 2010], однако, точные механизмы регуляции проницаемости эндотелия с участием Т-кадгерина до сих пор оставались невыясненными. В данной работе мы исследовали сигнальные пути и выявили биохимические механизмы регуляции барьерной функции эндотелия с участием Т-кадгерина. Полученные результаты при изучении экспрессии Т-кадгерина методом вестерн блоттинга в разных субклеточных фракциях, в частности, в ядерной, согласуются с данными конфокальной микроскопии и ядерного фракционирования, опубликованными ранее [Andreeva et al., 2010], в которых было показано, что в норме Т-кадгерин обнаруживается в ядрах первичной культуры эндотелиальных клеток, а также в ядрах линейных клеток HEK293. 300 Анализ первичной структуры Т-кадгерина показал, что его последовательность богата остатками основных аминокислот и содержит также лейцин-богатую последовательность на COOH-конце, что является сигналом ядерной локализации [Kutay et al., 2005]. Это может указывать на то, что локализация Т-кадгерина в ядерной фракции не является артефактом вследствие лентивирусной трансдукции, однако физиологическое значение данного феномена остается неясным. Локализация Т-кадгерина в ядрах клеток косвенно подтверждает предположение о том, что Т-кадгерин может участвовать в регуляции транскрипции генов, как было показано ранее для Е-кадгерина [Ferber et al., 2008] и урокиназы – другого GPI-заякоренного белка [Stepanova et al., 2008]. Об этом же свидетельствуют данные по повышению содержания циклина D1 в эндотелиальных клетках [Joshi et al., 2007] и активации SRE элемента в NIH3T3 фибробластах при гиперэкспрессии Т-кадгерина. Также с предположением о регуляторной функции Т-кадгерина коррелируют данные о том, что увеличение его экспрессии приводит к блокированию клеток в G 2/M фазе в клетках гепатоцеллюлярной карциномы in vitro [Chan et al., 2008]. При повреждении сосудов при таких патологических состояниях, как атеросклероз, артериальная гипертензия и сахарный диабет, барьерная функция эндотелия может быть нарушена вследствие разрушения межклеточных контактов, сокращения и ретракции эндотелиальных клеток. Многие вещества, такие как гистамин, тромбин, цитокины, антиоксиданты, VEGF и активированные нейтрофилы обратимо увеличивают проницаемость эндотелия [Dejana et al., 2008]. В основе механизма действия таких воспалительных агентов, как гистамин, лежит активация сигнальных каскадов с участием фосфолипазы С (PLC) и протеинкиназы С (PKC), мобилизация внутриклеточного кальция, активация MAPK киназ, формирование стресс-фибрилл и нарушение гомофильных взаимодействий между молекулами VE-кадгерина на соседних клетках [Guo et al., 2008]. Тромбин, хорошо изученный агент, вызывающий увеличение проницаемости эндотелия, вызывает ретракцию эндотелиальных клеток и формирование щелей между ними. В основе действия тромбина лежит активация протеинкиназы С (PKC), повышение содержания внутриклеточного Ca2+, активация G белков, PI3-киназы, MAPK киназы, и белков семейства Rho ГТФаз, что усиливает сборку стресс-фибрилл актина, взывает фосфорилирование лѐгких цепи миозина и сокращение актомиозинового комплекса [Vouret-Craviari et al., 1998]. В эндотелиальных клетках, цитоплазматический домен VE-кадгерина заякорен на цитоскелет через взаимодействие с бета-катенином, белком p120ctn, альфа-катенином. Прочная и стабильная межклеточная адгезия в эндотелии невозможна без связи кадгеринового комплекса с цитоскелетом, которую обеспечивают катенины [Dejana et al., 301 2008, Vestweber, 2008]. В нескольких публикациях было показано, что фосфорилирование VE-кадгерина или белков кадгеринового комплекса приводит к снижению барьерной функции эндотелия [Dejana et al., 2008, Vestweber, 2008] вследствие того, что это фосфорилирование приводит к нарушению взаимодействия VE-кадгерина с адаптерными белками катенинами и цитоскелетом с последующей разборкой адгезивных контактов и увеличением проницаемости эндотелия [Dejana et al., 2008]. Внутриклеточный домен VEкадгерина может фосфорилироваться по нескольким тирозиновым остаткам [Esser et al., 1998, Andriopoulou et al., 1999]. Так, было показано, что взаимодействие VEGF со своим рецептором VEGFR2 через Src-зависимый сигнальный путь приводит к активации малых ГТФаз Rac. Rac в свою очередь вызывает активацию прямого нисходящего сигнального белка-посредника р-21-активируемую киназу РАК, который стимулирует фосфорилирование VE-кадгерина по серину S665 [Gavard and Gutkind, 2006] или тирозину Y685 [Wallez et al., 2006, Wallez et al., 2007]. Фосфорилирование по этим остаткам приводит к сокращению актомиозинового комплекса и ретракции клеток. При фосфорилировании VE-кадгерина по S665 на мембране рекрутируется белок бета-аррестин-2; связывание этого белка с кадгериновым комплексом приводит к быстрой интернализации VE-кадгерина по клатрин-зависимому пути [Gavard and Gutkind 2006]. Кроме того, известно, что антигенактивированные лимфоциты адгезируют к эндотелию, это вызывает фосфорилирование VE-кадгерина по тирозиновым остаткам Y645, Y731 и Y733 с участием активированных Rho ГТФаз, с последующей реорганизацией актинового цитоскелета и разрушением межклеточных контактов [Turowski et al., 2008]. В настоящей работе показано, что гиперэкспрессия Т-кадгерина вызывает фосфорилирование VE-кадгерина именно по тирозину Y731, но не по тирозину Y658 (Рисунок 3.83), что приводит к клатринзависимому эндоцитозу VЕ-кадгерина с поверхности эндотелиальных клеток и увеличению проницаемости эндотелиального монослоя. Эксперименты по ко-локализации VEкадгерина и клатрина были также подтверждены ингибиторным анализом с использованием диназора. Данные по диназору, известному блокатору динамина, который необходим для отшнуровывания эндоцитозных пузырьков [Macia et al., 2006], подтвердили, что эндоцитоз происходит по клатрин-зависимому пути (Рисунок 3.80). Дальнейшие эксперименты показали ко-локализацию VE-кадгерина с маркѐром лизосом Lysotracker RED®, что означает, что интернализованный VE-кадгерин подвергается необратимой деградации в лизосомах, а не рециркулирует обратно на поверхность мембраны эндотелиальных клеток и не участвует в формировании адгезивных контактов [Xiao et al., 2003a, Xiao et al., 2003b]. 302 Как уже говорилось выше, внутриклеточный домен VE-кадгерина взаимодействует с цитоскелетом через адаптерные белки бета-катенин и белок р120сtn, что необходимо для прочной межклеточной адгезии. Тирозин 731 локализуется во внутриклеточном домене VEкадгерина в участке, ответственном за связывание с бета-катенином, а тирозин 658 - в участке связывания с р120сtn [Lampugnani et al., 2002, Allingham et al., 2007, Turowski et al., 2008]. Фосфорилирование по этим тирозинам приводит к нарушению взаимодействия VEкадгерина с бета-катенином и p120ctn, соответственно. Известно, что оба этих белка опосредуют не только связь кадгеринов с цитоскелетом, но также могут сами участвовать в проведении сигнала от факторов роста внутрь клетки: катенины могут проникать в ядро и участвовать в регуляции экспрессии генов [Behrens et al., 1996; Gumbiner, 1995; Mukherjee et al., 2006]. В ранее опубликованном исследовании было показано, что повышенная экспрессия Т-кадгерина может приводить к активации и стабилизации бета-катенина, который не подвергается деградации в протеосомах, а транслоцируется в ядро и участвует в регуляции экспрессии генов [Joshi et al., 2007]. В настоящей работе путѐм клеточного фракционирования и Вестерн блоттинга с последующим денситометрическим анализом полученных изображений не было обнаружено статистически значимых различий в содержании бета-катенина в ядерной, мембранной и цитозольной фракциях в контрольных, T-cad и si-Т-cad клетках. Тем не менее, наблюдалась тенденция к увеличению содержания бета-катенина в ядерной фракции клеток, гиперэкспрессирующих Т-кадгерин, по сравнению с контрольными и si-Т-cad клетками. Количество фосфорилированного/деградирующего бета-катенина было практически одинаковым как в контрольных клетках, так и в клетках, гиперэкспрессирующих Т-кадгерин. Эти данные позволяют предполагать, что высвобождаемый из кадгеринового комплекса бета-катенин не подвергается в дальнейшем деградации в протеосомах, однако, дальнейшая его судьба, а также точные механизмы его участия в регуляции экспрессии генов при гиперэкспрессии Т-кадгерина требуют дальнейшего изучения. В ряде опубликованных работ было показано, что белок p120ctn в связанном с VEкадгерином виде защищает последний от интернализации и деградации, увеличивая стабильность кадгерин-цитоскелетного комплекса [Davis et al., 2003, Xiao et al., 2003a]. В наших экспериментах было обнаружено, что в то время как повышение экспрессии Ткадгерина не оказывает влияния на экспрессию/локализацию p120ctn, в клетках с подавленной экспрессией Т-кадгерина содержание p120ctn в мембранной фракции увеличивалось. Это позволяет предполагать, что при подавлении Т-кадгерина содержание p120ctn в составе VE-кадгеринового комплекса увеличивается, что стабилизирует адгезивные контакты и усиливает барьерную функцию эндотелия (Рисунок 3.85). 303 Rho ГТФазы (RhoA, Rac1, и Cdc42) являются ключевыми регуляторами барьерной функции эндотелия, контролирующими уровень фосфорилирования белков кадгеринового комплекса, формирование актиновых стресс-фибрилл и регуляцию активности актомиозинового комплекса [Hall, 1998, Ponimashkin et al., 2002]. Активная ГТФаза RhoA стимулирует формирование стресс-фибрилл актина, а также активирует Rho киназы ROCK I и II типов, приводя к фосфорилированию легких цепей миозина и сокращению клеток [Arber et al., 1998, Essler et al., 1998, Hordijk et al., 1999]. Активные ГТФазы Cdc42 и Rac связываются и активируют киназу PAK1, которая контролирует сократимость клеток за счѐт фосфорилирования LIMK1 киназы [Maekawa et al., 1999, Dan et al., 2001]. В настоящей работе мы проверяли предположение о том, что Т-кадгерина может опосредовать свои эффекты в эндотелиальных клетках через активацию Rho ГТФаз. Действительно, сначала на линейных фибробластах NIH3T3 было обнаружено, что гиперэкспрессия Т-кадгерина приводит к активации сигнальных каскадов с участием Rac1 и Cdc42, но не RhoA, а в дальнейшем было показано, что гиперэкспрессия Т-кадгерина вызывает активацию RhoA, Rac1 и Cdc42 в эндотелиальных клетках. Известно, что точный паттерн активации конкретных Rho ГТФаз и последующая сигнализация может быть различна в разных типах исследуемых клеток [Yap and Kovacs, 2003]. Поэтому, в дальнейших экспериментах для изучения Т-кадгерин-зависимой внутриклеточной сигнализации мы использовали первичную культуру эндотелиальных клеток человека (HUVEC). Было обнаружено, что гиперэкспрессия Т-кадгерина вызывает активацию RhoA, Rac1 и Cdc42, в то время как в контрольных и si-Т-cad клетках подобных эффектов обнаружено не было. С помощью вестерн блоттинга и ингибиторного анализа было продемонстрировано, что гиперэкспрессия Т-кадгерина вызывает активацию прямых мишеней Rho ГТФаз: PAK1 и ROCK, а также LIMK1 киназы, что приводит к реорганизации микротрубочкового цитоскелета. Последующий анализ с помощью конфокального микроскопа и ингибиторного анализа подтвердил, что активация сигнального каскада с участием RhoA, Rac1 и Cdc42 приводит к активации адаптерных белков и вызывает сборку стресс-фибрилл актина. Известно, что помимо регуляции динамического состояния цитоскелета, активация Rac1/Cdc42 сигнального пути активирует митогенные киназы p38/MAPK и ERK1/2, которые влияют на активность генов, ответственных за клеточную подвижность, апоптоз, воспаление, а также пролиферацию клеток и дифференцировку [Zhang and Liu 2002, Stockton et al., 2004]. Результаты наших экспериментов указывают на то, что изменения экспрессии Т-кадгерина не влияют на уровень фосфорилирования p38 или ERK1/2 в эндотелиальных клетках. Ранее в ряде публикаций было показано, что активация Src 304 киназы приводит к фосфорилированию VE-кадгерина по тирозиновым остаткам Y658, Y685 [Wallez et al., 2007, Dejana et al., 2008] и Y731 [Allingham et al., 2007], что вызывает нарушение взаимодействия VE-кадгерина с катенинами и цитоскелетом. В настоящей работе связи между уровнем экспрессии Т-кадгерина и активацией/фосфорилированием киназы Src в эндотелиальных клетках, обнаружено не было. Эти результаты позволяют предположить, что в эндотелиальных клетках Т-кадгерин вызывает активацию именно Rhoопосредованного сигнального каскада, но не оказывает влияния на активность p38, Erk1/2 и Src. Таким образом, представленные данные указывают на то, что гиперэкспрессия Ткадгерина снижает барьерную функцию эндотелия за счет клатрин-опосредованного эндоцитоза основного белка адгезивных контактов VE-кадгерина с его последующей деградацией в лизосомах. Предположительно, в основе механизма интернализации VEкадгерина лежит Rho-зависимое фосфорилирование цитоплазматического домена VEкадгерина по тирозину в 731 положении. Активация Rho ГТФаз приводит также к активации нисходящих сигнальных посредников ROCK-II, LIMK и PAK1, вызывает сборку актиновых стресс-фибрилл и деполимеризацию микротрубочек. Патологические процессы, такие как воспаление, атеросклероз, артериальная гипертензия и сахарный диабет, вызывают нарушение барьерной функции эндотелия, которое связано с активацией/дисфункцией эндотелиальных клеток и сопровождается разборкой адгезивных контактов. Полученные результаты позволяют предполагать, что помимо хорошо известных агентов, влияющих на проницаемость эндотелия, Т-кадгерин является еще одним важным регулятором его барьерной функции (Рисунке 3.98). Наши результаты позволяют предположить возможную связь между повышением уровня Ткадгерина в эндотелиальных клетках кровеносных сосудов и прогрессированием сердечнососудистых заболеваний, характеризующихся ослаблением барьерной функции эндотелия. 305 Рисунок 3.98. Механизмы регуляции барьерной функции эндотелия с участием Ткадгерина. 306 3.6 Т-кадгерин при атеросклерозе Сердечно-сосудистые заболевания представляют собой глобальную проблему во всем мире и являются ведущей причиной смертности. В отличие от других сердечнососудистых заболеваний, атеросклероз прогрессирует достаточно медленно в течение нескольких десятилетий. Однако по непонятным причинам состояние сосудистой стенки в зоне атеросклеротического поражения может внезапно меняться, что может вызывать тромбоз и потенциально приводить к летальному исходу. Описано несколько типов атеросклеротических поражений, в зависимости от стадии и прогрессии заболевания: липидная полоска, фиброатерома и фиброзная бляшка, при этом структура и клеточный состав сосудистой стенки претерпевают значительные изменения. Формирование атеросклеротической бляшки сопровождается усиленной продукцией компонентов внеклеточного матрикса, увеличением содержания накапливающих липиды пенистых клеток, воспалением, некрозом и клеточной деградацией в зоне поражения [Stary et al., 1995]. На начальном этапе развития атеросклероза происходит формирование липидной полоски, характеризующееся появлением в интиме артерий пятен и полосок, содержащих липиды. Для этой стадии характерно повреждение эндотелия и возникновение эндотелиальной дисфункции, сопровождающееся повышением проницаемости эндотелиального барьера. В липидных полосках, главным образом, присутствуют пенистые клетки и Т-лимфоциты, а также макрофаги и гладкомышечные клетки. Позднее при прогрессировании атеросклероза в участках отложения липидов происходит разрастание соединительной ткани, что ведет к формированию фиброзной бляшки, в центре которых образуется липидное ядро. Вокруг липидного ядра формируется «покрышка», представляющая собой соединительнотканный каркас, отделяющий ядро от просвета сосуда. Параллельно происходит васкуляризация очага атеросклеротического поражения в силу присутствия большого количества факторов роста, стимулирующих неоангиогенез [Stary et al., 1995]. Вновь образующиеся сосуды (vasa vasorum) нестабильны, характеризуются повышенной проницаемостью, склонностью к образованию микротромбов и разрывам в сосудистой стенке. Это в свою очередь приводит к еще большему накоплению липидов и вторичному воспалению [Virmani et al., 2005]. Клиническое течение атеросклероза и прогноз зависит от структуры фиброзной покрышки и размеров липидного ядра. Даже на ранних стадиях формирования бляшки при хорошо выраженном липидном ядре и тонкой соединительнотканной капсуле, существует высокий риск возникновения повреждений и разрывов фиброзной капсулы под действием высокого 307 артериального давления с формированием «осложненной» атеросклеротической бляшки. В случае если фиброзная покрышка хорошо выражена, она меньше подвержена повреждению и разрывам [Hansson, 2005]. Механизмы возникновения и прогрессии атеросклероза, а также причины формирования стабильных и нестабильных бляшек до сих пор остаются неясны [Lloyd-Jones et al., 2009]. Принято считать, что значительную роль в развитии атеросклероза, (ре)стеноза, ишемии и инфаркта миокарда и др., играет изменение экспрессии белков межклеточной адгезии на поверхности клеток сосудов, включая резидентные эндотелиальные и гладкомышечные клетки, а также мигрирующие из крови иммунные клетки и клеткипредшественники. В норме барьерная функция эндотелия сосудов обеспечивается за счет межклеточных контактов [Dejana, 2004]. В этом процессе участвуют адгезивные межклеточные VE-кадгерин содержащие контакты, а также плотные контакты, содержащие белки окклюдины и клаудины [Vestweber, 2003; Lampugnani and Dejana, 1997]. Эндотелиальные клетки взаимодействуют с муральными клетками (гладкомышечными клетками (ГМК) и перицитами) за счет N-кадгерин содержащих адгезивных контактов, а с белками внеклеточного матрикса за счет белков семейства интегринов, что обеспечивает стабильность структуры и функции сосудов [Armulik et al., 2005]. В адвентиции атеросклеротических бляшек муральных клеток, которые формируются вокруг новообразованных vasa vasorum всегда недостаточно, поэтому характерной особенностью этих сосудов является повышенная проницаемость [Sluimer et al., 2009]. Активация эндотелиальных клеток [Rondaij et al., 2006]. При активации эндотелия нарушаются межклеточные взаимодействия, и происходит разрушение кадгерин-содержащих адгезивных контактов, что сопровождается экспрессией на поверхности эндотелия P- и Eселектинов, способствующих адгезии лейкоцитов, а также молекул адгезии семейства иммуноглобулинов ICAM-1 и VCAM-1, обеспечивающих прочную адгезию клеток воспаления [Imhof and Aurrand-Lions, 2006]. ГМК также претерпевают фенотипические изменения, приводящие к изменению секреторного профиля, активации их миграции и пролиферации [Thyberg et al., 1990; Ross 1993; Jang et al., 1994]. При этом у ГМК изменяется профиль экспрессии молекул адгезии: на поверхности клеток наблюдается экспрессия 1-интегрина [Boudreau et al., 1991] и VCAM-1 [Li et al., 1993; O‘Brien et al., 1993], способствующих прогрессии атеросклероза. Ранее в нашей лаборатории было обнаружено, что в сердечно-сосудистой системе человека и животных присутствует еще один представитель суперсемейства молекул адгезии, нетипичный GPI-заякоренный Ткадгерин [Ivanov et al., 2001]. 308 Наибольшая экспрессия Т-кадгерина во взрослом организме была обнаружена в аорте, сонных артериях, подвздошных, почечных артерий и сердце. Было показано, что Ткадгерин экспрессирован во всех слоях сосудистой стенки у человека, однако, наибольший уровень его экспрессии обнаружен в эндотелиальных клетках, перицитах и ГМК протеогликанового слоя интимы и vasa vasorum адвентиции [Ivanov et al., 2001]. Также было обнаружено, что экспрессия Т-кадгерина повышается при некоторых сердечнососудистых заболеваниях [Ivanov et al., 2001; Kudrjashova et al., 2002]. Для выявления возможной функции Т-кадгерина в сосудах во взрослом организме было проведено двойное иммуногистохимическое окрашивание аутопсийных образцов аорты здоровых доноров и больных атеросклерозом на разных стадиях. Окрашивание параллельных криосрезов проводили с использованием антител к Т-кадгерину и антигену ГМК α-SMA или антигену эндотелиальных клеток CD31. 3.6.1 Экспрессия Т-кадгерина в здоровых сосудах Иммуногистохимическое окрашивание криосрезов ткани выявило экспрессию Ткадгерина во всех слоях стенки аорты (Рисунок 3.99 и Рисунок 3.100): во внутреннем, интимальном слое сосуда, в медии, и в наружном адвентициальном слое сосудистой стенки. При этом в эндотелиальном слое, выстилающем просвет сосуда, экспрессия Т-кадгерина наблюдалась во всех клетках, позитивных по CD31 (Рисунок 3.99 А, Б). Также Т-кадгерин экспрессируется в некоторых CD31-позитивных клетках, расположенных в субэндотелиальным слое. Данные об экспрессии Т-кадгерина в эндотелии сосудов согласуются с данными, полученными нами ранее по экспрессии Т-кадгерина во всех клетках первичной культуры эндотелиальных клеток HUVEC (Рисунок 3.75). В медии Ткадгерин обнаруживается в ГМК, позитивных по α-SMA, а также в vasa vasorum (стрелка на Рисунке 3.100 Г). Однако в отличие от эндотелиальных клеток не все ГМК в аорте экспрессируют Т-кадгерин: в медии сосуда обнаруживаются α-SMA-позитивные клетки, не экспрессирующие Т-кадгерин. Во внутреннем интимальном слое присутствует незначительное количество ГМК, экспрессирующих Т-кадгерин. Стрелками на Рисунке 3.100 показаны двойные позитивные по Т-кадгерину и α-SMA клетки. 309 Рисунок 3.99. Двойное иммуногистохимическое окрашивание нормальной аорты человека антителами к Т-кадгерину (розовый цвет) и маркѐру эндотелиальных клеток CD31 (чѐрный цвет). Ядра клеток докрашены гематоксилином (синий цвет). (А, Б) – слой интимы (И) сосуда, (В, Г) – слой медии (М) сосуда. Стрелками указаны клетки, коэкспрессирующие Т-кадгерин и CD31 антиген эндотелиальных клеток. В здоровой ткани Т-кадгерин экспрессируется в эндотелиальных клетках, выстилающих просвет сосуда, а также в CD31-позитивных клетках, расположенных в субэндотелиальным слое (стрелки на рисунке Б), в медии сосуда – в vasa vasorum (стрелки на рисунке Г). Масштаб А, и В - 50 мкм, Б и Г – 20 мкм. 310 Рисунок 3.100. Двойное иммуногистохимическое окрашивание нормальной аорты человека антителами к Т-кадгерину (розовый цвет) и маркѐру гладкомышечных клеток α-SMA (чѐрный цвет). Ядра клеток докрашены гематоксилином (синий цвет). (А, Б) – слой интимы (И) сосуда, (В, Г) – слой медии (М) сосуда. Стрелками указаны клетки, коэкспрессирующие Т-кадгерин и a-SMA антиген ГМК. В здоровой ткани Т-кадгерин экспрессируется в ГМК, расположенных в субэндотелиальным слое (стрелки на рисунке Б) и в медии сосуда (стрелки на рисунке Г). Масштаб - 50 мкм. 311 Также Т-кадгерин присутствует в следовых количествах в протеогликановом слое сосудистой стенки, о чѐм свидетельствует положительное окрашивание внеклеточного матрикса, не содержащего клетки, антителами к Т-кадгерину (Рисунок 3.99 и Рисунок 3.100), что является либо неспецифическим окрашиванием, либо свидетельствует о возможном слущивании белков с поверхности клеток, в том числе и Т-кадгерина. Для подтверждения этого предположения необходимо проведение дальнейших исследований. На Рисунке 3.101 представлены параллельные криосрезы интимы и медии здорового сосуда, окрашенного красителем Oil Red, для выявления триглицеридов и липидов. Отсутствие Oil Red свидетельствует о том, что в стенке здорового сосуда накопления липидов не выявляется. Таким образом, полученные данные свидетельствуют о том, что в норме наибольшая экспрессия Т-кадгерина наблюдается в интиме аорты в эндотелиальных клетках, а в медии - в части ГМК и в vasa vasorum. Рисунок 3.101. Окрашивание Oil Red нормальной аорты человека. В стенке здорового сосуда накопления липидов не выявлено. И – интима сосуда, М - медия сосуда. Масштаб 25 мкм. 312 3.6.2 Экспрессия Т-кадгерина в сосудах при атеросклерозе Для изучения экспрессии Т-кадгерина в сосудах при атеросклерозе были использованы образцы ткани разных типов атеросклеротических поражений, а именно: липидная полоска, нестабильная фиброатерома и мягкий кальциноз. 3.6.2.1 Липидная полоска Липидная полоска была выявлена микроскопически при окрашивании красителем Oil Red как плоская желтоватая полоска в интимальной зоне сосуда (Рисунок 3.102). При окрашивании липидной полоски красителем Oil Red были выявлены типичные скопления пенистых клеток различного размера и плотности, расположенные в субинтимальном слое сосудистой стенки. При иммуногистохимическом окрашивании криосрезов аорты в липидной полоске были обнаружены патологические изменения, характерные для данного типа. Так, при двойном иммуногистохимическом окрашивании антителами к Т-кадгерину и антигену эндотелиальных клеток CD31 было обнаружено утолщение интимального слоя (Рисунок 3.103 А, Б, В). Как и в нормальной аорте экспрессия Т-кадгерин обнаруживалась во всех эндотелиальных клетках (Рисунок 3.103 А, Б). Экспрессия Т-кадгерина также была выявлена в некоторых CD31-позитивных клетках, расположенных в субэндотелиальным слое. Следует отметить, что в медиальном слое сосуда возрастает доля клеток, коэкспрессирующих CD31 - маркѐр эндотелиальных клеток и Т-кадгерин. Аналогичным образом в липидной полоске увеличивается количество клеток ко-экспрессирующих α-SMA и Т-кадгерин по сравнению с нормальным, здоровым сосудом, причем не только в медии, но и в интиме (Рисунок 3.103 Г, Д, Е). 