Ab initio
реклама

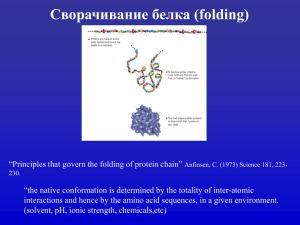
Предсказание пространственной структуры белков Иерархия структуры белков Первичная: белковая последовательность Вторичная: локальный фолдинг - α-спирали, β-листы, петли Третичная: пространственная организация полипептидной цепи Четвертичная: доменная организация белков Структура белков определяет их функцию Структура белка Регуляция Структура Сигнальные пути Функция Движение Катализ Транспорт Первичная структура: последовательность • Первичной структурой белка является его аминокислотная последовательность МОНОМЕР ПОЛИМЕР Аминокислота Полипептид Пептидная связь Пептидная связь Первичная структура: последовательность • Двадцать аминокислот, встречающихся в белках, имеют различные свойства. Первичная структура: последовательность Вторичная структура: α-спирали, β-листы, петли • α-спирали и β-листы формируются путем образования водородных связей между атомами кислорода и водорода главной цепи. Вторичная структура: α-спирали Вторичная структура: β-листы Антипараллельный β-лист Параллельный β-лист β-лист β-тяж Смешанный β-лист Структурные мотивы Третичная структура: домены Мозаичная структура белков Третичная структура: пространственная укладка белка (фолд) Четвертичная структура: мультимерные белки и белковые комплексы Гемоглобин - тетрамер Рибосома: РНК-белковый комплекс - синтез белка Реплисома - копирование ДНК Сворачивание белка (фолдинг) • Сворачивание белка - процесс укладки полипептидной цепи в компактную пространственную структуру. • Аминокислотная последовательность белка одназначно определяет его пространственную структуру [Anfinsen et al., 1950s]. • Пространственная структура белка определяет его функцию. Нековалентные (“слабые”) взаимодействия Водородные связи Ионные связи Гидрофобные взаимодействия Ван-дер-Ваальсовы взаимодействия Гидрофобность аминокислот • Гидрофобные эффекты играют важную роль в сворачивании белка Экспериментально измеренные уровни гидрофобности аминокислот Определение пространственной структуры белка • Экспериментальный подход - Рентгеновская кристаллография Спектроскопия ядерного магнитного резонанса • Вычислительный подход - Предсказание пространственной структуры белка на основе информации о его последовательности - нерешенная задача Предсказание пространственной структуры белка • Ab initio - моделирование укладки “из первых принципов” - без использования дополнительной информации о структурах схожих белков. • Предсказание на основе гомологии (homology modeling) моделирование на основе известных структур схожих белков. • Тридинг (Threading) - моделирование на основе слабой гомологии. Предсказание структуры ab initio • Функция потенциальной энергии - Модель водного раствора Оценка попарного взаимодействия между аминокислотами • Поиск в пространстве всевозможных конформаций - Модель на основе “решетки” Молекулярная динамика Использование библиотек известных 3D фрагментов • Предсказание вторичной структуры Предсказание структуры с использованием решетки • HP-модель (Hydrophobic-Polar) - рассматривает гидрофобные взаимодействия как наиболее важные. - Не существует эффективных алгоритмов Плохо отражает реальность Предсказание структуры с использованием решетки ROSETTA • Используются структурно консервативные фрагменты длиной 4-10 аминокислот • Поиск в пространстве конформаций осуществляется методом Монте Карло • Полученные структуры кластеризуются и в качестве результата выдаются наилучшие структуры для каждого кластера Предсказание структуры на основе гомологии • Выравнивание рассматриваемой последовательности с последовательностями белков с известной 3D структурой (обычно >30% сходства) • Наложение моделируемой последовательности на известную структуру согласно выравниванию • Локальное улучшение полученной пространственной структуры - Число уникальных укладок (фолдов), наблюдающихся в белках, ограничено (несколько тысяч) - 90% помещаемых в PDB структур имеют уже известные укладки (фолды) Примеры укладок (фолдов) Предсказание структуры на основе гомологии Raw model Loop modeling Side chain placement Refinement Тридинг (Threading) - предсказание структуры на основе слабой гомологии • Главное отличие от моделирования по гомологии поиск наилучшей структуры осуществляется с помощью выравнивания последовательности со структурой, а не с последовательностью. При этом используется специальным образом определенная весовая функция. MTYKLILN …. NGVDGEWTYTE Основные компоненты тридинга • библиотека уникальных укладок (фолдов) • функция, определяющая вес выравнивания последовательности со структурой • алгоритм нахождения наилучшего выравнивания CASP - конкурс методов предсказания структуры белков Предсказания вторичной структуры • Задача: отнести позиции заданой последовательности GHWIATRGQLIREAYEDYRHFSSECPFIP к одному из трех классов вторичной структуры - α-спиралям (H), β-листам (E) или петлям (C): CEEEEECHHHHHHHHHHHCCCHHCCCCCC Методы предсказания вторичной структуры • Статистические методы - • Chou-Fasman, GOR Ближайшего соседа • NNSSP, SSPAL Нейронные сети • PHD, Psi-Pred, J-Pred • Метод опорных векторов • Скрытые Марковские модели Точность предсказания вторичной структуры • Точность современных методов достигает 80% • Оценка различий во вторичной структуре для гомологичных белков ~ 12%. Таким образом, теоретический предел точности предсказания вторичной структуры ~ 88%. 25 PSIPRED SSpro PROF PHDpsi JPred2 PHD Percentage of all 150 proteins 20 15 10 5 0 30 40 50 60 70 80 90 Percentage correctly predicted residues per protein 100 Благодарности • При подготовке слайдов использовались материалы лекций: • Михаила Гельфанда (ИППИ) • Андрея Миронова (МГУ) • Serafim Batzoglou (Stanford) • Manolis Kellis (MIT) • Pavel Pevzner (UCSD)