313 Рисунок 3.102. Атеросклеротическое поражение аорты человека. Липидная полоска. Окрашивание Oil Red. И – интима сосуда, М - медия сосуда, А – адвентиция сосуда. Липидная полоска выявляется как плоская полоска желтоватого цвета в субинтимальной зоне сосуда. Стрелкой на рисунке А указано накопление липидов. Масштаб 25 мкм. 314 Рисунок 3.103. Атеросклеротическое поражение аорты человека. Липидная полоска. (А, Б, В, И) - Двойное иммуногистохимическое окрашивание антителами к Ткадгерину (розовый цвет) и маркѐру эндотелиальных клеток CD31 (чѐрный цвет). (Г Д, Е, К) - Двойное иммуногистохимическое окрашивание антителами к Т-кадгерину (розовый цвет) и антигену гладкомышечных клеток α-SMA (чѐрный цвет). Ядра клеток докрашены гематоксилином (синий цвет). И - интима сосуда, М – медия сосуда, А – адвентиция сосуда. Т-кадгерин экспрессируется в эндотелиальных клетках, выстилающих просвет сосуда, и в клетках, расположенных в субэндотелиальным слое. Экспрессия Т-кадгерина обнаруживается также в ГМК медии сосуда. При данном типе поражения аорты характерно утолщение интимы. Стрелками на рисунке И показаны vasa vasorum в адвентициальном слое сосуда, ко-экспрессирующие CD31 и Т-кадгерин. Масштаб 50 мкм. 315 3.6.2.2 Нестабильная фиброатерома Для этого типа атеросклеротического поражения характерно присутствие липидных капель в интимальном и медиальном слое сосуда, которое выявляли при окрашивании Oil Red. На Рисунке 3.104 и Рисунке 3.105 представлены криосрезы нестабильной фиброатеромы, характеризующейся разрастанием соединительной ткани в стенке сосуда. В отличие от нормы и липидной полоски, в образцах аорты с нестабильной фиброатеромой экспрессия Т-кадгерина была повышена (Рисунок 3.105). Целостность интимы при данном повреждении сосуда нарушается, о чѐм свидетельствует отсутствие экспрессии CD31 в слое клеток, выстилающих просвет (Рисунок 3.105 А, Б). В субинтимальном слое фиброатеромы аорты увеличивается количество клеток, экспрессирующих Т-кадгерин и интенсивность окрашивания на этот маркѐр. Во внутреннем слое сосуда, окрашенном антителами к CD31, маркѐру эндотелиальных клеток, экспрессия Т-кадгерина существенно увеличивается (стрелки на Рисунке 3.105). Т-кадгерин интенсивно окрашивается в ГМК медии, а также в vasa vasorum. Экспрессия Т-кадгерина также детектируется во внеклеточном матриксе медиального слоя сосуда, о чѐм свидетельствует положительное окрашивание на Т-кадгерин бесклеточных структур (стрелка на Рисунке 3.105 Б). В ряде случаев при формировании фиброатеромы наблюдается кальцификация в области липидного ядра и других частях атеросклеротического поражения. 316 Рисунок 3.104. Атеросклеротическое поражение аорты человека. Нестабильная фиброатерома. Окрашивание Oil Red. И - интима сосуда, М – медия сосуда. Стрелками обозначены накопления липидов в сосудистой стенке. Масштаб 25 мкм. Рисунок 3.105. Атеросклеротическое поражение аорты человека. Нестабильная фиброатерома, характеризующаяся разрастанием соединительной ткани в стенке сосуда. А - Двойное иммуногистохимическое окрашивание антителами к Т-кадгерину (розовый цвет) и маркѐру эндотелиальных клеток CD31 (чѐрный цвет). Б - Двойное иммуногистохимическое окрашивание антителами к Т-кадгерину (розовый цвет) и антигену гладкомышечных клеток α-SMA (чѐрный цвет). Ядра клеток докрашены гематоксилином (синий цвет). В Медиальном (М) слое сосуда наблюдается увеличение экспрессии Т-кадгерина в сосудах (стрелки на рисунке А). Кроме того, обнаруживается эктопическое внеклеточное окрашивание на Т-кадгерин. На рисунке Б в центре стрелкой указан некроз. Масштаб 50 мкм. 3.6.2.3 Мягкий кальциноз Мягкий кальциноз при атеросклеротическом поражении сосуда характеризуется массивным отложением солей кальция в атеросклеротической бляшке. При изучении экспрессии Т-кадгерина в данном типе поражения было обнаружено, что Т-кадгерин присутствует в интиме сосуда, но в то же время, количество эндотелиальных клеток, 317 экспрессирующих CD31, в интиме сосуда значительно снижено (Рисунок 3.106). Это свидетельствует о явном повреждении эндотелиального слоя, выстилающего просвет аорты. При этом по сравнению с нормой в медиальном слое поврежденного сосуда экспрессия Т-кадгерина резко возрастет, как в клетках, экспрессирующих CD31, так и в αSMA-позитивных гладкомышечных клетках (Рисунок 3.106 А, Б). Также в медиальном слое аорты увеличивается толщина коллагеновых волокон (Рисунок 3.106). Увеличение коллагена в медии, в данном случае, может свидетельствовать об активации ГМК, которые активно продуцируют компоненты внеклеточного матрикса. Рисунок 3.106. Атеросклеротическое поражение аорты человека. Мягкий кальциноз. (А, Б) - Двойное иммуногистохимическое окрашивание антителами к Ткадгерину (розовый цвет) и маркѐру эндотелиальных клеток CD31 (чѐрный цвет). (В, Г) - Двойное иммуногистохимическое окрашивание антителами к Т-кадгерину (розовый цвет) и антигену гладкомышечных клеток α-SMA (чѐрный цвет). Ядра клеток докрашены гематоксилином (синий цвет). И - интима сосуда, М – медия сосуда. Увеличивается количество ГМК, экспрессирующих Т-кадгерин. В медии увеличивается доля клеток, ко-экспрессирующих Т-кадгерин и CD31. Масштаб 50 мкм. 318 3.6.3 Обсуждение результатов Полученные результаты об экспрессии Т-кадгерина в интиме, медии и адвентиции нормальной аорты свидетельствуют о том, что этот белок экспрессируется во всех слоях сосуда в эндотелиальных и гладкомышечных клетках и в протеогликановом слое. Данные о повышении экспрессии Т-кадгерина в эндотелиальных клетках в сосуде при различных заболеваниях [Рисунки 3.103 - 3.106, а также Ivanov et al., 2001], и выявление Т-кадгерина на микрочастицах в крови пациентов с атеросклерозом [Philippova et al., 2011] предположительно свидетельствуют о происходящих процессах активации/дисфункции эндотелия. Активация/дисфункция эндотелия является характерной особенностью многих патологических состояний, таких как атеросклероз, и сопровождается нарушением межклеточных взаимодействий и, как следствие, увеличением проницаемости эндотелия [Frey et al., 2009]. Данные о повышении экспрессии Т-кадгерина при атеросклерозе коррелируют с данными, полученными в настоящей работе на культуре эндотелиальных клеток HUVEC о том, что гиперэкспрессия Т-кадгерина вызывает снижение барьерной функции эндотелия. Многочисленные патофизиологические исследования на животных и человеческом аутопсийном материале свидетельствуют о том, что исходно происходящее при атеросклерозе повреждение эндотелиального монослоя вызывает компенсаторную реакцию и активацию эндотелия, что в свою очередь приводит к нарушению нормального гомеостаза и функционирования эндотелиального монослоя [Du et al., 2012]. Активация/дисфункция эндотелия включает увеличение проницаемости эндотелиального монослоя, накопление липопротеидов в сосудистой стенке, увеличение продукции NO, эйкозаноидов, цитокинов, факторов роста, вазоактивных пептидов и фибринолитических факторов. Исследования последних лет показали, что из костного мозга и из периферической крови можно выделить популяцию CD34+VEGFR-2+ эндотелиальных прогениторных клеток (ЭПК), впервые обнаруженную и охарактеризованную в лаборатории Асахара [Asahara et al., 1997]. ЭПК можно культивировать в условиях in vitro и дифференцировать в клетки, имеющие свойства зрелого эндотелия (CD31+, E-селектин+, эндотелиальная NO синтаза (eNOS)+, и обладающие способностью эндоцитировать липопротеиды низкой плотности (ЛНП). Позднее было обнаружено, что эти ЭПК способны мобилизоваться в периферическую кровь из костного мозга в ответ на различные повреждения органов и тканей, в том числе и в ответ на повреждение сосудов [Du et al., 2012]. Мобилизованные 319 ЭПК мигрируют в зону повреждения, где они дифференцируются в зрелые эндотелиальные клетки in situ, а также оказывают паракринный стимулирующий регенерацию эффект [Du et al., 2012]. Также считается, что именно ЭПК вносят существенный вклад в постнатальный ангиогенез в норме и при патологическом неоангиогенезе, характерном для опухолевого роста и атеросклероза [Du et al., 2012]. Несмотря на значительный прогресс в области изучения возможного использования ЭПК для терапии сердечно-сосудистых заболеваний, до сих пор остается открытым вопрос о патофизиологической роли собственных эндогенных ЭПК в прогрессии атеросклероза или участии этих клеток в репарации сосудов при атеросклерозе [Du et al., 2012]. В настоящей работе было обнаружено увеличение количества CD31-позитивных клеток в субэндотелиальным слое и в медии при атеросклерозе, что, по всей видимости, отражает мобилизацию клеток костномозгового происхождения или активацию и миграцию резидентных прогениторных клеток сосудистой стенки в бляшку. Впервые в настоящем исследовании обнаружено, что эти клетки предшественники экспрессируют Ткадгерин. Работ об экспрессии Т-кадгерина в прогениторных клетках на настоящий момент в литературе нет, и выяснение роли Т-кадгерина в миграции и дифференцировке ЭПК из костного мозга или периферической крови или миграции резидентных прогениторных/столовых клеток сосудистой стенки в норме и при патологии нуждается в дальнейшем исследовании. Известно, что vasa vasorum нормальных артерий представляют собой функционально значимую микрососудистую сеть, локализующуюся в адвентиции и питающие сосуды. В атеросклеротической бляшке экспрессируется большое количество проангиогенных факторов, вызывающих активный ангиогенез, в результате которого вновь образующиеся vasa vasorum прорастают внутрь бляшки, что является причиной формирования нестабильной бляшки и увеличивает риск летального исхода [Kolodgie et al., 2010]. Высокая экспрессия Т-кадгерина в активно прорастающих в бляшку vasa vasorum, возможно, отражает навигационные свойства этого белка при таком патологическом ангиогенезе, которые были описаны выше. В отличие от эндотелиальных клеток не все ГМК в аорте экспрессируют Ткадгерин: в медии сосуда обнаруживаются α-SMA-позитивные клетки, не экспрессирующие Т-кадгерин. Ранее в нашей лаборатории было обнаружено, что уровень экспрессии Т-кадгерина в ГМК сосуда может варьировать в зависимости от их фенотипа [Ivanov et al., 2001]. Резидентные ГМК интимы, располагающиеся в субэндотелиальном слое, экспрессируют мало α-SMA и много Т-кадгерина, в то время как в более глубоких 320 слоях интимы ГМК, имеющие контрактильные свойства и экспрессирующие α-SMA, содержат меньшее количество Т-кадгерина. По всей видимости, экспрессия Т-кадгерина больше характерна для ГМК секреторного фенотипа, чем для контрактильного. Для медии же характерно присутствие ГМК, экспрессирующих большое количество α-SMA и имеющих сократительный фенотип, необходимый для поддержания тонуса сосуда [Ivanov et al., 2001]. Увеличение количества ГМК, экспрессирующих Т-кадгерин, возможно, отражает фенотипические изменения ГМК при атеросклерозе и увеличение количества клеток секреторного фенотипа, что коррелирует с патологическими процессами в бляшке. Повышенный уровень липопротеидов низкой плотности (ЛНП) в плазме крови служит фактором риска развития атеросклероза, так как приводит к накоплению липидов в сосудистой стенке [Goldstein et al., 1985]. ЛНП представляют собой макромолекулярные комплексы и служат для транспорта холестерина и триглицеридов к периферическим тканям в организме. Перенос ЛНП происходит путем рецептор-опосредованного эндоцитоза, в котором участвует «классический» апо В, Е рецептор [Brown and Goldstein, 1986]. ЛНП также способны влиять на функциональную активность целого ряда клеток, причем эти эффекты не связаны с эндоцитозом [Bochkov et al., 1991]. Помимо описанных эффектов, ЛНП способны активировать макрофаги [Kelley et al., 1988], вызывать изменение формы и агрегацию тромбоцитов, секрецию сурфактанта альвеолоцитами [VoynoYasenetskaya et al., 1993], миграцию и пролиферацию гладкомышечных клеток (ГМК) в сосудистой стенке [Resink et al., 1995], регулировать сокращение сосудов [Sachinidis et al., 1990]. Такие эффекты сходны с действием гормонов и медиаторов, и поэтому было предложено называть их «гормоноподобными» [Block et al., 1988]. Ряд данных позволяет предположить, что в основе «быстрых» клеточных эффектов липопротеидов лежит активация систем внутриклеточной сигнализации. В пользу этого предположения свидетельствуют данные о способности липопротеидов влиять на активность аденилатциклазы, изменять рН цитоплазмы и стимулировать фосфорилирование специфических клеточных белков [Pedreno et al., 2001]. Ранее в нашей лаборатории было показано, что ЛНП стимулируют фосфоинозититидный обмен, активируют фосфолипазу С и вызывают мобилизацию внутриклеточного Ca2+ в ГМК [Bochkov et al., 1993]. В отличие от хорошо изученного «метаболического» действия ЛНП, опосредуемого апо В,Е-рецептором, механизмы «гормоноподобных» эффектов мало известны. Характерной особенностью таких эффектов является их быстрота, насыщаемость и обратимость, что свидетельствует об участии специфического рецептора в передаче сигнала от ЛНП внутрь клетки, однако, ни природа предполагаемого рецептора, ни механизмы его сопряжения с системами вторичных посредников долгое время оставались 321 неясны. В нашей лаборатории был обнаружен атипичный участок специфического связывания ЛНП на культивируемых ГМК (гладкомышечных клетках) сосудов [Bochkov et al., 1994]. Анализ связывания 125 I-ЛНП с мембранами ГМК показал, что ГМК помимо высокоаффинного, «классического» участка связывания с Кd 1-2 мкг/мл экспрессируют низкоаффинный участок специфического связывания ЛНП с К d40-50 мкг/мл [Tkachuk et al, 1994]. Для выделения этого атипичного участка связывания был использован метод лигандного блоттинга. Используя мембраны ГМК медии аорты человека, в нашей лаборатории были выделен, очищен и охарактеризован белок р105/р130. Анализ лигандной селективности и чувствительности к различным ингибиторам показал, что р105/р130 является новым ЛНП-связывающим белком, отличным от всех известных липопротеидсвязывающих рецепторов [Bochkov et al., 1996]. Параметры связывания 125I-ЛНП с белками р105/р130 совпадают с параметрами ЛНП-опосредованной активации систем вторичных посредников [Stambolsky et al., 1999]. Сиквенирование очищенных р105/р130 показало, что последовательность этих белков соответствует последовательности Т-кадгерина, причем р105 представляет собой его зрелую форму, а р130 – предшественник [Tkachuk et al, 1996]. Связывание Т-кадгерина с ЛНП ингибируется антителами против ЛНП (антитела против апо В белка) и поликлональными антителами против Т-кадгерина, произведенными в нашей лаборатории [Bochkov et al., 1996]. Таким образом, ранее нами был получен ряд данных, позволяющих предположить, что Т-кадгерин является возможным «сигнальным» рецептором ЛНП [Tkachuk et al, 1996]. На основании полученных данных был сделан вывод о том, что экспрессия Ткадгерина повышается в сосудистых клетках при атеросклерозе (Рисунки 3.103 и 3.104, а также Ivanov et al., 2001). Однако до сих пор не ясно, какую функцию при этом выполняет Т-кадгерин, является ли Т-кадгерин при этом рецептором ЛНП, и, если да, то имеет ли это какое-либо физиологическое значение для клетки. Для того, чтобы доказать, что Т-кадгерин является рецептором ЛНП и способен опосредовать связывания и 125 изменение гормоноподобные эффекты ЛНП, мы проанализировали параметры I-ЛНП с мембранами линейных клеток, гиперэкспрессирующих Т-кадгерин, концентрации внутриклеточного кальция, отражающего внутриклеточной сигнализации, в культуре тех же клеток при добавлении ЛНП. активацию 322 3.7 Т-кадгерин-рецептор липопротеидов низкой плотности 3.7.1 Получение клонов клеток, гиперэкспрессирующих Т-кадгерин. Анализ экспрессии Т-кадгерина в клонах Для изучения Т-кадгерина как возможного рецептора липопротеидов низкой плотности мы получали клоны клеток, гиперэкспрессирующих Т-кадгерин, были использованы 2 линии клеток: линия почечного эпителия человека HEK293 и линия мышиных фибробластов L929. Для гиперэкспрессии Т-кадгерина клетки HEK293 котрансфицировали плазмидами pcDNA3.1/T-cad, кодирующей кДНК Т-кадгерина человека, и pcDNA3.1/GFP, кодирующей ген зеленого флуоресцентного белка. Контрольные клетки ко-трансфицировали плазмидами pcDNA3.1, кодирующей антисмысловую последовательность люциферазы, и pcDNA3.1/GFP. В результате трансфекции с последующим клонированием на селективной среде, содержащей антибиотик пуромицин, были отобраны два клона клеток, гиперэкспрессирующих Т-кадгерин (Th5 и Th8) и GFP, а также клон контрольных клеток, экспрессирующих GFP (GFP). Уровень экспрессии Ткадгерина в выбранных клонах оценивали методом иммуноблоттинга (Рисунок 3.107). В работе также использовали клоны клеток линии L929, клон ТС3, гиперэкспрессирующий Ткадгерин, и контрольный клон (К9), полученные и протестированные на экспрессию Ткадгерина аналогичным образом. Анализируя полученные результаты, мы обнаружили, что выбранные клоны Th5, Th8 и ТС3, также как и контрольные HUVEC, экспрессируют Т-кадгерин в двух формах – в виде зрелого белка с молекулярной массой 105 кДа, и в виде предшественника, с молекулярной массой 130 кДа. Клон GFP линии HEK293 Т-кадгерин не экспрессирует. 323 130 кДа 105 кДа GFP ТС3 К9 Th5 Th8 HUVEC Рисунок 3.107. Анализ экспрессии Т-кадгерина после трансфекции клеток плазмидой, содержащей ген человеческого Т-кадгерина в клонах клеточных линий НЕК293 и L929. Лизаты клеток (50 мкг белка на дорожку) были проанализированы методом электрофореза в 10% ПААГе с последующим Вестен блоттингом с использованием антител против ЕС1 домена Т-кадгерина. GFP - клон контрольных клеток HEK293 (1 дорожка); TC3 - клон клеток L929, гиперэскпрессирующий Т-кадгерин (2 дорожка); K9 - контрольный клон клеток L929 (3 дорожка); Th5 - клон клеток HEK293 (4 дорожка) и Th8 (5 дорожка) – клоны клеток НЕК293, гиперэкспрессирующих Т-кадгерин человека; в качестве положительного контроля использовали клетки HUVEC, нативно экспрессирующие Т-кадгерин (6 дорожка). 3.7.2 Связывание [125I]-ЛНП с мембранами клеток L929 и HEK293 Для изучения параметров связывания ЛНП с мембранами клеток, гиперэкспрессирующих Т-кадгерин, и мембранами контрольных клеток, использовали ЛНП, меченные радиоактивным 125I (125I-ЛНП). В эксперименте использовали клетки линии HEK293: Th5 и Th8, гиперэкспрессирующие Т-кадгерин, и контрольный клон GFP; а также клетки линии L929: ТС3, гиперэкспрессирующий Т-кадгерин, и контрольный клон К9. Количество связанных 125 I-ЛНП с мембранами клеток рассчитывали после измерения радиоактивности клеточных лизатов на γ-счетчике. При этом конечная радиоактивность йода (в пересчѐте на белок) составляла 450 cpm/нг. Полученные данные нормировали по общей концентрации белка. Для изучения связывания мембран клеток, гиперэкспрессирующих Т-кадгерин, с ЛНП мы использовали свежую донорскую плазму. Далее методом препаративного ультрацентрифугирования в градиенте плотности раствора NaBr выделяли и диализовали ЛНП. Метили ЛНП радиоактивной меткой, используя монохлорид йода ( 125ICl). На Рисунке 3.108 представлены данные, показывающие концентрационную зависимость связывания 125 I-ЛНП с мембранами клеток линии L929 (ТС3 и К9). Специфическое связывание 125I-ЛНП с мембранами клеток вычисляли как разность между величиной общего связывания и неспецифического связывания, которое определяли в присутствии избытка немеченных ЛНП (для этого клетки инкубировали с 200-кратным избытком немеченных ЛНП). Связывание 125 I-ЛНП проводили при температуре +40C для минимизации интернализации ЛНП [Tkachuk et al., 1994]. Связывание меченных 125 I-ЛНП 324 клетками анализировали с помощью линейной и нелинейной регрессии с использованием программы GraphPad PRISM 4.0. Полученные данные свидетельствуют о связывании лиганда с двумя участками. Прямые пересекают ось 0У в двух различных точках (Рисунок 3.108), что подтверждает наши ранее полученные данные о том, что помимо специфичного, высокоаффинного рецептора ЛНП с Кd 1-2 мкг/мл, присутствует низкоаффинный сайт связывания ЛНП, предположительно Т-кадгерин, с Кd 40-50 мкг/мл [Tkachuk et al, 1994]. Обе линии пересекают ось 0Х в отрицательных координатах, причѐм точка пересечения с осью 0Х для клеток ТС3 расположена ближе к началу координат, чем для К9. Эти результаты свидетельствует о том, что гиперэкспрессия Т-кадгерина приводит к увеличению числа низкоаффинных участков связывания (Bmax) без достоверного изменения константы связывания (Kd). 0.00012 1/cpm, имп/мин 0.0001 0.00008 0.00006 0.00004 0.00002 0 -0.1 -0.05 0 0.05 0.1 0.15 0.2 0.25 1/ [ЛНП], мкг Рисунок 3.108. График концентрационной зависимости связывания 125I -ЛНП с мембранами клеток линии L929. Связывание ЛНП, меченых 125I, с мембранами клеток линии L929. (♦) – Связывание ЛНП с мембранами клеток, гиперэкспрессирующих Ткадгерин (клон ТС3).(□) – Связывание ЛНП с мембранами контрольных клеток (клон К9). 325 Для высокоаффинного участка связывания ЛНП параметры составляли: для клеток Т-кадгерин гиперэкспрессирующих HEK293 клеток Bmax 18.73 ± 3.7 нг/10 мг при Kd 3.46 ± 1.9 мкг/мл, для контрольных клеток Bmax 18.73 ± 2.8 при Kd 1.65 ± 0.64. Для L929 были получены аналогичные результаты: для Т-кадгерин гиперэкспрессирующих клеток Bmax составляло 21.84 ± 3.29 при Kd 1.16 ± 0.32, а для контрольных клеток Bmax 19.76 ± 3.37 при Kd 0.61 ± 0.38 (данные не представлены). Параметры связывания для низкоаффинного участка, предположительно Ткадгерина, для клеток L929 и HEK293, представлены на Рисунке 3.109 и Рисунке 3.110, соответственно. Bmax в Т-кадгерин гиперэкспрессирующих L929 клетках составляло 113.6 ± 5.8 нг/10 мг белка при Kd 61.4 ± 7.51 мкг/мл, в то время как для контрольных клеток Bmax 88.7 ± 7.6 нг/10 мг белка при Kd 69.8 ± 5.0 мкг/мл. Для HEK293, гиперэкспрессирующих Т-кадгерин, Bmax составляет 315.5 ± 7.3 нг/10 мг белка при Kd 64.9 ± 4.1 мкг/мл, для контрольных клеток - Bmax 198.3 ± 12.9 нг/10 мг белка при Kd 64.1 ± 3.5 мкг/мл. Полученные результаты подтверждают нашу гипотезу о том, что Т-кадгерин является рецептором ЛНП, так как гиперэкспрессия Т-кадгерина в клетках мышиных фибробластов приводит к увеличению участков связывания ЛНП с мембранами клеток. Мы предположили, что гиперэкспрессия Т-кадгерина в клетках не только увеличивает связывание ЛНП на поверхности клеток, но, возможно, также активирует внутриклеточную сигнализацию, внутриклеточного кальция. проявляющуюся повышением концентрации 326 Рисунок 3.109. Изотерма связывания 125I-ЛНП с мембранами L929 клеток. Инкубацию клеток с 125I-ЛНП (концентрация 1-100 мкг/мл) проводили в течение 4 ч при +40С в среде культивирования ДМЕМ, содержащей 0,1% БСА. Специфическое связывание рассчитывали, вычитая из величин общего связывания значения неспецифического связывания, которые определяли в присутствии 50-ти ратного избытка немеченых ЛНП, р<0,05. Рисунок 3.110. Изотерма связывания 125I-ЛНП с мембранами HEK293 клеток. Инкубацию клеток с 125I-ЛНП (концентрация 1-100 мкг/мл) проводили в течение 4 ч при +40С в среде культивирования ДМЕМ, содержащей 0,1% БСА. Специфическое связывание рассчитывали, вычитая из величин общего связывания значения неспецифического связывания, которые определяли в присутствии 50-ти кратного избытка немеченых ЛНП, р<0,05. 327 3.7.3 Введение ЛНП в среду культивирования клеток, гиперэкспрессирующих Ткадгерин, повышает концентрацию внутриклеточного кальция Для того чтобы ответить на вопрос, имеет ли гиперэкспрессия Т-кадгерина физиологическое значение, то есть, вызывает ли гиперэкспрессия Т-кадгерина активацию внутриклеточной сигнализации, мы измеряли концентрацию кальция в клетках в ответ на добавление ЛНП в среду культивирования. Было проанализировано изменение внутриклеточной концентрации кальция [Ca 2+]in в ответ на добавление ЛНП в среду культивирования клеток, гиперэкспрессирующих Ткадгерин, и контрольных клеток двух клеточных линий: HEK293 (клоны Th5 и Th8, гиперэкспрессирующие Т-кадгерин, и контрольные клоны GFP) и L929 (контрольный клон К9 и ТС3, гиперэкспрессирующий Т-кадгерин). Для каждого типа клеток эксперименты повторяли по 3 раза, при этом в каждом эксперименте показания снимали с 20-30 клеток. Клетки предварительно инкубировали с двухволновым зондом Fura-2АМ, используемым для измерения [Ca2+]in (Рисунок 3.111). После насыщения клеток флуоресцентным красителем в среду добавляли раствор ЛНП (в конечной концентрации 60 мкг/мл, что соответствует Кd низкоаффинного участка связывания, предположительно Т-кадгерину), и измеряли концентрацию [Ca2+]in, исходя из интенсивности флуоресценции зонда. В каждом эксперименте одновременно регистрировали изображение флуоресценции всех выбранных клеток в поле зрения микроскопа. и изменение спектра 328 А Б B Г Рисунок 3.111. Фазово-контрастное изображение клеток до начала эксперимента по измерению [Ca2+] in (A, Б), и флуоресцентное изображение клеток после инкубации с двух волновым зондом Fura-2AM (B, Г). A, B – контрольный клон К9 линии L929; Б, Г – клон Th5, гиперэкспрессирующий Т-кадгерин линии HEK293. 329 При добавлении ЛНП в среду инкубирования L929 клеток было зарегистрировано изменение интенсивности флуоресценции зонда как в контрольных, так и в опытных клетках. При этом при увеличении связывания молекул зонда Fura-2AM с Са2+ спектр флуоресценции смещается в более коротковолновую область, и отношение F340/F380 (флуоресценция при длинах волн 340 нм и 380 нм, соответственно) увеличивалось, что регистрировали в виде пика на графике (Рисунок 3.112 и Рисунок 3.113). В конце каждого эксперимента в качестве положительного контроля добавляли иономицин, который является ионофором и вызывает увеличение концентрации [Ca2+] в цитоплазме более активное, чем выведение Ca2+ клеточными системами. Добавление иономицина в среду культивирования клеток после добавления ЛНП, сопровождалось повторным увеличением интенсивности флуоресценции зонда Fura-2AM, что свидетельствует об обратимости эффектов повышения внутриклеточного кальция, вызываемых ЛНП. При измерении [Ca2+]in в клетках линии L929 было обнаружено, что гиперэкспрессия Т-кадгерина вызывает увеличение кальциевого ответа в 2 раза по сравнению с контролем в ответ на добавление ЛНП в среду культивирования (Рисунок 3.112 и Рисунок 3.113). Исходная концентрация [Ca2+]in в клетках L929 (контрольных и опытных) составляла около 200 нМ. В клетках ТС3, гиперэкспрессирующих Т-кадгерин, разница между исходной концентрацией [Ca2+]in и концентрацией после добавления ЛНП составила около 1200 нМ, а в контрольных клетках К9 – 570 нМ. Длительность кальциевого ответа в клетках, гиперэкспрессирующих Т-кадгерин, и в контрольных клетках была соизмерима и составляла приблизительно 120 сек. Результаты, полученные при измерении кальциевого ответа в клетках линии HEK293, гиперэкспрессирующих Т-кадгерин (клоны Th5 и Th8), и контрольных были аналогичны результатам, полученным для клеток L929 (Рисунок 3.114 и Рисунок 3.115). 330 A введение ЛНП (60 мкг/мл) 1.00 B введение иономицина введение ЛНП (60 мкг/мл) введение иономицина 0.75 F340/F380 F340/F380 1.00 0.50 0.75 0.50 0.25 0.25 0 100 200 300 400 500 600 700 0 Время (сек) 100 200 300 400 500 600 700 800 Время (сек) Рисунок 3.112. Изменение интенсивности флуоресценции зонда Fura-2АМ (F340/F380) в клетках линии L929 при добавлении ЛНП в среду культивирования (одновременная запись в 22 клетках); А – изменение флуоресценции в клетках К9 (контроль); B - изменение флуоресценции в клетках, гиперэкспрессирующих Т-кадгерин (клон ТС3). A B введение ЛНП (60 мкг/мл) 1500 введение ЛНП (60 мкг/мл) 2250 2000 1250 [Ca2+]in нМ [Ca2+]in нМ 1750 1000 750 500 1500 1250 1000 750 500 250 250 0 0 0 100 200 300 Время (сек) 400 500 0 100 200 300 400 500 600 Время (сек) Рисунок 3.113. Изменение концентрации внутриклеточного кальция в ответ на введение ЛНП в среду культивирования в клетках линии L929 (одновременная запись в 22 клетках); А – изменение [Ca2+] in в контрольных клетках (К9); B - изменение [Ca2+] in в клетках, гиперэкспрессирующих Т-кадгерин (ТС3). 331 С D введение иономицина введение ЛНП (60 мкг/мл) 1.50 1.50 1.25 F340/F380 F340/F380 1.25 1.00 0.75 1.00 0.75 0.50 0.50 0.25 0.25 0 100 200 300 400 500 0 100 200 Время (сек) 400 500 B A B введение ЛНП (60 мкг/мл) 1.25 введение иономицина введение иономицина введение ЛНП (60 мкг/мл) 1.25 1.00 F340/F380 1.00 F340/F380 300 Время (сек) A 0.75 0.75 0.50 0.50 0.25 0.25 0 100 200 300 400 500 600 700 800 900 0 Время (сек) C введение иономицина введение ЛНП (60 мкг/мл) 100 200 300 400 500 600 Время (сек) D Рисунок 3.114. Изменение интенсивности флуоресценции зонда Fura-2АМ (F340/F380) в клетках линии HEK293 при добавлении ЛНП в среду культивирования. А, B – изменение интенсивности флуоресценции в контрольных клетках (клон GFP, одновременная запись в 21 и 6 клетках, соответственно); C, D - изменение интенсивности флуоресценции в клетках, гиперэкспрессирующих Т-кадгерин (клоны Th8 и Th5, одновременная запись в 24 и 28 клетках, соответственно). 332 C 700 700.0 600.0 500 500.0 [Ca ]in нМ 600 400.0 2+ 400 2+ [Ca ]in нМ D введение ЛНП (60 мкг/мл) введение ЛНП (60 мкг/мл) 300 300.0 200 200.0 100 100.0 0 0 100 200 300 400 0 100 Время (сек) 200 300 400 Время (сек) A B A B введение ЛНП (60 мкг/мл) введение ЛНП (60 мкг/мл) 700 1000 900 600 [Ca2+]in нМ 400 2+ [Ca ]in нМ 800 500 300 700 600 500 400 300 200 200 100 100 0 0 0 100 200 300 400 500 600 0 Время (сек) C 100 200 300 400 500 Время (сек) D Рисунок 3.115. Изменение концентрации внутриклеточного кальция в ответ на введение ЛНП в среду культивирования в клетках линии HEK293. A, B - изменение [Ca2+] in в контрольных клетках (клон GFP, одновременная запись в 21 и 6 клетках соответственно); C, D - изменение [Ca2+] in в клетках, гиперэкспрессирующих Ткадгерин (клоны Th8 и Th5, одновременная запись в 24 и 28 клетка, соответственно). 333 Исходная концентрация Ca2+ в клетках HEK293, контрольных (GFP) и гиперэкспрессирующих Т-кадгерин (Th5 и Th8), была одинаковая, и составила ≈150 нМ. При добавлении ЛНП происходило резкое увеличение [Ca2+]in, разница в амплитуде кальциевом ответе составила (в среднем) - 330 нМ в контроле, и 520 нМ в клетках, гиперэкспрессирующих Т-кадгерин. Статистический анализ полученных результатов показал, что в клетках Th5 и Th8 линии HEK293, гиперэкспрессирующих Т-кадгерин, введение ЛНП достоверно повышает [Ca2+]in по сравнению с контрольными GFP клетками этой же линии. Следует отметить, что при измерении [Ca2+]in в контрольных клонах клеток HEK293 (GFP), большая часть клеток отреагировала небольшим кальциевым скачком в ответ на введение ЛНП, однако, в нескольких клетках ответа не было. Также примечательно то, что в большинстве клеток линии HEK293 наблюдался следующий эффект: чем выше было увеличение [Ca2+]in в ответ на добавление ЛНП, тем больше времени требовалось для восстановления исходного уровня кальция. Средние значения увеличения [Ca2+]in, суммированные по трѐм экспериментам для каждого типа клеток (L929 и HEK293), приведены на Рисунке 3.116 и Рисунке 3.117. Увеличение [Ca2+]in в ответ на введение ЛНП в клетках линии L929, гиперэкспрессирующих Т-кадгерин, составило 120% по сравнению с контрольными клетками (100%); в клонах линии HEK293, гиперэкспрессирующих Т-кадгерин (Th5 и Th8) - составило 240% и 115%, соответственно; разница была статистически достоверна. Таким образом, связывание ЛНП с мембранами клеток сопровождается активацией внутриклеточной сигнализации: при добавлении ЛНП в среду культивирования клеток L929 и клеток НЕК293 наблюдается повышение внутриклеточной концентрации Ca2+, амплитуда кальциевого ответа в клетках, гиперэкспрессирующих Т-кадгерин, достоверно превышает аналогичный ответ в контроле. 334 Изменение [Ca2+]in в клетках L929 при добавлении ЛНП 1600 * [Ca2+]in, нМ 1400 1200 1000 800 600 400 200 0 контроль TC3 Рисунок 3.116. Изменение уровня кальция в клетках L929, контрольных и в клетках, гиперэкспрессирующих Т-кадгерина (TC3 клон) в ответ на добавление ЛНП (N=3, p<0,05 по сравнению с контролем). Изменение [Ca2+]in в клетках HEK293 при добавлении ЛНП 1600 [Ca2+]in, нМ 1400 1200 1000 800 * 600 * 400 200 0 контроль Th8 Th5 Рисунок 3.117. Изменение уровня кальция в клетках HEK293, контрольных и в клетках, гиперэкспрессирующих Т-кадгерин (Th5 и Th8 клоны) в ответ на добавление ЛНП (N=3, p<0,05 по сравнению с контролем). 335 Увеличение внутриклеточного кальция в ответ на введение ЛНП в среду культивирования может быть вызвано поступлением Ca2+ извне (из межклеточного пространства), либо за счет мобилизации кальция из внутриклеточных депо. Для выяснения механизмов, обуславливающих увеличение концентрации внутриклеточного Ca 2+ при действии ЛНП, нами были использованы два подхода: ингибирование Ca 2+-зависимых каналов эндоплазматического ретикулума (ЭПР), и измерение [Ca2+]in в бескальциевой среде. Известно, что ЭПР играет важную роль в регуляции концентрации 2+ внутриклеточного Ca , он является основным внутриклеточным кальциевым депо. Сигнал от цитоплазматических рецепторов посредством системы вторичных посредников активирует кальциевые каналы ЭПР, в результате чего происходит выход ионов кальция из ЭПР и, как следствие, активация целого ряда биохимических процессов. Кальциевые насосы (SERCA pumps - sarco(endo)plasmic reticulum Ca2+-ATPase) регулируют концентрацию Ca2+ в цитоплазме: увеличение содержания [Ca2+]in вызывает активацию Ca2+ АТФазы ЭПР (SERCA) и захват ионов [Ca2+] в ЭПР. Если ЛНП вызывают увеличение [Ca2+]in в клетках за счет его выхода из ЭПР, то при блокировании кальциевых насосов ЭПР при последующем добавлении ЛНП кальциевого сигнала наблюдаться не будет. В качестве ингибитора Ca2+ АТФазы ЭПР была использована циклопиазоновая кислота (СРА, cyclopiazonic acid) [Beck et al., 2004], которая без повышения внутриклеточного содержания инозитола (1, 4, 5) селективно блокирует SERCA, что вызывает выход Ca2+ из ЭПР и приводит к полному опустошению агонист-чувствительных Ca2+ депо [Demaurex et al., 1992; Uyama et al., 1992]. Эксперименты проводили в среде, содержащей Ca2+. Добавление СРА в концентрации 20 мкМ в течение 5 мин к контрольным и Т-кадгерин гиперэкспрессирующим клеткам вызывало временное увеличение [Ca2+]in в силу выхода Ca2+ из внутриклеточных депо. Последующее добавление ЛНП на фоне действия СРА не оказывало никакого эффекта на [Ca2+]in ни в контрольных, ни в Т-кадгерин гиперэкспрессирующих клетках. Для доказательства обратимости эффекта СРА в эксперименте использовался иономицин, который добавляли в среду культивирования клеток сразу после добавления ЛНП (в конечной концентрации 60 мкг/мл) в (Рисунок 3.118). 336 A CPA 1.25 B введение введение ЛНП (60 мкг/мл) иономицина 1.25 1.00 F340/F380 1.00 F340/F380 введение введение ЛНП (60 мкг/мл) иономицина CPA 0.75 0.50 0.75 0.50 0.25 0.25 0.00 0.00 0 100 200 300 400 500 600 700 800 900 1000 0 100 200 300 Время (сек) C 1.00 D введение введение ЛНП (60 мкг/мл) иономицина CPA 1.00 0.75 0.75 F340/F380 F340/F380 700 800 900 1000 Время (сек) введение введение ЛНП (60 мкг/мл) иономицина CPA 400 500 600 0.50 0.50 0.25 0.25 0.00 0.00 0 100 200 300 400 500 Время (сек) 600 700 800 900 0 100 200 300 400 500 600 700 800 900 Время (сек) Рисунок 3.118. Изменение интенсивности флуоресценции зонда Fura-2АМ (F340/F380) в ответ на введение СРА с последующим добавлением ЛНП (60 мкг/мл) в среду культивирования клеток линий L929 и HEK293. A, B – изменение флуоресценции в клетках L929, гиперэкспрессирующих Т-кадгерин (TC3), и контрольных (К9), соответственно. C, D – изменение флуоресценции в клетках НЕК293, гиперэкспрессирующих Т-кадгерин (Th5), и контрольных (GFP), соответственно. Одновременная запись в 39 (А), 28 (B), 23 (C) и 29 (D) клетках. 337 Этот эксперимент был проведен на обоих типах клеточных линий, и результаты были схожими. Полученные данные позволяют предположить, что ЛНП стимулируют повышение [Ca2+]in в значительной степени за счет его выхода из ЭПР. Для подтверждения этого предположения был использован второй подход, при котором оценивали изменение концентрации [Ca2+]in в бескальциевой среде. При этом использовали буфер для измерения кальция, содержащий 100 мкМ ЭГТА. Было обнаружено, что добавление ЛНП в бескальциевую среду культивирования клеток приводило к существенно меньшему повышению внутриклеточного [Ca2+]in, чем в предыдущем эксперименте. В клетках линии НЕК293, гиперэкспрессирующих Т-кадгерин (Тh8), разница между исходной 2+ концентрацией [Ca ]in и концентрацией кальция после введения ЛНП составила всего 250 нМ, в контрольных клетках (GFP) - 25 нМ (Рисунок 3.119). Аналогичные результаты были получены для клеточной линии L929. Добавление ЛНП (60 мкг/мл) в бескальциевую среду культивирования приводило к слабому повышению [Ca2+]in. В клетках, гиперэкспрессирующих Т-кадгерин (TC3) разница между исходной концентрацией [Ca2+]in и концентрацией кальция после введения ЛНП составила всего 400 нМ, в контрольных клетках - 100 нМ. Таким образом, связывание ЛНП с мембранами клеток, гиперэкспрессирующих Т-кадгерин, вызывает повышение внутриклеточного кальция в значительной мере за счет его выхода из ЭПР. Результаты, полученные в экспериментах в бескальциевой среде, дают основание предполагать небольшое участие в этом процессе кальция, находящегося в межклеточном пространстве. Изменение[Ca2+]in в клетках HEK293 при добавлении ЛНП в среду культивирования, не содержащей ионов кальция 1600 [Ca2+]in, нМ 1400 1200 1000 800 600 * 400 200 0 контроль Th8 Рисунок 3.119. Изменение [Ca2+] in в клетках НЕК293, контрольных и гиперэкспрессирующих Т-кадгерин (Тh8), в ответ на добавление ЛНП в бескальциевой среде. 338 3.7.4 ЛНП стимулируют миграцию клеток, гиперэкспрессирующих Т-кадгерин Результаты, полученные ранее в нашей лаборатории [Bochkov et al., 1993; Bochkov et al., 1994; Tkachuk et al, 1994; Bochkov et al., 1996] и в данной работе, показывают, что Ткадгерин опосредует сигнальные эффекты ЛНП в различных типах клеток. Мы предположили, что ЛНП способны не только активировать внутриклеточную сигнализацию, но также влиять на функциональное состояние клеток, усиливая их миграцию. Для изучения влияния гиперэкспрессии Т-кадгерина на миграционную способность клеток в ответ на добавление ЛНП в качестве хемоаттрактанта была использована камера Бойдена. В верхнюю камеру вносили клетки L929, гиперэкспрессирующие Т-кадгерин (ТС3), или контрольные клетки (К9), в концентрации при которой клетки формировали субконфлюентный монослой, а в нижнюю камеру добавляли ЛНП в концентрации 60 мкг/мл, что соответствует Kd низкоаффинного участка связывания, предположительно Т-кадгерину. Гиперэкспрессия Т-кадгерина сама по себе увеличивала способность клеток к миграции по сравнению с контрольными клетками (в контрольной среде, содержащей 0.1% БСА). Добавление ЛНП в нижнюю камеру дозозависимо стимулировало миграцию как контрольных, так и опытных клеток по сравнению с контрольными условиями (среда с 0.1% БСА), однако, миграция Т-кадгерин гиперэкспрессирующих клеток была значительно выше, чем контроле (среда с 0.1% БСА с ЛНП в концентрации 60 мкг/мл) (Рисунок 3.120). Также, в качестве контроля в нижнюю камеру добавляли ЛНП, предварительно инкубированные с антителами против ЛНП (ЛНП+Fab) (в концентрации 100 мкг/мл), а также антитела против ЛНП (100 мкг/мл) в качестве негативного контроля (Fab). Изучая миграцию клеток было обнаружено, что добавление ЛНП в нижнюю камеру в 1,5 раза усиливает миграцию Т-кадгерин гиперэкспрессирующих клеток (ТС3) по сравнению с контрольными клетками (К9) (Рисунок 3.120). Преинкубирование ЛНП с антителами к ЛНП блокирует стимулирующий эффект ЛНП на миграцию клеток; раствор антител (Fab) сам по себе не оказывает влияния на миграцию, и при этом миграция соответствует базовому уровню. В данном эксперименте не использовали клеточную линию HEK293, так как эти клетки не обладают необходимым миграционным потенциалом. Полученные данные свидетельствуют о том, что гиперэкспрессия Т-кадгерина увеличивает способность клеток к миграции по градиенту БСА (положительного контроля) и значительно увеличивает способность клеток мигрировать по градиенту хемоаттрактанта - ЛНП. 339 ** БСА ЛНП * 70 ЛНП+Fab Интенсивн. миграции, усл.ед. 60 Fab 50 * 40 30 20 10 0 контроль TC3 Рисунок 3.120. Гиперэкспрессия Т-кадгерина усиливает миграцию L929 клеток в ответ на введение ЛНП. Миграцию клеток, гиперэкспрессирующих Т-кадгерин (ТС3) или контрольных, оценивали в камере Бойдена с добавлением в нижнюю камеру: ЛНП в качестве хемоаттрактанта; ЛНП, преинкубированные с антителами против ЛНП, (ЛНП+Fab); Fab - антитела против ЛНП, БСА - негативный контроль. N=3, *,** р<0,05. 340 Таким образом, полученные результаты по изучению влияния ЛПН на [Ca2+]in в клетках, гиперэкспрессирующих Т-кадгерин, а также по изучению влияния ЛНП на миграционную активность этих клеток, подтверждают ранее выдвинутое предположение о том, что Т-кадгерин является низкоаффинным рецептором ЛНП, опосредующим сигнальные эффекты ЛНП в клетках и стимулирующим клеточную миграцию. 3.7.5 Обсуждение результатов Ранее в нашей лаборатории было показано, что связывание ЛНП на поверхности гладкомышечных клеток стимулирует фосфоинозитидный обмен, активируют фосфолипазу С и вызывают мобилизацию внутриклеточного Ca2+ [Bochkov et al., 1993]. Этот эффект ЛНП по кинетике напоминал действие классического Ca2+-мобилизующего гормона ангиотензина II и развивался гораздо быстрее, чем эффект другого индуктора мобилизации Ca2+ фактора роста PDGF-BB. Подобным механизмом действия обладают все Ca2+мобилизующие гормоны. Сходство с действием гормонов усиливалось и способностью ЛНП индуцировать осцилляции внутриклеточной концентрации Ca 2+ ([Ca2+]in) в одиночных клетках, а также тем, что повышение [Ca2+]in ингибируется активаторами протеинкиназы С и сАМР-зависимой протеинкиназы [Bochkov et al., 1992]. Кроме того, ЛНП активируют в ГМК фосфатидилинозитольный обмен, что также характерно для Ca2+-мобилизующих гормонов [Bochkov et al., 1992]. Сигнальные эффекты ЛНП были зарегистрированы в диапазоне концентраций, не превышающем их физиологический уровень в крови и стенке сосуда [Bochkov et al., 1993]. В поиске нового рецептора, опосредующего эти сигнальные гормоноподобные эффекты ЛНП, методом лигандного блоттинга в нашей лаборатории был выделен Т-кадгерин [Tkachuk et al., 1996]. Для того, чтобы доказать участие Т-кадгерина в индукции внутриклеточной сигнализации под действием ЛНП, мы провели стабильную гиперэкспрессию Т-кадгерина в клетках линий эпителия почки человека НЕК293 и мышиных фибробластов L929. Анализ специфического связывание 125I-ЛНП с мембранами клеток показал, что гиперэкспрессия Ткадгерина приводит к увеличению числа низкоаффинных участков связывания (Bmax) (примерно на 40%) по сравнению с контролем без достоверного изменения константы связывания (Kd) (Рисунок 3.109 и Рисунок 3.110). Таким образом, гиперэкспрессия Ткадгерина в клетках HEK293 и L929 увеличивала специфическое связывание ЛНП (Bmax) с поверхностью мембран клеток, что свидетельствует о том, что ЛНП является специфическим лигандом Т-кадгерина. Поскольку значение Kd достоверно не изменялось, был сделан вывод о том, что новых сайтов связывания обнаружено не было. Полученные данные хорошо согласуются с результатами, полученными ранее в нашей лаборатории, о 341 присутствии специфического низкоаффинного участка связывания на ГМК [Tkachuk et al, 1994), а также с данными других авторов о том, что эффект увеличения связывания ЛНП с мембранами клеток гиперэкспрессирующими HEK293, Т-кадгерин, полностью отсутствовал после обработки GPI-специфической фосфолипазой С [Resink et al., 1999; Niermann et al., 2000]. Для ответа на вопрос, является ли Т-кадгерин физиологически значимым рецептором ЛНП или имеет ли связывание ЛНП с Т-кадгерином какое-либо физиологическое значение, мы измеряли концентрацию кальция в клетках в ответ на добавление ЛНП в среду культивирования в концентрации, соответствующей Kd низкоаффинного участка связывания на ГМК. Амплитуда [Ca2+]in ответа была в 2 - 2,5 раз больше в клетках, гиперэкспрессирующих Т-кадгерину, по сравнению с контрольными. После этого [Ca2+]in концентрация постепенно возвращалась к исходному уровню, по всей видимости, за счет активности Ca2+-АТФазы плазматической мембраны или в силу секвестрирования ионов Ca2+ из цитоплазмы за счет работы митохондриального унитранспортѐра [Berridge et al., 2003]. Эффект ЛНП был целиком заблокирован в присутствии циклопиазоновой кислоты - специфического ингибитора Ca2+-зависимой ATPазы саркоплазматического ретикулума. Эти данные указывают на то, что связывание ЛНП с Т-кадгерином вызывает мобилизацию Ca2+ из эндоплазматического ретикулума [Demaurex et al., 1992; Uyama et al., 1992] за счет активации внутриклеточных сигнальных каскадов, подобных тем, которые опосредуют эффекты других Ca2+-мобилизующих гормонов. Связывание Т-кадгерина с ЛНП ингибируется антителами против ЛНП (антитела против апо В белка) и поликлональными антителами против Т-кадгерина, произведѐнными в нашей лаборатории [Bochkov et al., 1996]. Эти данные позволяют утверждать, что Ткадгерина является рецептором ЛНП, опосредующим активацию внутриклеточной сигнализации и сигнальные эффекты ЛНП. Известно, что нарушения Ca2+ сигнализации наблюдаются при патогенезе таких заболеваний как гипертония, ишемическая болезнь сердца и диабет (цитируется в Kudrjashova et al., продолжительность) 2002). со Изменение временем может Ca2+ характеристик приводить к ответа активации (амплитуда, транскрипции определенных генов, экспрессия которых приводит к гипертрофии миокарда [Berridge et al., 2003]. При наступлении застойной сердечной недостаточности амплитуда Ca 2+ ответа значительно уменьшается в связи с подавлением сигнализации с участием системы адренергических рецепторов и снижения активности кальциевых насосов (SERCA) [Berridge et al., 2003]. Хотя достаточно давно известно, что Т-кадгерин высоко экспрессирован в сердце и сосудах во взрослом организме [Ivanov et al., 2001], его функция 342 в сердечно-сосудистой системе только сейчас начинает проясняться. Необходимы дальнейшие исследования для того, чтобы выявить физиологическую роль Т-кадгерина в ЛНП-индуцированной Ca2+-сигнализации в сердечно-сосудистой системе в норме и при патологии. Используя камеру Бойдена, мы показали, что ЛНП стимулируют Т-кадгеринзависимую миграцию клеток по градиенту БСА как в контроле, так и в Т-кадгерин экспрессирующих клетках, что соответствует ранее опубликованным данным, полученным на HUVEC и ГМК [Ivanov et al., 2004]. Однако миграция Т-кадгерин экспрессирующих клеток по градиенту ЛНП значительно превышала аналогичные показатели в контрольных клетках, и это эффект полностью блокировался добавлением Fab фрагментов антител против ЛНП (Рисунок 3.120) и ЕС1 домена Т-кадгерина [Bochkov et al., 1996]. Полученные результаты свидетельствуют о том, что Т-кадгерин является рецептором ЛНП, вызывающим активацию внутриклеточной сигнализации и клеточную миграцию. Данные литературы подтверждают специфичность полученных эффектов в настоящей работе, и роль Т-кадгерина как сигнальной молекулы. Так, при поляризации Т-кадгерин локализуется на лидирующем крае мигрирующих эндотелиальных клеток и ГМК. В отличие от Ткадгерина «классический» VE-кадгерин не обнаруживается на лидирующем крае мигрирующих клеток HUVEC и локализуется либо в зоне клеточных контактов, либо в цитоплазме [Philippova, et al., 2003]. In vivo Т-кадгерин локализуется на апикальной поверхности кишечного эпителия цыпленка, а в культуре MDCК клеток, трансфицированных плазмидой, содержащей ген Т-кадгерина, Т-кадгерин обнаруживается вне зоны клеточных контактов на апикальной поверхности, в то время как N-кадгерин располагается базолатерально в адгезивных контактах [Koller and Ranscht, 1996]. Т-кадгерин является GPI-заякоренным белком и поэтому отличается структурно и функционально от других членов суперсемейства кадгеринов [Cebecauer et al., 1998]. Ткадгерин, хотя и опосредует слабую гомофильную адгезию [Vestal and Ranscht, 1992], по всей видимости, в первую очередь выполняет функцию сигнальной молекулы и/или навигационного рецептора. Известно, что GPI белки участвуют в различных сигнальных каскадах, таких как Ca2+ сигнализация или тирозиновое фосфорилирование. Т-кадгерин присутствует в липидных плотах, в которых в большом количестве обнаруживаются другие GPI-заякоренные белки и сигнальные молекулы, такие как субъединицы G белков (Gas), SRC киназы и uPAR (рецептор урокиназы) [Philippova et al., 1998]. Таким образом, в данной части работы мы показали, что Т-кадгерин является рецептором ЛНП, опосредующим сигнальные эффекты и миграцию клеток по градиенту ЛНП. 343 ГЛАВА 4. ЗАКЛЮЧЕНИЕ В нашей работе получены свидетельства того, что Т-кадгерин участвует в процессах регуляции роста не только нервов, но и сосудов. Мы обнаружили также, что Ткадгерин регулирует проницаемость эндотелиального монослоя и установили молекулярные механизмы внутриклеточной сигнализации с участием Т-кадгерина. Очевидно, что в процессах ангиогенеза помимо факторов роста, цитокинов и хемокинов важную роль играют навигационные рецепторы и их лиганды. Навигационные рецепторы обеспечивают направленный рост сосудов и нервов [Melani and Weinstein, 2010]. К навигационным рецепторам относят Эфрины и их рецепторы, Нейропилины и Плексины рецепторы Семафоринов, Robo - рецепторы Слит белков, и UNC5B рецепторы, связывающие Нетрины (Рисунок 4.1). Известно, что регуляция направленного роста аксонов и сосудов имеет сходные молекулярные механизмы [Carmeliet, 2003]. Одни и те же молекулы (пары лиганд-рецептор) определяют направление роста аксонов и сосудов. При выборе траектории роста сосудов и нервов взаимодействие Эфринов с рецепторами, Семафоринов с рецепторами, а также Т-кадгеринов, как правило, вызывает отталкивание. При росте аксонов и миграции эндотелиальных «концевых» клеток Нетрины могут служить как сигналом отталкивания (при взаимодействии с UNC5), так аттрактивным стимулом (при взаимодействии с DCC), что зависит от типа клеток и рецептора. NRP не сигнализируют самостоятельно, а являются ко-рецепторами Плексинов в нервной системе, где вызывают отталкивание, или с ко-рецепторами VEGFR2 в эндотелии, где они служат аттрактивным стимулом. Robo 1 рецепторы связываются со Слит лигандами и являются рецепторами, передающими аттрактивный сигнал, как в нервной системе, так и в сосудах, в то время как Robo 4 подавляет миграцию эндотелиальных клеток и участвует в поддержании целостности сосудистой стенки. На сегодняшний день к навигационным молекулам относят также урокиназную систему, активность которой обеспечивает направленный протеолиз на лидирующем крае клетки и способствует клеточной миграции, и Т-кадгерин (Рисунок 1.9 и Рисунок 1.10) [Rubina et al., 2007; Ткачук и др., 2013]. До исследований, проведенных в настоящей работе, о Т-кадгерине было известно, что он экспрессируется в развивающейся нервной системе цыпленка на ранних этапах эмбриогенеза, где он функционирует как навигационная «молекула отталкивания»: Ткадгерин подавляет рост аксонов и миграцию клеток нервного гребня через ткани, клетки которых экспрессируют Т-кадгерин, участвует в объединении аксонов в пучки и формировании синапса в эмбриогенезе. 344 Рисунок 4.1. Одни и те же молекулы определяют направление роста аксонов (а) и сосудов (б). При выборе траектории роста взаимодействие Эфринов с рецепторами, Семафоринов с рецепторами, а также Т-кадгеринов, как правило, вызывает отталивание, как в случае роста аксонов, так и при миграции эндотелиальных «концевых» клеток. Нетрины могут вызывать как отталкивание, так и служить аттрактивным стимулом для конуса роста аксона и эндотелиальных «концевых» клеток, что зависит от рецептора, с которым они связываются, а также от типа клеток. NRP вызывают отталкивание в нервной системе, а в эндотелии служат аттрактивным стимулом. Robo 1 вызывают аттракцию в нервной системе и в сосудах, Robo 4 подавляет миграцию эндотелиальных клеток. Урокиназный рецептор, связываясь с урокиназой, облегчает миграцию клеток за счет направленного протеолиза на лидирующем крае. Рецептор DСС (deleted in colorectal cancer); Рецептор NRP Нейропилин (Neuropilin); Рецептор Roundabout (ROBO1 и ROBO4); рецептор UNC-5 (Uncoordinated-5); VEGFR - рецептор VEGF; uPAR – рецептор урокиназы (Адаптировано из Melani and Weinstein, 2010). 345 О роли Т-кадгерина в формировании сердечно-сосудистой системы в эмбриогенезе млекопитающих данных в литературе до сих пор не было. Поскольку ранее в нашей лаборатории были получены результаты о том, что во взрослом максимальная экспрессия Т-кадгерина организме наблюдается в нервной и сердечно-сосудистой системах, мы мы предположили, что и в эмбриогенезе Т-кадгерин может играть роль не только в развивающейся нервной системе, но и в сердечнососудистой. В настоящем исследовании впервые получены результаты, свидетельствующие о том, что в эмбриогенезе Т-кадгерин экспрессируется в сердечно-сосудистой системе и головном мозге васкуляризации; на стадии активного морфологически формирования паттерн экспрессии органных структур Т-кадгерина и их соответствует формирующимся сосудам. Мы предполагаем, что Т-кадгерин функционирует при этом как «молекула отталкивания», которая направляет рост сосудов в формирующемся мозге и сердце, ограничивая прорастание в зоны, где клетки экспрессируют Т-кадгерин, также как это происходит при росте аксонов в нервной системе. Поскольку рост сосудов и нервов происходит параллельно, то нельзя исключить того, что Т-кадгерин также ограничивает прорастание нервов через сосуды и наоборот. Для определения конкретной роли Ткадгерина в морфогенезе формирующихся структур сердца и мозга и их васкуляризации необходимы дополнительные исследования. Для выявления возможной роли Т-кадгерина в процессах ангиогенеза в настоящей работе был использован ряд экспериментальных подходов. Данные, полученные на моделях in vivo и in vitro, позволяют утверждать, что Т-кадгерин действительно является «молекулой отталкивания» и ингибирует ангиогенез на уровне подавления миграции эндотелиальных клеток и формирования мелких сосудов и капилляров. По аналогии с нервными волокнами в основе такого подавления лежит гомофильное взаимодействие между молекулами Т-кадгерина на мигрирующих эндотелиальных клетках и на клетках ткани, через которые они мигрируют [Rubina et al., 2007]. Данные об участии Т-кадгерина в процессах ангиоенеза в настоящей работе получены впервые. Во взрослом организме процессы ангиогенеза строго регулируются, потеря контроля приводит патологические к развитию состояния патологий. сосудов Мы предположили, (избыточный, что недостаточный различные ангиогенез, патологическое ремоделирование сосудов) во взрослом организме могут быть связаны с нарушением экспрессии или функционирования Т-кадгерина. Обнаруженная нами способность Т-кадгерина подавлять ангиогенез делает его привлекательной мишенью для противоопухолевой терапии, основанной на антиангиогенном подходе. В литературе существуют противоречивые данные о роли Т-кадгерина в опухолевом росте и 346 метастазировании. Проведенное в настоящей работе исследование на биопсийном материале предраковых состояний кожи, рака кожи и меланомы человека свидетельствуют о частичной или полной потере экспрессии Т-кадгерина в опухолевых клетках, а также об изменении экспрессии стандартных маркеров сосудистых клеток и Т-кадгерина в опухолевых сосудах. Кроме того, было обнаружено снижение экспрессии Т-кадгерина и аберрантная экспрессия маркеров клеток сосудов в образцах, что коррелирует со степенью злокачественности новообразований. Это означает, что потеря экспрессии Т-кадгерина в клетках неопластических образований может способствовать прорастанию в них сосудов, экспрессирующих Т-кадгерин, или в случае сохранения экспрессии Т-кадгерина в опухолевых клетках васкуляризация происходит сосудами, не экспрессирующими Ткадгерин [Rubina et al., 2012; Rubina et al., 2013]. Эти результаты и ряд данных литературы позволил высказать предположение о том, что Т-кадгерин является опухолевым супрессором [Takeuchi et al., 2002a; Andreeva and Kutuzov, 2010]. Для установления не только корреляционной, но и возможной причинноследственной связи между экспрессией Т-кадгерина и опухолевой прогрессией мы использовали модель роста агрессивной меланомы у мышей, метастазирующей в легкие. Полученные результаты подтверждают тот факт, что Т-кадгерин действительно является антиангиогенной молекулой и достоверно подавляет опухолевый неоангиогенез [Yurlova et al., 2010; Rubina et al., 2013]. Однако по нашим данным Т-кадгерин не может считаться фактором опухолевой супрессии, поскольку клетки меланомы при гиперэкспрессии Ткадгерина активируют компенсаторные механизмы: они «включают» гены, способствующие их большей выживаемости, миграции и инвазии, а также начинают секретировать хемокины, молекулы адгезии, протеазы и факторы роста, способствующие привлечению мезенхимных стромальных клеток, что способствует опухолевому росту и метастазированию. Кроме того, известно, что клетки агрессивной меланомы человека способны формировать васкулогенные лакуны в процессе «васкулогенной мимикрии» [Maniotis et al., 1999], что обеспечивает дополнительные возможности для их роста, питания и метастазирования. Полученные нами данные на первичных опухолевых узлах и метастазах свидетельствуют о том, что клетки меланомы человека, формирующие лакуны, экспрессируют Т-кадгерин, что, несмотря на антиангиогенные эффекты Т-кадгерина, может обеспечивать питание опухоли альтернативным путем (Рисунок 3.15 и Рисунок 3.16). Таким образом, Т-кадгерин, по всей видимости, играет двойную роль: с одной стороны, он действительно обладает антиангиогенными свойствами и способен подавлять неоангиогенез в первичных опухолях, с другой, увеличивает способность клеток к инвазии и метастазированию. Противоречивость данных литературы о роли Т-кадгерина в 347 онкологии, возможно, связана с разной степенью зависимости различных опухолей от васкуляризации, а также с вариативностью компенсаторных механизмов, которые способны включать разные типы опухолевых клеток. Полученные данные имеют фундаментальное значение для понимания биологии опухолевых клеток и механизмов опухолевой прогрессии, лежащих в основе опухолевого роста и метастазирования, а также низкой чувствительности к лекарственной терапии. Возможно, неудачи, связанные с применением антиангиогенных препаратов в клинике, направленных на подавление опухолевого неоангионенеза, например, блокаторов VEGF, связаны с тем, что опухолевые клетки могут инициировать другие способы формирования сосудов помимо собственно ангиогенеза (Рисунок 1.2). Опухолевые клетки могут использовать альтернативные факторы роста (не VEGF) и хемокины для формирования сосудистых структур, активировать окружающую строму, индуцировать рекрутирование эндотелиальных клеток из костного мозга, оккупировать и использовать предсуществующие сосуды (co-option), стимулировать ветвление и расщепление существующих сосудов (intussusception), формировать сосудистые структуры в процессе «васкулогенной мимикрии», кроме того, «раковые стволовые клетки» способны сами дифференцироваться в эндотелиальные клетки [Carmeliet and Jain, 2011]. Некоторые из перечисленных механизмов обнаружены в настоящей работе. Ранее в нашей лаборатории было показано, что экспрессия Т-кадгерина повышается в эндотелиальных клетках и ГМК сосудов при атеросклерозе [Ivanov et al., 2001, Kudrjashova et al., 2002]. В настоящем исследовании мы подтвердили тот факт, что прогрессирование атеросклероза сопровождается увеличением экспрессии Т-кадгерина в сосудистых клетках. Помимо этого, при атеросклерозе мы обнаружили увеличение количества CD31/Т-кадгерин двойных позитивных клеток в субэндотелиальным слое и в медии. Эти клетки, возможно, являются клетками-предшественниками, мигрировавшими из костного мозга, сосудистой или стенки. активированными Возможно, резидентными Т-кадгерин, является прогениторными навигационной клетками молекулой, участвующей в миграции стволовых/прогениторных клеток в сосудистую стенку в норме и/или при патологии. Данных о роли Т-кадгерина в направленной миграции стволовых/прогениторных клеток во взрослом организме в литературе нет. Это направление исследований является перспективным с точки зрения изучения механизмов регенерации и морфогенеза и роли навигационных молекул в этих процессах. Известно, что высокий уровень ЛНП в крови приводит к накоплению липидов в сосудистой стенке и способствует развитию атеросклероза. Кроме того, ЛНП 348 потенциируют действие адреналина и ангиотензина II, что способствует повреждению сосудистой стенки и прогрессии сердечно-сосудистых заболеваний. В нашей лаборатории было показано, что помимо «классического» апо В/Е рецептора ЛНП, участвующего в эндоцитозе ЛНП в сосудистых клетках, на поверхности целого ряда клеток присутствует еще низкоаффинный рецептор, опосредующий быстрые «гормоноподобные» эффекты ЛНП (активацию фосфолипазы С и фосфоинозитидного обмена, увеличение концентрации цитоплазматического Ca2+, изменение активности аденилатциклазы и рН цитоплазмы, фосфорилирование белков) [Tkachuk et al, 1994; Tkachuk et al, 1996; Kuzmenko et al, 1994]. Этот рецептор был выделен и идентифицирован как Т-кадгерин. Однако физиологическая роль Т-кадгерина в сосудах долгое время оставалась неизвестна. В настоящей работе установлено, что Т-кадгерин является рецептором ЛНП, опосредующим активацию внутриклеточной сигнализации c участием [Ca2+]in и миграцию клеток по градиенту ЛНП [Rubina et al., 2005]. В последние годы появились данные о том, что Т-кадгерин является рецептором не только ЛНП, но способен специфически связываться с высокомолекулярными комплексами адипонектина. Адипонектин является адипокином, секретируемым жировой тканью и регулирующим обмен липидов и глюкозы. Именно высокомолекулярные комплексы адипонектина обладают защитным антиатерогенным действием в сосудистой стенке и кардиопротективным в миокарде, подавляя воспалительную реакцию, развитие гипертрофии миокарда и ишемическое повреждение сердца [Denzel et al., 2010; ParkerDuffen et al., 2013; Parker-Duffen and Walsh, 2014]. Известно, что адипонектин также предотвращает связывание ЛНП с протеогликанами, что препятствует накоплению ЛНП в сосудистой стенке. Физиологическая концентрация ЛНП в плазме крови человека составляет 1000 мкг/мл и выше, размер ЛНП примерно 512 кДа, Кd связывания Т-кадгерина с ЛНП составляет 40-60 мкг/мл. Концентрация адипонектина в плазме крови составляет 515 мкг/мл при размере высокомолекулярного адипонектина 360–540 кДа, Кd связывания Ткадгерина с адипонектином по предварительным неопубликованным данным, полученным в нашей лаборатории, составляет 3 мкг/мл. Существование двух лигандов Т-кадгерина с такими характеристиками позволяет предположить конкурентное связывание между высокомолекулярным адипонектином и ЛНП. Известно, что Т-кадгерин в сосудах не участвует в межклеточной адгезии, не располагается в межклеточных контактах (Рисунок 1.1), а выполняет сигнальную функцию. Можно предположить, что у взрослых здоровых людей Т-кадгерин в сосудах и в сердце постоянно связан с адипонектином, который опосредует свое защитное действие через активацию внутриклеточной сигнализации с 349 участием AMP киназы. Возможно, адипонектин конкурентно препятствует связыванию ЛНП с Т-кадгерином, что предотвращает «гормоноподобные» негативные эффекты ЛНП в сосудах. При повышении концентрации ЛНП в крови может происходить вытеснение адипонектина из связывания с Т-кадгерином, в результате чего устраняется защитное действие адипонектина и происходит активация [Ca2+]in сигнализации в сосудистых клетках и ремоделирование сосудистой стенки, как это происходит при атеросклерозе. Известно также, что атеросклеротические бляшки достаточно сильно васкуляризированы vasa vasorum, которые обладают повышенной проницаемостью, что добавляет нестабильности атеросклеротической бляшке. Возможно, при повышенном содержании ЛНП в бляшке Т-кадгерин оккупирован ЛНП и не может выполнять функцию «молекулы отталкивания» для прорастающих в бляшку vasa vasorum. Многие патологические состояния, в том числе атеросклероз, сопровождаются эндотелиальной дисфункцией, которая физиологически характеризуется изменением фенотипа эндотелиальных клеток, разрушением межклеточных контактов и увеличением проницаемости эндотелия. Ранее в нашей лаборатории было показано, что гиперэкспрессия Т-кадгерина приводит к изменению фенотипа эндотелиальных клеток с покоящего на промиграторный, что сопровождается снижением клеточной адгезии на субстрате [Philippova et al., 2005]. В настоящей работе мы обнаружили, что Т-кадгерин является фактором, влияющим на барьерную функцию эндотелия (гиперэкспрессия приводит к снижению барьерной функции эндотелия, а подавление Т-кадгерина - наоборот), и впервые раскрыли биохимические и молекулярно биологические механизмы участия Т-кадгерина в регуляции проницаемости эндотелия. На уровне внутриклеточной сигнализации эффекты Т-кадгерина связаны с активацией малых Rho ГТФаз, их нисходящих сигнальных посредников ROCK-II, LIMK и PAK1, перестройкой актинового цитоскелета и микротрубочек, интернализацией VE-кадгерина с поверхности мембраны и его деградацией в лизосомах. Следует отметить, что факты участия навигационных молекул не только в направленном росте сосудов в эмбриогенезе и при патологическом ангиогенезе, но и в регуляции функционирования зрелых сосудов, известны [Adams and Eichmann, 2010; Melani and Weinstein, 2010]. Так, было обнаружено, что Robo 4 участвует в поддержании целостности сосудистой стенки. Robo 4 по принципу обратной связи нейтрализует ангиогенные эффекты VEGF, а именно миграцию эндотелиальных клеток, формирование капилляро-подобных трубочек и увеличение проницаемости эндотелия. Таким образом 350 Robo 4 предотвращает избыточный ангиогенез, как это было показано на животных моделях патологического ангиогенеза в сетчатке [Jones et al., 2009]. Кроме того, существует предположение, что Robo 4 выполняет свою функцию не столько направляя рост сосудов, сколько участвуя в регуляции клеточной специализации - формировании ―tip‖ и ―stalk‖ клеточного фенотипа [Jones et al., 2008]. Поскольку в настоящей работе было обнаружено антиангиогенной влияние Т-кадгерина именно на начальные этапы ангиогенеза, было бы интересно проверить экспрессию Т-кадгерина в ―tip‖ и ―stalk‖ клетках и ответить на вопрос о его возможной роли в специализации эндотелиальных клеток. Известно, что экспрессия Т-кадгерина и адипонктина взаимно регулируется по принципу обратной связи: при снижении уровня адипонектина экспрессия Т-кадгерина повышается, а при его увеличении – наоборот [Parker-Duffen and Walsh, 2014]. Мы предполагаем, что в норме в ответ на различные повреждения сосудов может происходит увеличение экспрессии Т-кадгерина как рецептора адипонектина, опосредующего его защитное действие в сосудах. У людей, страдающих ожирением, уровень адипонектина в крови оказывается снижен, что, возможно, вызывает увеличение экспрессии Т-кадгерина, который сам по себе или опосредовано за счет связывания с ЛНП может вызывать увеличение проницаемости эндотелия и негативные эффекты ЛНП в сосудистой стенке. Дальнейшее изучение роли Т-кадгерина с использованием трансгенных животных и других животных моделей сердечно-сосудистых заболеваний позволит прояснить роль Ткадгерина в функционировании системы кровообращения, а также в развитии сердечнососудистых заболеваний. Таким образом, Т-кадгерин является многофункциональной молекулой. Гомофильное взаимодействие между молекулами Т-кадгерина на контактирующих клетках лежит в основе регуляции процессов ангиогенеза. В зрелых сосудах Т-кадгерин функционирует как сигнальная молекула. Т-кадгерин участвует в регуляции проницаемости эндотелиального монослоя. Т-кадгерин является рецептором ЛНП, опосредующим их сигнальные эффекты и миграцию клеток по градиенту ЛНП. 351 ВЫВОДЫ 1. В эмбриогенезе у мыши начало экспрессия мРНК и белка Т-кадгерина в головном мозге и в сердечно-сосудистой системе коррелирует с процессами активного формирования и роста сосудов. 2. Снижение уровня экспрессии Т-кадгерина коррелирует с малигнизацией таких новообразований кожи человека, как псориаз, кератоз, кератоакантома, базалиома, плоскоклеточный и метатипический рак и меланома. 3. Экспрессия Т-кадгерина в опухолевых клетках приводит к подавлению неоангионенеза в первичном узле меланомы у мышей. Однако Т-кадгерин не может считаться опухолевым супрессором, поскольку клетки меланомы, экспрессирующие Ткадгерин, «включают» гены, способствующие их большей выживаемости, миграции, инвазии и метастазированию. 4. Т-кадгерин является навигационной молекулой не только в нервной системе, но и в кровеносных сосудах. Экспрессия Т-кадгерина в клетках стромы подавляет прорастание в нее кровеносных сосудов. В основе эффектов Т-кадгерина лежит гомофильное взаимодействие, подавление миграции эндотелиальных клеток и формирования капилляроподобных структур. 5. Т-кадгерин участвует в регуляции барьерной функции эндотелия: экспрессия Ткадгерина в клетках повышает проницаемость эндотелиального монослоя, а подавление Ткадгерина - снижает. Регуляция проницаемости эндотелия Т-кадгерином осуществляется при участии Rho белков (RhoA, Cdc42, Rac1), ROCK-II, LIMK и PAK1, и сопровождается эндоцитозом VE- кадгерина, перестройкой как актинового, так и тубулинового цитоскелета. 6. Т-кадгерин экспрессируется в интиме, медии и адвентиции нормальной аорты во всех слоях; его экспрессия повышается при атеросклерозе в липидной полоске, нестабильной фиброатероме и при мягком кальцинозе. При атеросклерозе отмечается увеличение количества двойных позитивных по CD31 и Т-кадгерину клеток в субэндотелиальным слое и в медии сосудов. 7. Т-кадгерин является рецептором ЛНП, опосредующим Са2+ сигнализацию и направленную миграцию клеток по градиенту концентрации ЛНП. 352 СПИСОК ЛИТЕРАТУРЫ 1. Abdel-Rahman M, Boru G, Massengill J, Salem M, Davidorf F. MET oncogene inhibition as a potential target of therapy for uveal melanomas. // Investigative Ophthalmology & Visual Science. 2010. Vol. 51, №7. P. 3333-3339. 2. Ackerman A, Mones J. Solar (actinic) keratosis is squamous cell carcinoma. // British Journal of Dermatology. 2006. Vol. 155, №1. P. 9-22. 3. Adachi Y, Takeuchi T, Sonobe H, Ohtsuki Y. An adiponectin receptor, T-cadherin, was selectively expressed in intratumoral capillary endothelial cells in hepatocellular carcinoma: possible cross talk between T-cadherin and FGF-2 pathways. // Virchows Archiv. 2006. Vol. 448, №3. P. 311-318. 4. Adams R, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. // Nature reviews Molecular cell biology. 2007. Vol. 8, №6. P. 464-478. 5. Adams R, Diella F, Hennig S et al. The cytoplasmic domain of the ligand ephrinB2 is required for vascular morphogenesis but not cranial neural crest migration. // Cell. 2001. Vol. 104, №1. P.157–69. 6. Adams R, Eichmann A. Axon guidance molecules in vascular patterning. // Cold Spring Harbor perspectives in biology. 2010. Vol. 2, №5. P. 001875. 7. Addison C, Daniel T, Burdick M et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. // J Immunol 2000. №165. P. 5269–5277. 8. Aguirre-Ghiso J, Liu D, Mignatti A. et al. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. // Mol. Biol. Cell. 2001. Vol. 12, №4. P. 863-879. 9. Aidoudi S, Bujakowska K, Kieffer N, Bikfalvi A. The CXC-chemokine CXCL4 interacts with integrins implicated in angiogenesis. // PLoS One. 2008. Vol. 3, №7, P. 1-14. 10. Albelda S, Muller W, Buck C, Newman P. Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a novel vascular cell-cell adhesion molecule. // The Journal of cell biology. 1991. Vol. 114, №5. P. 1059-1068. 11. Allingham M, van Buul J, Burridge K. ICAM-1-mediated, Src-and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte 353 transendothelial migration. // The Journal of Immunology. 2007. Vol. 179, №6. P. 40534064 12. Andreeva A, Han J, Kutuzov M, Profirovic J, Tkachuk V, Voyno-Yasenetskaya T. Tcadherin modulates endothelial barrier function. // Journal of cellular Physiology. 2010. Vol. 223, №1. P. 94-102. 13. Andreeva A, Kutuzov M, Tkachuk V, Voyno-Yasenetskaya T. T-cadherin is located in the nucleus and centrosomes in endothelial cells. // American Journal of Physiology-Cell Physiology. 2009. Vol. 297, №5. P. 1168-1177. 14. Andriopoulou P, Navarro P, Zanetti A, Lampugnani M, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. // Arteriosclerosis, thrombosis, and vascular biology. 1999. Vol. 19, №10. P. 2286-2297. 15. Angelini D, Hyun S, Grigoryev D et al. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. // American Journal of Physiology-Lung Cellular and Molecular Physiology. 2006. Vol. 291, №6. P. 1232-1245. 16. Angiolillo A, Sgadari C, Taub D et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. // The Journal of experimental medicine. 1995. Vol. 182, №1. P. 155-162 17. Angst B, Marcozzi C, Magee A. The cadherin superfamily: diversity in form and function. // Journal of cell science. 2001. Vol. 114, №4. P. 629-641. 18. Appleton B, Wu P, Maloney J et al. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. // EMBO J. 2007. №26. P. 4902– 4912 19. Ara T, Tokoyoda K, Okamoto R et al. The role of CXCL12 in the organ- specific process of artery formation. // Blood. 2005. №105. P. 3155–3161. 20. Arber S, Barbayannis F, Hanser H et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. // Nature. 1998. Vol. 393, №6687. P. 805-809. 21. Arenberg D, Kunkel S, Polverini P et al. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. // J Clin Invest. 1996. № 97. P. 2792– 2802. 354 22. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. // Circulation research. 2005. Vol. 97, №6. P. 512-523. 23. Asahara T, Murohara T, Sullivan A et al. Isolation of putative progenitor endothelial cells for angiogenesis. // Science. 1997. Vol. 275, №5302. P. 964-966. 24. Augustin H, Koh G, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. // Nature reviews Molecular cell biology. 2009. Vol. 10, №3. P. 165-177. 25. Bagri A, Tessier-Lavigne M, Watts R. Neuropilins in tumor biology. // Clin Cancer Res. 2009. №15. P. 1860–1864. 26. Balestrieri M, Balestrieri A, Mancini F, Napoli C Understanding the immunoangiostatic CXC chemokine network. // Cardiovasc Res. 2008. №78. P. 250–256. 27. Banks E, Frierson Jr H, Mills S, George E, Zarbo R, Swanson P. Basaloid squamous cell carcinoma of the head and neck: a clinicopathologic and immunohistochemical study of 40 cases. // The American journal of surgical pathology. 1992. Vol. 16, №10. P. 939-940. 28. Barcelos L, Coelho A, Russo R et al. Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. // Microvasc Res. 2009. № 78. P. 148–154. 29. Bar-Eli M. Molecular mechanisms of melanoma metastasis. // Journal of cellular physiology. 1997. Vol. 173, №2. P. 275-278 30. Bauer E, Uitto J, Walters R, Eisen A. Enhanced collagenase production by fibroblasts derived from human basal cell carcinomas. // Cancer research. 1979. Vol. 39, №11. P. 4594-4599. 31. Beck A, Nieden R, Schneider H, Deitmer J. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. // Cell Calcium. 2004. Vol. 35, №1. P. 47-58. 32. Beckermann B, Kallifatidis G, Groth A et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. // British journal of cancer. 2008. Vol. 99, №4. P. 622-631. 33. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. // Nature reviews Drug discovery. 2009. Vol. 8, №3. P. 235-253. 355 34. Behrens J, von Kries J, Kohl M et al. Functional interaction of beta-catenin with the transcription factor LEF-1. // Nature. 1996. Vol. 382, №6592. P. 638-642 35. Belloni D, Scabini S, Foglieni C et al. The vasostatin-I fragment of chromogranin A inhibits VEGF-induced endothelial cell proliferation and migration. // The FASEB Journal. 2007. Vol. 21, №12. P. 3052-3062. 36. Bergers G, Brekken R, McMahon G et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. // Nature cell biology. 2000. Vol. 2, №10. P. 737-744. 37. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. // Nature Reviews Cancer. 2008. Vol. 8, №8. P. 592-603. 38. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. // Journal of Clinical Investigation. 2003. Vol. 111, №9. P. 1287-1295. 39. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. // Neuro-oncology. 2005. Vol. 7, №4. P. 452-464. 40. Bernardini G, Spinetti G, Ribatti D et al. I-309 binds to and activates endothelial cell functions and acts as an angiogenic molecule in vivo. // Blood. 2000. №96. P. 4039–4045. 41. Berridge M, Bootman M, Roderick H. Calcium signalling: dynamics, homeostasis and remodelling. // Nature Reviews Molecular Cell Biology. 2003. Vol. 4, №7. P. 517-529. 42. Berx G, Van Roy F. Involvement of members of the cadherin superfamily in cancer. // Cold Spring Harbor perspectives in biology. 2009. Vol. 1, №6. P. 003129. 43. Blashuk O W, Rowlands T M. Cadherins as modulators of angiogenesis and structural integrity of blood vessels. // Cancer and Metastasis Reviews. 2000. №9. P. 1-5. 44. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. // Nature reviews Molecular cell biology. 2002. Vol. 3, №12. P. 932-943. 45. Block L, Knorr M, Vogt E. et al. Low density lipoprotein causes general cellular activation with increased phosphatidylinositol turnover and lipoprotein catabolism. // Proceedings of the National Academy of Sciences. 1988. Vol. 85, №3. P. 885-889. 46. Bobik A, Tkachuk V. Metalloproteinases and plasminogen activators in vessel remodeling. // Current hypertension reports. 2003. Vol. 5, №6. P. 466-472. 356 47. Bochkov V, Rozhkova T, Matchin Y et al. LDL-and agonist-induced Ca2+-mobilization in platelets of healthy subjects and in patients with familial hyperlipoproteinemia type II. // Thrombosis research. 1991. Vol. 61, №4. P. 403-409. 48. Bochkov V, Tkachuk V, Buhler F, Resink T. Phosphoinositide and calcium signalling responses in smooth muscle cells: comparison between lipoproteins, Ang II, and PDGF. // Biochemical and biophysical research communications. 1992. Vol. 188, №3. P. 12951304. 49. Bochkov V, Tkachuk V, Hahn A, Bernhardt J, Buhler F, Resink T. Concerted effects of lipoproteins and angiotensin II on signal transduction processes in vascular smooth muscle cells. // Arteriosclerosis, Thrombosis, and Vascular Biology. 1993. Vol. 13, №9. P. 12611269. 50. Bochkov V, Tkachuk V, Kuzmenko Y, Borisova Y, Buhler F, Resink T. Characteristics of low and high density lipoprotein binding and lipoprotein-induced signaling in quiescent human vascular smooth muscle cells. // Molecular pharmacology. 1994. Vol. 45, №2. P. 262-270. 51. Bochkov V, Tkachuk V, Philippova M, Stambolsky D, Buhler F, Resink T. Ligand selectivity of 105 kDa and 130 kDa lipoprotein-binding proteins in vascular-smoothmuscle-cell membranes is unique. // Biochem J. 1996. Vol. 317 P. 297-304. 52. Bodnar R J, Yates C C, Rodgers M E, Du X, Wells A. IP-10 induces dissociation of newly formed blood vessels. // J Cell Sci. 2009. №122. P. 2064–2077. 53. Bodnar R, Yates C, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. // Circulation research. 2006. Vol. 98, №5. P. 617-625. 54. Bohuslav J, Horejsi V, Hansmann C et al. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. // The Journal of experimental medicine. 1995. Vol. 181, №4. P. 1381-1390. 55. Bosserhoff A, Ellmann L, Kuphal S. Melanoblasts in culture as an in vitro system to determine molecular changes in melanoma. // Experimental dermatology. 2011. Vol. 20, №5. P. 435-440. 357 56. Boudreau N, Turley E, Rabinovitch M. Fibronectin, hyaluronan, and a hyaluronan binding protein contribute to increased ductus arteriosus smooth muscle cell migration. // Dev Biol. 1991. Vol. 143, №2. P. 235-47. 57. Brakenhielm E, Veitonmaki N, Cao R et al. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. // PNAS 2004. Vol.101, №8. P. 2476-2481. 58. Breier G, Breviario F, Caveda L et al. Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. // Blood. 1996. Vol. 87, №2. P. 630-641. 59. Breier G. Angiogenesis in embryonic development—a review. // Placenta. 2000. Vol. 21 P. 11-15 60. Briggs M W, Sacks D B. IQGAP1 as a signal integrater: Ca2+, calmodulin, Cdc42 and the cytoskeleton. // FEBS Letters. 2003. №542. P. 7-11. 61. Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. // EMBO Rep. 2003. Vol. 4, №6. P. 571-574. 62. Brown M, Goldstein J. A receptor-mediated pathway for cholesterol homeostasis. // Science. 1986. Vol. 232, №4746. P. 34-47. 63. Bruses J. Cadherin-mediated adhesion at the interneuronal synapse. // Current opinion in cell biology. 2000. Vol. 12, №5. P. 593-597. 64. Buechner S, Philippova M, Erne P, Mathys T, Resink T. High T-cadherin expression is a feature of basal cell carcinoma. // British Journal of Dermatology. 2009. Vol. 161, №1. P. 199-202 65. Buhler F, Tkachuk V, Hahn A, Resink T. Low-and high-density lipoproteins as hormonal regulators of platelet, vascular endothelial and smooth muscle cell interactions: relevance to hypertension. // Journal of hypertension. 1991. №9. P.28-36. 66. Burger J A, Burkle A. The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. // Br J Haematol. 2007. №137. P. 288-296 67. Burggren W, Keller B. Development of cardiovascular systems. Cambridge, UK: Cambridge University Press, 1997. 358 68. Busso N, Masur S K, Lazega D Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. // JCB. 1994. Vol. 126, №1. P. 259-270. 69. Cao Y, Sonveaux P, Liu S et al. Systemic overexpression of angiopoietin-2 promotes tumor microvessel regression and inhibits angiogenesis and tumor growth. // Cancer research. 2007. Vol. 67, №8. P. 3835-3844. 70. Cao Y, Sun Z, Liao L et al. Human adipose tissue-derived stem cells differentiate into endothelial cells in vitro and improve postnatal neovascularization in vivo. // Biochem Biophys Res Commun. 2005. Vol. 332, №2. P. 370-379. 577. Carlson B. Human embryology & developmental biology. St. Louis: Mosby, 1999 С. 209-247. 71. Carmeliet P, Jain R. Molecular mechanisms and clinical applications of angiogenesis. // Nature. 2011. Vol. 473, №7347. P. 298-307. 72. Carmeliet P. Angiogenesis in health and disease. // Nature Medicine. 2003. Vol. 9, №6. P. 653-660. 73. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. // Nature Medicine. 2000. Vol. 6, №4. P. 389-395. 74. Cassarino D, DeRienzo D, Barr R. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. // Journal of Cutaneous Pathology. 2006. Vol. 33, №4. P. 261-279. 75. Cavallaro U, Liebner S, Dejana E. Endothelial cadherins and tumor angiogenesis. // Experimental cell research. 2006. Vol. 312, №5. P. 659-667. 76. Cebecauer M, Cerny J, Horejsi V. Incorporation of leucocyte GPI-anchored proteins and protein tyrosine kinases into lipid-rich membrane domains of COS-7 cells. // Biochemical and biophysical research communications. 1998. Vol. 243, №3. P. 706-710. 77. Ceradini D, Kulkarni A, Callaghan M et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. // Nat Med. 2004. №10. P. 858–864. 78. Chan D, Lee J, Chan P, Ng I. Genetic and epigenetic inactivation of T-cadherin in human hepatocellular carcinoma cells. // International Journal of Cancer. 2008. Vol. 123, №5. P. 1043-1052. 359 79. Chaplin D, Pettit G, Parkins C, Hill S. Antivascular approaches to solid tumour therapy: evaluation of tubulin binding agents. // The British journal of cancer Supplement. 1996. № 27. P. 86. 80. Chappuis-Flament S, Wong E, Hicks L, Kay C, Gumbiner B. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. // The Journal of cell biology. 2001. Vol. 154, №1. P. 231-243. 81. Charalambous C, Pen L, Su Y et al. Interleukin-8 differen- tially regulates migration of tumor-associated and normal human brain endothelial cells. // Cancer Res. 2005. №65. P. 10347–10354. 82. Charo I, Taubman M. Chemokines in the pathogenesis of vascular disease. // Circ Res. 2004. № 95. P. 858–866. 83. Chiesa A, Rapizzi E, Tosello V et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. // Biochemical Journal. 2001. Vol. 355 P. 1-12 84. Clark Jr W, Elder D, Guerry IV D, Epstein M, Greene M, Van Horn M. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. // Human pathology. 1984. Vol. 15, №12. P. 1147-1165. 85. Clark W. Tumour progression and the nature of cancer. // British journal of cancer. 1991. Vol. 64, №4. P. 631. 86. Clowes A, Clowes M, Au Y et al. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid artery. // Circ. Res. 1990. №67. P. 61–67. 87. Collen D. The plasminogen (fibrinolytic) system. // Thromb Haemost. 1999. Vol. 82, №2. P. 259-270. 88. Dan C, Kelly A, Bernard O, Minden A. Cytoskeletal changes regulated by the PAK4 serine/threonine kinase are mediated by LIM kinase 1 and cofilin. // Journal of Biological Chemistry. 2001. Vol. 276, №34. P. 32115-32121. 89. Danen E, Ten Berge P, Van Muijen G, Van't Hof-Grootenboer A, Brocker E, Ruiter D. Emergence of alpha 5 beta 1 fibronectin- and alpha v beta 3 vitronectin-receptor expression in melanocytic tumour progression. // Histopathology. 1994. Vol. 24, №3. P. 249-256. 360 90. Davis M, Ireton R, Reynolds A. A core function for p120-catenin in cadherin turnover. // The Journal of cell biology. 2003. Vol. 163, №3. P. 525-534. 91. Davy A, Soriano P. Ephrin signaling in vivo: look both ways. // Developmental dynamics. 2005. Vol. 232, №1. P. 1-10. 92. De Smet F, Segura I, De Bock K. et al. Mechanisms of Vessel Branching: Filopodia on Endothelial Tip Cells Lead the Way. // Arterioscler Thromb Vasc Biol. 2009. №29. P. 639649. 93. Deanfield J, Halcox J, Rabelink T. Endothelial function and dysfunction testing and clinical relevance. // Circulation. 2007. Vol. 115, №10. P. 1285-1295. 94. Defilippi P, Olivo C, Venturino M et al. Actin cytoskeleton organization in response to integrin-mediated adhesion. // Microscopy Res Tech. 1999. №47. P. 67–78. 95. Dejana E, Lampugnani M, Martinez-estrada O, Bazzoni G. The molecular organization of endothelial junctions and their functional role in vascular morphogenesis and permeability. // International Journal of Developmental Biology. 2000. Vol. 44, №6. P. 743-748. 96. Dejana E, Orsenigo F, Lampugnani M. The role of adherens junctions and VE-cadherin in the control of vascular permeability. // Journal of cell science. 2008. Vol. 121, №13. P. 2115-2122. 97. Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. // Journal of Clinical Investigation. 1996. Vol. 98, №9. P. 1949. 98. Dejana E. Endothelial cell-cell junctions: happy together. // Nature Reviews Molecular Cell Biology. 2004. Vol. 5, №4. P. 261-270. 99. Delcourt N, Bockaert J, Marin P. GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. // Trends Pharmacol Sci. 2007. Vol. 28, №12. P. 602-607 100.Demaurex N, Lew D, Krause K. Cyclopiazonic acid depletes intracellular Ca2+ stores and activates an influx pathway for divalent cations in HL-60 cells. // Journal of Biological Chemistry. 1992. Vol. 267 P. 2318-2324. 101.Denekamp J. Review article: angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. // Br J Radiol. 1993. Vol. 66, №783. P. 181196. 361 102.Denzel M, Scimia M, Zumstein P et al. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. // J Clin Invest. 2010. Vol. 120, №12. P. 4342-4352. 103.Desgrosellier J, Cheresh D. Integrins in cancer: biological implications and therapeutic opportunities. // Nature Reviews Cancer. 2010. Vol. 10, №1. P. 9-22. 104.Deshane J, Chen S, Caballero S et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. // J Exp Med. 2007. №204. P. 605–618. 105.Devalaraja R, Nanney L, Du J et al. Delayed wound healing in CXCR2 knockout mice. // J Invest Dermatol. 2000. №115. P. 234–244. 106.Deveza L, Choi J, Yang F. Therapeutic Angiogenesis for Treating Cardiovascular Diseases. // Theranostics. 2012. Vol. 2, №8. P. 801-814. 107.Dinehart S, Nelson-Adesokan P, Cockerell C, Russell S, Brown R. Metastatic cutaneous squamous cell carcinoma derived from actinic keratosis. // Cancer. 1997. Vol. 79, №5. P. 920-923. 108.Dinehart S, Pollack S. Metastases from squamous cell carcinoma of the skin and lip: an analysis of twenty-seven cases. // Journal of the American Academy of Dermatology. 1989. Vol. 21, №2. P. 241-248. 109.Distler J, Hirth A, Kurowska-Stolarska M, Gay R, Gay S, Distler O. Angiogenic and angiostatic factors in the molecular control of angiogenesis. // The quarterly journal of nuclear medicine: official publication of the Italian Association of Nuclear Medicine (AIMN) and the International Association of Radiopharmacology (IAR). 2003. Vol. 47, №3. P. 149-161. 110.Doyle D, Goings G, Upshaw-Earley J, Page E, Ranscht B, Palfrey H. T-cadherin is a major glycophosphoinositol-anchored protein associated with noncaveolar detergent-insoluble domains of the cardiac sarcolemma. // Journal of Biological Chemistry. 1998. Vol. 273, №12. P. 6937-6943. 111.Du F, Zhou J, Ren Gong X et al. Endothelial progenitor cells in atherosclerosis. // Frontiers in bioscience: a journal and virtual library. 2012. Vol. 17 P. 2327. 112.Duda D et al. CXCL12 (SDF1?) — CXCR4/CXCR7 pathway inhibition: anemerging sensitizer for anti-cancer therapies? // Clin. Cancer Res. 2011. №17. P. 2074–2080. 362 113.Dumler I, Stepanova V, Jerke U et al. Urokinase-induced mitogenesis is mediated by casein kinase 2 and nucleolin. // Curr Biol. 1999. Vol. 9, №24. P. 1468-1476. 114.Dutt P, Wang J, Groopman J. Stromal cell - derived factor-1 alpha and stem cell factor/kit ligand share signaling pathways in hemopoietic progenitors: a potential mechanism for cooperative induction of chemotaxis. // Journal of Immunology. 1998. Vol. 161, №7. P. 3652–3658. 115.Efimenko A, Starostina E, Rubina K, Kalinina N, Parfenova E. Viability and angiogenic activity of mesenchymal stromal cells from adipose tissue and bone marrow in hypoxia and inflammation in vitro. // Tsitologiia. 2009. Vol. 52, №2. P. 144-154. 116.Eichmann A, Le Noble F., Autiero M, Carmeliet P. Guidance of vascular and neural network formation. // Curr Opin Neurobiol. 2005. №15. P. 108-115. 117.Ellis L. The role of neuropilins in cancer. // Mol. Cancer Ther. 2006. Vol. 5, №5. P. 10991107. 118.Epstein E. Quantifying actinic keratosis: assessing the evidence. // American Journal of Clinical Dermatology. 2004. Vol. 5, №3. P. 141-144. 119.Erez N, Zamir E, Gour B, Blaschuk O, Geiger B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: involvement of fibroblast growth factor receptor-mediated signalling. // Experimental cell research. 2004. Vol. 294, №2. P. 366378. 120.Esser S, Lampugnani M, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. // Journal of Cell Science. 1998. Vol. 111, №13. P. 1853-1865. 121.Essler M, Amano M, Kruse H, Kaibuchi K, Weber P, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. // Journal of Biological Chemistry. 1998. Vol. 273, №34. P. 2186721874. 122.Evers E, Zondag G, Malliri A. et al. Rho family proteins in cell adhesion and cell migration. // European journal of cancer. 2000. Vol. 36, №10. P. 1269-1274. 363 123.Fagotto F, Rohani N, Anne-Sophie Touret, Rui Li. A Molecular Base for Cell Sorting at Embryonic Boundaries: Contact Inhibition of Cadherin Adhesion by Ephrin/EphDependent Contractility. // Developmental Cell. 2013. №27. P. 72–87. 124.Falcon B, Hashizume H, Koumoutsakos P et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. // The American journal of pathology. 2009. Vol. 175, №5. P. 2159-2170. 125.Farmer E, Helwig E. Metastatic basal cell carcinoma: a clinicopathologic study of seventeen cases. // Cancer. 1980. Vol. 46, №4. P. 748-757. 126.Feng Y, Sun X, Yang H, Teitelbaum D. Dissociation of E-cadherin and catenin in a mouse model of total parenteral nutrition: a mechanism for the loss of epithelial cell proliferation and villus atrophy. // The Journal of Physiology. 2009. Vol. 587, №3. P. 641-654. 127.Ferber E, Kajita M, Wadlow A et al. A role for the cleaved cytoplasmic domain of Ecadherin in the nucleus. // Journal of biological chemistry. 2008. Vol. 283, №19. P. 1269112700. 128. Ferrara N. VEGF-A: a critical regulator of blood vessel growth. // European cytokine network. 2009. Vol. 20, №4. P. 158-163 129.Filardo E, Brooks P, Deming S, Damsky C, Cheresh D. Requirement of the NPXY motif in the integrin beta 3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. // The Journal of cell biology. 1995. Vol. 130, №2. P. 441-450. 130.Finne E, Munthe E, Aasheim H. A new ephrin- A1 isoform (ephrin-A1b) with altered receptor binding properties abrogates the cleavage of ephrin-A1a. // Biochem. J. 2004. №379. P. 39–46. 131.Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? // Nature Reviews Cancer. 2008. Vol. 8, №12. P. 942-956. 132.Fleming T, Papenbrock T, Fesenko I et al. Assembly of tight junctions during early vertebrate development. // Cell and Development Biology. 2000. №11. P. 291-299. 133.Folberg R, Hendrix M, Maniotis A. Vasculogenic mimicry and tumor angiogenesis. // The American journal of pathology. 2000. Vol. 156, №2. P. 361-381. 134.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. // Nature medicine. 1995. Vol. 1, №1. P. 27-30. 364 135.Folkman J. Angiogenesis: an organizing principle for drug discovery? // NatureRev. Drug Discov. 2007. №6. P. 273–286. 136.Folkman J. Tumor angiogenesis: from bench to bedside. // Springer. 2008. P. 3-28. 137.Folkman J. Tumor angiogenesis: therapeutic implications. // The New England journal of medicine. 1971. Vol. 285 P. 1182-1186. 138.Fournier-Thibault C, Blavet C, Jarov A, Bajanca F, Thorsteinsdottir S, Duband J. Sonic hedgehog regulates integrin activity, cadherin contacts, and cell polarity to orchestrate neural tube morphogenesis. // The Journal of Neuroscience. 2009. Vol. 29, №40. P. 1250612520. 139.Fowler S, Berberian P, Shio H et al. Characterization of cell populations isolated from aortas of rhesus monkeys with experimental atherosclerosis. // Circ Res. 1980. №46. P. 520– 530. 140.Fredette B, Miller J, Ranscht B. Inhibition of motor axon growth by T-cadherin substrata. // Development. 1996. Vol. 122, №10. P. 3163-3171. 141.Fredette B, Ranscht B. T-cadherin expression delineates specific regions of the developing motor axon-hindlimb projection pathway. // The Journal of neuroscience. 1994. Vol. 14, №12. P. 7331-7346. 142.Freeman R, Barr R, Elder D. What is the boundary that separates a thick actinic keratosis from a thin squamous-cell carcinoma? // The American Journal of Dermatopathology. 1984. Vol. 6 P. 301–306. 143.Frenzel E, Johnson R. Gap junction formation between cultured embryonic lens cells is inhibited by antibody to N-cadherin. // Developmental biology. 1996. Vol. 179, №1. P. 116 144.Frey R, Ushio-Fukai M, Malik A. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. // Antioxidants & redox signaling. 2009. Vol. 11, №4. P. 791-810. 145.Frixen U, Behrens J, Sachs M et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. // The Journal of cell biology. 1991. Vol. 113, №1. P. 173-185. 365 146.Fujii T, Yonemitsu Y, Onimaru M et al. Nonendothelial mesenchymal cell-derived MCP-1 is required for FGF-2-mediated therapeutic neovascularization: critical role of the inflammatory/arteriogenic pathway. // Arterioscler Thromb Vasc Biol. 2006. №26. P. 2483–2489. 147.Fuller T, Korff T, Kilian A et al. Forward EphB4 signaling in endothelial cells controls cellular repulsion and segregation from EphrinB2 positive cells. // J Cell Sci. 2003. №116. P. 2461-2470. 148.Gabri M, Mazorra Z, Ripoll G et al. Complete antitumor protection by perioperative immunization with GM3/VSSP vaccine in a preclinical mouse melanoma model. // Clinical cancer research. 2006. Vol. 12, №23. P. 7092-7098. 149.Gaengel K, Genov'e G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. // Arteriosclerosis, thrombosis, and vascular biology. 2009. Vol. 29, №5. P. 630-638. 150.Galvez B, Genis L, Matias-Roman S et al. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoat- tractant protein-1/ccl2 and interleukin8/CXCL8 in endothelial cells during angiogenesis. // J Biol Chem. 2005. №280. P. 1292– 1298. 151.Ganju R, Brubaker S, Meyer J et al. The alpha-chemokine, stromal cell-derived factor1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. // Journal of Biological Chemistry. 1998. Vol. 273, №36. P. 23169–23175. 152.Ganzler S, Redies C. R-cadherin expression during nucleus formation in chicken forebrain neuromeres. // The Journal of neuroscience. 1995. Vol. 15, №6. P. 4157-4172. 153.Garcia C, Poletti E, Crowson A. Basosquamous carcinoma. // Journal of the American Academy of Dermatology. 2009. Vol. 60, №1. P. 137-143. 154.Garkavtsev I, Kozin S, Chernova O et al. The candidate tumour suppressor protein ING4 regu- lates brain tumour growth and angiogenesis. // Nature. 2004. №428. P. 328–332. 155.Gavard J, Gutkind J. VEGF controls endothelial-cell permeability by promoting the betaarrestin-dependent endocytosis of VE-cadherin. // Nature cell biology. 2006. Vol. 8, №11. P. 1223-1234. 366 156.Gengrinovitch S, Greenberg S, Cohen T et al. Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. // J Biol Chem. 1995. №270. P. 15059–15065. 157.George S, Beeching C. Cadherin: catenin complex: a novel regulator of vascular smooth muscle cell behaviour. // Atherosclerosis. 2006. Vol. 188, №1. P. 1-11. 158.Geretti E, Shimizu A, Klagsbrun M. Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. // Angiogenesis. 2008. №11. P. 31–39. 159.Gerety S, Wang H, Chen Z, Anderson D. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. // Mol. Cell. 1999. №4. P. 403–414. 160.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. // Cell Tissue Res. 2003. №314. P. 15-23. 161.Gerhardt H, Semb H. Pericytes: gatekeepers in tumour cell metastasis? // Journal of molecular medicine. 2008. Vol. 86, №2. P. 135-144. 162.Ghosh S, Joshi M, Ivanov D et al. Use of multicellular tumor spheroids to dissect endothelial cell-tumor cell interactions: a role for T-cadherin in tumor angiogenesis. // FEBS letters. 2007. Vol. 581, №23. P. 4523-4528. 163.Ghosh S., Brown R., Jones J. C. et al. Urinary-type plasminogen activator (uPA) expression and uPA receptor localization are regulated by alpha 3beta 1 integrin in oral keratinocytes.l. // J. Biol. Chem. 2000. Vol. 275, №31. P. 23869–23876. 164.Gilbert S, Singer S. Developmental Biology, Eighth Edition. Sinauer Associates, 2006. 165.Gloushankova N. Changes in regulation of cell—cell adhesion during tumor transformation. // Biochemistry (Moscow). 2008. Vol. 73, №7. P. 742-750. 166.Godic A. New approaches to psoriasis treatment. // Acta Dermatovenerologica. 2004. Vol. 13, №2. P. 50-57. 167.Godt D, Tepass U. Drosophila oocyte localization is mediated by differential cadherinbased adhesion. // Nature. 1998. Vol. 395, №6700. P. 387-391. 367 168.Goldstein J, Brown M, Anderson R, Russell D, Schneider W. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. // Annual review of cell biology. 1985. Vol. 1, №1. P. 1-39. 169.Gomes D, Rodrigues M, Leite M et al. c-Met must translocate to the nucleus to initiate calcium signals. // Journal of biological chemistry. 2008. Vol. 283, №7. P. 4344-4351. 170.Gorovoy M, Niu J, Bernard O et al. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. // Journal of biological chemistry. 2005. Vol. 280, №28. P. 26533-26542. 171.Gory-Faure S, Prandini M, Pointu H et al. Role of vascular endothelial-cadherin in vascular morphogenesis. // Development. 1999. Vol. 126, №10. P. 2093-2102. 172.Gotsch U, Borges E, Bosse R et al. VE-cadherin antibody accelerates neutrophil recruitment in vivo. // Journal of cell science. 1997. Vol. 110, №5. P. 583-588. 173.Gotthardt M, Trommsdorff M, Nevitt M. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. // J.Biol.Chem. 2000. Vol. 275, №33. P. 25616-25624. 174.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. // Nature. 2007. Vol. 445, №7130. P. 851-857. 175.Green A, Harwood C, Lear J, Proby C, Sinnya S, Soyer H. Skin cancer prevention: recent evidence from randomized controlled trials. // Current dermatology reports. 2012. Vol. 1, №3. P. 123-130. 176.Grunewald M, Avraham I, Dor Y et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. // Cell. 2006. №124. P. 175–189. 578. Grynkiewicz G, Poenie M, Tsien R. A new generation of Ca2+ indicators with greatly improved fluorescence properties. // Journal of biological chemistry. 1985. Vol. 260, №6. P. 3440-3450 177.Guleng B, Tateishi K, Ohta M et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. // Cancer Res. 2005. №65. P. 5864–5871. 178.Gumbiner B. Regulation of cadherin adhesive activity. // The Journal of cell biology. 2000. Vol. 148, №3. P. 399-404. 368 179. Gumbiner B. Regulation of cadherin adhesive activity. // The Journal of cell biology. 2000. Vol. 148, №3. P. 399-404 180.Gumbiner B. Regulation of cadherin-mediated adhesion in morphogenesis. // Nature reviews Molecular cell biology. 2005. Vol. 6, №8. P. 622-634. 181. Gumbiner B. Regulation of cadherin-mediated adhesion in morphogenesis. // Nature reviews Molecular cell biology. 2005. Vol. 6, №8. P. 622-634 182. Gumbiner B. Signal transduction of beta-catenin. // Current opinion in cell biology. 1995. Vol. 7, №5. P. 634-640 183.Guo M, Breslin J, Wu M, Gottardi C, Yuan S. VE-cadherin and beta-catenin binding dynamics during histamine-induced endothelial hyperpermeability. // American journal of physiology-cell physiology. 2008. Vol. 294, №4. P. 977-984. 184.Guo T, McCay J, Zhang L et al. Genistein modulates immune responses and increases host resistance to B16F10 tumor in adult female B6C3F1 mice. // The Journal of nutrition. 2001. Vol. 131, №12. P. 3251-3258. 185.Gupta S, Lysko P, Pillarisetti K et al. Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. // J Biol Chem. 1998. №273. P. 4282–4287. 186.Guthrie S. Neuronal development: sorting out motor neurons. // Current biology. 2002. Vol. 12, №14. P. 488-490. 187.Gutmann D, Wu Y, Hedrick N, Zhu Y, Guha A, Parada L. Heterozygosity for the neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes. // Human molecular genetics. 2001. Vol. 10, №26. P. 3009-3016. 188.Haass N, Herlyn M. Normal human melanocyte homeostasis as a paradigm for understanding melanoma. // The journal of investigative dermatology SP. 2005. Vol. 10, №2. P. 153-163. 189. Halaban R, Rubin J S, White W Met and HGF-SF in normal melanocytes and melanoma cells. // EXS. 1993. Vol. 65. P. 329-339. 190.Hall A. Rho GTPases and the actin cytoskeleton. // Science. 1998. Vol. 279, №5350. P. 509-514. 369 191.Hamada K, Oike Y, Ito Y et al. Distinct roles of ephrin-B2 forward and EphB4 reverse signaling in endothelial cells. // Arterioscler. Thromb. Vasc. Biol. 2003. №23. P. 190–197. 192.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. // Cell. 1996. Vol. 86, №3. P. 353-364. 193.Hanahan D, Weinberg R. The hallmarks of cancer. // Cell. 2000. Vol. 100, №1. P. 57-70 194.Hansell P, Maione T, Borgstrom P. Selective binding of platelet factor 4 to regions of active angiogenesis in vivo. // Am J Physiol. 1995. №269. P. 829–836. 195.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. // N Engl J Med. 2005. Vol. 352, № 16. P. 1685-95. 196.Hasan J, Byers R, Jayson G. Intra-tumoural microvessel density in human solid tumours. // British journal of cancer. 2002. Vol. 86, №10. P. 1566-1577. 197.Havel R, Eder H, Bragdon J. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. // Journal of clinical investigation. 1955. Vol. 34, №9. P. 1345. 198.Hebbard L, Garlatti M, Young L, Cardiff R, Oshima R, Ranscht B. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. // Cancer research. 2008. Vol. 68, №5. P. 1407-1416. 199.Hedgecock E, Culotti J, Hall D. The unc-5, unc-6, and unc-40 genes guide circumferential migrationsof pioneer axons and mesodermal cells on the epidermis in C.elegans. // Neuron. 1990. №4. P. 61–85. 200.Helbling P, Saulnier D, Brandli A. The receptor tyrosine kinase EphB4 and ephrin-B ligands restrict angiogenic growth of embryonic veins in Xenopus laevis. Development. 2000. №127. P. 269–278. 201.Hellberg C, Ostman A, Heldin C. PDGF and vessel maturation. // Recent results cancer research. 2010. Vol.180. P. 103-114. 202.Hendrix M, Seftor E, Hess A, Seftor R. Molecular plasticity of human melanoma cells. // Oncogene. 2003. Vol. 22, №20. P. 3070-3075. 370 203.Hendrix M, Seftor E, Meltzer P et al. Expression and functional significance of VEcadherin in aggressive human melanoma cells: role in vasculogenic mimicry. // Proceedings of the national academy of sciences. 2001. Vol. 98, №14. P. 8018-8023. 204.Heymans S, Luttun A, Nuyens D et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. // Nat Med. 1999. Vol. 5, №10. P. 1135-1142. 205.Hibi K, Nakayama H, Kodera Y, Ito K, Akiyama S, Nakao A. CDH13 promoter region is specifically methylated in poorly differentiated colorectal cancer. // British journal of cancer. 2004. Vol. 90, №5. P. 1030-1033. 206.Hill C, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hsregulate transcriptional activation by SRF. // Cell. 1995. Vol. 81, №7. P. 11591170. 207.Hoang S, Liauw J, Choi M et al. Netrin-4 enhances angiogenesis and neurologic outcome after cerebral ischemia. // J. Cereb. Blood Flow Metab. 2008. №29. P. 385-397. 208.Hodivala-Dilke K. alphavbeta3 integrin and angiogenesis: a moody integrin in a changing environment. // Current opinion in cell biology. 2008. Vol. 20, №5. P. 514-519. 209.Holash J, Wiegand S, Yancopoulos G. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. // Oncogene. 1999. Vol. 18, №38. P. 5356-5362. 210.Holmgren L, O'Reilly M, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. // Nature medicine. 1995. Vol. 1, №2. P. 149-153. 211.Honda H, Wakamatsu B, Goldhaber J, Berliner J, Navab M, Weiss J. High-density lipoprotein increases intracellular calcium levels by releasing calcium from internal stores in human endothelial cells. // Atherosclerosis. 1999. Vol. 143, №2. P. 299-306. 212.Hordijk P, Anthony E, Mul F, Rientsma R, Oomen L, Roos D. Vascular-endothelialcadherin modulates endothelial monolayer permeability. // Journal of cell science. 1999. Vol. 112, №12. P. 1915-1923. 371 213.Horton L, Yu Y, Zaja-Milatovic S et al. Opposing roles of murine duffy antigen receptor for chemokine and murine CXC chemokine receptor-2 receptors in murine melanoma tumor growth. // Cancer Res. 2007. №67. P. 9791–9799. 214.Hotary K, Allen E, Brooks P, Datta N, Long M, Weiss S. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. // Cell. 2003. Vol. 114, №1. P. 33-45. 215.Hsu M, Meier F, Nesbit M et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. // The American journal of pathology. 2000. Vol. 156, №5. P. 15151525. 216.Hsu M, Shih D, Meier F et al. Adenoviral Gene Transfer of beta3 Integrin Subunit Induces Conversion from Radial to Vertical Growth Phase in Primary Human Melanoma. // The American journal of pathology. 1998. Vol. 153, №5. P. 1435-1442. 217.Huang Z, Wu Y, Hedrick N, Gutmann D. T-cadherin-mediated cell growth regulation involves G2 phase arrest and requires p21CIP1/WAF1 expression. // Molecular and cellular biology. 2003. Vol. 23, №2. P. 566-578. 218.Huber A, Kolodkin A, Ginty D, Cloutier J. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. // Annu Rev Neurosci. 2003. №26. P. 509 –563. 219.Huber T, Kouskoff V, Fehling H, Palis J, Keller G. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. // Nature. 2004. Vol. 432, №7017. P. 625-630. 220. Hudry-Clergeon H, Stengel D, Ninio E, Vilgrain I. Platelet-activating factor increases VE-cadherin tyrosine phosphorylation in mouse endothelial cells and its association with the PtdIns3'-kinase. // The FASEB journal. 2005. Vol. 19, №6. P. 512-520. 221.Hug C, Wang J, Ahmad N, Bogan J, Tsao T, Lodish H. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. // Proceedings of the National Academy of Sciences of the United States of America. 2004. Vol. 101, №28. P. 10308-10313. 372 222.Hung S, Pochampally R, Chen S et al. Angiogenic effects of human multipotent stromal cell conditioned medium activates the PI3K-Akt pathway in hypoxic endothelial cells to inhibit apoptosis, increase survival, and stimulate angiogenesis. // Stem Cells. 2007. Vol. 25, №9. P. 2363-2370. 223.Hwang J, Kim C, Son K et al. Angiogenic activity of human CC chemokine CCL15 in vitro and in vivo. // FEBS Lett. 2004. №570. P. 47–51. 224.Hwang J, Son K, Kim C et al. Human CC chemokine CCL23, a ligand for CCR1, induces endothelial cell migration and promotes angiogenesis. // Cytokine. 2005. №30. P. 254–263 225.Imhof B, Aurrand-Lions M. Angiogenesis and inflammation face off. // Nature Medicine. 2006. Vol. 12, №2. P. 171-172. 226.Iruela-Arispe M, Davis G. Cellular and molecular mechanisms of vascular lumen formation. // Developmental cell. 2009. Vol. 16, №2. P. 222-231. 227.Ishihara K, Saida T, Yamamoto A. Updated statistical data for malignant melanoma in Japan. // International journal of clinical oncology. 2001. Vol. 6, №3. P. 109-116. 228.Ito H. Chemokines in mesenchymal stem cell therapy for bone repair: a novel concept of recruiting mesenchymal stem cells and the possible cell sources. // Modern rheumatology. 2011. Vol. 21, №2. P. 113-121. 229.Ivanov D, Philippova M, Allenspach R, Erne P, Resink T. T-cadherin upregulation correlates with cell-cycle progression and promotes proliferation of vascular cells. // Cardiovascular research. 2004. Vol. 64, №1. P. 132-143. 230.Ivanov D, Philippova M, Antropova J et al. Expression of cell adhesion molecule Tcadherin in the human vasculature. // Histochemistry and cell biology. 2001. Vol. 115, №3. P. 231-242. 231.Jain R, Carmeliet P. Vessels of death or life. // Scientific American. 2001. Vol. 285, №6. P. 38. 232.Jain R. Molecular regulation of vessel maturation. // Nature medicine. 2003. Vol. 9, №6. P. 685-693. 233. Jang Y, Lincoff A, Plow E, Topol E. Cell adhesion molecules in coronary artery disease. // Journal of the American college of cardiology. 1994. Vol. 24, №7. P. 1591-1601 373 234.Johnson J. Cell adhesion molecules in the development and progression of malignant melanoma. // Cancer and Metastasis Reviews. 1999. Vol. 18, №3. P. 345-357. 235.Jones B, McTaggart S. Immunosuppression by mesenchymal stromal cells: from culture to clinic. // Exp Hematol. 2008. Vol. 36, №6. P. 733-741. 236.Jones C, Nishiya N, London N et al. Slit2-Robo4 signaling promotes vascular stability by blocking Arf6 activity. // Nat. Cell Biol. 2009. №11. P. 1325–1331. 237.Joris I, Zand T, Nunnari J et al. Studies on the pathogenesis of atherosclerosis. I. Adhesion and emigration of mono- nuclear cells in the aorta of hypercholesterolemic rats. // Am J Pathol. 1983. №113. P. 341–358. 238.Jorizzo J, Carney P, Ko W, Robins P, Weinkle S, Werschler W. Treatment options in the management of actinic keratosis. // Cutis. 2004. Vol. 74 P. 9-17. 239.Joshi M, Ivanov D, Philippova M, Erne P, Resink T. Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3 in endothelial cells. // The FASEB Journal. 2007. Vol. 21, №12. P. 3083-3095. 240.Joshi M, Philippova M, Ivanov D, Allenspach R, Erne P, Resink T. T-cadherin protects endothelial cells from oxidative stress-induced apoptosis. // The FASEB journal. 2005. Vol. 19, №12. P. 1737-1739. 241.Kalwitz G, Andreas K, Endres M et al. Chemokine profile of human serum from whole blood: migratory effects of CXCL-10 and CXCL-11 on human mesenchymal stem cells. // Connective tissue research. 2010. Vol. 51, №2. P. 113-122. 242.Kandimalla K, Scott O, Fulzele S, Davidson M, Poduslo J. Mechanism of neuronal versus endothelial cell uptake of Alzheimer's disease amyloid ? protein. // PLoS ONE. 2009. Vol. 4, №2. P. 4627. 243.Kaplan D. Distinct regions of the cadherin cytoplasmic domain are essential for functional interaction with galpha 12 and beta -catenin. // Journal of biological chemistry. 2001. Vol. 276, №47. P. 44037-44043. 244.Kaplan R et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the premetastatic niche. // Nature. 2005. №438, P. 820–827. 374 245.Kapustin A, Stepanova V, Aniol N et al. Fibulin-5 binds urokinase-type plasminogen activator and mediates urokinase-stimulated beta1-integrin-dependent cell migration. // Biochemical Journal. 2012. Vol. 443, №2. P. 491-503. 246.Kasama T, Muramatsu M, Kobayashi K et al. Interaction of monocytes with vascular endothelial cells synergisti- cally induces interferon gamma-inducible protein 10 expression through activation of specific cell surface molecules and cytokines. // Cell Immunol. 2002. №219. P. 131–139. 247.Kawakami M, Staub J, Clibi W et al. Involvement of H-cadherin (CDH13) on 16q in the region of frequent deletion in ovarian cancer. // Int. J. Oncol. 1999. №15. P. 715-720. 248.Keehn C, Smoller B, Morgan M. Ets-1 immunohistochemical expression in non-melanoma skin carcinoma. // Journal of cutaneous pathology. 2004. Vol. 31, №1. P. 8-13 249.Keeley EC, Mehrad B, Strieter RM Chemokines as mediators of neovascularization. // Arterioscler Thromb Vasc Biol. 2008. №28. P. 1928–1936. 250.Kelley J, Rozek M, Suenram C, Schwartz C. Activation of human peripheral blood monocytes by lipoproteins. // The American journal of pathology. 1988. Vol. 130, №2. P. 223-231. 251.Kenessey I, Keszthelyi M, Kramer Z et al. Inhibition of c-Met with the specific small molecule tyrosine kinase inhibitor SU11274 decreases growth and metastasis formation of experimental human melanoma. // Current cancer drug targets. 2010. Vol. 10, №3. P. 332342. 252.Keynes R, Stern C, others. Segmentation in the vertebrate nervous system. // Nature. 1984. Vol. 310, №5980. P. 786-789. 253.Kiedrowski L N-Methyl-d-aspartate excitotoxicity: Relationships among plasma membrane potential, Na+/Ca2+ exchange, mitochondrial Ca2+ overload and cytoplasmic concentrations of Ca2+, H+ and K+. // Mol Pharmacol. 1999. Vol. 56. P. 619–632. 254.Kim J, Han J, Shim Y et al. Abberant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. // Cancer. 2005. Vol. 104, №9. P. 1825-1833. 375 255.Kimura Y, Matsunami H, Takeichi M. Expression of cadherin-11 delineates boundaries, neuromeres, and nuclei in the developing mouse brain. // Developmental dynamics. 1996. Vol. 206, №4. P. 455-462. 256.Kinnaird T, Stabile E, Burnett M et al. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. // Circulation research. 2004. Vol. 94, №5. P. 678-685. 257.Kirchheimer J, Wojta J, Christ G et al. Functional inhibition of endogenously produced urokinase decreases cell proliferation in a human melanoma cell line. // Proc. Natl. Acad. Sci. USA. 1989. № 86. P. 5424-5428. 258.Klopp A, Gupta A, Spaeth E, Andreeff M, Marini F. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? // Stem cells. 2011. Vol. 29, №1. P. 11-19. 259.Knudson W, Biswas C, Toole B. Interactions between human tumor cells and fibroblasts stimulate hyaluronate synthesis. // Proceedings of the National Academy of Sciences. 1984. Vol. 81, №21. P. 6767-6771. 260.Kobayashi K, Inoguchi T, Sonoda N, Sekiguchi N, Nawata H. Adiponectin inhibits the binding of low-density lipoprotein to biglycan, a vascular proteoglycan. // Biochemical and biophysical research communications. 2005. Vol. 335, №1. P. 66-70. 261.Koch A, Fong T, Volpert O et al. Interleukin-4 is an inhibitor of angiogenesis. // Arthtritis Rheum. 1996. №39. P. S304. 262.Koch A, Polverini P, Kunkel S et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. // Science. 1992. Vol. 258, №5089. P. 1798-1801. 263.Koch A, Volin M, Woods J et al. Regulation of angiogenesis by the C-X-C chemokines interleukin-8 and epithelial neutrophil activating peptide 78 in the rheumatoid joint. // Arthritis Rheum. 2001. №44. P. 31–40. 264.Kohgo Y, Lynch M, Rueda B, Chung D. Induction of interleukin-8 preserves theangiogenic response in HIF-1alpha-deficient colon cancer cells. // Nat Med. 2005. №11. P. 992-997. 376 265.Koller E, Ranscht B. Differential targeting of T-and N-cadherin in polarized epithelial cells. // Journal of Biological Chemistry. 1996. Vol. 271, №47. P. 30061-30067. 266.Kolodgie F, Finn A, Narula J, Virmani R. Atherosclerotic plaque angiogenesis as a mechanism of intraplaque hemorrhage and acute coronary rupture. // Therapeutic angiogenesis for vascular diseases. 2010. P. 213-236. 267.Kolodkin A, Matthes D, O‘Connor T et al. Fasciclin IV: sequence, expression, and function during growth cone guidance in the grasshopper embryo. // Neuron. 1992. №9. P. 31–45. 268.Komarova Y, Malik A. Regulation of endothelial permeability via paracellular and transcellular transport pathways. // Annual review of physiology. 2010. Vol. 72 P. 463493. 269.Kondratiev S, Gnepp D, Yakirevich E et al. Expression and prognostic role of MMP2, MMP9, MMP13, and MMP14 matrix metalloproteinases in sinonasal and oral malignant melanomas. // Human pathology. 2008. Vol. 39, №3. P. 337-343. 270.Korematsu K, Redies C. Restricted expression of cadherin-8 in segmental and functional subdivisions of the embryonic mouse brain. // Developmental dynamics. 1997. Vol. 208, №2. P. 178-189. 271.Kouklis P, Konstantoulaki M, Malik A. VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. // Journal of biological chemistry. 2003. Vol. 278, №18. P. 16230-16236. 272.Koutsouki E, Beeching C, Slater S, Blaschuk O, Sala-Newby G, George S. N-cadherindependent cell-cell contacts promote human saphenous vein smooth muscle cell survival. // Arteriosclerosis, thrombosis, and vascular biology. 2005. Vol. 25, №5. P. 982-988. 273.Kovacs E, Ali R, McCormack A, et al. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. // Journal of biological chemistry. 2002. Vol. 277, №8. P. 6708-6718. 274.Kronstein R, Seebach J, Grossklaus S et al. Caveolin-1 opens endothelial cell junctions by targeting catenins. // Cardiovascular research. 2012. Vol. 93, №1. P. 130-140. 377 275.Kuchelmeister C, Schaumburg-Lever G, Garbe C. Acral cutaneous melanoma in caucasians: clinical features, histopathology and prognosis in 112 patients. // British journal of dermatology. 2000. Vol. 143, №2. P. 275-280. 276.Kudrjashova E, Bashtrikov P, Bochkov V et al. Expression of adhesion molecule Tcadherin is increased during neointima formation in experimental restenosis. // Histochemistry and cell biology. 2002. Vol. 118, №4. P. 281-290. 277.Kuijper S, Turner C, Adams R. Regulation of angiogenesis by Eph-ephrin interactions. // Trends in cardiovascular medicine. 2007. Vol. 17, №5. P. 145-151. 278.Kuphal S, Martyn A, Pedley J et al. H-Cadherin expression reduces invasion of malignant melanoma. // Pigment cell & melanoma research. 2009. Vol. 22, №3. P. 296-306. 279.Kurzen H, Munzing I, Hartschuh W. Expression of desmosomal proteins in squamous cell carcinomas of the skin. // Journal of cutaneous pathology. 2003. Vol. 30, №10. P. 621-630. 280.Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. // Trends in cell biology. 2005. Vol. 15, №3. P. 121-124. 281.Kuzmenko Y, Bochkov V, Philippova M, Tkachuk V, Resink T. Characterization of an atypical lipoprotein-binding protein in human aortic media membranes by ligand blotting. // Biochemistry journal. 1994. Vol. 303 P. 281-287. 282.Kuzmenko Y, Kern F, Bochkov V, Tkachuk V, Resink T. Density-and proliferation statusdependent expression of T-cadherin, a novel lipoprotein-binding glycoprotein: a function in negative regulation of smooth muscle cell growth? // FEBS letters. 1998. Vol. 434, №1. P. 183-187. 283.Kuzuya M, Iguchi A Role of matrix metalloproteinases in vascular remodeling. // J. Atheroscler. Thromb. 2003. Vol. 10, № 5. P. 275-282. 284.Lackmann M. Mechanisms and functions of Eph receptor signaling. Handbook of Cell Signaling, Three Volume Set 2 ed. 2010. Elsevier. Chapter 62. P.443-449. 285.Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. // Circ Res. 2007. Vol. 100, №6. P. 782-794. 286.Lampugnani M, Dejana E. Interendothelial junctions: structure, signalling and functional roles. // Current opinion in cell biology. 1997. Vol. 9, №5. P. 674-682 378 287.Lampugnani M, Zanetti A, Breviario F et al. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. // Molecular biology of the cell. 2002. Vol. 13, №4. P. 1175-1189. 288.Lanahan A, Hermans K, Claes F et al. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. // Developmental cell. 2010. Vol. 18, №5. P. 713-724. 289.Larrivee B, Freitas C, Suchting S et al. Guidance of vascular development: lessons from the nervous system. // Circ Res. 2009. Vol. 104, №4. P. 428-441. 290.Lasagni L, Francalanci M, Annunziato F et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. // J Exp Med. 2003. №197. P. 1537–1549. 291.Lebwohl M, Dinehart S, Whiting D et al. Imiquimod 5% cream for the treatment of actinic keratosis: results from two phase III, randomized, double-blind, parallel group, vehiclecontrolled trials. // Journal of the American academy of dermatology. 2004. Vol. 50, №5. P. 714-721. 292.Lee H, Blasco M, Gottlieb G et al. Essential role of mouse telomerase in highly proliferative organs. // Nature. 1998. Vol. 392, №6676. P. 569-574 293.Lee S, Chen T, Barber C et al. Autocrine VEGF signaling is required for vascular homeostasis. // Cell. 2007. Vol. 130, №4. P. 691-703. 294.Lee S. H-cadherin, a novel cadherin with growth inhibitory functions and diminished expression in human breast cancer. // Nature medicine. 1996. Vol. 2, №7. P. 776-782. 295.Lee Y, Kim D, Lee S, Kim D, Nam H, Cho M. Expression of the c-Met proteins in malignant skin cancers. // Annals of dermatology. 2011. Vol. 23, №1. P. 33-38. 296.Leong D., Hutmacher D., Chew F., and Lim T. Viability and adipogenic potential of human adipose tissue processed cell population obtained from pump-assisted and syringeassisted liposuction // Journal of Dermatological Science, 2005. Vol. 37, №3. P. 169-176. 297.Li A, Dubey S, Varney M et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. // J Immunol. 2003. №170. P. 3369–3376. 379 298.Li G, Herlyn M. Dynamics of intercellular communication during melanoma development. // Molecular medicine today. 2000. Vol. 6, №4. P. 163-169. 299.Li H, Cybulsky M, Gimbrone M, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. // Arteriosclerosis, thrombosis, and vascular biology. 1993. Vol. 13, №2. P. 197-204. 300.Li M, Ransohoff R. The roles of chemokine CXCL12 in embryonic and brain tumor angiogenesis. // Semin Cancer Biol. 2009. №19. P. 111–115. 301.Lijnen H, Maquoi E, Hansen L et al. Matrix metalloproteinase inhibition impairs adipose tissue development in mice. // Arterioscler. Thromb. Vasc. Biol. 2002. Vol. 22, № 3. P. 374-379 302.Lijnen H, Van Hoef B, Nelles L et al. Plasminogen activation with single-chain urokinasetype plasminogen activator (scu-PA). Studies with active site mutagenized plasminogen (Ser740-Ala) and plasmin-resistant scu-PA (Lys158-Glu). // J. Biol. Chem. 1990. Vol. 265, № 9. P. 5232–5236. 303.Lilien J, Balsamo J, Arregui C, Xu G. Turn-off, drop-out: Functional state switching of cadherins. // Developmental dynamics. 2002. Vol. 224, №1. P. 18-29. 304.Linask K, Lash J. Precardiac cell migration: fibronectin localization at mesodermendoderm interface during directional movement. // Developmental biology. 1986. Vol. 114, №1. P. 87-101. 305.Lloyd-Jones D, Adams R, Carnethon M et al. Heart disease and stroke statistics - 2009 update a report from the American heart association statistics committee and stroke statistics subcommittee. // Circulation. 2009. Vol. 119, №3. P. 21-181. 306.Losordo D, Dimmeler S. Therapeutic angiogenesis and vasculogenesisfor ischemic disease. Part I: angiogenic cytokines. // Circulation. 2004. №109. P. 2487-2491. 307. Lozano R, Naghavi M, Foreman K et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. // The Lancet. 2013. Vol. 380, №9859. P. 2095-2128 380 308.Lund H. Metastasis from sun-induced squamous-cell carcinoma of the skin: an uncommon event. // The Journal of dermatologic surgery and oncology. 1984. Vol. 10, №3. P. 169174. 309.Luo Y, Raible D, Raper J. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. // Cell. 1993. №75. P. 217–227. 310.Ma J, Wang Q, Fei T et al. MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. // Blood. 2007. №109. P. 987–994. 311.Ma Z, Webb D, Jo M, Gonias S. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. // J. Cell Sci. 2001. №114. P. 3387-3396. 312.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cellpermeable inhibitor of dynamin. // Developmental cell. 2006. Vol. 10, №6. P. 839-850. 313.Maekawa H, Oike Y, Kanda S et al. Ephrin-B2 induces migration of endothelial cells through the phosphatidylinositol-3 kinase pathway and promotes angiogenesis in adult vasculature. // Arterioscler. Thromb. Vasc. Biol. 2003. №23. P. 2008–2014. 314.Maekawa M, Ishizaki T, Boku S et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. // Science. 1999. Vol. 285, №5429. P. 895-898. 315.Maione T, Gray G, Petro J et al. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. // Science. 1990. Vol. 247, №4938. P. 77-79. 316.Maisonpierre P, Suri C, Jones P et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. // Science. 1997. Vol. 277, №5322. P. 55-60. 317.Maloney M. Histology of basal cell carcinoma. // Clinics in dermatology. 1995. Vol. 13, №6. P. 545-549. 318.Maniotis A, Folberg R, Hess A et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. // The American journal of pathology. 1999. Vol. 155, №3. P. 739-752. 381 319.Martin D, Galisteo R, Gutkind J. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. // J Biol Chem. 2009. №284. P. 6038–6042. 320.Martin-Rendon E, Hale S, Ryan D et al. Transcriptional profiling of human cord blood CD133 + and cultured bone marrow mesenchymal stem cells in response to hypoxia. // Stem Cells. 2007. Vol. 25, №4. P. 1003-1012. 321.Marumo T, Schini-Kerth V, Busse R. Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. // Diabetes. 1999. №48. P. 1131–1137. 322.Maruyama R, Toyooka S, Toyooka K et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. // Cancer research. 2001. Vol. 61, №24. P. 8659-8663. 323.Matsumoto K, Tajima H, Nakamura T. Hepatocyte growth factor is a potent stimulator of human melanocyte DNA synthesis and growth. // Biochemical and biophysical research communications. 1991. Vol. 176, №1. P. 45-51. 324.Matsumura T, Sakai M, Kobori S et al. Two intracellular signaling pathways for activation of protein kinase C are involved in oxidized low-density lipoprotein-induced macrophage growth. // Arteriosclerosis, thrombosis, and vascular biology. 1997. Vol. 17, №11. P. 30133020 325.Matsunami H, Takeichi M. Fetal brain subdivisions defined by R-and E-cadherin expressions: evidence for the role of cadherin activity in region-specific, cell-cell adhesion. // Developmental biology. 1995. Vol. 172, №2. P. 466-478. 326.Matteucci E, Bendinelli P, Desiderio M. Nuclear localization of active HGF receptor Met in aggressive MDA-MB231 breast carcinoma cells. // Carcinogenesis. 2009. Vol. 30, №6. P. 937-945. 327.Mc Gary E, Lev D, Bar-Eli M. Cellular adhesion pathways and metastatic potential of human melanoma. // Cancer biology & therapy. 2002. Vol. 1, №5. P. 459-465. 328.Mehrad B, Keane M P, Strieter R. Chemokines as mediators of angiogenesis. // Thromb Haemost. 2007. №97. P. 755–762. 382 329.Meilhac S, Esner M, Kerszberg M, Moss J, Buckingham M. Oriented clonal cell growth in the developing mouse myocardium underlies cardiac morphogenesis. // The Journal of cell biology. 2004. Vol. 164, №1. P. 97-109. 330.Mekkawy A, Pourgholami M, Morris D. Involvement of Urokinase-Type Plasminogen Activator System in Cancer: An Overview. // Med Res Rev. 2014. Vol. 34, №5. P. 918956 331.Melani M, Weinstein B. Common Factors RegulatingPatterning of the Nervousand Vascular Systems. // Annu. Rev. Cell Dev. Biol. 2010. №26. P. 639-665. 332.Mellado M, Rodriguez-Frade J, Aragay A et al. The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. // Journal of Immunology. 1998. Vol. 161, №2. P. 805–813. 333.Mendez-Ferrer S, Frenette P. Hematopoietic stem cell trafficking: regulated adhesion and attraction to bone marrow microenvironment. // Ann N Y Acad Sci. 2007. №1116. P. 392–413. 334.Menshikov M, Plekhanova O, Cai H et al. Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways. // Arterioscler Thromb Vasc Bio. 2006. Vol. 26, №4. P. 801-807. 335.Mestas J, Burdick M, Reckamp K et al. The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. // J Immunol. 2005. №175. P. 5351–5357. 579. Miller A, Mihm M. Melanoma. // New England journal of medicine. 2006. Vol. 355, №1. P. 51-65 336.Mishra P, Mishra P, Humeniuk R et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. // Cancer research. 2008. Vol. 68, №11. P. 4331-4339. 337.Morrison S, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. // Nature. 2006. Vol. 441, №7097. P. 1068-1074. 338.Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. // Circulation research. 2006. Vol. 98, №6. P. 743-756. 383 339.Mukhina S, Stepanova V, Traktouev D et al. The chemotactic action of urokinase on smooth muscle cells is dependent on its kringle domain characterization of interactions and contribution to chemotaxis. // Journal of biological chemistry. 2000. Vol. 275, №22. P. 16450-16458. 340.Mukoyama Y, Utani A, Matsui S, Zhou S, Miyachi Y, Matsuyoshi N. T-cadherin enhances cell-matrix adhesiveness by regulating beta1 integrin trafficking in cutaneous squamous carcinoma cells. // Genes to Cells. 2007. Vol. 12, №6. P. 787-796. 341.Mukoyama Y, Zhou S, Miyachi Y, Matsuyoshi N. T-cadherin negatively regulates the proliferation of cutaneous squamous carcinoma cells. // Journal of investigative dermatology. 2005. Vol. 124, №4. P. 833-838. 342.Munro J, Cotran R. The pathogenesis of atherosclerosis: ather- ogenesis and inflammation. // Lab Invest. 1988. №58. P. 249–261. 343.Murakami M et al. The FGF system has a key role in reg integrity. // J. Clin. Invest. 2008. Vol. 118, №10. P. 3355–3366. 344.Nagy J, Dvorak A, Dvorak H. VEGF-A and the induction of pathological angiogenesis. // Annual revew of pathology. 2007. Vol. 2 P. 251-275. 345.Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. // Development. 1995. Vol. 121, №5. P. 1321-1332. 346.Naldini L, Tamagnone L, Vigna E et al. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. // EMBO J. 1992. Vol. 11, №13. P. 4825-4833. 347.Natali P, Nicotra M, Di Renzo M et al. Expression of the c-Met/HGF receptor in human melanocytic neoplasms: demonstration of the relationship to malignant melanoma tumour progression. // British journal of cancer. 1993. Vol. 68, №4. P. 746-750. 348.Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. // Nature reviews cancer. 2008. Vol. 8, №8. P. 632-645. 349.Nguyen A, Cai H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. // Proc Natl Acad Sci U S A. 2006. №103. P. 6530–6535. 384 350.Nickoloff B, Mitra R, Varani J et al. Aberrant production of interleukin-8 and thrombospondin-1 by psoriatic keratinocytes mediates angiogenesis. // Am J Pathol. 1994. №144. P. 820–828. 351.Nicosia R, Ottinetti A. Modulation of microvascular growth and morphogenesis by reconstituted basement membrane gel in three-dimensional cultures of rat aorta: a comparative study of angiogenesis in matrigel, collagen, fibrin, and plasma clot. // In vitro cellular & developmental biology. 1990. Vol. 26, №2. P. 119-128. 352.Niermann T, Kern F, Erne P, Resink T. The glycosyl phosphatidylinositol anchor of human T-cadherin binds lipoproteins. // Biochemical and biophysical research communications. 2000. Vol. 276, №3. P. 1240-1247. 353.Niessen C, Gumbiner B. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. // The Journal of cell biology. 2002. Vol. 156, №2. P. 389-400. 354.Nieto M. The early steps of neural crest development. // Mechanisms of development. 2001. Vol. 105, №1. P. 27-35. 355.Niu J, Azfer A, Zhelyabovska O et al. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein(MCPIP). // JBC. 2008. №283. P. 14542-14551. 356.Noh H, Hong S, Huang S. Role of urokinase receptor in tumor progression and development. // Theranostics. 2013. Vol. 3, №7. P. 487-495. 357.Numasaki M, Watanabe M, Suzuki T et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2- dependent angiogenesis. // J Immunol. 2005. №175. P. 6177-6189. 358.O'Brien K, Allen M, McDonald T et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. // Journal of Clinical Investigation. 1993. Vol. 92,№2. P. 945. 359.Odekon L, Sato Y, Rifkin D. Urokinase-type plasminogen activator mediates basic fibroblast growth factor-induced bovine endothelial cell migration independent of its proteolytic activity. // J. Cell Physiol. 1992. Vol. 150. №2. P. 258-263. 385 360.Oh C, Penneys N. P27 and mib1 expression in actinic keratosis, Bowen disease, and squamous cell carcinoma. // The American journal of dermatopathology. 2004. Vol. 26, №1. P. 22-26 361.Okada S, Grobmyer S, Barnathan E. Contrasting effects of plasminogen activators, urokinase receptor, and LDL receptor-related protein on smooth muscle cell migration and invasion. // Arterioscler. Thromb. Vasc. Biol. 1996. Vol. 16, №10. P. 1269-1276. 362.Okamoto Y, Kihara S, Ouchi N et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. // Circulation. 2002. Vol. 106, №22. P. 2767-2770. 363.Ong L, Kim N, Mima T, Cohen-Gould L, Mikawa T. Trabecular myocytes of the embryonic heart require N-cadherin for migratory unit identity. // Developmental biology. 1998. Vol. 193, №1. P. 1-9. 364.O'Reilly M, Boehm T, Shing Y et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. // Cell. 1997. Vol. 88, №2. P. 277-285. 365.Orimo A, Gupta P, Sgroi D et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. // Cell. 2005. Vol. 121, №3. P. 335-348. 366.Ozawa M, Ringwald M, Kemler R. Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. // Proceedings of the national academy of sciences. 1990. Vol. 87, №11. P. 4246-4250. 367.Palmer A, Klein R. Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. // Genes Dev. 2003. Vol. 17,№12. P. 1429-1450. 368.Parangi S, O'Reilly M, Christofori G et al. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. // Proceedings of the national academy of sciences. 1996. Vol. 93, №5. P. 2002-2007. 369.Pardali E, Goumans M, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. // Trends in cell biology. 2010. Vol. 20, №9. P. 556567. 370.Parfyonova Y, Plekhanova O, Solomatina M et al. Contrasting effects of urokinase and tissue-type plasminogen activators on neointima formation and vessel remodelling after arterial injury. // J Vasc Res. 2004. Vol. 41, №3. P. 268-276. 386 371.Park K, Crouse D, Lee M et al. The axonal attractant Netrin-1 is an angiogenic factor. // PNAS. 2004. №101. P. 16210 –16215. 372.Park K, Morrison C, Sorensen L et al. Robo4 is a vascular-specific receptor that inhibits endothelial migration. // Dev Biol. 2003. №261. P. 251–267. 373.Parker M, Xu P, Guo H, Vander Kooi C. Mechanism of selective VEGF-A binding by neuropilin-1 reveals a basis for specific ligand inhibition. // PLoS One. 2012. Vol. 7,№11. e49177 374.Parker-Duffen J, Nakamura K, Silver M et al. Molecular basis of disease; T-cadherin is essential for adiponectin-mediated revascularization. // JBC. 2013. Vol. 288, №34. P. 24886-24897. 375.Parker-Duffen J, Walsh K. Cardiometabolic effects of adiponectin. // Best Pract Res Clin Endocrinol Metab. 2014. Vol. 28, №1. P. 81-91. 376.Pasquale EB. Eph-Ephrin bidirectional signaling in physiology and disease. // Cell. 2008. Vol. 133, №1. P. 38-52. 377.Passaniti A, Taylor R, Pili R et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. // Laboratory Investigation; a Journal Of Technical Methods and Pathology. 1992. Vol. 67, №4. P. 519-528. 378.Pedreno J, Hurt-Camejo E, Wiklund O, Badimon L, Masana L. Low-density lipoprotein (LDL) binds to a G-protein coupled receptor in human platelets: Evidence that the proaggregatory effect induced by LDL is modulated by down-regulation of binding sites and desensitization of its mediated signaling. // Atherosclerosis. 2001. Vol. 155, №1. P. 99-112. 379.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. // Cell. 2003. Vol. 112, №4. P. 535-548. 380.Perollet C, Han Z, Savona C et al. Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2 dimerization. // Blood. 1998. №91. P. 3289–3299. 381.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. // Trends Immunol. 2007. №28. P. 299–307. 387 382.Petreaca M, Yao M, Liu Y, DeFea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL8/CXCL8-induced endothelial permeability. // Molecular biology of the cell. 2007. Vol. 18, №12. P. 5014-5023. 383.Pfaff D, Philippova M, Buechner S et al. T-cadherin loss induces an invasive phenotype in human keratinocytes and squamous cell carcinoma (SCC) cells in vitro and is associated with malignant transformation of cutaneous SCC in vivo. // British journal of dermatology. 2010. Vol. 163, №2. P. 353-363. 384.Pfaff D, Philippova M, Kyriakakis E et al. Paradoxical effects of T-cadherin on squamous cell carcinoma: up-and down-regulation increase xenograft growth by distinct mechanisms. // The Journal of pathology. 2011. Vol. 225, №4. P. 512-524. 385.Philippova M, Banfi A, Ivanov D et al. Atypical GPI-anchored T-cadherin stimulates angiogenesis in vitro and in vivo. // Arteriosclerosis, thrombosis, and vascular biology. 2006. Vol. 26, №10. P. 2222-2230. 386.Philippova M, Bochkov V, Stambolsky D, Tkachuk V, Resink T. T-cadherin and signaltransducing molecules co-localize in caveolin-rich membrane domains of vascular smooth muscle cells. // FEBS letters. 1998. Vol. 429, №2. P. 207-210. 387.Philippova M, Danila D, Allenspach R et al. RhoA and Rac mediate endothelial cell polarization and detachment induced by T-cadherin. // FASEB J. 2005. №19. P. 588-590. 388.Philippova M, Inavov D, Tkachuk V et al. Cell adhesion molecule T-cadherin polarizes to the leading edge of migrating vascular cells. // Histochem. Cell Biol. 2003. №120. P. 353360. 389.Philippova M, Ivanov D, Joshi M et al. Identification of proteins associating with glycosylphosphatidylinositol-anchored T-cadherin on the surface of vascular endothelial cells: role for Grp78/BiP in T-cadherin-dependent cell survival. // Molecular and cellular biology. 2008. Vol. 28, №12. P. 4004-4017. 390.Philippova M, Joshi M, Kyriakakis E, Pfaff D, Erne P, Resink T. A guide and guard: the many faces of T-cadherin. // Cellular signalling. 2009. Vol. 21, №7. P. 1035-1044. 388 391.Philippova M, Suter Y, Toggweiler S et al. T-cadherin is present on endothelial microparticles and is elevated in plasma in early atherosclerosis. // European heart journal. 2011. Vol. 32, №6. P. 760-771. 392.Phillips R, Burdick M, Lutz M et al. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. // Am J Respir Crit Care Med. 2003. Vol. 167, №12. P. 1676–1686. 393.Pinon P, Wehrle-Haller B. Integrins: versatile receptors controlling melanocyte adhesion, migration and proliferation. // Pigment cell & melanoma research. 2011. Vol. 24, №2. P. 282-294. 394.Pitulescu M, Adams R. Eph/ephrin molecules—a hub for signaling and endocytosis. // Genes & development. 2010. Vol. 24, №22. P. 2480-2492. 395.Plekhanova O, Berk B, Bashtrykov P et al. Oligonucleotide microarrays reveal regulated genes related to inward arterial remodeling induced by urokinase plasminogen activator. // J Vasc Res. 2009. Vol. 46, №3. P. 177-187. 396.Plekhanova O, Parfyonova Y, Bibilashvily R et al. Urokinase plasminogen activator enhances neointima growth and reduces lumen size in injured carotid arteries. // J Hypertens. 2000. Vol. 18, №8. P. 1065-1069. 397.Plekhanova O, Parfyonova Y, Bibilashvily R et al. Urokinase plasminogen activator augments cell proliferation and neointima formation in injured arteries via proteolytic mechanisms. // Atherosclerosis. 2001. Vol. 159, №2. P. 297-306. 398.Plouet J, Moro F, Bertagnolli S et al. Extracellular cleavage of the vascular endothelial growth factor 189-amino acid form by urokinase is required for its mitogenic effect. // J Biol Chem. 1997. Vol. 272,№20. P. 13390-13396. 399.Poliakov A, Cotrina M, Wilkinson D. Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. // Developmental cell. 2004. Vol. 7, №4. P. 465-480. 400.Pollard T, Borisy G. Cellular motility driven by assembly and disassembly of actin filaments. // Cell. 2003. №112. P. 453– 465. 389 401.Ponimashkin EG, Profirovich J, Vaskunaite R et al. 5-Hydroxytryptamine 4(a) receptor is coupled to the Galpha subunit of heterotrimeric G13 protein. // JBC. 2002. Vol. 277. P. 20812-20819. 402.Potapova I, Gaudette G, Brink P et al. Mesenchymal stem cells support migration, extracellular matrix invasion, proliferation, and survival of endothelial cells in vitro. // Stem Cells. 2007. Vol. 25, №7. P. 1761-1768. 403.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. // Cell. 2011. №146. P. 873-887. 404.Prager G, Breuss J, Steurer S et al. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. // Blood. 2004. Vol. 103, №3. P. 955-962. 405.Prahst C, Heroult M, Lanahan A et al. Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. // J Biol Chem. 2008. №283. P. 25110 –25114. 406.Qian F, Zhang Z, Wu X, Li Y, Xu Q. Interaction between integrin a5 and fibronectin is required for metastasis of B16F10 melanoma cells. // Biochemical and biophysical research communications. 2005. Vol. 333, №4. P. 1269-1275. 407.Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. // Neuron. 2011 Vol. 71, №3. P. 406424 408.Quinn AG, Perkins W. Non-melanoma skin cancer and other epidermal skin tumors, In: Rook‘s Textbook of Dermatology, Burns, T., Breathnach, S., Cox, N. & Griffiths, C, Blackwell Publishing Ltd, ISBN 978-1-405-16169-5, Chichester, United Kingdom. 2010. 409.Ranscht B, Bronner-Fraser M. T-cadherin expression alternates with migrating neural crest cells in the trunk of the avian embryo. // Development. 1991. Vol. 111, №1. P. 15-22. 410.Ranscht B, Dours-Zimmermann M. T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. // Neuron. 1991. Vol. 7, №3. P. 391-402. 411.Rasmussen A, Cullen K. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. // Breast cancer research and treatment. 1998. Vol. 47, №3. P. 219-233. 390 412.Resink T, Bochkov V, Hahn A, Philippova M, Buhler F, Tkachuk V. Low-and highdensity lipoproteins as mitogenic factors for vascular smooth muscle cells: individual, additive and synergistic effects. // Journal Of Vascular Research. 1995. Vol. 32, №5. P. 328-338. 413.Resink T, Kuzmenko Y, Kern F et al. LDL binds to surface-expressed human T-cadherin in transfected HEK293 cells and influences homophilic adhesive interactions. // FEBS letters. 1999. Vol. 463, №1. P. 29-34. 414.Resnati M, Pallavicini I, Wang J et al. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. // PNAS. 2002. № 99. P. 13591364. 415.Riener M, Nikolopoulos E, Herr A et al. Microarray comparative genomic hybridization analysis of tubular breast carcinoma shows recurrent loss of the CDH13 locus on 16q. // Human pathology. 2008. Vol. 39, №11. P. 1621-1629. 416.Riou P, Saffroy R, Chenailler C et al. Expression of T-cadherin in tumor cells influences invasive potential of human hepatocellular carcinoma. // The FASEB journal. 2006. Vol. 20, №13. P. 2291-2301. 417.Rippey J. Why classify basal cell carcinomas. // Histopathology. 1998. Vol. 32, №5. P. 393-398. 418.Risau W, others. Mechanisms of angiogenesis. // Nature. 1997. Vol. 386, №6626. P. 671674. 419.Rivera LB, Bergers G. Angiogenesis. Targeting vascular sprouts. // Science. 2014. Vol. 27, №344. P. 1449-1450. 420.Rollins B. Chemokines. // Blood. 1997. Vol. 90, №3. P. 909-928. 421.Rolny C et al. HRG inhibits tumor growth and metastasis by inducingmacrophage polarization and vessel normalization through downregulation of PlGF. // Cancer Cell. 2011. №19. P. 31–44. 422.Romagnani P, Annunziato F, Lasagni L et al. Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. // J Clin Invest. 2001. №107. P. 53–63. 391 423.Rondaij M, Bierings R, Kragt A, van Mourik J, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. // Arteriosclerosis, thrombosis, and vascular biology. 2006. Vol. 26, №5. P. 1002-1007. 424.Rose O, Grund C, Reinhardt S, Starzinski-Powitz A, Franke W. Contactus adherens, a special type of plaque-bearing adhering junction containing M-cadherin, in the granule cell layer of the cerebellar glomerulus. // Proceedings of the National Academy of Sciences. 1995. Vol. 92, №13. P. 6022-6026. 425. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. // Nature. 1993. Vol.362, №6423. P. 801-809. 426.Rossi D, Zlotnik A. The biology of chemokines and their receptors. // Annual review of immunology. 2000. Vol. 18, №1. P. 217-242. 427.Rubina K, Kalinina N, Bochkov V, Parfyonova Y, Tkachuk V. T-cadherin as an antiadhesive and guidance molecule interacting with low density lipoproteins. Annals EAS. Liege: European Academy of Sciences. 2013. P. 1-14. 428.Rubina K, Kalinina N, Potekhina A et al. T-cadherin suppresses angiogenesis in vivo by inhibiting migration of endothelial cells. // Angiogenesis. 2007. Vol. 10, №3. P. 183-195. 429.Rubina K, Sysoeva V, Semina E, et al. Malignant transformation in skin is associated with the loss of T-cadherin expression in human keratinocytes and heterogeneity in T-cadherin expression in tumor vasculature // Tumor Angiogenesis, ed. by Ran S. Rijeka: InTech. 2012. P. 135-166. 430.Rubina K, Talovskaya E, Cherenkov V et al. LDL induces intracellular signaling and cell migration via atypical LDL-binding protein T-cadherin. // Mol Cell Biochem. 2005. Vol.273. P. 33-41. 431.Ryschich E, Lizdenis P, Ittrich C et al. Molecular fingerprinting and autocrine growth regulation of endothelial cells in a murine model of hepatocellular carcinoma. // Cancer Res. 2006. №66. P. 198–211. 432.Ryu J, Lee C, Hong K et al. Activation of fractalkine/CX3CR1 by vascular endothelial cells induces angiogenesis through VEGF-A/KDR and reverses hindlimb ischaemia. // Cardiovasc Res. 2008. №78. P. 333–340. 392 433.Sachinidis A, Mengden T, Locher R, Brunner C, Vetter W. Novel cellular activities for low density lipoprotein in vascular smooth muscle cells. // Hypertension. 1990. Vol. 15 P. (6 Pt 2): 704-711. 434.Saijo Y, Tanaka M, Miki M et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. // J Immunol. 2002. №169. P. 469–475. 435.Sainson R, Johnston D, Chu H et al. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. // Blood. 2008. №111. P. 4997–5007. 436.Sakai M, Hibi K, Koshikawa K. et al. Frequent promoter methylation and gene silencing of CDH13 in pancreatic cancer. // Cancer science. 2004. Vol. 95, №7. P. 588-591. 437.Saksela O, Rifkin D. Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. // JCB. 1990. Vol. 110, №3. P. 767-775. 438.Salanga C, O‘Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. // Cell Mol Life Sci. 2009. №66. P.1370–1386. 439.Salcedo R, Ponce M, Young H et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. // Blood. 2000. №96. P. 34–40. 440.Salcedo R, Wasserman K, Young H et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascular- ization induced by stromal-derived factor-1alpha. // Am J Pathol. 1999. №154. P. 1125–1135. 441.Salcedo R, Young H, Ponce M et al. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. // J Immunol. 2001. №166. P. 7571–7578. 442.Salvucci O, Yao L, Villalba S et al. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. // Blood. 2002. № 99. P. 2703–2711. 443.Santiago A, Erickson C. Ephrin-B ligands play a dual role in the control of neural crest cell migration. // Development. 2002. №129. P. 3621–3632. 393 444.Sarbia M, Verreet P, Bittinger F et al. Basaloid squamous cell carcinoma of the esophagus: diagnosis and prognosis. // Cancer. 1997. Vol. 79, №10. P. 1871-1878. 445.Sato M, Mori Y, Sakurada A, Fujimura S, Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. // Human genetics. 1998. Vol. 103, №1. P. 96-101. 446.Sato Y, Abe M, Takaki R. Platelet factor 4 blocks the binding of basic fibroblast growth factor to the receptor and inhibits the spontaneous migration of vascular endothelial cells. // Biochem Biophys Res Commun. 1990. №172. P. 595–600. 447. Schaper W. Collateral circulation: Past and present (INVITED EDITORIAL). // Basic research in cardiology. 2008. Vol. 104, №1. P. 5-21 448.Schioppa T, Uranchimeg B, Saccani A. et al. Regulation of the chemokine receptor CXCR4 by hypoxia. // J Exp Med. 2003. №198. P. 1391–1402. 449.Schraufstatter I, Chung J, Burger M. IL8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. // American Journal of Physiology, Lung Cellular and Molecular Physiology. 2001. Vol. 280, №6. P. 1094–1103. 450.Scortegagna M, Cataisson C, Martin R et al. HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. // Blood. 2008. №111. P. 3343–3354. 451.Scutti J, Matsuo A, Pereira F et al. Role of SOCS-1 gene on melanoma cell growth and tumor development. // Translational oncology. 2011. Vol. 4, №2. P. 101. 452.Seed M, Walsh D. Angiogenesis in Inflammation: Mechanisms and Clinical Correlates. Springer. 2008. 453.Semenza G. Oxygen homeostasis. // Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2010. Vol. 2, №3. P. 336-361. 454.Semenza G. Vasculogenesis, Angiogenesis, and Arteriogenesis: Mechanisms of Blood Vessel Formation and Remodeling. // J Cell Biochem. 2007. №102. P. 840–847. 455.Semina E, Rubina K, Rutkevich P et al. T-cadherin activates Rac1 and Cdc42 and changes endothelial permeability. // Biochemistry 2009. Vol. 74, №4. P. 362-370. 456.Sgadari C, Angiolillo A, Tosato G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. // Blood. 1996. №87. P. 3877–3882. 394 457.Shan W, Tanaka H, Phillips G et al. Functional cis-heterodimers of N-and R-cadherins. // The Journal of cell biology. 2000. Vol. 148, №3. P. 579-590. 458. Shapiro L, Colman D. Structural biology of cadherins in the nervous system. // Current opinion in neurobiology. 1998. Vol. 8, №5. P. 593-599 459.Shapiro L, Fannon A, Kwong P et al. Structural basis of cell-cell adhesion by cadherins. // Nature. 1995. Vol. 374, №6520. P. 327-337. 460.Shibuya M. Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. // BMB Rep. 2008. Vol. 41, №4. P. 278-286. 461.Shimoyama Y, Tsujimoto G, Kitajima M, Natori M. Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. // Biochem J. 2000. Vol. 349 P. 159-167. 462.Shiraishi I, Takamatsu T, Fujita S. 3-D observation of N-cadherin expression during cardiac myofibrillogenesis of the chick embryo using a confocal laser scanning microscope. // Anatomy and embryology. 1993. Vol. 187, №2. P. 115-120. 463.Sierro F, Biben C, Martinez-Munoz L et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/ SDF-1 receptor, CXCR7. // Proc Natl Acad Sci USA. 2007. №104. P. 14759–14764. 464.Simonini A, Moscucci M, Muller DW et al. IL-8 is an angiogenic factor in human coronary atherectomy tissue. // Circulation. 2000. №101. P. 1519–1526. 465.Singh S, Nannuru K, Sadanandam A, Varney M, Singh R. CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. // British journal of cancer. 2009. Vol. 100, №10. P. 1638-1646. 466.Singh S, Sadanandam A, Rakesh K. Singh Chemokines in tumor angiogenesis and metastasis. // Cancer Metastasis Rev. 2007. Vol. 26, №3-4. P. 453-467. 467.Sitrin R, Pan P, Harper H et al. Clustering of urokinase receptors (uPAR; CD87) induces proinflammatory signaling in human polymorphonuclear neutrophils. // J. Immunol. 2000. № 165. P. 3341-3349. 468.Sluimer JC, Kolodgie FD, Bijnens AP et al. Thin-walled microvessels in human coronary atherosclerotic plaques shows incomplete endothelial junctions. Relevance of 395 compromised structural integrity for intraplaque microvascular leakage. // Journal of the American College of Cardiology. 2009. Vol. 53, № 17. P. 1517–1527. 469.Smith J, Irons G. Metastatic basal cell carcinoma: review of the literature and report of three cases. // Annals of plastic surgery. 1983. Vol. 11, №6. P. 551-553. 470.Song H, Kwon K, Lim S et al. Transfection of mesenchymal stem cells with the FGF-2 gene improves their survival under hypoxic conditions. // Mol Cells. 2005. Vol. 19, №3. P. 402-407. 471.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. // Gene therapy. 2008. Vol. 15, №10. P. 730-738. 472.Spring H, Schuler T, Arnold B et al. Chemokines direct endothelial progenitors into tumor neovessels. // Proc Natl Acad Sci USA. 2005. №102. P. 18111–18116. 473.Stamatovic S, Keep R, Mostarica-Stojkovic M, Andjelkovic A. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. // J Immunol. 2006. №177. P. 2651–2661. 474.Stambolsky D, Kuzmenko Y, Philippova M et al. Identification of 130 kDa cell surface LDL-binding protein from smooth muscle cells as a partially processed T-cadherin precursor. // Biochimica et Biophysica Acta (BBA) Biomembranes. 1999. Vol. 1416, №1. P. 155-160. 475.Stary H, Chandler A, Dinsmore R et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. // Arteriosclerosis, thrombosis, and vascular biology. 1995. Vol. 15, №9. P. 1512-1531. 580. Staton C, Stribbling S, Tazzyman S, Hughes R, Brown N, Lewis C. Current methods for assaying angiogenesis in vitro and in vivo. // International journal of experimental pathology. 2004. Vol. 85, №5. P. 233-248 476.Stein I, Itin A, Einat P et al. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. // Mol Cell Biol. 1998. Vol. 18, №6. P. 3112-3119. 396 477.Steinberg M, McNutt P. Cadherins and their connections: adhesion junctions have broader functions. // Current opinion in cell biology. 1999. Vol. 11, №5. P. 554-560. 478.Stepanova V, Lebedeva T, Kuo A. et al. Nuclear translocation of urokinase-type plasminogen activator. // Blood. 2008. Vol. 112, №1. P. 100-110. 479.Stieger S, Bloch S, Foreman O, Wisner E, Ferrara K, Dayton P. Ultrasound assessment of angiogenesis in a matrigel model in rats. // Ultrasound in medicine & biology. 2006. Vol. 32, №5. P. 673-681. 480.Stockton R, Reutershan J, Scott D, Sanders J, Ley K, Schwartz M. Induction of Vascular Permeability: betaPIX and GIT1 Scaffold the Activation of Extracellular Signal-regulated Kinase by PAK. // Molecular Biology of the Cell. 2007. Vol. 18, №6. P. 2346-2355. 481.Stockton R, Schaefer E, Schwartz M. p21-activated kinase regulates endothelial permeability through modulation of contractility. // Journal of Biological Chemistry. 2004. Vol. 279, №45. P. 46621-46630. 482.Strasly M, Cavallo F, Geuna M et al. IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. // J Immunol. 2001. №166. P. 3890–3899. 483.Strasly M, Doronzo G, Cappello P et al. CCL16 activates an angiogenic program in vascular endo- thelial cells. // Blood. 2004. №103. P. 40–49. 484.Strieter R, Burdick M, Gomperts B et al. CXC chemokines in angiogenesis. // Cytokine Growth Factor Rev. 2005. Vol. 16,№6. P. 593-609. 485.Strieter R, Kunkel S, Arenberg D, Burdick M, Polverini P. Interferon [gamma]-inducible protein-10 (IP-10), a member of the CXC chemokine family, is an inhibitor of angiogenesis. // Biochemical and biophysical research communications. 1995. Vol. 210, №1. P. 51-57. 486.Strieter R, Kunkel S, Elner V et al. Interleukin-8. A corneal factor that induces neovascularization. // Am J Pathol. 1992. №141. P. 1279–1284. 487.Struyf S, Burdick M, Peeters E et al. Platelet factor-4 variant chemokine CXCL4L1 inhibits melanoma and lung carci- noma growth and metastasis by preventing angiogenesis. // Cancer Res. 2007. №67. P. 5940–5948. 397 488.Struyf S, Burdick M, Proost P et al. Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. // Circ Res. 2004. №95. P. 855-857. 489.Suchting S, Heal P, Tahtis K et al. Soluble Robo4 receptor inhibits in vivo angiogenesis and endothelial cell migration. // FASEB J. 2005. №19. P. 121-123. 490.Suehiro Y, Okada T, Okada T et al. Aneuploidy predicts outcome in patients with endometrial carcinoma and is related to lack of CDH13 hypermethylation. // Clinical Cancer Research. 2008. Vol. 14, №11. P. 3354-3361. 491.Suzuki S. Structural and functional diversity of cadherin superfamily: are new members of cadherin superfamily involved in signal transduction pathway? // Journal of cellular biochemistry. 1996. Vol. 61, №4. P. 531-542. 492.Swift M, Weinstein B. Arterial-venous specification during development. // Circulation research. 2009. Vol. 104, №5. P. 576-588. 493.Tachibana K, Hirota S, Iizasa H et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. // Nature. 1998. №393. P. 591-594. 494.Takami M, Terry V, Petruzzelli L. Signaling pathways involved in IL-8-dependent activation of adhesion through Mac-1. // Journal of Immunology. 2002. Vol. 168, №9. P. 4559–4566. 495.Takeuchi T, Liang SB, Matsuyoshi N et al. Loss of T-cadherin (CDH13, H-cadherin) expression in cutaneous squamous cell carcinoma. // Laboratory Investigation. 2002a. Vol. 82, №8. P. 1023-1029. 496.Takeuchi T, Liang SB, Ohtsuki Y. Downregulation of expression of a novel cadherin molecule, T-cadherin, in basal cell carcinoma of the skin. // Molecular Carcinogenesis. 2002b. Vol. 35, №4. P. 173-179. 497.Takeuchi T, Misaki A, Fujita J et al. T-cadherin (CDH13, H-cadherin) expression downregulated surfactant protein D in bronchioloalveolar cells. // Virchows Arch. 2001. № 438. P. 370-375. 498.Takeuchi T, Misaki A, Liang S et al. Expression of T-Cadherin (CDH13, H-Cadherin) in Human Brain and Its Characteristics as a Negative Growth Regulator of Epidermal Growth 398 Factor in Neuroblastoma Cells. // Journal of neurochemistry. 2000. Vol. 74, №4. P. 14891497. 499.Takeuchi T, Ohtsuki Y. Recent progress in T-cadherin (CDH13, H-cadherin) research. // Histol Histopathol. 2001. Vol. 16, №4. P. 1287-1293. 500.Tanaka T, Manome Y, Wen P et al. Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. // Nat Med. 1997. №3. P. 437–442. 501.Taub D. Cytokine, growth factor, and chemokine ligand database. // Curr Protoc Immunol. 2004. №6.29, P. 1-89 502.Teicher B. Andews P. Anticancer drug development guide. 2d edition. Totowa, N.J.: Humana Press, 2004. P. 451. 503.Teillet M, Kalcheim C, Le Douarin N. Formation of the dorsal root ganglia in the avian embryo: segmental origin and migratory behavior of neural crest progenitor cells. // Developmental biology. 1987. Vol. 120, №2. P. 329-347. 504.Teng M, von Scheidt B, Duret H, Towne J, Smyth M. Anti-IL-23 monoclonal antibody synergizes in combination with targeted therapies or IL-2 to suppress tumor growth and metastases. // Cancer research. 2011. Vol. 71, №6. P. 2077-2086. 505.Thelen M. Dancing to the tune of chemokines. //Nature Immunology. 2001. Vol. 2, №2. P. 129-134. 506. Thyberg J, Hedin U, Sj"olund M, Palmberg L, Bottger B. Regulation of differentiated properties and proliferation of arterial smooth muscle cells. // Arteriosclerosis, Thrombosis, and Vascular Biology. 1990. Vol. 10, №6. P. 966-990. 507.Tkachuk V, Kuzmenko Y, Resink T, Stambolsky D, Bochkov V. Atypical low density lipoprotein binding site that may mediate lipoprotein-induced signal transduction. // Molecular pharmacology. 1994. Vol. 46, №6. P. 1129-1137. 508.Tkachuk V, Stepanova V, Little P et al. Regulation and role of urokinase plasminogen activator in vascular remodelling. // Clin Exp Pharmacol Physiol. 1996. Vol. 23, №9. P. 759-765. 509.Tlsty T, Coussens L. Tumor stroma and regulation of cancer development. // Annu Rev Pathol Mech Dis. 2006. Vol. 1 P. 119-150. 399 510.Toyooka K, Toyooka S, Virmani A et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. // Cancer research. 2001. Vol. 61, №11. P. 4556-4560. 511.Traktuev D, Tsokolaeva Z, Shevelev A et al. Urokinase gene transfer augments angiogenesis in ischemic skeletal and myocardial muscle. // Mol Ther. 2007. Vol. 15, №11. P. 1939-1946. 512.True L, Zhang H, Ye M et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. // Modern Pathology. 2010. Vol. 23, №10. P. 1346-1356. 513.Turowski P, Martinelli R, Crawford R et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. // Journal of cell science. 2008. Vol. 121, №1. P. 29-37. 514.Tvorogov D, Anisimov A, Zheng W et al. Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. // Cancer cell. 2010. Vol. 18, №6. P. 630-640. 515.Uhrin P, Breuss J. uPAR: a modulator of VEGF-induced angiogenesis. // Cell Adh Migr. 2013. Vol. 7, №1. P. 23-26. 516.Uyama Y, Imaizumi Y, Watanabe M. Effects of cyclopiazonic acid, a novel Ca(2+)ATPase inhibitor, on contractile responses in skinned ileal smooth muscle. // Br J Pharmacol. 1992. Vol. 106, №1. P. 208-214. 517.Van den Hooff A. Stromal involvement in malignant growth. // Adv Cancer Res. 1988. Vol. 50, № 159. P. 96. 518.Varon SS, Somjen GG. Neuron-glia interactions. // Neurosci Res Program Bull. 1979. Vol. 1 P. 1-239. 519.Vassalli J, Baccino D, Belin D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. // J.Cell Biol. 1985. №100. P. 86-92. 520.Vasudevan A, Bhide P. Angiogenesis in the embryonic CNS: a new twist on an old tale. // Cell adhesion & migration. 2008. Vol. 2, №3. P. 167-169. 521.Verdolini R, Amerio P, Goteri G et al. Cutaneous carcinomas and preinvasive neoplastic lesions. Role of MMP-2 and MMP-9 metalloproteinases in neoplastic invasion and their 400 relationship with proliferative activity and p53 expression. // Journal of cutaneous pathology. 2001. Vol. 28, №3. P. 120-126. 522.Verin A, Birukova A, Wang P et al. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. // American Journal of PhysiologyLung Cellular and Molecular Physiology. 2001. Vol. 281, №3. P. 565-574. 523.Veschini L, Crippa L, Dondossola E, Doglioni C, Corti A, Ferrero E. The vasostatin-1 fragment of chromogranin A preserves a quiescent phenotype in hypoxia-driven endothelial cells and regulates tumor neovascularization. // The FASEB Journal. 2011. Vol. 25, №11. P. 3906-3914. 524.Vestal D, Ranscht B. Glycosyl phosphatidylinositol-anchored T-cadherin mediates calcium-dependent, hom.ophilic cell adhesion. // The Journal of cell biology. 1992. Vol. 119, №2. P. 451-461. 525.Vestweber D. Commentary Lymphocyte trafficking through blood and lymphatic vessels: more than just selectins, chemokines and integrins. // European journal of immunology. 2003. Vol. 33, №5. P. 1361-1364. 526.Vestweber D. VE-cadherin the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. // Arteriosclerosis, thrombosis, and vascular biology. 2008. Vol. 28, №2. P. 223-232. 527.Vieira C, Pombero A, Garcia-Lopez R et al. Molecular mechanisms controlling brain development: an overview of neuroepithelial secondary organizers. // Int J Dev Biol. 2010. Vol. 54, №1. P. 7-20. 528.Virmani R, Kolodgie F, Burke A et al. Atherosclerotic plaque progression and vulnerability to rupture angiogenesis as a source of intraplaque hemorrhage. // Arteriosclerosis, thrombosis, and vascular biology. 2005. Vol. 25, №10. P. 2054-2061. 529.Visentin B, Vekich J, Sibbald B et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. // Cancer cell. 2006. Vol. 9, №3. P. 225-238. 530.Volin M, Woods J, Amin M et al. Fractalkine: a novelangiogenic chemokine in rheumatoid arthritis. // Am J Pathol. 2001. №159. P. 1521–1530. 401 531.Voura E, Sandig M, Siu C. Cell-cell interactions during transendothelial migration of tumor cells. // Microscopy research and technique. 1998. Vol. 43, №3. P. 265-275. 532.Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. // Molecular biology of the cell. 1998. Vol. 9, №9. P. 26392653. 533.Voyno-Yasenetskaya T, Dobbs L, Erickson S, Hamilton R. Low density lipoprotein-and high density lipoprotein-mediated signal transduction and exocytosis in alveolar type II cells. // Proceedings of the National Academy of Sciences. 1993. Vol. 90, №9. P. 42564260. 534.Wain S, Kier R, Vollmer R, Bossen E. Basaloid-squamous carcinoma of the tongue, hypopharynx, and larynx: Report of 10 cases. // Human pathology. 1986. Vol. 17, №11. P. 1158-1166. 535.Wakazono Y, Kurahashi T, Nakahira K et al. Appearance of a fast inactivating voltagedependent K currents in developing cerebellar granule cells in vitro. // Neuroscience research. 1997. Vol. 29, №4. P. 291-301. 536.Wallez Y, Cand F, Cruzalegui F et al. Src kinase phosphorylates vascular endothelialcadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. // Oncogene. 2007. Vol. 26, №7. P. 1067-1077. 537.Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. // Trends in cardiovascular medicine. 2006. Vol. 16, №2. P. 55-59. 538.Wang B, Xiao Y, Ding B et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. // Cancer Cell. 2003. №4. P. 19-29. 539.Wang G, Wang J, Mancianti M, Epstein Jr E. Basal Cell Carcinomas Arise from Hair Follicle Stem Cells in Ptch1+/- Mice. // Cancer cell. 2011. Vol. 19, №1. P. 114-124. 540.Wang J, Dai J, Jung Y et al. A glycolytic mechanism regulating an angiogenic switch inprostate cancer. // Cancer Res. 2007. №67. P. 149–159. 541.Wang M, Monticone R, Lakatta E. Arterial aging: a journey into subclinical arterial disease. // Curr Opin NEphrol Hypertens. 2010. Vol. 19, №2. P. 201-207. 402 542.Wang M, Zhang J, Spinetti G et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. // Am J Pathol. 2005. Vol. 167, №5. P. 1429-1442. 543.Weber K, Nelson P, Grone H, Weber C. Expression of CCR2 by endothelial cells: implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation ofendothelium. // Arterioscler Thromb Vasc Biol. 1999. №19. P. 2085–2093. 544.Weigelt B, Peterse J, Van't Veer L. Breast cancer metastasis: markers and models. // Nature reviews cancer. 2005. Vol. 5, №8. P. 591-602. 545.Weinstein B. Vessels and nerves: marching to the same tune. // Cell. 2005. Vol. 120, №3. P. 299-302. 546.Weis S, Cheresh D. av Integrins in Angiogenesis and Cancer. // Cold Spring Harb Perspect Med. 2011.Vol. 1, №1. a006478. 547.Weis S, Cheresh D. Pathophysiological consequences of VEGF-induced vascular permeability. // Nature. 2005. Vol. 437, №7058. P. 497-504. 548.Wilson B, Ii M, Park K et al. Netrins promote developmental and therapeutic angiogenesis. // Science. 2006. №313. P.640 – 644. 549.Winters R, Naud S, Evans MF, Trotman W, Kasznica P, Elhosseiny A. Ber-EP4, CK1, CK7 and CK14 are Useful Markers for Basaloid Squamous Carcinoma: A Study of 45 Cases. // Head Neck Pathol. 2008. Vol. 2, № 4. P. 265–271. 550.Winzenburg S, Niehans G, Evan G, Kathleen D, Adams G. Basaloid squamous carcinoma: a clinical comparison of two histologic types with poorly differentiated squamous cell carcinoma. // Otolaryngology-Head and Neck Surgery. 1998. Vol. 119, №5. P. 471-475. 551.Wu Y, Li Y, Matsushima K et al. CCL3-CCR5 axis regulates intratumoralaccumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. // J Immunol. 2008. №181. P. 6384–6393. 552.Wyder L, Vitaliti A, Schneider H et al. Increased expression of H/T-cadherin in tumorpenetrating blood vessels. // Cancer research. 2000. Vol. 60, №17. P. 4682-4688. 553.Xiao K, Allison D, Buckley K et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. // The Journal of cell biology. 2003. Vol. 163, №3. P. 535-545. 403 554. Xiao K, Allison D, Kottke M et al. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. // Journal of Biological Chemistry. 2003. Vol. 278, №21. P. 19199-19208. 555.Xu F, Shi J, Yu B, Ni W, Wu X, Gu Z. Chemokines mediate mesenchymal stem cell migration toward gliomas in vitro. // Oncology reports. 2010. Vol. 23, №6. P. 1561. 556.Xu W, Huang C, Wang J et al. Comparison of the effects of recombinant human endostatin and docetaxel on human umbilical vein endothelial cells in different growth states. // Chinese medical journal. 2011. Vol. 124, №18. P. 2583-2889. 557. Yamada M, Kim S, Egashira K et al. Molecular mechanism and role of endothelial monocyte chemoattractant protein-1 induction by vascular endothelial growth factor. // Arterioscler Thromb Vasc Biol. 2003. №23. P. 1996–2001 558.Yancopoulos G, Davis S, Gale N, Rudge J, Wiegand S, Holash J. Vascular-specific growth factors and blood vessel formation. // Nature. 2000. Vol. 407, №6801. P. 242-248. 559.Yang Y, Zou L, Wang Y et al. Axon guidance cue Netrin-1 has dual function in angiogenesis. // Cancer Biol Ther. 2007. №6. P. 743–748. 560.Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. // Journal of Skin Cancer. 2011. Vol. 2011. P. 1-13. 561.Yap A. Kovacs E. Direct cadherin-activated cell signaling: a view from the plasma membrane. // J.Cell.Biol. 2003. №160. P. 11-16. 562.Yoon Y, Liang Z, Zhang X et al. CXC chemokine receptor-4 antagonist blocks both growth of primary tumor andmetastasis of head and neck cancer in xenograft mouse models. // Cancer Res. 2007. №67. P. 7518–7524. 563.You J, Yang C, Huang J et al. Fractalkine, a CX3C chemokine, as amediator of ocular angiogenesis. // Invest Ophthalmol Vis Sci. 2007. №48. P. 5290–5298. 564.Yu T, Rahman Z, Ross B. Actinic keratoses-surgical and physical therapeutic modalities. // Cutis; cutaneous medicine for the practitioner. 2003. Vol. 71, №5. P. 381-384. 565.Yurlova, E., Rubina, K., Sysoeva, V., Sharonov, G., Semina, E., Parfenova, E., Tkachuk, V.T-cadherin suppresses the cell proliferation of mouse melanoma B16F10 and tumor angiogenesis in the model of chorioallantoic membrane // Russian Journal of Developmental Biology. 2010. Vol. 41. №4. P. 217-226. 404 566.Zalaudek I, Bonifazi E, Ferrara G, Argenziano G. Keratoacanthomas and Spitz Tumors: Are They Both ‗Self-Limiting‘Variants of Malignant Cutaneous Neoplasms? // Dermatology. 2009. Vol. 219, №1. P. 3-6. 567.Zbaren P, Nuyens M, Stauffer E. Basaloid squamous cell carcinoma of the head and neck. // Current opinion in otolaryngology & head and neck surgery. 2004. Vol. 12, №2. P. 116121. 568. Zhang W, Liu H. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. // Cell research. 2002. Vol. 12, №1. P. 9-18 569.Zhou S, Matsuyoshi N, Liang S, Takeuchi T, Ohtsuki Y, Miyachi Y. Expression of Tcadherin in basal keratinocytes of skin. // Journal of investigative dermatology. 2002. Vol. 118, №6. P. 1080-1084. 570.Zhou S, Matsuyoshi N, Takeuchi T et al. Reciprocal altered expression of T-cadherin and P-cadherin in psoriasis vulgaris. // British J. Dermatol. 2003. №149. P. 268-273. 571.Zucchini C, Bianchini M, Valvassori L et al. Identification of candidate genes involved in the reversal of malignant phenotype of osteosarcoma cells transfected with the liver/bone/kidney alkaline phosphatase gene. // Bone. 2004. Vol. 34, №4. P. 672-679. 572.ДИРЕКТИВА 2010/63/EU ЕВРОПЕЙСКОГО ПАРЛАМЕНТА И СОВЕТА ЕВРОПЕЙСКОГО СОЮЗА. Международные рекомендации по проведению медикобиологических исследований с использованием животных. Основные принципы. Страсбург: Ланималогия. 2010. С. 29. 573.Манк М. Биологи развития млекопитающих. Методы. Москва: Мир, 1990 С. 406. 574.Парфенова Е, Плеханова О, Меньшиков М и др. Регуляция роста и ремоделирования кровеносных сосудов: уникальная роль урокиназы. // Рос.физиол. журн. им. И. М. Сеченова. 2009. Том 95, №5. С. 442-464. 575.Парфенова Е, Ткачук В. Терапевтический ангиогенез: достижения, проблемы, перспективы// Кардиологический вестник. 2007. №2. С. 5-15. 576.Рубина К, Ткачук В. Т-кадгерин как антиадгезивная молекула и возможный рецептор липопротеидов низкой плотности в клетках кровеносных сосудов. // Рос. Физиол. Ж. 2004. Том 90, №8, С. 968-986. 405 581. Ткачук В. А., Плеханова О. С., Белоглазова И. Б., Парфенова Е. В.Роль мультидоменной структуры урокиназы в регуляции роста и ремоделирования сосудов. // Укр. біохім. журн. 2013. Том 85, № 6. С. 18-45.